Back to Journals » Cancer Management and Research » Volume 11
Natural gypenosides: targeting cancer through different molecular pathways
Authors Ahmad B ![]() , Khan S
, Khan S ![]() , Nabi G
, Nabi G ![]() , Gamallat Y
, Gamallat Y ![]() , Su P, Jamalat Y, Duan P, Yao L
, Su P, Jamalat Y, Duan P, Yao L
Received 24 August 2018
Accepted for publication 8 February 2019
Published 27 March 2019 Volume 2019:11 Pages 2287—2297
DOI https://doi.org/10.2147/CMAR.S185232
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Rituraj Purohit
Bashir Ahmad,1,2 Suliman Khan,1,3,4 Ghulam Nabi,4,5 Yaser Gamallat,2 Pengyu Su,2 Yazeed Jamalat,2 Pengfei Duan,1,3 Lunguang Yao1,3
1Henan Provincial Engineering Research Center for Health Products of Livestock and Poultry, Nanyang Normal University, Nanyang, People’s Republic of China; 2College of Basic Medical Sciences, Dalian Medical University, Dalian City, People’s Republic of China; 3Collaborative Innovation Centre of Water Security for Water Source Region of Mid-line of South-to-North Diversion Project of Henan Province/Key Laboratory of Ecological Security for Water Source Region of Mid-line of South-to-North Diversion Project of Henan Province, School of Agricultural Engineering, Nanyang Normal University, Nanyang, People’s Republic of China; 4University of Chinese Academy of Sciences, Beijing, People’s Republic of China; 5Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, Hubei, People’s Republic of China
Abstract: The second foremost cause of mortality around the word is cancer. Conventional therapies, such as radiation, surgery, and chemotherapy have limited accessibility owing to secondary resistance. Therefore, convenient, safe, and nonresistant drugs are urgently needed. Plant-derived natural products have attracted considerable interest owing to their high efficacy, low toxicity, and convenience. Gypenosides (Gyp) inhibit invasion, migration, metastasis, and proliferation and induce apoptosis in different cancers, including oral, lung, colorectal, hepatocellular, and leukemic cancers through different molecular pathways. This review summarizes Gyp studies on cancer to serve as a reference for further research and clinical trials.
Keywords: cancer, surgery, chemotherapy, radiation therapy, Gyp, oral, lung, leukemic, colorectal, hepatocellular
Introduction
According to the WHO, cancer causes more deaths than stroke or any coronary heart disease.1 Cancer cases increase because of numerous risk factors, aging, and population growth.2 The continuous global epidemiologic and demographic transition signal shows that cancer will increase in the next decades, particularly in low- and middle-income countries and is expected to have an annual record of 20 million new cases in 2025.3 In a 2012 cancer report, approximately 14.1 million new cases and 8.2 million deaths were recorded from 20 large areas of the world, and lung cancer was the most common (1.82 million), followed by breast cancer (1.67 million), and colorectal cancer (1.36 million). The major cause of death was also lung cancer (1.36 million), followed by liver cancer (0.745 million) and stomach cancer (0.723 million).4 Available clinical treatment for cancer includes surgery, chemotherapy, and radiotherapy.5 Furthermore, radiotherapy and chemotherapy can develop gradual resistance against therapy in cancer cells.6 Therefore, novel, affordable, and effective anticancer drugs must be developed.7 Medicinal plants provide a common alternative treatment for cancer in various countries.8,9 Numerous natural products (NPs) were approved by the Food and Drug Administration for the treatment of cancer.10 Many researchers reported new antitumor NPs, but their main molecular mechanisms remain incompletely understood. NP function contains different natural compounds and thus function through different pathways.11 Systematic biology is an emerging approach that focuses on molecular interactions in biological systems.12 Cancer is subject to complicated cell transformation processes that result in changes at the genetic, epigenetic, and cellular levels.13 Therefore, systematic biology can help us understand the molecular mechanisms of NPs and ultimately uncover the new window to cancer treatment.
Gynostemma pentaphyllum (GpM)-derived extracts and fractions and its derivative compounds show anticancer activity in vivo and in vitro. Several clinical trials showed that GpM has a potential curative effect on cancer.14 GpM (Thunb) Makino, known in China as Jiaogulan, belongs to the plant family Cucurbitaceae that abundantly grows in Korea, Japan, and southern China. It is a well-known folk medicine in China; in fact, it was reported for the first time as a vegetable in a book named “Herbs for Famine,” which was published in 1368–1644 AD during the Ming Dynasty.15 Various dammarane-type gypenosides (Gyp; >170 types), which have been isolated from GpM since 1976, have attracted interest because of its potential in treating wheeze, cough, hepatitis, bronchitis, and cancer.16–18 The major component of the GpM Makino extract is Gyp, which is a popular folk medicine in China and is often used for treating several diseases, including hyperlipoproteinemia,19 cardiovascular diseases,20 and hepatitis.21 Moreover, the extract has antioxidative,22 anti-inflammatory,22 and anticancer properties.14,23,24 Recently, its anticancer activities against different cancer cell lines, including esophageal cancer Eca-109 cells, human colon cancer SW620,25 oral cancer SAS cells,26 and cervical epidermoid carcinoma cells, have been reported.27 However, further investigation is needed for the satisfactory definition of antitumor mechanisms of GpM28 and its derivative (Gyp). Li et al reviewed the anticancer studies on the entire plant of GpM (Thunb);14 however, they did not explain the anticancer effect of the compound they extracted from GpM. Gyp derived from GpM is an active compound for cancer treatment. Therefore, in this review, we summarize available studies on Gyp to provide a comprehensive reference for further research and clinical trials.
Targeting cancer with Gyp through oxidative stress
In biological systems, oxidative stress refers to the physiological disbalance in ROS level, such as H2O2 or O2, as well as the capability of the body to remove it. Furthermore, oxidative stress can be defined as the disturbance in redox signaling and control.29 ROS produced throughout the body are the by-products of cellular aerobic metabolism, ongoing stress, and exposure to UV light or X-ray.30 It plays an important role in cell signaling and in the regulation of cytokine, growth factor, hormone action, transcription, ion transport, neuromodulation, immunomodulation, and apoptosis.30,31 Furthermore, ROS has a fundamental role in different types of cellular processes, such as gene expression, cell survival, differentiation, proliferation, enzyme regulation, and elimination of foreign particles and pathogens.32,33 Multiple studies showed that oxidative stress in cancer cells is high, which increases cell proliferation, survival, metastasis, and angiogenesis and disrupts cell death signaling and drug resistance.34–36 ROS promotes tumor proliferation, although a recent research confirms that ROS is useful in cancer treatment. The phytochemical induces ROS generation in cancer cells above a threshold level, thereby killing these cells.34,36–38 Gyp induces ROS generation in various types of cancers. In SW-480, oesophageal cancer Eca-109, SW-620, Caco2, WEHI-3, SAS, human hepatoma HepG-2, and Huh-7 cells, Gyp can induce apoptosis and inhibit cell growth and proliferation through oxidative stress and by increasing ROS generation and mitochondrial membrane potential (MMP) depolarization. Furthermore, the ROS generation can be reversed through N-acetyl-Lc-cysteine pre-treatment.24,25,39–45 However, the exact molecular mechanism is unexplored in cancer cells and needs further clarification. Once the oxidative stress is generated, it activates several apoptotic pathways, including mitochondrial-dependent pathways (MDPs).
Targeting cancer with Gyp through intrinsic apoptosis pathway
Mitochondrial-dependent apoptosis is an important pathway for the induction of apoptosis, and disturbance in this pathway can inhibit apoptosis. The intrinsic pathway is controlled through B-cell lymphoma 2 (Bcl-2) family protein, which either increases or decreases the mitochondrial membrane permeability for the release of cytochrome-c (Cyt-c) and other apoptotic proteins.46 A group of antiapoptotic proteins, including Bcl-2, B-cell lymphoma-extra-large (BclxL), Bcl-w, Bcl-2 related protein A1, and myeloid cell leukemia 1, possess sequence similarity in its all Bcl2-homology 1–4 domains and increase cell survival. Proapoptotic proteins include multidomain Bcl-2-associated X (BAX), Bcl-2 homologous killer (BAK), and BH3-only protein. These proteins function as receptor mediators that induce endoplasmic reticulum (ER) or mitochondrial stress-dependent apoptosis.47 BH3-only proteins have two subclasses, one of which one is an “activator’’ and includes total BH3 interacting-domain death antagonist and Bcl-2-like protein 11. This subclass directly activates the BAX/BAK to cause MMP depolarization. The second subclass includes “sensitizers/derepressors,” such as Bcl-2 interacting killer, Bcl-2-associated death promoter, Bcl-2-modifying factor, phorbol-12-myristate-13-acetate-induced protein 1, harakiri, and p53 upregulated modulator of apoptosis. This subclass neutralizes antiapoptotic proteins instead of directly activating BAX/BAK.48,49 Meanwhile, antiapoptotic proteins block death signaling by directly inhibiting the activation of BAX/BAK or activator BH3-only proteins.50 Antiapoptotic proteins, including Bcl-2 and Bclxl, are involved in cancer progression51 and thereby induce the resistance of tumor cells to many types of apoptotic stimuli, including cytotoxic anticancer drugs.49 Gyps target MDPs through pro- and antiapoptotic protein modulation and promote apoptosis. In Colo 205, WEHI-3, HL-60 cells, SCC-4, SAS, and human hepatoma Huh-7 and A549 cells, Gyps induce morphological changes and apoptosis and inhibit cell proliferation by targeting MDPs.40,41,43–45,52–54 Moreover, Gyps inhibit Bcl-xl and Bcl-2 and upregulate BAX, thus promoting the release of Cyt-c and Endo G from the mitochondria. Upon Cyt-c release, caspase-3,9 is activated, and poly (ADP-ribose) polymerases (PARP) are subsequently upregulated. The PARPs then enter the nucleus and cause DNA damage, alter cell morphology, inhibit proliferation, and induce apoptotic death.40,41,43–45,52–54
Targeting cancer with Gyp through extrinsic apoptosis pathway
The extrinsic pathway is activated through tumor necrosis factor (TNF) family proteins, including Fas or TNF receptor-1 [TNFR1]). These proteins engage the death domain (DD)-containing receptors and activate the death effector domain, which contains capases. Moreover, the death legends expressed on cytotoxic T cells, natural killer cells (NKs), and other types of relevant cells eradicate transforming cells.55 Fas or TNFR1 activates caspase-8 through the Fas-associated death domain protein and forms a death-inducing signaling complex that activates caspase-3 and promote cell death.56,57 Gyp modulates the extrinsic apoptosis pathway. In SAS cells, Gyp activates the extrinsic pathway and then caspase-8 through the activation of Fas/FasL. It also activates caspase-3 and PARP and damages the DNA and induces apoptosis.44,53
Targeting cancer with Gyp through ER stress
The ERis involved in sensing, synthesis, and signaling in eukaryotic cells. The ER must tightly regulate oxidizing and Ca2+-rich folding environments to perform these functions. Protein folding and Ca2+ buffering in the ER are regulated by several chaperones, including calreticulin, calnexin, protein disulfide isomerases, and glucose-regulated protein GRP78 (BiP). Several pathophysiological conditions, such as hypoxia, ER-Ca2+ depletion, hypoglycemia, viral infections, and oxidative injury affect the homeostasis of ER and disrupt protein folding load and capacity, thereby causing ER stress. The ER responds to these changes by activating an integrated signal transduction pathway, and this process is called unfolded protein response (UPR).58 The UPR regulates ER homeostasis by coordinating the complex processes of gene transcription, activates ER folding machinery components, and controls ER quality and ER-associated degradation (ERAD) pathway. However, as ER stress intensifies, the UPR consequently changes from prosurvival to prodeath response and usually ends in the activation of intrinsic apoptosis.59 In mammals, protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor-6 (ATF6), and inositol-requiring enzyme 1 are ER stress transducer proteins that activate survival and apoptotic pathways. When the UPR exhibits prosurvival response, it activates ER chaperones, translational attenuation, and ERAD; conversely, it activates C/EBP homologous protein 10 (CHOP)/GADD153 and caspase-12 when the response is proapoptosis.60 In cancer, ER stress apoptotic proteins are usually downregulated, and Gyp increases ER stress and consequently induces cell apoptosis. In HL-60, WEHI-3 cells Gyp ROS mediate ER stress by increasing GADD153, GRP78, PERK, and ATF6-α ATF4-α levels, thereby activating caspase-12, which in turn activates caspase-3,7 and PARP; these processes instigate DNA fragmentation and compel the cells toward apoptosis.39,40,41,52,54
Targeting cancer with Gyp through the cell cycle pathway
Cell growth is regulated by a major process called the cell cycle and at different checkpoints by different cyclin interactions among specific cyclin-dependent kinases (CDKs) that form active complexes. The process at each checkpoint completes before the progression to the next phase of the cell cycle.61 Moreover, different CDK inhibitors negatively regulate CDKs. Among CDKs, p21 regulates cell cycle at different checkpoints.62,63 The failure of the checkpoints induces mutation and genomic rearrangements, causing genetic disturbance, and ultimately cancer.63 Meanwhile, p53 has a key component role in cell cycle regulation. It becomes active to a wide range of damage and stresses.64,65 When activated by genotoxic stress, p53 regulates the p21WAF1/CIP1/SDI1 gene encoding CDK universal inhibitors that inhibit cell cycle progression.66 Many studies suggest that anticancer compounds arrest the cell cycle selective checkpoints and cause death to cancer cells through apoptosis.67 Gyp induces cell cycle arrest in different types of cancer cells. Gyp causes DNA damage in SCC-4, SW-620, Caco2, SW-480, and rat hepatic stellate cells. DNA damage activates checkpoint protein 2 (Chk2), which in turn activates p53 and p21, inhibits Cdk2 and cyclin E, and promotes G1S and G0G1 phase cell cycle arrest. Furthermore, Gyp activates p27, p21, and p16, which inhibit Cdk2, cyclin E, and cyclin D1/3K, CDK4/6 and promote G1S and G0G1 phase cell cycle arrest.39–42,52–54,68
Targeting cancer with Gyp through DNA repairing pathway
Cell homeostasis is maintained through the preservation of its genomic integrity. DNA damage response (DDR) reverses intrinsic and extrinsic DNA damage and transmits the genome to new dividing cells, which are required for cell survival during replication. Genotoxic drugs and radiations are used for treating cancer, but the DNA repair mechanism contributes to resistance to chemotherapy and radiotherapy. Resistance can be prevented, and the efficacy of cancer therapies can be increased by using inhibitors against DDR major components, including ataxia telangiectasia mutated (ATM), ATM and Rad-related (ATR), DNA-dependent protein kinase, catalytic subunit (DNA-PK), and checkpoint protein 1 and 2 to confer chemo- and radiosensitivity in cancer cells.69 Gyp has an important role in DNA-repairing gene regulation for overcoming cancer. Specifically, Gyp decreases cell viability and induces death in SAS cells and human oral cancer, and these processes are correlated with inreased DNA migration and decreased expression levels of 14-3-3σ, DNA-PK, ATM, ataxia-telangiectasia, p53, ATR, and breast cancer gene 1 at mRNA. Furthermore, Gyp induces DNA damage in SAS cells and inhibits the expression of DNA-repairing genes.70
Targeting cancer with Gyp through the PI3K/AKT/mTOR pathway
Phosphatidylinositol-3-kinase, protein kinase B, and the mammalian target of the rapamycin signaling pathway increase cell survival and growth through different mechanisms.71,72 In different types of human cancers, the PI3K/AKT pathway is overexpressed through different mechanisms.73–76 The phosphorylation of two AKT residues, including serine 473 and threonine 308, leads to AKT activation.77 Subsequently, AKT enters the nucleus and modulates the activities of several factors regulating transcription. The mammalian target of rapamycin (mTOR) becomes phosphorylated because of the PI3K/AKT signaling, and its overexpression is associated with poor recovery. NPs have attracted considerable interest because they potentially kill cancer cells through different mechanisms. For example, Gyp inhibits the proliferation of SAS, SCC-4, and PDGF-induced rat hepatic stellate cells through the PI3K/AKT pathway and by downregulating PI3K, AKT, and P70SK phosphorylation.26,68,78 Furthermore, in SAS cells and SCC-4 cells, Gyp targets the PI3K pathway through downregulation of son of sevenless (SOS), RAS, urokinase-type plasminogen activator (uPA), and focal adhesion kinase (FAK), which further inhibit PI3K and Rho-A. As a result, they inhibit MMP-2,7,9 and ultimately inhibit cell invasion, migration, and metastasis, as shown in Figure 2 and Table 1.26,78
 | Table 1 Effect of Gyp on different cancer cell lines, proteins or genes involved and its mechanism |
Targeting cancer with Gyp through nuclear factor-kB (NF-kB) pathway
The NF-kB is a transcription factor complex consisting of hetero- and homodimers of five members of a Rel family, such as RelA (p65), RelB, c-Rel, NFkB1 (p50/p105), and NF-kB2 (p52/p100.79 The functions of NF-kB are mostly deregulated in cancer.80 NF-kB activation, which has been found in a variety of cancers, including leukemia, lymphoma, colon, breast, liver, prostate, pancreas, and ovarian cancers, is associated with aggressiveness, tumorigenesis, poor survival, and chemoresistance.81–83 NF-kB activation occurs in response to DNA damage, which consequently activates various NF-kB target genes, including COX-284 and iNOS.85 These genes have a pivotal role in prosurvival antiapoptosis. Therefore, NF-kB is the candidate of therapeutic resistance in different cancers. Gyp targets the NF-kB pathway to cure cancer. In SAS cells and SCC-4 cells, Gyp targets NF-kB pathway through downregulation of SOS, RAS, uPA, and FAK. These genes further downregulate AKT, NF-kB, iNOS, and COX-2, which activate p53. As a result, MMP-2,7,9 is inhibited, which decreases cell invasion, migration, and metastasis as depicted in Figure 2 and Table 1.26,78
Targeting cancer with Gyp through mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) (Ras-Raf-MEK-ERK) pathway
MAPK/ERK pathway, also known as the Ras-Raf-MEK-ERK pathway, possesses several cascades and is mostly deregulated in human cancers.86 It regulates many cell functions, including apoptosis, differentiation, cell growth, proliferation, senescence, and migration.87 The MAPK/ERK pathway molecules are activated through its phosphorylation. When ERK is activated, it enters the nucleus where transcription factor phosphorylation occurs due to it. When these transcription factors phosphorylate, they bind to the promoter region of various genes including cytokines and growth factors. Such genes are responsible for the reduction in apoptosis and elevation in cell proliferation.88 When the normal signaling of this pathway is disturbed, they cause senescence, drug resistance, and tumorigenesis.87,89,90 In many human cancers, failure is detected in this pathway.91,92 Therefore, targeting the MAPK/ERK pathway, especially with NP including Gyp, may open a new window for cancer treatment. In SAS cells and SCC-4 cells, Gyp targets the ERK1/2 pathway through downregulation of SOS, RAS, uPA, and FAK). These genes further downregulate side ERK1/2, and directly downregulate the matrix metalloproteinase-2,7,9, thereby inhibiting cell invasion, migration, and metastasis as illustrated in Figure 2 and Table 1.26,78
Conclusions
The studies indicated that Gyp has therapeutic potential in the treatment of different cancers owing to its low toxicity owing and a long history of human use. Furthermore, it induces apoptosis through different molecular pathways and can thus be used in combination with other drugs to overcome resistance to available targeted drugs. More preclinical and clinical studies are needed to design and conduct a definite dose of Gyp for various types of cancer and for specific pathways or genes. Available anticancer information about Gyp is summarized in Table 1 and Figures 1 and 2.
 | Figure 1 Gyp induces apoptosis, causes cell cycle arrest, and inhibits cell proliferation and DNA repair. (A). Gyp increases Ca+ and ROS generation. ROS inhibits MMP and modulates the mitochondrial proteins directly or through the activation of ERK1/2. Consequently, the rate of cytochrome C and AIF translocation from the mitochondria to the cytoplasm increases, and activated caspase-9, in turn, activates caspase-3,7. However, ROS generation induces endoplasmic reticulum stress by activating oxidation-inducing proteins GADD153, PERK, ATF6, and IRE-α. Consequently, they activate caspase-12, which further activates caspase-3,7. Similarly, caspase-3,7 is activated by activating Fas, Fasl, and caspase-8. Activated caspase-3,7 causes DNA damage in cancer cells, leads to cell apoptosis and (B) activates chk-2, p53, and p21, which inhibit CDK2 and cyclin E. However, Gyp inhibits CDK2 and cyclin E by activating p16, p21, and p27. Then, the cells undergo S and G0G1 phase cell cycle arrest. Furthermore, the activated p16, p21, and p27 inhibit the cyclin D1/3k and CDK4/6, leading to S phase cell cycle arrest. (C) Gyp inhibits cancer cell proliferation through two mechanisms. First, when the cell cycle arrest occurs, cell proliferation is inhibited. Second, Gyp inhibits PI3K, AKT, and p70S6K and cell proliferation. (D). Gyp inhibits DNA-repairing genes including MGMT, DNAPK, ATM/ATR, p53, BRCA1, and 14–3–3σ at mRNA level and inhibits DNA repair. Abbreviations: Gyp, gypenosides; ROS, reactive oxygen species; MMP, mitochondrial membrane potential; ATF6, activating transcription factor-6; BRCA1, breast cancer gene 1; IRE, inositol-requiring enzyme; ATM, ATM, ataxia telangiectasia mutated; ATR, ATR, ATM and Rad-related. |
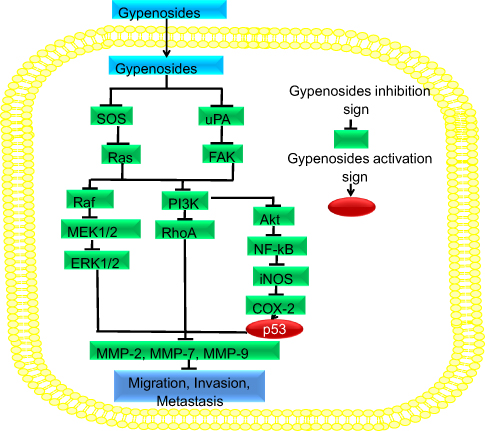 | Figure 2 Gyp inhibits migration, invasion, and metastasis. Gyp downregulates SOS, RAS, uPA, and FAK, which in turn downregulates Raf, MEK1/2, and ERK1/2. These proteins directly downregulate MMP-2,7,9. On the other side, they inhibit PI3K, which downregulates Rho-A, as well as Akt, NF-kB, COX-2, which further inhibit MMP-2,7,9 and invasion, migration, and metastasis. Abbreviations: Gyp, gypenosides; MMP, mitochondrial membrane potential. |
Abbreviation list
GpM, Gynostemma pentaphyllum; Gyp, gypenosides MMP, mitochondrial membrane potential; Bcl-2, B-cell lymphoma 2; Cyt-c, cytochrome-c; BclxL, B-cell lymphoma-extra-large; Bfl-1/A1, Bcl-2-related protein A1; Mcl-1, myeloid cell leukemia 1; BH1-Bh4, Bcl2-homology 1–4; BAX, Bcl-2-associated X; BAK, Bcl-2 homologous killer; tBid, total BH3 interacting domain death antagonist; Bim, Bcl-2-like protein 11; Bik, Bcl-2 interacting killer; Bad, Bcl-2-associated death promoter; BmF, Bcl-2-modifying factor; Noxa, phorbol-12-myristate-13-acetate-induced protein 1; Hrk, harakiri; Puma, p53 upregulated modulator of apoptosis; MDP, mitochondrial-dependent pathway; PARP, poly (ADP-Ribose) polymerases; DNA, deoxyribonucleic acid; TNF, tumor necrosis factor; TNFR1, TNF receptor-1; ER, endoplasmic reticulum; ; UPR, unfolded protein response; ERAD, ER-associated degradation; PERK, protein kinase RNA-like endoplasmic reticulum kinase; ATF6, activating transcription factor-6; IRE1, inositol-requiring enzyme 1; CHOP, C/EBP homologous protein 10; CDKs, cyclin-dependent kinases; DDR, DNA damage response; ATM, ataxia telangiectasia mutated; ATR, ATM and Rad-related; DNA-PK, DNA-dependent protein kinase, catalytic subunit; Chk1, Chk2, checkpoint protein 1 and 2; BRCA1, breast cancer gene one; mTOR, mammalian target of rapamycin; NF-kB, nuclear factor-kB; MAPK/ERK, mitogen-activated protein kinase/extracellular signal regulated kinases; uPA, urokinase type plasminogen activator; FAK, focal adhesion kinase.
Acknowledgment
The work presented here was funded by the National Science Foundation of China (Grant no. 31870917) and the Scientific and Technological Research Project of Henan Province (Grant no. 182102110084).
Disclosure
The authors report no conflicts of interest in this work.
References
1.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21294
3. Bray F. Transitions in human development and the global cancer burden. World Cancer Rep. 2014;54–68.
4. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
5. Qi F, Li A, Inagaki Y, et al. Chinese herbal medicines as adjuvant treatment during chemo- or radio-therapy for cancer. Biosci Trends. 2010;4(6):297–307.
6. Pereira DM, Valentao P, Correia-da-Silva G, Teixeira N, Andrade PB. Plant secondary metabolites in cancer chemotherapy: where are we? Curr Pharm Biotechnol. 2012;13(5):632–650. doi:10.2174/138920112799857530
7. Coseri S. Natural products and their analogues as efficient anticancer drugs. Mini Rev Med Chem. 2009;9(5):560–571. doi:10.2174/138955709788167592
8. Tascilar M, de Jong FA, Verweij J, Mathijssen RH. Complementary and alternative medicine during cancer treatment: beyond innocence. Oncologist. 2006;11(7):732–741. doi:10.1634/theoncologist.11-7-732
9. Wang CZ, Calway T, Yuan CS. Herbal medicines as adjuvants for cancer therapeutics. Am J Chin Med. 2012;40(4):657–669. doi:10.1142/S0192415X12500498
10.
11. Yin SY, Wei WC, Jian FY, Yang NS. Therapeutic applications of herbal medicines for cancer patients. Evid Based Complement Alternat Med. 2013;2013:302426. doi:10.1155/2013/302426
12. Kitano H. Systems biology: a brief overview. Science. 2002;295(5560):1662–1664. doi:10.1126/science.1069492
13. Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81(4):303–311. doi:10.1111/j.1399-0004.2011.01809.x
14. Li Y, Lin W, Huang J, Xie Y, Ma W. Anti-cancer effects of Gynostemma pentaphyllum (Thunb.) Makino (Jiaogulan). Chin Med. 2016;11:43. doi:10.1186/s13020-016-0114-9
15. Yang F, Shi H, Zhang X, Yu LL. Two novel anti-inflammatory 21-nordammarane saponins from tetraploid Jiaogulan (Gynostemma pentaphyllum). J Agric Food Chem. 2013;61(51):12646–12652. doi:10.1021/jf404726z
16. Wu PK, Liu X, Hsiao WW-l. The assessment of anti-cancer activities and saponin profiles of Gynostemma pentaphyllum saponins obtained from different regions of China. J Biotechnol. 2008;136:S85. doi:10.1016/j.jbiotec.2008.07.192
17. Qian H, Fu S, Jiang G, Wang L, Fu X, Ye M. Protective effect of jiaogulan on cellular immunity of the patients with primary lung cancer treated by radiotherapy plus chemotherapy. Lung Cancer. 1996;1(14):156.
18. Tai WC-S, Wong WY, Wang Y, Hsiao W-LW. The anti-cancer and anti-hyperlipidemia effects of triterpenoids from Gynostemma pentaphyllum in the Apc min/+ mouse model. AACR. 2010.
19. Yang YH, Yang J, Jiang QH. Hypolipidemic effect of gypenosides in experimentally induced hypercholesterolemic rats. Lipids Health Dis. 2013;12:154. doi:10.1186/1476-511X-12-154
20. Yu H, Guan Q, Guo L, et al. Gypenosides alleviate myocardial ischemia-reperfusion injury via attenuation of oxidative stress and preservation of mitochondrial function in rat heart. Cell Stress Chaperones. 2016;21(3):429–437. doi:10.1007/s12192-016-0705-5
21. Chen MH, Wang QF, Chen LG, et al. The inhibitory effect of Gynostemma pentaphyllum on MCP-1 and type I procollagen expression in rat hepatic stellate cells. J Ethnopharmacol. 2009;126(1):42–49. doi:10.1016/j.jep.2009.08.012
22. Zhao J, Ming Y, Wan Q, et al. Gypenoside attenuates hepatic ischemia/reperfusion injury in mice via anti-oxidative and anti-apoptotic bioactivities. Exp Ther Med. 2014;7(5):1388–1392. doi:10.3892/etm.2014.1569
23. Tai WC, Wong WY, Lee MM, Chan BD, Lu C, Hsiao WL. Mechanistic study of the anti-cancer effect of Gynostemma pentaphyllum saponins in the Apc(Min/+) mouse model. Proteomics. 2016;16(10):1557–1569. doi:10.1002/pmic.201500293
24. Yan H, Wang X, Niu J, Wang Y, Wang P, Liu Q. Anti-cancer effect and the underlying mechanisms of gypenosides on human colorectal cancer SW-480 cells. PLoS One. 2014;9(4):e95609. doi:10.1371/journal.pone.0095609
25. Yan H, Wang X, Wang Y, Wang P, Xiao Y. Antiproliferation and anti-migration induced by gypenosides in human colon cancer SW620 and esophageal cancer Eca-109 cells. Hum Exp Toxicol. 2014;33(5):522–533. doi:10.1177/0960327113493302
26. Lu KW, Chen JC, Lai TY, et al. Gypenosides inhibits migration and invasion of human oral cancer SAS cells through the inhibition of matrix metalloproteinase-2–9 and urokinase-plasminogen by ERK1/2 and NF-kappa B signaling pathways. Hum Exp Toxicol. 2011;30(5):406–415. doi:10.1177/0960327110396521
27. Chiu TH, Chen JC, Chung JG. N-acetyltransferase is involved in gypenosides-induced N-acetylation of 2-aminofluorene and DNA adduct formation in human cervix epidermoid carcinoma cells (Ca Ski). In Vivo. 2003;17(3):281–288.
28. Zhao Y, Niu Y, Xie Z, Shi H, Chen P, Yu LL. Differentiating leaf and whole-plant samples of di-and tetraploid Gynostemma pentaphyllum (Thunb.) Makino using flow-injection mass spectrometric fingerprinting method. J Funct Foods. 2013;5(3):1288–1297. doi:10.1016/j.jff.2013.04.013
29. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1865–1879. doi:10.1089/ars.2006.8.1865
30. Mittler R, Vanderauwera S, Suzuki N, et al. ROS signaling: the new wave? Trends Plant Sci. 2011;16(6):300–309. doi:10.1016/j.tplants.2011.03.007
31. Gloire G, Legrand-Poels S, Piette JN. F-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72(11):1493–1505. doi:10.1016/j.bcp.2006.04.012
32. Gorlach A, Dimova EY, Petry A, et al. Reactive oxygen species, nutrition, hypoxia and diseases: problems solved? Redox Biol. 2015;6:372–385. doi:10.1016/j.redox.2015.08.016
33. Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. doi:10.1016/j.redox.2015.08.010
34. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi:10.1038/nrd2998
35. Hong YH, Uddin MH, Jo U, et al. ROS accumulation by PEITC selectively kills ovarian cancer cells via UPR-mediated apoptosis. Front Oncol. 2015;5:167. doi:10.3389/fonc.2015.00167
36. Zhu L, Ren L, Chen Y, Fang J, Ge Z, Li X. Redox status of high-mobility group box 1 performs a dual role in angiogenesis of colorectal carcinoma. J Cell Mol Med. 2015;19(9):2128–2135. doi:10.1111/jcmm.12658
37. Wei C, Xiao Q, Kuang X, Zhang T, Yang Z, Wang L. Fucoidan inhibits proliferation of the SKM-1 acute myeloid leukaemia cell line via the activation of apoptotic pathways and production of reactive oxygen species. Mol Med Rep. 2015;12(5):6649–6655. doi:10.3892/mmr.2015.4252
38. Seo KH, Ryu HW, Park MJ, et al. A furanoxanthone from garciana mangostana, induces reactive oxygen species-mediated apoptosis in lung cancer cells and decreases xenograft tumor growth. Phytother Res. 2015;29(11):1753–1760. doi:10.1002/ptr.5428
39. Kong L, Wang X, Zhang K, et al. Gypenosides synergistically enhances the anti-tumor effect of 5-fluorouracil on colorectal cancer in vitro and in vivo: a role for oxidative stress-mediated DNA damage and p53 activation. PLoS One. 2015;10(9):e0137888. doi:10.1371/journal.pone.0137888
40. Hsu HY, Yang JS, Lu KW, et al. An experimental study on the antileukemia effects of gypenosides in vitro and in vivo. Integr Cancer Ther. 2011;10(1):101–112. doi:10.1177/1534735410377198
41. Lin JJ, Hsu HY, Yang JS, et al. Molecular evidence of anti-leukemia activity of gypenosides on human myeloid leukemia HL-60 cells in vitro and in vivo using a HL-60 cells murine xenograft model. Phytomedicine. 2011;18(12):1075–1085. doi:10.1016/j.phymed.2011.03.009
42. Liu J, Peng WX, Mo YY, Luo D. MALAT1-mediated tumorigenesis. Front Biosci (Landmark Ed). 2017;22:66–80. doi:10.2741/4472
43. Chen JC, Lu KW, Tsai ML, et al. Gypenosides induced G0/G1 arrest via CHk2 and apoptosis through endoplasmic reticulum stress and mitochondria-dependent pathways in human tongue cancer SCC-4 cells. Oral Oncol. 2009;45(3):273–283. doi:10.1016/j.oraloncology.2008.05.012
44. Lu KW, Chen JC, Lai TY, et al. Gypenosides suppress growth of human oral cancer SAS cells in vitro and in a murine xenograft model: the role of apoptosis mediated by caspase-dependent and caspase-independent pathways. Integr Cancer Ther. 2012;11(2):129–140. doi:10.1177/1534735411403306
45. Wang QF, Chiang CW, Wu CC, et al. Gypenosides induce apoptosis in human hepatoma Huh-7 cells through a calcium/reactive oxygen species-dependent mitochondrial pathway. Planta Med. 2007;73(6):535–544. doi:10.1055/s-2007-967200
46. Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013;19(6):546–558. doi:10.1089/ars.2012.4905
47. Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006;13(8):1378–1386. doi:10.1038/sj.cdd.4401975
48. Letai A. BCL-2: found bound and drugged! Trends Mol Med. 2005;11(10):442–444. doi:10.1016/j.molmed.2005.08.007
49. Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–1358. doi:10.1038/ncb1499
50. Green DR. At the gates of death. Cancer Cell. 2006;9(5):328–330. doi:10.1016/j.ccr.2006.05.004
51. Zhu MY, Chen F, Niyazi M, Sui S, Gao DM. Variation in apoptotic gene expression in cervical cancer through oligonucleotide microarray profiling. J Low Genit Tract Dis. 2015;19(1):46–54. doi:10.1097/LGT.0000000000000030
52. Chen JC, Lu KW, Lee JH, Yeh CC, Chung JG. Gypenosides induced apoptosis in human colon cancer cells through the mitochondria-dependent pathways and activation of caspase-3. Anticancer Res. 2006;26(6b):4313–4326.
53. Liu JS, Chiang TH, Wang JS, et al. Induction of p53-independent growth inhibition in lung carcinoma cell A549 by gypenosides. J Cell Mol Med. 2015;19(7):1697–1709. doi:10.1111/jcmm.12658
54. Lu HF, Chen YS, Yang JS, et al. Gypenosides induced G0/G1 arrest via inhibition of cyclin E and induction of apoptosis via activation of caspases-3 and −9 in human lung cancer A-549 cells. In Vivo. 2008;22(2):215–221.
55. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. doi:10.1016/S0092-8674(01)00237-9
56. Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19(3–4):325–331. doi:10.1016/j.cytogfr.2008.04.001
57. Fulda S. Targeting extrinsic apoptosis in cancer: challenges and opportunities. Semin Cell Dev Biol. 2015;39:20–25. doi:10.1016/j.semcdb.2015.01.006
58. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi:10.1146/annurev.biochem.73.011303.074134
59. Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65(6):862–894. doi:10.1007/s00018-008-7565-9
60. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7(4):335–345. doi:10.1023/A:1016175429877
61. Khan M, Rasul A, Yi F, Zhong L, Ma T. Jaceosidin induces p53-dependent G2/M phase arrest in U87 glioblastoma cells. Asian Pac J Cancer Prev. 2011;12(12):3235–3238.
62. Lu MC, Yang SH, Hwang SL, et al. Induction of G2/M phase arrest by squamocin in chronic myeloid leukemia (K562) cells. Life Sci. 2006;78(20):2378–2383. doi:10.1016/j.lfs.2005.09.048
63. Yang G, Chang B, Yang F, et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res. 2010;16(12):3171–3181. doi:10.1158/1078-0432.CCR-10-0613
64. Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26(9):1306–1316. doi:10.1038/sj.onc.1210263
65. Bourougaa K, Naski N, Boularan C, et al. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell. 2010;38(1):78–88. doi:10.1016/j.molcel.2010.01.041
66. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi:10.1038/35042675
67. Khan M, Zheng B, Yi F, et al. Pseudolaric Acid B induces caspase-dependent and caspase-independent apoptosis in u87 glioblastoma cells. Evid Based Complement Alternat Med. 2012;2012:957568. doi:10.1155/2012/957568
68. Chen MH, Chen SH, Wang QF, et al. The molecular mechanism of gypenosides-induced G1 growth arrest of rat hepatic stellate cells. J Ethnopharmacol. 2008;117(2):309–317. doi:10.1016/j.jep.2008.02.009
69. Furgason JM, Bahassi EM. Targeting DNA repair mechanisms in cancer. Pharmacol Ther. 2013;137(3):298–308. doi:10.1016/j.pharmthera.2012.10.009
70. Lu KW, Chen JC, Lai TY, et al. Gypenosides causes DNA damage and inhibits expression of DNA repair genes of human oral cancer SAS cells. In Vivo. 2010;24(3):287–291.
71. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–1083. doi:10.1200/JCO.2009.25.3641
72. Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany, NY). 2011;3(3):192–222. doi:10.18632/aging.100296
73. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87–90. doi:10.1016/j.gde.2009.11.002
74. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi:10.1126/science.1096502
75. Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3(10):1221–1224. doi:10.4161/cc.3.10.1164
76. Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103(5):1289–1294. doi:10.1073/pnas.0510772103
77. Vincent EE, Elder DJ, Thomas EC, et al. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br J Cancer. 2011;104(11):1755–1761. doi:10.1038/bjc.2011.132
78. Lu KW, Tsai ML, Chen JC, et al. Gypenosides inhibited invasion and migration of human tongue cancer SCC4 cells through down-regulation of NFkappaB and matrix metalloproteinase-9. Anticancer Res. 2008;28(2a):1093–1099.
79. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. doi:10.1016/0092-8674(86)90346-6
80. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. doi:10.1038/nrm2083
81. Arkan MC, Greten FR. IKK- and NF-kappaB-mediated functions in carcinogenesis. Curr Top Microbiol Immunol. 2011;349:159–169.
82. Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830. doi:10.1038/sj.onc.1209942
83. Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336(1–2):25–37. doi:10.1007/s11010-009-0267-2
84. Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. 1995;270(52):31315–31320. doi:10.1074/jbc.270.52.31315
85. Park JH, Jeong YJ, Won HK, Choi SY, Park JH, Oh SM. Activation of TOPK by lipopolysaccharide promotes induction of inducible nitric oxide synthase through NF-kappaB activity in leukemia cells. Cell Signal. 2014;26(5):849–856. doi:10.1016/j.cellsig.2014.01.004
86. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103–119. doi:10.1517/14728222.2011.645805
87. Chang F, Steelman LS, Lee JT, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–1293. doi:10.1038/sj.leu.2402637
88. McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22(4):708–722. doi:10.1038/sj.leu.2404889
89. Martelli AM, Evangelisti C, Chiarini F, et al. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim Biophys Acta. 2010;1803(9):991–1002. doi:10.1016/j.bbamcr.2010.04.005
90. Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1(2):89–103.
91. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–942. doi:10.1038/nrd4281
92. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.