")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Near-infrared quantum dots for HER2 localization and imaging of cancer cells
Authors Rizvi S, Rouhi S, Taniguchi S, Yang SY, Green M, Keshtgar M, Seifalian A
Received 15 July 2013
Accepted for publication 26 September 2013
Published 11 March 2014 Volume 2014:9(1) Pages 1323—1337
DOI https://doi.org/10.2147/IJN.S51535
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Video abstract presented by Sarwat Rizvi
Views: 1027
Sarwat B Rizvi,1 Sepideh Rouhi,1 Shohei Taniguchi,2 Shi Yu Yang,1 Mark Green,2 Mo Keshtgar,1,3 Alexander M Seifalian1,3
1UCL Centre for Nanotechnology and Regenerative Medicine, University College London, 2Department of Physics, King's College London, 3Royal Free London NHS Foundation Trust Hospital, London, UK
Background: Quantum dots are fluorescent nanoparticles with unique photophysical properties that allow them to be used as diagnostic, therapeutic, and theranostic agents, particularly in medical and surgical oncology. Near-infrared-emitting quantum dots can be visualized in deep tissues because the biological window is transparent to these wavelengths. Their small sizes and free surface reactive groups that can be conjugated to biomolecules make them ideal probes for in vivo cancer localization, targeted chemotherapy, and image-guided cancer surgery. The human epidermal growth factor receptor 2 gene (HER2/neu) is overexpressed in 25%–30% of breast cancers. The current methods of detection for HER2 status, including immunohistochemistry and fluorescence in situ hybridization, are used ex vivo and cannot be used in vivo. In this paper, we demonstrate the application of near-infrared-emitting quantum dots for HER2 localization in fixed and live cancer cells as a first step prior to their in vivo application.
Methods: Near-infrared-emitting quantum dots were characterized and their in vitro toxicity was established using three cancer cell lines, ie, HepG2, SK-BR-3 (HER2-overexpressing), and MCF7 (HER2-underexpressing). Mouse antihuman anti-HER2 monoclonal antibody was conjugated to the near-infrared-emitting quantum dots.
Results: In vitro toxicity studies showed biocompatibility of SK-BR-3 and MCF7 cell lines with near-infrared-emitting quantum dots at a concentration of 60 µg/mL after one hour and 24 hours of exposure. Near-infrared-emitting quantum dot antiHER2-antibody bioconjugates successfully localized HER2 receptors on SK-BR-3 cells.
Conclusion: Near-infrared-emitting quantum dot bioconjugates can be used for rapid localization of HER2 receptors and can potentially be used for targeted therapy as well as image-guided surgery.
Keywords: anti-HER2 antibody, HER2 localization, quantum dots, in vitro imaging, nanotechnology, cancer
Introduction
Among the many potential applications of nanotechnology in medicine, cancer diagnosis and therapy remains the most significant and has led to the development of a new discipline of nano-oncology.1–13 Nanoparticles or nanomaterials can be broadly classified as organic and inorganic. Organic nanoparticles include nanoliposomes, dendrimers, amphiphilic block copolymer micelles, and carbon nanotubes. Inorganic nanoparticles can be metallic (eg, gold, silver, superparamagnetic iron oxide) or composed of semiconductor material, eg, quantum dots (QDs). Each type of nanoparticle has unique characteristics based on its composition, size, and surface chemistry, that determine its stability, biocompatibility, and interaction with the surrounding environment. Nanoparticles are actively being developed as probes for in vivo tumor targeting, biomolecular profiling of cancers, nanovectors for drug delivery, and various theranostic applications.2,4
Of all the different types of nanoparticles, semiconductor nanocrystals, or QDs, have gained significant attention due to their unique photophysical properties as the next-generation fluorophores. QDs are fluorescent 2–10 nm nanocrystals that can be used as alternative probes for a host of diagnostic, therapeutic, and theranostic applications in medical and surgical oncology.14 Their broad absorption and narrow symmetric emission spectra, large molar extinction coefficients, high quantum yield, and enhanced photostability give them tremendous advantages over traditional fluorophores for biomolecular and cellular imaging in vitro and in vivo.15 QDs can be size-tuned to emit at near-infrared (NIR, 650–1,000 nm) wavelengths, which are ideal for deep tissue imaging because the biological window is transparent to these wavelengths. This is based on the fact that tissue chromophores like hemoglobin absorb light in the visible spectrum (400–700 nm), leading to scattering, diffraction, and poor penetration through the skin. Light in the NIR range does not undergo this absorption and scattering effect, allowing deeper penetration and visibility.16–20 Multiple QDs of different colors can be excited by a single wavelength of light, a concept of multiplexed imaging that is a certain advantage in biological imaging where multiple targets can be detected at a single point in time. Also, their small size and free surface reactive groups allow conjugation to various biomolecules for targeted localization, making them potential candidates for various applications, like cancer localization, detection of micrometastasis, image-guided drug delivery, and image-guided cancer surgery.16,20–28
The human epidermal growth factor receptor 2 gene ErbB2 (commonly referred to as HER2/neu) is a v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 gene29 that may be overexpressed in a number of cancers, including breast, colorectal, non-small-cell lung, and head and neck cancer.30–32 The gene encodes a member of the epidermal growth factor (EGF) family of transmembrane receptors with tyrosine kinase activity, including EGFR (also called HER1 or ErbB1), HER2 (ErbB2 or neu), HER3 (ErbB3), and HER4 (ErbB4). The HER2/neu gene is overexpressed in 25%–30% of breast cancers,33,34 and the primary mechanism of overexpression is amplification, which leads to increased tyrosine kinase activity and dysregulated growth of cells. The significance of determining HER2 status in breast cancer treatment relates to the fact that HER2-positive tumors are associated with higher aggressiveness, recurrence, and increased mortality among newly diagnosed cases that do not receive systemic chemotherapy.29 Hence targeting and blocking the HER2 receptor using a humanized monoclonal antibody called trastuzumab (Herceptin®; Genentech Inc, South San Francisco, CA, USA) leads to improved response rates and delays the time to disease progression, and hence prolongs survival either alone or in combination with other chemotherapies in metastatic disease.35 However, the use of trastuzumab is associated with a risk of cardiotoxicity36,37 and this, coupled with the high costs of the drug and therapies of up to 9–12 months, demands that HER2 testing has high sensitivity to identify all patients with HER2 positivity as well as high specificity to detect all the negative patients who would not benefit from the therapy and hence not suffer the side effects.29
The current methods for detecting HER2 status include immunohistochemistry and fluorescence in situ hybridization (FISH), and both have problems based on variability and consistency of results.29,38 Immunohistochemistry is used to detect protein expression and FISH to detect gene amplification. The disadvantages of immunohistochemistry include it being prone to interference factors, having unstable sensitivity, and showing a high discrepancy between laboratories; further, it has a subjective interpretation and is a semiquantitative technique. FISH, on the other hand, although highly sensitive, is complex, expensive, labor-intensive, time-consuming, and requires special equipment, including manual expertise.29,39–42 The FISH assay is technically more reproducible and is currently considered the “gold standard” for HER2 testing. However, it may cause difficulty in assessment of the morphological features of the tumor along with decay in the fluorescence signal, which leads to loss of results after a few weeks. To overcome these problems, chromogen in situ hybridization has been used as an alternative method because its signals are permanent and samples can be assessed in the light of morphological features.43 However, there are mixed reports regarding the sensitivity of chromogen in situ hybridization in comparison with FISH, particularly in low amplification tumors.43–45 Also, all the above methods can be used for ex vivo detection of HER2 status, and to date there is no means of detecting HER2 status in vivo.
NIR-emitting QDs (NIR-QDs) have been investigated as promising probes for in vitro and in vivo imaging.19,24 The potential application of QDs as molecular probes for the detection of breast cancer has already been described.38,46–50 Many researchers have suggested the application of QDs as fluorescent probes for immunohistochemistry based on their advantages over traditional fluorophores38,46–48 Chen et al demonstrated that QD immunohistochemistry can be used for quantitative determination of the HER2 load which may better reveal breast cancer heterogeneity.49 In another study, the same group looked at simultaneous detection of HER2 and the estrogen receptor in breast cancer using QD immunohistochemistry, and showed that multiplexed imaging of the HER2 and estrogen receptor would enhance the understanding of their interaction during evolution of breast cancer.51 In a similar context, Liu et al used QD-based multiplexed imaging to demonstrate that high HER2 expression is associated with increased destruction of the extracellular matrix and vascular invasion of breast cancer.52 In this study, we explored direct conjugation of the anti-HER2 antibody with the QD surface using carbodiimide surface chemistry for direct detection of HER2 receptors without the need for a secondary antibody, as is required for immunohistochemistry. We have previously reported the aqueous synthesis of a core/shell/shell QD based on CdTe/CdSe/ZnSe coated with mercaptoundecanoic acid (MUA) as an NIR probe for deep tissue imaging.53 Here we have demonstrated the localization of HER2 receptors in both fixed and live cells, examined two protocols of QD bioconjugation, and also evaluated the in vitro toxicity of QDs as a first step towards their eventual application for in vivo imaging.
Materials and methods
Materials
McCoy’s 5A (modified) Medium was obtained from the American Type Culture Collection (Manassas, VA, USA); a penicillin-streptomycin mixture, trypsin/ethylenediamine tetraacetic acid, phosphate-buffered saline (without Ca2+, Mg2+, or phenol red), and Dulbecco’s Modified Eagle Medium (DMEM) and F12 in a 1:1 mixture with HEPES and L-glutamate was sourced from Lonza (Walkersville, MD, USA); DMEM and mouse antihuman unconjugated monoclonal antibody erbB-2 (HER2) was sourced from Invitrogen (Carlsbad, CA, USA); fetal bovine serum, Tween 20, N-ethyl-N’-(3-dimethylaminopropyl)carbodiimide (EDC) ≥98.0% was purchased from Fluka (St Louis, MO, USA); and N-hydroxysuccinimide (NHS) 98% was obtained from Sigma-Aldrich (St Louis, MO, USA). A Cell Titer-Blue® cell viability assay was obtained from Promega (Madison, WI, USA). CdTe/CdSe/ZnSe quantum dots coated with MUA were synthesized in the laboratory according to our published protocols.53
Methods
Preparation of QDs
Core/shell/shell CdTe/CdSe/ZnSe QDs were aqueously synthesized by a previously described one pot method.53 The QDs had an MUA coating and were soluble in aqueous medium. After synthesis, the QDs were purified by addition of isopropanol/butanol (1:1) mixture followed by centrifugation and redispersion in deionized water or phosphate-buffered saline. The QD concentration was calculated by assessment of the dry weight of QDs per mL of growth solution. We noticed that, after vacuum-drying, the QDs could not be redispersed in water. Therefore, after centrifugation, the wet pellet was dispersed in phosphate-buffered saline and, based on calculations from the amount of dry QDs per mL of growth solution, QD concentrations were prepared for the in vitro cytotoxicity assessments. The concentration of elemental cadmium was calculated using inductively coupled plasma mass spectrometry.
Characterization
Absorption spectroscopy measurements were taken using a U-4100 ultraviolet-visible NIR spectrophotometer (Hitachi Ltd, Tokyo, Japan) over a wavelength of 300–1,100 nm. The samples were measured in a quartz cuvette with a 1 cm path length and using an aqueous solvent (deionized water or phosphate-buffered saline) as a reference. Photoluminescence spectra were obtained using an LS 50B spectrometer (PerkinElmer, Boston, MA, USA) in the same cuvette as absorption spectroscopy (excitation wavelength being 400 nm). The diameter of the QD cores was assessed using a Tecnai™ 20 transmission electron microscope (FEI Co, Hillboro, OR, USA) with an acceleration voltage of 200 kV for normal and high resolution images. Samples were dropped onto a copper grid with an amorphous carbon film and left to evaporate under ambient conditions. Dynamic light scattering studies were performed to establish the hydrodynamic diameter of the MUA-coated QDs using a Delsa™ nano submicron particle size and zeta potential particle analyzer (Beckman Coulter Inc, Pasadena, CA, USA). All samples were thoroughly sonicated at 37°C for 15 minutes prior to measurements. A FT/IR 6300 Fourier transform infrared (FTIR) spectroscope (Jasco Analytical Instruments, Easton, MD, USA) with an MCT detector was used for FTIR analysis with phosphate-buffered saline as a background reference. FTIR of the MUA powder was done to demonstrate attachment of MUA to the QD surface. A few drops of unconjugated and conjugated QDs were placed separately on the ATR prism and spectra were obtained in solution.
Cell culture
SK-BR-3 cells (a human breast adenocarcinoma cell line overexpressing HER2) were obtained and cultured in McCoy’s 5A Medium with 10% fetal bovine serum and 1% penicillin/streptomycin formulated by the American Type Culture Collection. MCF7 cells (a human breast ductal carcinoma cell line not overexpressing HER2) were sourced from the European Collection of Cell Cultures (Salisbury, UK) and cultured in DMEM:F12 in a 1:1 mixture (with HEPES and L-glutamine) and 10% fetal bovine serum and 1% penicillin/streptomycin. HepG2 cells were supplied at passage number 50 from the Liver Group at the Centre for Hepatology, Department of Medicine, Royal Free Hospital and Medical School, University College London. Once received, the medium was changed to DMEM (+4.5 g/L glucose) and supplemented with fetal bovine serum 10% and penicillin/streptomycin 1%, which was then replaced a few times over a period of a week to 10 days. All cells were cultured and passaged in 75 cm2 cell culture flasks up to 90% confluence with complete cell culture medium (CCM) in a humidified chamber at 37°C with 5% CO2. The CCM was changed every 2–3 days and both cell lines were used at passage 4 and 5.
The methodology for testing of in vitro cytotoxicity was established using the Cell Titre Blue assay. To establish the best seeding density for in vitro cytotoxicity in a 96-well plate, SK-BR-3, MCF7, and HepG2 cells were laid down at densities of 40 × 103, 20 × 103, 10 × 103, 5 × 103, and 2.5 × 103 cells in 100 μL of CCM per well. The cells were incubated in a humidified chamber at 37°C with 5% CO2 overnight, following which the CCM was removed, the cells were washed with phosphate-buffered saline, and fresh CCM was replaced. Next, 20 μL of Cell Titre Blue assay was added to each well and the plates were incubated in a humidified chamber at 37°C with 5% CO2 for 4 hours. The fluorescence of the 96-well plates was read using a Fluoroskan Ascent microplate fluorometer (Thermo Scientific, Baltimore, MD, USA). The fluorescence was measured with excitation at 530 nm and emission at 620 nm. For both SK-BR-3 and MCF7 cells, the best seeding density in a 96-well plate was 20,000 cells per well and for HepG2 was 40,000 cells per well.
In vitro cytotoxicity
For in vitro toxicity, the cells were seeded in 96-well plates at the established density of 20,000–40,000 cells per well in 100 μL of CCM and incubated overnight in a humidified chamber at 37°C with 5% CO2. Following this, the CCM was removed, the cells were washed with phosphate-buffered saline and then exposed to variable concentrations of QDs (1.25–60 μg/mL) in CCM, where the volume of CCM was kept constant and the concentrations of QDs were altered using variable volumes of phosphate-buffered saline. The plates were incubated in a humidified chamber at 37°C with 5% CO2 for one hour and 24 hours. After the desired exposure times, the CCM with QDs was removed and replaced with fresh CCM after washing the cells twice with phosphate-buffered saline. Next, 20 μL of Cell Titre Blue assay was added to each well, the plates were incubated in a humidified chamber at 37°C with 5% CO2 for 4 hours, and cell viability was established by determining fluorescence using the Fluoroskan Ascent microplate fluorometer filter sets for excitation (Ex) and emission (Em) 530Ex/620Em. QD concentrations alone without cells were also read by the plate reader to establish that the fluorescence measurements represented cell viability and not QD fluorescence. For comparison, all cell viability (CV) data were presented as the percentage of treated cells (exposed to QDs) to untreated cells (not exposed to QDs), which was calculated using the following formula:
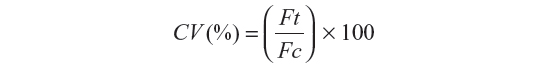
where Ft represents the fluorescence reading for treated cells and Fc represents the fluorescence reading for untreated cells in the cell viability assay. All assays were performed in triplicate and the results averaged.
In vitro imaging
For assessing the morphology of the cells after QD exposure, both cell types were cultured in glass-bottomed 96-well plates. After QD exposure for one hour and 24 hours, the cells were washed with phosphate-buffered saline and fixed with glutaraldehyde before imaging with a light microscope. An EC-1 confocal microscope from Nikon (Tokyo, Japan) was used to assess fluorescence.
Bioconjugation of QDs
A solution of QDs with a concentration of 0.5 mg/mL was prepared in phosphate-buffered saline and the anti-HER2 antibody was diluted in phosphate-buffered saline to a concentration of 100 μg/mL. Two different protocols were examined for bioconjugation based on the use of both EDC and NHS or EDC alone for activation of the QDs. For the first protocol, 1 mg of EDC powder and 1 mg of NHS powder were added to 0.5 mL of 0.5 mg/mL CdTe/CdSe/ZnSe-MUA QDs. For the second protocol, only 1 mg of EDC powder was added to the same concentration of QDs. To activate the QDs, the materials were incubated at room temperature in an orbital rotator for 40 minutes. After the activating process, all of the antibody solution (100 μg/mL) was added and left for one hour at room temperature on the orbital rotator, following which it was purified by centrifugal filtration using an Amicon Ultra-0.5 centrifugal filter unit with an Ultracel-30 membrane (Millipore, Billerica, MA, USA) for 10 minutes at a speed of 10 g. This removed all the unconjugated QDs and unreacted materials as a filtrate. The antibody-QD conjugate retained by the filtration membrane was diluted 50 times in phosphate-buffered saline and characterized using FTIR to confirm bioconjugation. Figure 1 is a schematic diagram that demonstrates the methodology used for bioconjugation and targeted localization of the surface receptors.
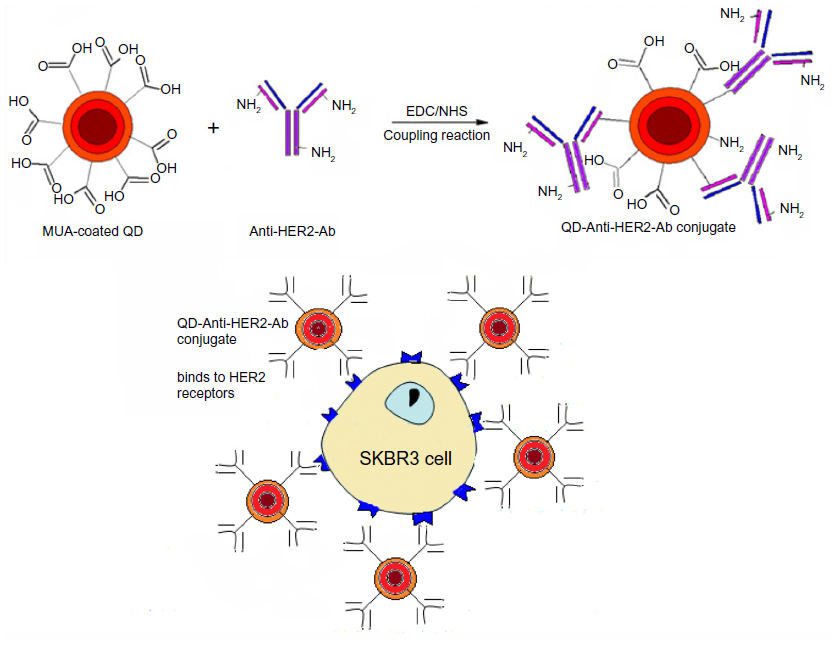 | Figure 1 Methodology of bioconjugation of mercaptoundecanoic acid-coated QDs and application for localization of HER2 receptors. Anti-HER2-Ab conjugates to the QD surface through an amide linkage. The QD-Anti-HER-Ab conjugate then targets and localizes to the HER2 receptors overexpressed on SK-BR-3 cells. |
HER2 localization using the QD-antibody bioconjugate
SK-BR-3 cells and MCF7 cells were cultured on glass chamber slides in McCoy’s 5A Medium and DMEM:F12 with 10% fetal bovine serum and 5% penicillin/streptomycin overnight. When they reached approximately 80% confluence, the CCM was removed and the cells were washed thoroughly with phosphate-buffered saline and fixed with glutaraldehyde. For live cell imaging, each chamber was washed and replenished with 1 mL of fresh CCM. Next, 1 mL of QD-antibody conjugate prepared using EDC/NHS or EDC alone was added to each chamber of fixed and live cells. The treated plates were placed on a gentle shaker for one hour at room temperature, following which the QD-antibody solution was removed and the cells were washed thoroughly with 0.05% phosphate-buffered saline and Tween 20 for 10 minutes to remove all unbound QD-antibody conjugates. The slides were visualized using an EC1 confocal microscope from Nikon.
Statistical analysis
The statistical analysis was performed using the Prism software and nonparametric one-way analysis of variance with Dunnett’s and Bonferroni multiple comparisons test. A P-value <0.05 was considered to be statistically significant.
Results
QD characterization
The QDs demonstrated an absorption onset (700 nm) and emission peak (720 nm) in the NIR range (Figure 2A). Transmission electron microscopy showed that the QDs were mostly spherical in shape with a core diameter of 4±0.8nm (Figure 2B). Dynamic light scattering studies showed that MUA-coated QDs had a hydrodynamic diameter of 19.8±5 nm. FTIR confirmed the presence of the MUA coating, showing peaks at 998 cm−1 from O–H bending of the carboxylic acid, as well as peaks at 1,259 cm−1 and 1,742 cm−1 from stretching of C−O and C=O groups respectively, showing that the MUA had bonded to the QD surface (Figure 6A). The dry weight of QDs in the growth solution was 0.4 mg/mL and this equaled an elemental cadmium concentration of 148 μg/mL by inductively coupled plasma mass spectrometry. The molar concentration of QDs calculated based on Beer Lamberts Law was 8.08 μM.
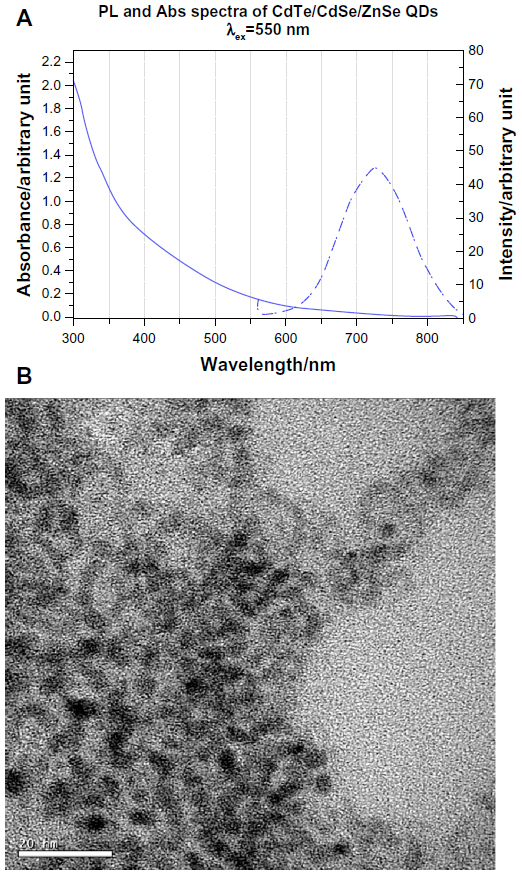 | Figure 2 Characterization of the QDs. |
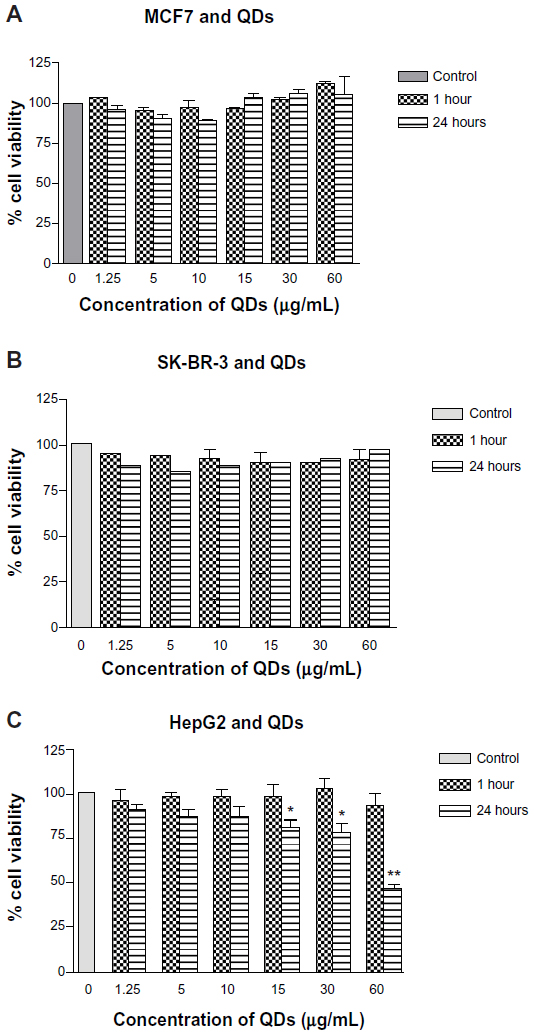 | Figure 3 In vitro cytotoxicity of CdTe/CdSe/ZnSe mercaptoundecanoic acid-coated QDs in MCF7, SK-BR-3, and HepG2 cells. |
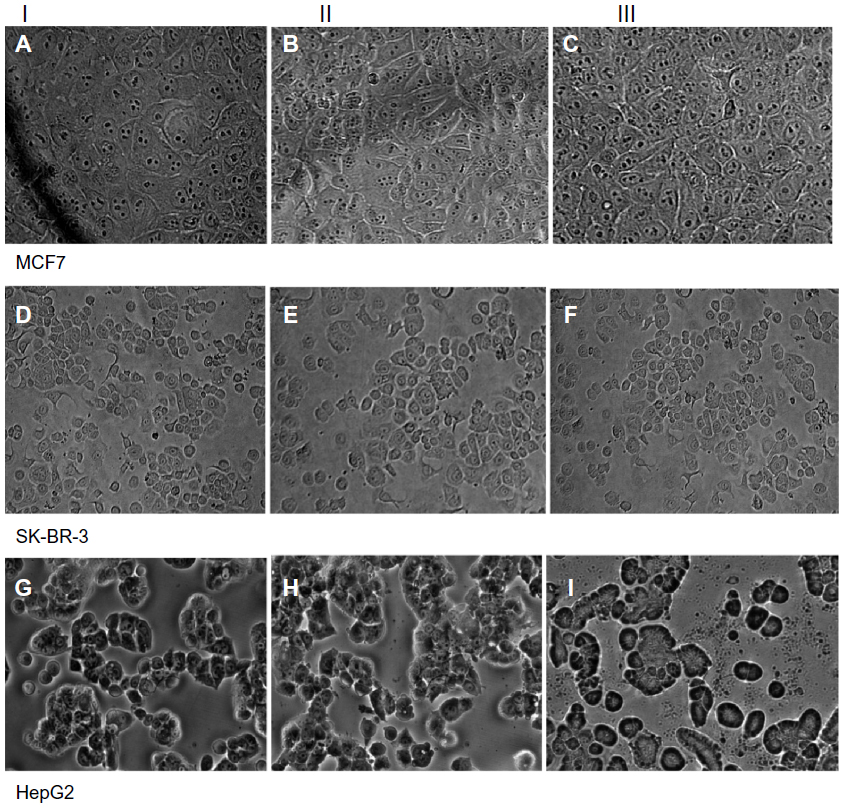 | Figure 4 Light microscopy images of MCF7 (A–C) SK-BR-3 (D–F), and HepG2 (G–I) cells exposed to 60 μg/mL of QDs for one hour and 24 hours. Column I shows the control cells; column II shows cells after one hour of exposure; and column III shows cells after 24 hours of exposure. (B and C) MCF7 cells and (E and F) SK-BR-3 cells show no change in cell morphology or cell numbers at either time point. (H and I) HepG2 cells show no evidence of change in cell morphology at one hour (G) but there is evidence of cellular breakdown with loss of polygonal architecture at 24 hours (I). |
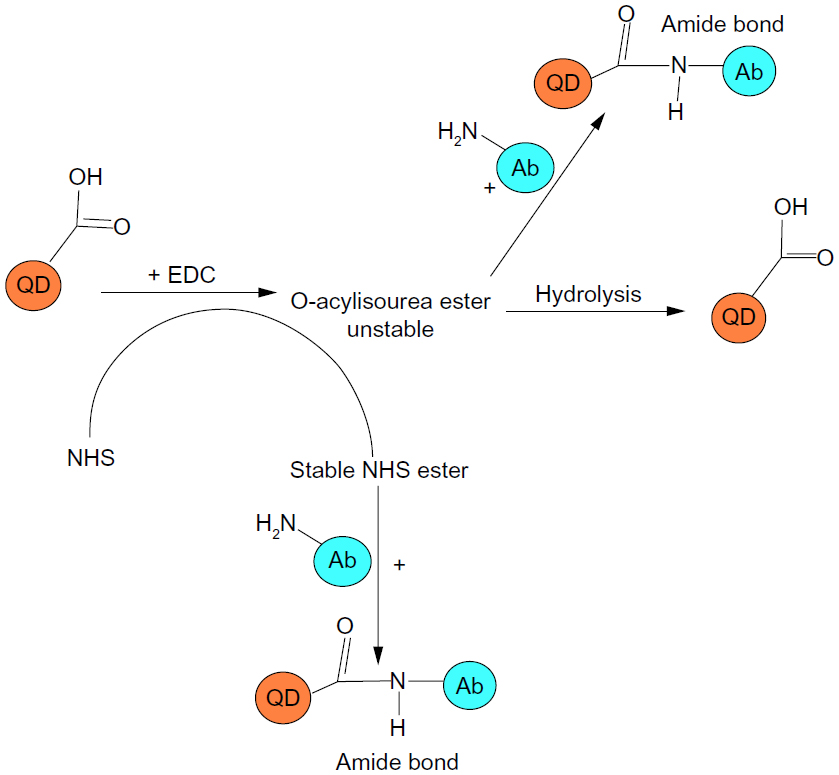 | Figure 5 Carbodiimide-based coupling reaction. The -COOH groups of mercaptoundecanoic acid on the QD surface are activated by EDC to form an unstable intermediate O-acylisourea which may react with the NH2 groups on the Ab to form an amide bond or rapidly hydrolyze to yield -COOH groups. Addition of NHS yields a stable reactive NHS ester intermediate that reacts with NH2 groups on the Ab to yield a stable amide bond. |
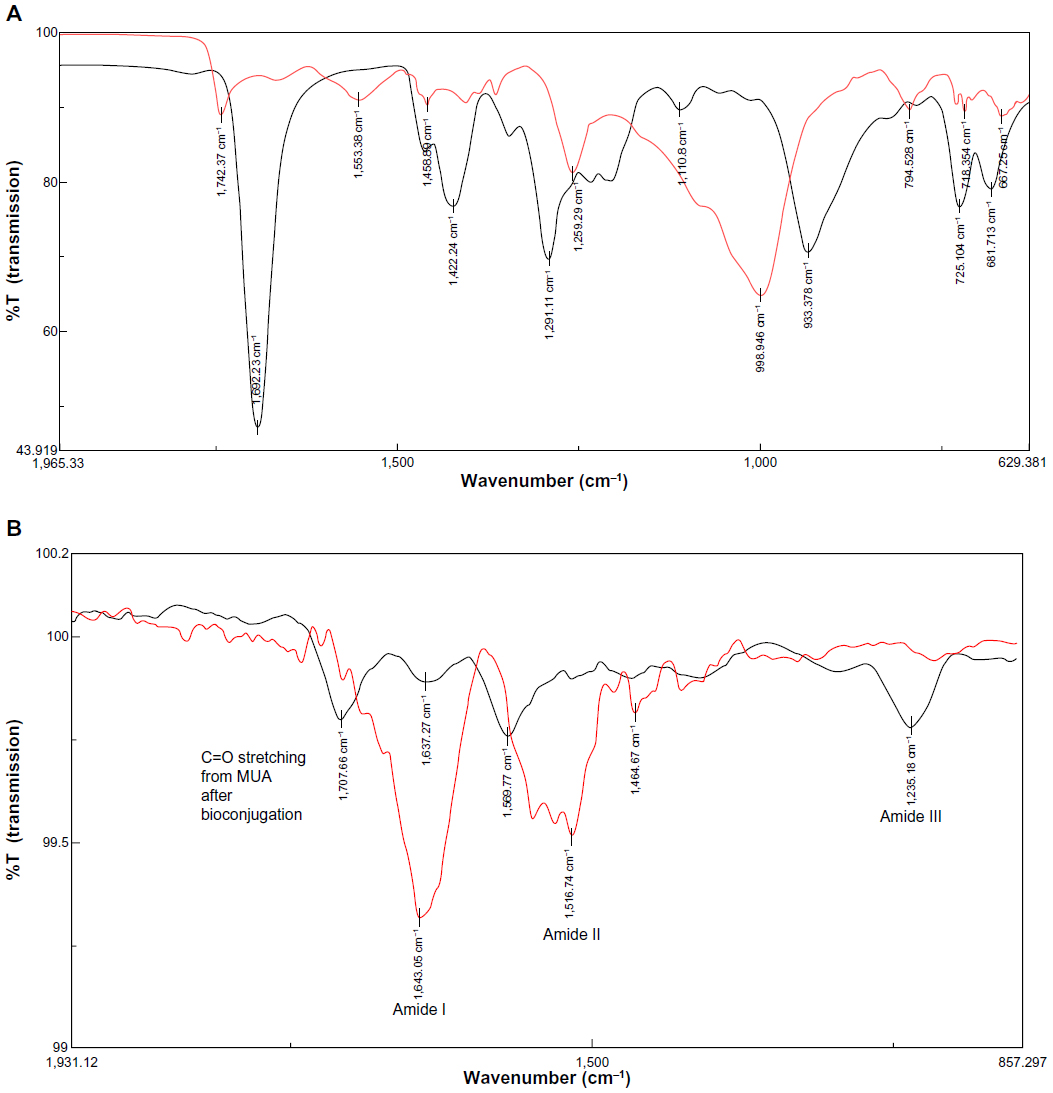 | Figure 6 Fourier transform infrared spectra of (A) CdTe/CdSe/ZnSe MUA QDs (red) compared with MUA (black); (B) anti-HER2-antibody (red) compared with bioconjugated QD-antibody (black). |
In vitro cytotoxicity
There was no significant difference in the viability of MCF7 cells on exposure to a QD concentration of up to 60 μg/mL for one hour and 24 hours compared with the control group. The cell viabilities at 24 hours appeared marginally lower when compared with the one-hour samples for QD concentrations at 1.25–10 μg/mL and 60 μg/mL. However, this was not statistically significant. Similarly, there was no evidence of a significant decrease in viability of SK-BR-3 cells at any concentration (0–60 μg/mL) at one hour and 24 hours of exposure (Figure 3). HepG2 cells did not show any significant difference in cell viability compared with the control at one hour. However, at 24 hours, there was a significant concentration-dependent decrease in cell viability evident at 15, 30, and 60 μg/mL (P<0.05, P<0.01, and P<0.01, respectively). In comparison with one hour, the cell viability decreased significantly to ≤75% at 30 μg/mL and <50% at 60 μg/mL (P<0.05 and P<0.001, respectively).
Cell morphology
There was no evidence of changes in cellular morphology and architecture in MCF7 (Figure 4A–C) and SK-BR-3 cells (Figure 4D–F) at one hour and 24 hours when compared with the control group. However, HepG2 cells (Figure 4G–I) showed evidence of cellular breakdown and loss of polygonal architecture, with reduced cell density at 24 hours of exposure at higher concentrations of 30–60 μg/mL in comparison with the control group. On a one-hour exposure, HepG2 cells showed no evidence of any morphological changes (Figure 4H).
Bioconjugation of QDs
The EDC and NHS powder readily dissolved in 1 mL of 0.5 mg/mL QD solution. EDC is a water-soluble carbodiimide used for immunoconjugate preparations, crosslinking of protein to nucleic acids, and peptide synthesis (Figure 5). It is used for activating the carboxylic end of the amino acid for the coupling of primary amines to form an amide bond. NHS is used as a catalyst to increase the efficiency of the coupling reaction. While a reaction with amines and a normal carboxylic acid would just make a salt, activation by NHS would prime the acid to make an amide bond.
After activation, 1 mL of antibody solution (100 μg/mL) was added to the activated QDs and the solution was left at room temperature for one hour. The QDs glowed a deep red on ultraviolet excitation, confirming that fluorescence was not quenched during the bioconjugation process. Centrifugal filtration was used to remove any unbound QDs, as the filtration membrane had a cutoff of 30 kDa and the size of the antibody was 185 kDa. Because only a very small amount of antibody was added (10 μg) with an excess of QDs (500 μg), we assumed that all the antibody would conjugate to the QDs and any excess unbound QDs would pass out through the filtration membrane. After filtration, the centrifugate of the sample (EDC + NHS) retained by the filtration membrane glowed a deep red, with minimal or no fluorescence in the filtrate, indicating that all the QDs had conjugated to the antibody. However, the EDC-only sample showed evidence of minimal fluorescence in the centrifugate, indicating that the majority of the QDs were not bound and the coupling reaction was inefficient.
Characterization of QD-antibody bioconjugate
FTIR was used to confirm the efficiency of the coupling reaction. Three samples were analyzed, including QD-antibody conjugated by the EDC/NHS coupling method, anti-HER2 antibody, and CdTe/CdSe/ZnSe MUA QDs alone. The sample of QD conjugated to the antibody using EDC did not show any prominent peaks and is therefore not shown. The graphs were overlaid to compare the absorption peaks (Figure 6B). There were four significant peaks in the QD-antibody conjugated using the EDC/NHS coupling method that were not detected in the other samples. The peak at 1,707.66 cm−1 corresponds to the C=O stretching from carboxylic acid on the QDs. In the QD sample, the C=O peak occurs at 1,742 cm−1 and a shift to a lower wave number of 1,707.66 cm−1 may correspond to QD conjugation with the antibody. The second peak at 1,637.27 cm−1 corresponds to stretching vibrations in C=O and C−N from an amide I linkage. The prominent peak at 1,569.66 cm−1 was closest to amide II and corresponded to NH bending and stretching vibrations from C−N. The infrared absorption peak at 1,235.18 cm−1 was close to amide III (1,200–1,305 cm−1) and was related to CN stretching and NH bending. Amide III is a complex band dependent on the details of the force field, nature of the side chains, and hydrogen bonding. The antibody on its own showed prominent peaks at 1,643 cm−1 and 1,516 cm−1, representing amide I and amide II bonds. The shift in the peak at 1,516 cm−1 to 1,569 cm−1 may indicate bioconjugation. Also, the prominent peak at 998 cm−1 from the O–H groups on the MUA disappeared, indicating formation of an amide link. The FTIR results showed carbodiimide-activated coupling between the amine groups on the antibody and the carboxylic groups on the QD. This was not observed for the EDC-only sample. The QDs were still glowing a deep red on ultraviolet excitation after the conjugation reaction.
Detection of cancer marker HER2 with QD-anti-HER2-antibody conjugate
The QD-anti-HER2-antibody conjugate was used to localize HER2 receptors on two breast cancer cell lines, including SK-BR-3 cells which overexpress HER2 receptors and MCF7 cells that do not (negative control). Both fixed cells (Figure 7) and live cells (Figure 8) were exposed to the QD-anti-HER2-antibody conjugate for a period of one hour. Confocal laser scanning microscopy images were taken and compared. The blue color shows the cell autofluorescence and the red color shows the QDs. Only cell autofluorescence was seen in the confocal images of unexposed SK-BR-3 and MCF7 cells. Fixed SK-BR-3 cells exposed to QD-anti-HER2-antibody (EDC and NHS) clearly showed evidence of fluorescence as small red dots on the cell surface, unlike the MCF7 cells, that did not show this pattern, with only occasional sites of nonspecific uptake (Figure 7). When live cells were exposed to the QD-anti-HER2-antibody conjugate (EDC and NHS), the SK-BR-3 cells clearly showed evidence of intracellular uptake with marked red fluorescence, unlike MCF7 cells, that showed a significantly lower uptake due to reduced expression of HER2 receptors (Figure 8). Two different protocols of QD conjugation were compared for both fixed and live cells. We found that QDs activated by EDC alone showed minimal HER2 localization because the efficiency of the reaction was low compared with use of EDC and NHS. This may be due to the instability of the bioconjugation intermediate at physiological pH when EDC is used on its own.
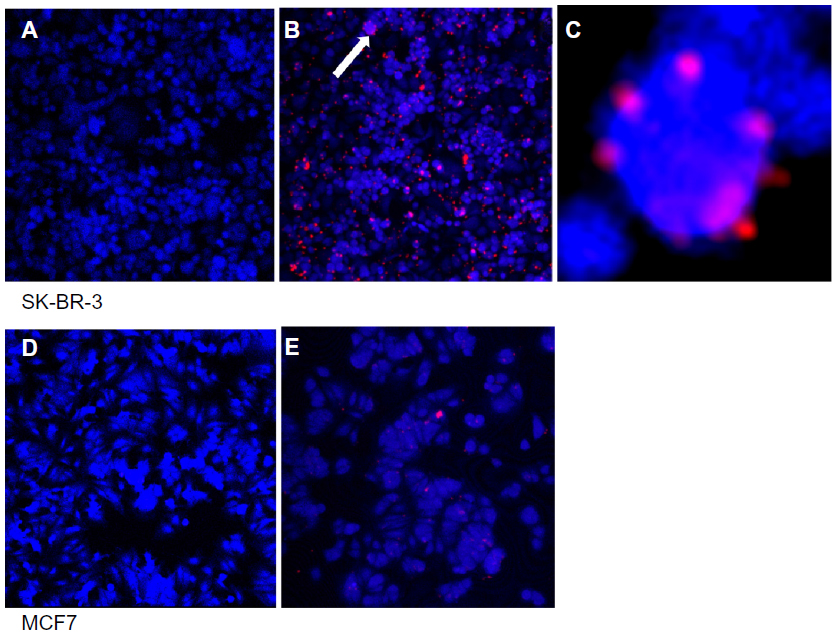 | Figure 7 Fixed cell imaging of HER2 receptors in SK-BR-3 (A–C) and MCF7 (D and E) cell lines. (A) Control SK-BR-3 and (B) SK-BR-3 cells exposed to QD-anti-HER2-antibody. (C) Magnified inset of (B) showing ring-like fluorescence of QDs on the cell surface. (D) Control MCF7 and (E) MCF7 exposed to the QD-antiHER2-antibody bioconjugate. |
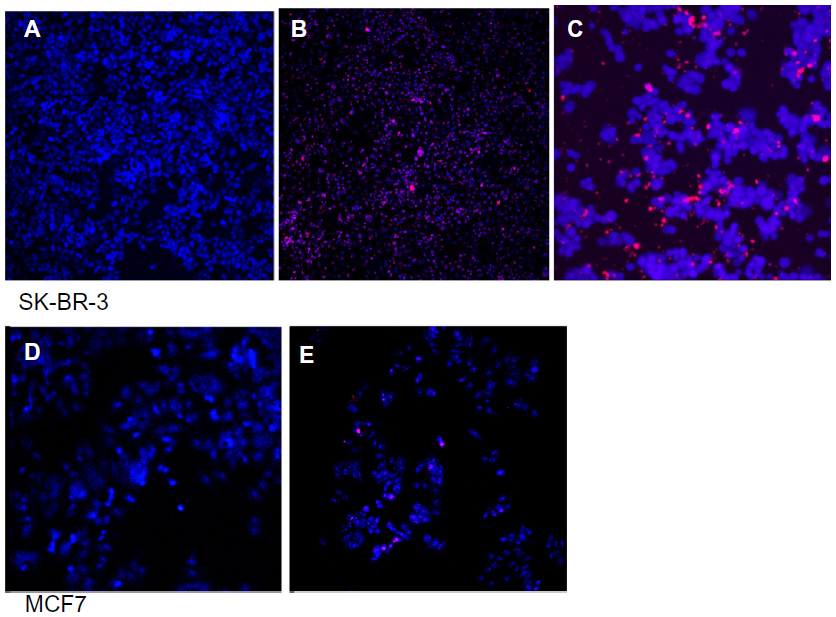 | Figure 8 Live cell imaging of SK-BR-3 (A–C) and MCF7 (D and E) cells with the QD-anti-HER2-antibody probe. (A) Control SK-BR-3 cells. (B) Significantly higher uptake of QD-antibody bioconjugate by HER2-overexpressing cells. (C) SK-BR-3 cells stained with the nuclear stain diamidino-2-phenylindole (DAPI) show localization of the QDs to the perinuclear region on live cell imaging (D) MCF7 control and (E) MCF7 cells show lower uptake of the QD-antibody probe because of their lower expression of the HER2 receptor. |
Discussion
In this study, we established the application of aqueously synthesized NIR-QDs based on CdTe/CdSe/ZnSe for localization of HER2 receptors in vitro for both fixed and live cells. The NIR-QDs were coated with MUA and showed an absorption onset and emission in the NIR range (Figure 2A). This means that, on in vivo injection, NIR light could be used to penetrate tissues to excite QDs which emitted at a slightly shifted NIR wavelength and could be detected by an NIR-sensitive camera. Characterization of the QDs by transmission electron microscopy showed that they were approximately spherical in shape with a diameter of 4±0.8 nm (Figure 2B). FTIR analysis of the QDs compared with the spectra from MUA alone confirmed the presence of an MUA coating on the QD surface through a visible shift in the peak at 933 cm−1 on MUA to 998 cm−1 from O–H bending of carboxylic acid when immobilized on the QD surface. The carbonyl peak at 1,692 cm−1 in the MUA also shifted to 1,742 cm−1 on the MUA coated QDs (Figure 6).
The main limitation to biomedical application of QDs is their toxicity, given that most QDs are based on heavy metals including cadmium, tellurium, and selenium. QD toxicity has been attributed to a range of factors, including surface coating, charge, chemistry, size, and mechanical and photochemical stability in the physiological environment.54 While some studies have shown the biocompatibility of QDs,55–57 others have demonstrated evidence of in vitro toxicity at concentrations as low as 10 μg/mL.58–63 We examined the toxicity of MUA-QDs in three different cell lines, including SK-BR-3, MCF7, and HepG2, and found that the QDs at concentrations of 60 μg/mL were biocompatible with SK-BR-3 and MCF7 cell lines at one hour and 24 hours. However, HepG2 cells showed evidence of toxicity at QD concentrations ≥15 μg/mL at 24 hours (Figure 3). This demonstrates a differential toxicity of QDs in different cell lines and may suggest different effects at the tissue and organ levels. It is likely that because the liver is a key site of metabolism, QDs are exposed to metabolic degradation after uptake by hepatocytes, leading to release of their toxic core components and reactive oxygen species. Therefore, hepatotoxicity is a considerable concern given that most in vivo studies demonstrate maximum uptake by organs of the reticuloendothelial system, including the liver, spleen, lymph nodes, and bone marrow.64–67 However, various in vivo studies report that the time to maximum uptake by the liver is 4–6 hours, allowing enough time for an actively targeted probe to home in to its molecular destination.68–71
While the toxicity of QDs is a definite concern for various biological applications, cancer therapy is one area where this toxicity may be utilized to aid killing cancer cells. For example, QDs can potentially be used as agents for photodynamic therapy in the management of cancer, when conjugated with photoactive dyes that can be activated with light to generate free radicals leading to localized cell death.15 Bioconjugated targeted NIR-QDs visible in deep tissues would be ideal for this purpose because they would accumulate at the site of the cancer which can then be ablated by light activation under image guidance.
Carbodiimide chemistry is commonly used to conjugate proteins. Figure 5 shows a schematic diagram explaining the coupling reaction between MUA-QDs and the anti-HER2 antibody. A carbodiimide compound facilitates the direct conjugation of -COOH groups with primary amines. EDC is a zero-length crosslinker because it does not become part of the final crosslink between molecules. Also, because proteins and peptides contain both carboxyl and amino groups, direct EDC-mediated crosslinking may cause random polymerization of the polypeptides. EDC activates the carboxyl group on the MUA to form an active O-acylisourea intermediate that reacts with the amine on the protein which forms an amide bond with the -COOH and an EDC byproduct is released as a soluble urea derivative. However, the O-acylisourea intermediate is unstable in aqueous solution, and failure to react with an amine results in hydrolysis of the intermediate and regeneration of the carboxyl group. EDC crosslinking is most efficient at an acidic pH, and while a physiological pH is compatible with the reaction, the coupling efficiency is lower and requires a higher EDC concentration. NHS, or its water-soluble analog (Sulfo-NHS) is often included in EDC coupling protocols to improve efficiency. EDC couples NHS to carboxylic groups, resulting in formation of an NHS ester that is considerably more stable than the O-acylisourea intermediate while allowing for efficient conjugation to primary amines at physiological pH. Hence, in summary, EDC reacts with a carboxyl group on the MUA-QD to form an amine-reactive O-acylisourea intermediate. However, this intermediate is unstable, and may either react with an amine on the antibody to yield a bioconjugate joined by a stable amide bond or may hydrolyze in aqueous solution to reform the -COOH group. Given that the efficiency of the reaction is low at physiological pH, addition of NHS stabilizes the amine-reactive intermediate by converting it to an amine-reactive NHS ester which then reacts with the amine group on the antibody to yield a stable amide bond along with increasing the efficiency of the EDC-mediated coupling reaction.72,73
The QD-antibody conjugate was characterized by FTIR and showed successful conjugation in the EDC/NHS mediated coupling reaction (Figure 6). However, the EDC-only protocol did not lead to bioconjugation, probably because the reaction was carried out at physiological pH and had lower efficiency. Addition of NHS increased the coupling efficiency by stabilizing the amine-reactive intermediate, allowing the reaction to proceed to completion. The EDC/NHS coupling protocol showed three bands in the FTIR spectra, representing amide I features (1,600–1,700 cm−1) from the stretching vibrations of the C=O and C–N groups, and amide II features between 1,510–1,580 cm−1 from in plane N–H bending as well as from C–N stretching vibrations. The slight shift at 1,707 cm−1 may represent stretching of the carbonyl groups of MUA. When compared with FTIR of the unconjugated antibody, there was a shift in the amide II band at 1,516 cm−1 in the unconjugated antibody to 1,569 cm−1 after conjugation, as well as formation of a new band at 1,225 cm−1 from an amide III linkage.
We demonstrated application of the anti-HER2-QD-antibody conjugate for in vitro imaging of fixed and live cells. In fixed cell imaging, we found that the QD-antibody conjugate attached to the extracellular domain of the HER2 receptor, with overall uptake of QDs being significantly higher in HER2-overexpressing SK-BR-3 cells (Figure 7A–C) as compared with the HER2 under-expressting MCF7 cell line (Figure 7D and E). Xiao et al reported that SK-BR-3 cells express 15 times higher amounts of HER2 protein and a ten times greater number of copies of genes than MCF7 cells, making these cells suitable reference materials for HER2 testing.74 The lower uptake of the QD-antibody probe by MCF7 cells may indicate that the QD-antibody probe is highly sensitive and detected low amounts of HER2 receptors as well. Previous studies have demonstrated a complete outline of the cells because the QD-antibody conjugate attaches to the extracellular receptors. However, most of these studies used QD-based immunohistochemistry via the primary and secondary antibody to localize the HER2 receptors. We directly conjugated the antibody to the QD surface, and it is likely that because the antibody concentration was fairly low, a ring-like outline of cells was only occasionally visualized (Figure 7C). We noted that some amount of QD aggregation occurred during bioconjugation, and this may be due to instability of the MUA coating as small molecular thiol ligands may desorb from the QD surface, leading to colloidal instability. Further refinement of the procedure is therefore required with the use of biocompatible and biostable surface coatings. When exposed to live cells, the QD-antibody conjugate was rapidly taken up by the SK-BR-3 cells, as evident from the increased intracellular fluorescence (Figure 8A and B) in comparison with MCF7 cells (Figure 8D and E). Nuclear staining with diamidino-2-phenylindole (DAPI) demonstrated that QDs localized to the perinuclear region in live cells exposed to SK-BR-3 (Figure 8C). Our results are in agreement with previous studies demonstrating the molecular pathway of HER2 receptors and indicating that the HER2 receptor is endocytosed and migrates from the cytoplasm to the perinuclear region.75 In a study evaluating the molecular mechanisms of movement within cells, Watanabe et al demonstrated that, after endocytosis, HER2 receptors moved along the membrane by transferring actin filaments and were then rapidly transported towards the nucleus via microtubules.76
The overall size of the QD-antibody bioconjugate can influence its application for cancer localization in vivo. IgG is a large antibody of 185 kDa. When conjugated to a QD, a larger complex is formed. Smaller antibody fragments would increase extravasation into solid tumors in vivo compared with larger antibodies.77 The application of small proteins as molecular probes for cancer targeting has various advantages, including high affinity and specificity, small size, and rapid clearance.78 Gao et al showed the application of affibody-based QD probes for HER2 localization and targeted in vivo imaging.79 The main features of affibody molecules are their significantly smaller size and molecular weight (7 kDa), high affinity, and rapid tumor targeting. Similarly, Barat et al demonstrated the use of small bivalent antibody fragments called cys-diabodies for QD bioconjugation and targeted cancer localization.80 They conjugated amino-poly(ethylene glycol)-coated CdTe/ZnS QDs to anti-HER2 cys-diabodies by EMCS (N-epsilon-Malemidocaproyl-oxysuccinimide ester) coupling to form an anti-HER2 iQdot. The conjugate showed no spectral differences from the unconjugated QDs, and immunofluorescence imaging showed homogenous surface labeling of cell membranes with the QD conjugate. There was no change in the photoluminescence of the QDs after conjugation. They demonstrated that cys-diabodies retained the antigen reorganization sites of the antibody and yet had a small size, making them more favorable for in vivo imaging applications.
Apart from elucidating the biology, pathology, and evolution of breast cancer, QD-based molecular probes have been investigated for targeted in vivo imaging of cancer.67,70 The real challenge in breast cancer therapy is the application of bioconjugated QD probes for in vivo targeting of HER2 receptors as a means for image-guided therapy and cancer surgery. Optical imaging is a valuable tool for live imaging and particularly tumor targeting using QD probes.81 NIR-QDs can be a valuable probe for deep tissue imaging applications. This is based on the fact that NIR light can penetrate deep tissues without being scattered or absorbed, compared with visible light. Hence targeted localization of tumors using NIR-QD bioconjugates would allow better resection of tumor margins under optical guidance. QDs have been demonstrated to be alternative probes for sentinel lymph node localization in breast cancer surgery. NIR-QDs would be ideal for this application because the NIR fluorescence would be detectable in deep tissues, allowing accurate localization of the sentinel lymph node prior to the surgical incision. Based on the localization of HER2 receptors in vitro, it is reasonable to predict that NIR-QD bioconjugates could potentially be used for detecting micrometastasis in the sentinel lymph nodes. This may prove invaluable in breast cancer surgery, whereby the presence or absence of micrometastasis in the sentinel lymph nodes may allow the surgeon to bypass the sentinel lymph node biopsy procedure.
The most important step in the clinical translation of this QD-based molecular targeting technology is determination of its toxicity in vivo. While a considerable amount of research has focused on determining the in vitro toxicity of QDs, the in vivo behavior of these novel probes needs to be elaborated further. Tiwari et al82 reported no significant toxicity of QDs conjugated to the anti-HER2 antibody compared with unconjugated QDs, indicating that the bioconjugated probes were biocompatible and therefore suitable for breast cancer imaging and surgery.
Conclusion and future directions
We have demonstrated the bioconjugation of NIR-QDs to an anti-HER2 antibody using an EDC/NHS coupling method. Addition of NHS increases the efficiency of the coupling reaction, leading to a stable QD-antibody conjugate. HER2 receptors were successfully localized in both fixed and live cancer cells. Given that the NIR-QDs can be visualized in deep tissues, this property can be used for image-guided delivery of chemotherapeutic agents to cancer sites as well as sites of micrometastasis which would otherwise not be detected. NIR optical imaging can potentially be used to guide the surgeon to an adequate tumor resection margin, based on uptake of the NIR-QD antibody probe. While the toxicity of the QD-antibody probe is a definite concern, active targeting would allow considerably lower doses to be used for tumor localization. At the same time, eventual resection of the tumor would remove the major bulk of the QDs, leading to minimal toxicity. Further studies to investigate the in vivo behavior of the QD-antibody bioconjugate are essential prior to clinical translation.
Acknowledgment
The authors acknowledge the financial support of the Javon Charitable Trust, London, UK, for research and development work on detection and treatment of cancer using nanoparticles.
Disclosure
The authors report no conflicts of interest in this work.
References
Schroeder A, Heller DA, Winslow MM, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2011;12(1):39–50. | |
Fernandez-Fernandez A, Manchanda R, McGoron AJ. Theranostic applications of nanomaterials in cancer: drug delivery, image-guided therapy, and multifunctional platforms. Appl Biochem Biotechnol. 2011;165(7–8):1628–1651. | |
Monsky WL, Vien DS, Link DP. Nanotechnology development and utilization: a primer for diagnostic and interventional radiologists. Radiographics. 2011;31(5):1449–1462. | |
Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Multistage nanovectors: from concept to novel imaging contrast agents and therapeutics. Acc Chem Res. 2011;44(10):979–989. | |
Singh R, Nalwa HS. Medical applications of nanoparticles in biological imaging, cell labeling, antimicrobial agents, and anticancer nanodrugs. J Biomed Nanotechnol. 2011;7(4):489–503. | |
Kievit FM, Zhang M. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv Mater. 2011;23(36):H217–H247. | |
Solomon M, D’Souza GG. Recent progress in the therapeutic applications of nanotechnology. Curr Opin Pediatr. 2011;23(2):215–220. | |
Jain AK, Das M, Swarnakar NK, Jain S. Engineered PLGA nanoparticles: an emerging delivery tool in cancer therapeutics. Crit Rev Ther Drug Carrier Syst. 2011;28(1):1–45. | |
Grimm J, Scheinberg DA. Will nanotechnology influence targeted cancer therapy? Semin Radiat Oncol. 2011;21(2):80–87. | |
Grobmyer SR, Morse DL, Fletcher B, et al. The promise of nanotechnology for solving clinical problems in breast cancer. J Surg Oncol. 2011;103(4):317–325. | |
Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Nanomedicine for targeted cancer therapy: towards the overcoming of drug resistance. Drug Resist Updat. 2011;14(3):150–163. | |
Kim PS, Djazayeri S, Zeineldin R. Novel nanotechnology approaches to diagnosis and therapy of ovarian cancer. Gynecol Oncol. 2011;120(3):393–403. | |
Wesselinova D. Current major cancer targets for nanoparticle systems. Curr Cancer Drug Targets. 2011;11(2):164–183. | |
Tan A, Yildirimer L, Rajadas J, De La Peña H, Pastorin G, Seifalian A. Quantum dots and carbon nanotubes in oncology: a review on emerging theranostic applications in nanomedicine. Nanomedicine (Lond). 2011;6(6):1101–1114. | |
Rizvi SB, Ghaderi S, Keshtgar M, Seifalian AM. Semiconductor quantum dots as fluorescent probes for in vitro and in vivo bio-molecular and cellular imaging. Nano Rev. August 16, 2010. [Epub ahead of print.] | |
Gu YP, Cui R, Zhang ZL, Xie ZX, Pang DW. Ultrasmall near-infrared ag2se quantum dots with tunable fluorescence for in vivo imaging. J Am Chem Soc. 2011;134(1):79–82. | |
Leung K. 64Cu-Tetraazacyclododecane-N,N’,N’’,N’’’-tetraacetic acid-quantum dot-c(Arg-Gly-Asp-D-Tyr-Lys). Molecular Imaging and Contrast Agent Database. Bethesda, MD, USA: National Center for Biotechnology Information; 2004–2013. | |
Chen H, Cui S, Tu Z, Ji J, Zhang J, Gu Y. Characterization of CdHgTe/CdS QDs for near infrared fluorescence imaging of spinal column in a mouse model. Photochem Photobiol. 2011;87(1):72–81. | |
Pic E, Pons T, Bezdetnaya L, et al. Fluorescence imaging and whole-body biodistribution of near-infrared-emitting quantum dots after subcutaneous injection for regional lymph node mapping in mice. Mol Imaging Biol. 2010;12(4):394–405. | |
Kim S, Lim YT, Soltesz EG, et al. Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat Biotechnol. 2004;22(1):93–97. | |
Samia AC, Dayal S, Burda C. Quantum dot-based energy transfer: perspectives and potential for applications in photodynamic therapy. Photochem Photobiol. 2006;82(3):617–625. | |
Nida DL, Rahman MS, Carlson KD, Richards-Kortum R, Follen M. Fluorescent nanocrystals for use in early cervical cancer detection. Gynecol Oncol. 2005;99(3 Suppl 1):S89–S94. | |
Samia AC, Chen X, Burda C. Semiconductor quantum dots for photodynamic therapy. J Am Chem Soc. 2003;125(51):15736–15737. | |
Yong KT, Roy I, Ding H, Bergey EJ, Prasad PN. Biocompatible near-infrared quantum dots as ultrasensitive probes for long-term in vivo imaging applications. Small. 2009;5(17):1997–2004. | |
Mahmoud W, Sukhanova A, Oleinikov V, et al. Emerging applications of fluorescent nanocrystals quantum dots for micrometastases detection. Proteomics. 2010;10(4):700–716. | |
Manabe N, Hoshino A, Liang YQ, Goto T, Kato N, Yamamoto K. Quantum dot as a drug tracer in vivo. IEEE Trans Nanobioscience. 2006;5(4):263–267. | |
Zrazhevskiy P, Sena M, Gao X. Designing multifunctional quantum dots for bioimaging, detection, and drug delivery. Chem Soc Rev. 2010;39(11):4326–4354. | |
Gao X, Cui Y, Levenson RM, Chung LW, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22(8):969–976. | |
Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25(1):118–145. | |
John M. Anti-epidermal growth factor receptor monoclonal antibodies as potential anti-cancer agents. J Steroid Biochem Mol Biol. 1990;37(6):889–892. | |
Grunwald V, Hidalgo M. Developing inhibitors of the epidermal growth factor receptor for cancer treatment. J Natl Canc Inst. 2003;95(12):851–867. | |
Yasui W, Sumiyoshi H, Hata J, et al. Expression of epidermal growth factor receptor in human gastric and colonic carcinomas. Cancer Res. 1988;48(1):137–141. | |
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. | |
Press MF, Pike MC, Chazin VR, et al. Her-2/neu expression in node-negative breast cancer: direct tissue quantitation by computerized image analysis and association of overexpression with increased risk of recurrent disease. Cancer Res. 1993;53(20):4960–4970. | |
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. | |
Hayes DF, Picard MH. Heart of darkness: the downside of trastuzumab. J Clin Oncol. 2006;24(25):4056–4058. | |
Goldenberg MM. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin Ther. 1999;21(2):309–318. | |
Chen C, Peng J, Xia HS, et al. Quantum dots-based immunofluorescence technology for the quantitative determination of HER2 expression in breast cancer. Biomaterials. 2009;30(15):2912–2918. | |
Ginestier C, Charafe-Jauffret E, Penault-Llorca F, et al. Comparative multi-methodological measurement of ERBB2 status in breast cancer. J Pathol. 2004;202(3):286–298. | |
Payne SJ, Bowen RL, Jones JL, Wells CA. Predictive markers in breast cancer – the present. Histopathology. 2008;52(1):82–90. | |
Taylor CR, Levenson RM. Quantification of immunohistochemistry – issues concerning methods, utility and semiquantitative assessment II. Histopathology. 2006;49(4):411–424. | |
Yaziji H, Goldstein LC, Barry TS, et al. HER-2 testing in breast cancer using parallel tissue-based methods. JAMA. 2004;291(16):1972–1977. | |
Tanner M, Gancberg D, Di Leo A, et al. Chromogenic in situ hybridization: a practical alternative for fluorescence in situ hybridization to detect HER-2/neu oncogene amplification in archival breast cancer samples. Am J Pathol. 2000;157(5):1467–1472. | |
Arnould L, Denoux Y, MacGrogan G, et al. Agreement between chromogenic in situ hybridisation (CISH) and FISH in the determination of HER2 status in breast cancer. Br J Cancer. 2003;88(10):1587–1591. | |
Bhargava R, Lal P, Chen B. Chromogenic in situ hybridization for the detection of HER-2/neu gene amplification in breast cancer with an emphasis on tumors with borderline and low-level amplification: does it measure up to fluorescence in situ hybridization? Am J Clin Pathol. 2005;123(2):237–243. | |
Wu X, Liu H, Liu J, et al. Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat Biotechnol. 2003;21(1):41–46. | |
Xing Y, Chaudry Q, Shen C, et al. Bioconjugated quantum dots for multiplexed and quantitative immunohistochemistry. Nat Protoc. 2007;2(5):1152–1165. | |
Chen C, Xia HS, Gong YP, et al. The quantitative detection of total HER2 load by quantum dots and the identification of a new subtype of breast cancer with different 5-year prognosis. Biomaterials. 2010;31(33):8818–8825. | |
Chen C, Sun SR, Gong YP, et al. Quantum dots-based molecular classification of breast cancer by quantitative spectroanalysis of hormone receptors and HER2. Biomaterials. 2011;32(30):7592–7599. | |
Xiao Y, Gao X, Gannot G, et al. Quantitation of HER2 and telomerase biomarkers in solid tumors with IgY antibodies and nanocrystal detection. Int J Cancer. 2008;122(10):2178–2186. | |
Chen C, Peng J, Xia H, et al. Quantum-dot-based immunofluorescent imaging of HER2 and ER provides new insights into breast cancer heterogeneity. Nanotechnology. 2010;21(9):095101. | |
Liu XL, Peng CW, Chen C, et al. Quantum dots-based double-color imaging of HER2 positive breast cancer invasion. Biochem Biophys Res Commun. 2011;409(3):577–582. | |
Taniguchi S, Green M, Rizvi SB, Seifalian A. The one-pot synthesis of core/shell/shell CdTe/CdSe/ZnSe quantum dots in aqueous media for in vivo deep tissue imaging. J Mater Chem. 2011;21(9):2877–2882. | |
Hardman R. A toxicologic review of quantum dots: toxicity depends on physicochemical and environmental factors. Environ Health Perspect. 2006;114(2):165–172. | |
Hanaki K, Momo A, Oku T, et al. Semiconductor quantum dot/albumin complex is a long-life and highly photostable endosome marker. Biochem Biophys Res Commun. 2003;302(3):496–501. | |
Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Long-term multiple color imaging of live cells using quantum dot bioconjugates. Nat Biotechnol. 2003;21(1):47–51. | |
Voura EB, Jaiswal JK, Mattoussi H, Simon SM. Tracking metastatic tumor cell extravasation with quantum dot nanocrystals and fluorescence emission-scanning microscopy. Nat Med. 2004;10(9):993–998. | |
Chan WH, Shiao NH, Lu PZ. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol Lett. 2006;167(3):191–200. | |
Kirchner C, Liedl T, Kudera S, et al. Cytotoxicity of colloidal CdSe and CdSe/ZnS nanoparticles. Nano Lett. 2005;5(2):331–338. | |
Shiohara A, Hoshino A, Hanaki K, Suzuki K, Yamamoto K. On the cyto-toxicity caused by quantum dots. Microbiol Immunol. 2004;48(9):669–675. | |
Lovric J, Bazzi HS, Cuie Y, Fortin GR, Winnik FM, Maysinger D. Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots. J Mol Med. 2005;83(5):377–385. | |
Chang E, Thekkek N, Yu WW, Colvin VL, Drezek R. Evaluation of quantum dot cytotoxicity based on intracellular uptake. Small. 2006;2(12):1412–1417. | |
Derfus AM, Chan WCW, Bhatia SN. Probing the cytotoxicity of semiconductor quantum dots. Nano Lett. 2004;4(1):11–18. | |
Fischer HC, Liu L, Pang KG, Chan WG. Pharmacokinetics of nanoscale quantum dots: In vivo distribution, sequestration, and clearance in the rat. Adv Funct Mater. 2006;16(10):1299–1305. | |
Schipper ML, Cheng Z, Lee SW, et al. microPET-based biodistribution of quantum dots in living mice. J Nucl Med. 2007;48(9):1511–1518. | |
Schipper ML, Iyer G, Koh AL, et al. Particle size, surface coating, and PEGylation influence the biodistribution of quantum dots in living mice. Small. 2009;5(1):126–134. | |
Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Noninvasive imaging of quantum dots in mice. Bioconjug Chem. 2003;15(1):79–86. | |
Kato S, Itoh K, Yaoi T, et al. Organ distribution of quantum dots after intraperitoneal administration, with special reference to area-specific distribution in the brain. Nanotechnology. 2010;21(33):335103. | |
Su Y, Peng F, Jiang Z, et al. In vivo distribution, pharmacokinetics, and toxicity of aqueous synthesized cadmium-containing quantum dots. Biomaterials. 2011;32(25):5855–5862. | |
Diagaradjane P, Orenstein-Cardona JM, Colon-Casasnovas NE, et al. Imaging epidermal growth factor receptor expression in vivo: pharmacokinetic and biodistribution characterization of a bioconjugated quantum dot nanoprobe. Clin Cancer Res. 2008;14(3):731–741. | |
Chen Z, Chen H, Meng H, et al. Bio-distribution and metabolic paths of silica coated CdSeS quantum dots. Toxicol Appl Pharmacol. 2008;230(3):364–371. | |
Updegrove TB, Correia JJ, Chen Y, Terry C, Wartell RM. The stoichiometry of the Escherichia coli Hfq protein bound to RNA. RNA. 2011;17(3):489–500. | |
Naue N, Fedorov R, Pich A, Manstein DJ, Curth U. Site-directed mutagenesis of the chi subunit of DNA polymerase III and single-stranded DNA-binding protein of E. coli reveals key residues for their interaction. Nucleic Acids Res. 2011;39(4):1398–1407. | |
Xiao Y, Gao X, Maragh S, Telford WG, Tona A. Cell lines as candidate reference materials for quality control of ERBB2 amplification and expression assays in breast cancer. Clin Chem. 2009;55(7):1307–1315. | |
Tada H, Higuchi H, Wanatabe TM, Ohuchi N. In vivo real-time tracking of single quantum dots conjugated with monoclonal anti-HER2 antibody in tumors of mice. Cancer Res. 2007;67(3):1138–1144. | |
Watanabe TM, Higuchi H. Stepwise movements in vesicle transport of HER2 by motor proteins in living cells. Biophys J. 2007;92(11):4109–4120. | |
Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52(12):3402–3408. | |
Miao Z, Levi J, Cheng Z. Protein scaffold-based molecular probes for cancer molecular imaging. Amino Acids. 2011;41(5):1037–1047. | |
Gao J, Chen K, Miao Z, et al. Affibody-based nanoprobes for HER2-expressing cell and tumor imaging. Biomaterials. 2011;32(8):2141–2148. | |
Barat B, Sirk SJ, McCabe KE, et al. Cys-diabody quantum dot conjugates (immunoQdots) for cancer marker detection. Bioconjug Chem. 2009;19;20(8):1474–1481. | |
Park JM, Ikeda DM. Promising techniques for breast cancer detection, diagnosis, and staging using non-ionizing radiation imaging techniques. Phys Med. 2006;21 Suppl 1:7–10. | |
Tiwari DK, Jin T, Behari J. Bio-distribution and toxicity assessment of intravenously injected anti-HER2 antibody conjugated CdSe/ZnS quantum dots in Wistar rats. Int J Nanomedicine. 2011;6:463–475. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.