Back to Journals » Nature and Science of Sleep » Volume 13
Narcolepsy Type 1: A Remitting Disease? An Unusual Case Report
Authors Vellieux G, Frija-Masson J, Rouvel-Tallec A, Drouot X, d'Ortho MP
Received 16 March 2021
Accepted for publication 16 June 2021
Published 24 September 2021 Volume 2021:13 Pages 1669—1673
DOI https://doi.org/10.2147/NSS.S311283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sarah L Appleton
Geoffroy Vellieux,1,2 Justine Frija-Masson,1,2 Anny Rouvel-Tallec,1,2 Xavier Drouot,3 Marie-Pia d’Ortho1,2
1Université de Paris, NeuroDiderot, Inserm, Paris, F-75019, France; 2Centre du sommeil, Service de Physiologie – Explorations Fonctionnelles, AP-HP, Hôpital Bichat-Claude Bernard, Paris, F-75018, France; 3Neurophysiologie Clinique et Explorations Fonctionnelles, CHU de Poitiers, Poitiers, France
Correspondence: Geoffroy Vellieux
Université de Paris, NeuroDiderot, Inserm, Paris, F-75019, France
Tel +33 140258401
Fax +33 140257181
Email [email protected]
Abstract: We describe the case of a male patient who was diagnosed with narcolepsy type 1 on the basis of sleep and wake symptoms, and the results of investigations including video-polysomnography, multiple sleep latency test, human leukocyte antigen status and orexin level in cerebrospinal fluid. During the first years after disease onset, the patient did not show any significant improvement despite treatment with a variety of stimulant and anti-cataplectic drugs. However, spontaneous remission of disease occurred after 15 years.
Keywords: narcolepsy, cataplexy, excessive daytime sleepiness, orexin, hypocretin, remission
Introduction
Narcolepsy type 1 (NT1) is considered to be a rare, lifelong neurological disorder related to selective loss of lateral hypothalamic orexin neurons due to a possible autoimmune process.1 NT1 is associated with a variety of sleep and wake symptoms. The main clinical features are severe, and include irresistible excessive daytime sleepiness (EDS), cataplexy, sleep hallucinations, sleep paralysis and fragmented nocturnal sleep.2 Pharmacological treatment of patients with NT1 relies mainly on stimulant and anti-cataplectic drugs.3 However, available treatment options are not disease modifying and NT1 is usually considered as a non-remitting chronic disease with a variable course over the years. Patients may experience an improvement in symptoms with advancing age, although a complete remission actually is a really uncommon phenomenon.4–6
We describe the unusual case of a man with NT1 with a disease course that evolved towards the remission of narcolepsy symptoms at 15 years after disease onset, allowing termination of all treatments.
Case Report
A 35-year-old man began to experience excessive daytime sleepiness (EDS) with irresistible sleep attacks and involuntary napping, usually with two short (5–10 minutes) and refreshing naps in the afternoon. He was working as an operator of construction machines and heavy goods vehicles. His medical history included right-shoulder surgery for multiple dislocations. He was an active smoker, reported occasional alcoholic consumption, and denied any drug use. His body mass index was 30 kg/m2. Simultaneous with the development of EDS, the patient displayed daily cataplexy triggered by laughter up to several times a day. No triggering events (eg infection or vaccination) preceding the onset of symptoms were identified. He did not mention the occurrence of sleep paralysis, sleep hallucinations or parasomnias. There was no personal history of head trauma or family history of narcolepsy. Furthermore, clinical data did not support a diagnosis of circadian rhythm sleep-wake disorder, sleep deprivation or mood disorder. On the first presentation at a sleep center, a few months after the symptoms onset, the Epworth Sleepiness Scale (ESS) score was 19. Baseline sleep tests were recorded without any ongoing medications. The overnight video-polysomnography (v-PSG) revealed the following: total sleep time (TST) of 476 minutes; sleep onset latency of 4 minutes; rapid eye movement (REM) sleep latency of 96 minutes; wake time after sleep onset (WASO) of 70 minutes; arousal and awakening index of 20/hour; mild obstructive sleep apnea syndrome (OSAS) with an apnea–hypopnea index (AHI) of 8/hour; and a periodic limb movement (PLM) during sleep index of 24/hour (including 5/hour that were associated with micro-arousals). Multiple sleep latency test (MSLT) showed a mean sleep latency (mSL) of 8 minutes: sleep latency, respectively, of 7.5, 5.5, 14, 3.5 and 8.5 minutes for each test, with two sleep-onset on REM sleep periods (SOREMP) during the fourth (REM sleep latency of 10.5 minutes) and fifth (REM sleep latency of 1 minute) naps. A brain MRI returned to normal. Urine toxicology screening was not performed. He tested positive for the narcolepsy associated human leukocyte antigen (HLA) DQB1*06:02 allele, and the orexin level detected with radioimmunological assay in cerebrospinal fluid (CSF) was decreased (<100 pg/mL). NT1 was therefore diagnosed according to the criteria of the International Classification of Sleep Disorders – 3rd edition.7
In 2005, treatment with modafinil and clomipramine was initiated. Maintenance of wakefulness test (MWT) performed during the therapy with modafinil 400 mg/day and clomipramine 50 mg/day showed that the treatment effectiveness was insufficient (mSL 10.8 minutes). Therefore, modafinil was substituted for methylphenidate. Moreover, since cataplexy persisted despite treatment with clomipramine, this was replaced by fluoxetine. Due to residual EDS, an add-on therapy with methylphenidate and modafinil was started. MWT during the treatment with methylphenidate 60 mg/day + modafinil 400 mg/day + fluoxetine 40 mg/day showed even worse results compared to the previous test, with a mSL of 3.4 minutes. Treatment with these two stimulant drugs was stopped, and fenethylline monotherapy was initiated. At the same time, fluoxetine at a dosage of up to 40 mg/day had no effect on cataplexy and was associated with diffuse myalgias; therefore, fluoxetine was discontinued and treatment with a higher dosage of clomipramine (100 mg/day) was started. This resulted in a decrease in the frequency of cataplexy. The fenethylline dosage was increased to 200 mg/day; then, MWT was repeated and a mSL of 24.4 minutes was observed, consistent with an improvement in patient’s alertness. A few months later, the patient reported EDS again and fragmented nocturnal sleep, therefore sodium oxybate was added to fenethylline. With fenethylline 200 mg/day + sodium oxybate 2.5 g/day + clomipramine 100 mg/day, the mSL on MWT was 1.3 minutes. The sodium oxybate dosage was then increased by up to 5 g/day and, due to cessation of commercialization in France, the patient stopped taking fenethylline. At a higher dosage of sodium oxybate, still associated with clomipramine 100 mg/day, the mSL on MWT was 13.5 minutes. The patient was then lost to follow-up for 6 years.
When the patient presented again to the sleep center, he was not taking medications regularly for several months despite persistent EDS with refreshing naps, some cataplexy, sleep paralysis and sleep hallucinations. Sleep investigations showed a mSL of 6.4 minutes on MSLT (sleep latencies and REM sleep latencies were not available) with four SOREMP and a mSL of 13.8 minutes on MWT. Treatment with modafinil was started again. At follow-up six and twelve months later, MWT during treatment with modafinil 100 mg/day showed a mSL of 34 and 31 minutes, respectively. Over this year, cataplexy, sleep paralysis and sleep hallucinations disappeared. The patient then interrupted his medical follow-up, this time for 3 years.
When the patient eventually returned to the sleep center, he admitted that he had stopped taking modafinil two years previously because all symptoms of narcolepsy, especially EDS and cataplexy, had disappeared. New sleep investigations were performed, while the patient was off all treatments. A 24-hour v-PSG was performed with the following results: TST of 423 minutes; sleep onset latency of 23 minutes; REM sleep latency of 6 minutes; WASO of 74 minutes; arousal and awakening index of 12.5/h; mild OSAS with AHI of 8/hour; PLM during sleep index of 15.7/h (including 9.4/h associated with micro-arousals); and moderate REM-sleep without atonia (RWSA) and no REM-sleep behavior disorders. MSLT found a normal mSL of 15 minutes: sleep latency, respectively, of 8, 15.5, 19, 12 and 20 minutes for each test, with two SOREMP (during the second and third naps, with, respectively, a REM sleep latency of 0 and 2.5 minutes), and MWT showed a mSL of 40 minutes. At a final visit six months later, the patient stated that he had no complaints relating to wakefulness and sleep quality, and did not report cataplexy, sleep paralysis nor sleep hallucinations. Again, without any treatment, MWT showed a mSL of 40 minutes. Blood and urine toxicology screening was negative. He refused a second lumbar puncture to determine orexin levels in CSF. The patient continued without any treatment given the absence of symptoms, including EDS, and resumed the job that he had stopped 15 years earlier, without any changes in his lifestyle and habits. Sleep history stages for this patient are summarized in Figure 1.
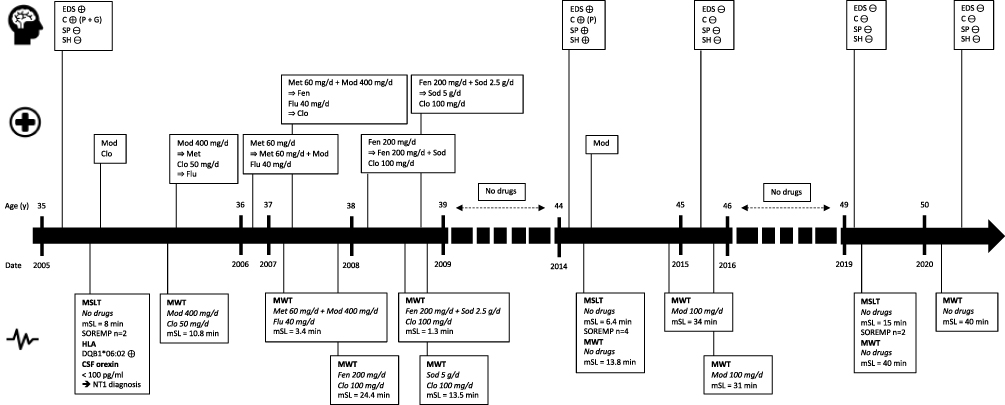 |
Figure 1 Main sleep history stages of reported patient. Abbreviations: C, cataplexy; Clo, clomipramine; CSF, cerebrospinal fluid; EDS, excessive daytime sleepiness; Fen, fenethylline; Flu, fluoxetine; G, generalized; Met, methylphenidate; Mod, modafinil; mSL, mean sleep latency; MSLT, multiple sleep latency test; MWT, maintenance of wakefulness test; NT1, narcolepsy type 1; P, partial; SH, sleep hallucinations; Sod, sodium oxybate; SOREMP, sleep-onset on rapid eye movement periods; SP, sleep paralysis; ⊕, present; ⊖, absent. |
Discussion
In our patient, the presence of EDS and multiple daily episodes of cataplexy contributed to the definitive diagnosis of NT1, along with specialized wake and sleep evaluation including v-PSG, MSLT, HLA status and CSF orexin assay. The diagnosis was made just a few months after disease onset, which is much faster than the usual delay between disease onset and the diagnosis of narcolepsy.8 However, this case has some atypical features. First, symptoms of NT1 started when the patient was 35 years of age. Although NT1 can certainly be present at this age, most patients have a disease onset during the adolescence with a peak occurring at 14.7 years.9 Initial MSLT revealed only mild EDS with a mSL of 8 minutes. This contrasts with significant subjective EDS, given that the ESS score at the same time was 19. Moreover, sleep paralysis and sleep hallucinations only appeared a few years after disease onset. However, results of repeated wake and sleep evaluations (including MSLT, performed in 2005, 2014 and 2019, which revealed each time at least two SOREMP) and decreased levels of orexin in the CSF supported the diagnosis of NT1. The association of NT1 with other sleep disorders such as OSAS, sleep parasomnias and PLM has been described in the literature.10–12 The presence of OSAS and PLM could be a confounding factor that might, at least in part, explain the EDS in our patient. However, OSAS and PLM indexes were always of mild severity when assessed using v-PSG in this case. The treatment of a moderate-severe OSAS and assessment of its effectiveness on EDS might be considered before initiating pharmacotherapy for narcolepsy.12 Furthermore, there was no history of head injury that may have caused post-traumatic narcolepsy or any other extra-neurologic symptoms indicative of secondary narcolepsy. At the end of the 15-year disease course in our patient, all subjective narcolepsy symptoms had disappeared and the mSL on MSLT was found to be in the normal range. However, some abnormal features characteristic of narcolepsy persisted, such as SOREMP, fragmented nocturnal sleep, PLM during sleep and RWSA. The progressive increase in the mSL and decrease in the number of SOREMP on MSLT as age increased suggest that the current criteria used to diagnose narcolepsy may be too stringent for new diagnoses in older patients. The important influence of age on MSLT results should therefore be taken into account when diagnosing a patient with narcolepsy.13 Repetitive assessment of patients with MSLT seems essential to evaluate the lifelong course of the disease.
Although the course and severity of narcolepsy vary considerably between subjects, NT1 is believed to have a stable non-remitting evolution. It is usually acknowledged that EDS with irresistible sleep episodes and nocturnal sleep disturbance persist or even worsen throughout the lifetime, whereas cataplexy, sleep hallucinations and sleep paralysis can improve or even disappear with advancing age.14–18 Almeneessier et al showed that anti-cataplectic medications were stopped without recurrence of cataplexy at 10 years of follow-up in 42% of their cohort of 38 patients with NT1.19 Regarding EDS, a recent retrospective study on 29 patients with NT1 reported no remission on clinical evaluation after a mean follow-up of 6.1 years, contrasting with patients who had narcolepsy without cataplexy or those with idiopathic hypersomnia.20 Other recent studies of patients with NT1 did not report any significant differences in the mSL and the number of SOREMP on MSLT performed at various follow-up times compared with baseline.21–23 However, a previous isolated report suggested that chronic persistence of symptoms is not necessarily universal. Büchele et al reported four cases of orexin-deficient patients with NT1 who showed spontaneous improvement, including sustained clinical remission that persisted after treatment withdrawal in three individuals.24 The CSF orexin level remained markedly decreased or became undetectable between the first evaluation and the follow-up visit in two of these patients, one of whom could stay awake on MWT in drug-free conditions.
Although the above four cases and our patient represent a very low proportion of the total NT1 population, they highlight important questions about the precise pathophysiology of this neurological degenerative condition due to the loss of a single neurotransmitter, which, though, produces effects on other neurotransmitters involved in wake promotion.25 The reversible disease course in these few cases may be in favor of the immunological hypothesis in NT1 pathophysiology, that is also supported by comorbid immunopathological diseases in narcoleptic patients and some reports of narcolepsy symptoms’ improvement after immunomodulatory therapy such as intravenous immunoglobulin therapy.26–28 As the destruction of orexin neurons seems irreversible, precise compensative mechanisms are currently unknown, and it may be that loss of orexin is not the only biological factor contributing to the symptoms of narcolepsy. Unfortunately, in the present case, the patient refused a second lumbar puncture for orexin assay in CSF. Despite the lack of follow-up data on orexin levels, our case adds important information about the course of narcolepsy. However, many questions remain unanswered, including the role of other neurotransmitters in promoting wakefulness, especially histamine, and the implications for cases with long-term improvement regarding emerging treatment options.29,30 Repetitive long-term follow-up with MSLT and MWT over decades seems essential to monitor the disease course and also to evaluate a possible recurrence of symptoms after a primary remission.
Conclusion
The current case showed an unusual disease course for a patient with NT1, displaying disease remission after 15 years, despite confirmation of the initial diagnosis based on orexin assay in CSF. Our data add to a few similar previously described cases and emphasize the need for data from prospective cohorts of patients with NT1 to better document the long-term evolution of this sleep-wake disorder that was previously assumed to be incurable.
Ethical Standard Statement
The patient’s written consent was obtained for publication of this case report. No institutional approval is required to publish the case details.
Acknowledgments
English language editing assistance was provided by Nicola Ryan, an independent medical writer.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Prof. Dr. Xavier Drouot reports grants from SOSoxygene and ISIS Atlantique, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kornum BR. Narcolepsy type 1: what have we learned from immunology? Sleep. 2020;43:zsaa055.
2. Kornum BR, Knudsen S, Ollila HM, et al. Narcolepsy. Nat Rev Dis Primers. 2017;3(1):16100. doi:10.1038/nrdp.2016.100
3. Barateau L, Lopez R, Dauvilliers Y. Management of narcolepsy. Curr Treat Options Neurol. 2016;18(10):43. doi:10.1007/s11940-016-0429-y
4. Bassetti CLA, Adamantidis A, Burdakov D, et al. Narcolepsy — clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nat Rev Neurol. 2019;15(9):519–539.
5. Barker EC, Flygare J, Paruthi S, et al. Living with narcolepsy: current management strategies, future prospects, and overlooked real-life concerns. Nat Sci Sleep. 2020;12:453–466. doi:10.2147/NSS.S162762
6. Furuta H, Thorpy MJ, Temple HM. Comparison in symptoms between aged and younger patients with narcolepsy. Psychiatry Clin Neurosci. 2001;55(3):241–242. doi:10.1046/j.1440-1819.2001.00841.x
7. American Academy of Sleep Medicine. International Classification of Sleep Disorders. Darien, IL: American Academy of Sleep Medicine; 2014.
8. Taddei RN, Werth E, Poryazova R, Baumann CR, Valko PO. Diagnostic delay in narcolepsy type 1: combining the patients’ and the doctors’ perspectives. J Sleep Res. 2016;25(6):709–715. doi:10.1111/jsr.12420
9. Dauvilliers Y, Montplaisir J, Molinari N, et al. Age at onset of narcolepsy in two large populations of patients in France and Quebec. Neurology. 2001;57(11):2029–2033. doi:10.1212/WNL.57.11.2029
10. Black J, Reaven NL, Funk SE, et al. Medical comorbidity in narcolepsy: findings from the Burden of Narcolepsy Disease (BOND) study. Sleep Med. 2017;33:13–18. doi:10.1016/j.sleep.2016.04.004
11. de Biase S, Gigli GL, Valente M. Important decisions in choosing the pharmacotherapy for narcoleptics. Expert Opin Pharmacother. 2019;20(5):483–486. doi:10.1080/14656566.2018.1561861
12. Sansa G, Iranzo A, Santamaria J. Obstructive sleep apnea in narcolepsy. Sleep Med. 2010;11(1):93–95. doi:10.1016/j.sleep.2009.02.009
13. Dauvilliers Y, Gosselin A, Paquet J, et al. Effect of age on MSLT results in patients with narcolepsy-cataplexy. Neurology. 2004;62(1):46–50. doi:10.1212/01.WNL.0000101725.34089.1E
14. Desforges JF, Aldrich,MS. Narcolepsy. N Engl J Med. 1990;323(6):389–394. doi:10.1056/NEJM199008093230606
15. Dauvilliers Y, Billiard M, Montplaisir J. Clinical aspects and pathophysiology of narcolepsy. Clin Neurophysiol. 2003;114(11):2000–2017. doi:10.1016/S1388-2457(03)00203-7
16. Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369(9560):499–511. doi:10.1016/S0140-6736(07)60237-2
17. Dauvilliers Y, Barateau L. Narcolepsy and other central hypersomnias. Continuum. 2017;23(4, Sleep Neurology):989–1004.
18. Parkes JD, Baraitser M, Marsden CD, Asselman P. Natural history, symptoms and treatment of the narcoleptic syndrome. Acta Neurol Scand. 1975;52(5):337–353. doi:10.1111/j.1600-0404.1975.tb05830.x
19. Almeneessier AS, Alballa NS, Alsalman B, Alessie S, Olaish AH, BaHammam AS. A 10-year Longitudinal Observational Study of cataplexy in a cohort of narcolepsy type 1 patients. Nat Sci Sleep. 2019;11:231–239. doi:10.2147/NSS.S229105
20. Kim T, Lee JH, Lee CS, Yoon IY. Different fates of excessive daytime sleepiness: survival analysis for remission. Acta Neurol Scand. 2016;134(1):35–41. doi:10.1111/ane.12504
21. Pizza F, Franceschini C, Peltola H, et al. Clinical and polysomnographic course of childhood narcolepsy with cataplexy. Brain. 2013;136(12):3787–3795. doi:10.1093/brain/awt277
22. Huang Y-S, Guilleminault C, Lin C-H, Chen C-H, Chin W-C, Chen T-S. Multiple sleep latency test in narcolepsy type 1 and narcolepsy type 2: a 5-year follow-up study. J Sleep Res. 2018;27(5):e12700. doi:10.1111/jsr.12700
23. Um YH, Kim T-W, Jeong J-H, et al. A Longitudinal Follow-Up Study on multiple sleep latency test and body mass index of patients with narcolepsy type 1 in Korea. J Clin Sleep Med. 2017;13(12):1441–1444. doi:10.5664/jcsm.6844
24. Büchele F, Baumann CR, Poryazova R, Werth E, Valko PO. Remitting narcolepsy? Longitudinal observations in a hypocretin-deficient cohort. Sleep. 2018;41:9. doi:10.1093/sleep/zsy118
25. Liblau RS, Vassalli A, Seifinejad A, Tafti M. Hypocretin (orexin) biology and the pathophysiology of narcolepsy with cataplexy. Lancet Neurol. 2015;14(3):318–328. doi:10.1016/S1474-4422(14)70218-2
26. Dauvilliers Y, Carlander B, Rivier F, Touchon J, Tafti M. Successful management of cataplexy with intravenous immunoglobulins at narcolepsy onset. Ann Neurol. 2004;56(6):905–908. doi:10.1002/ana.20339
27. Martínez-Orozco FJ, Vicario JL, Villalibre-Valderrey I, et al. Narcolepsy with cataplexy and comorbid immunopathological diseases. J Sleep Res. 2014;23(4):414–419. doi:10.1111/jsr.12143
28. Ruppert E, Zagala H, Chambe J, et al. Intravenous immunoglobulin therapy administered early after narcolepsy type 1 onset in three patients evaluated by clinical and polysomnographic follow-up. Behav Neurol. 2018;2018:1671072.
29. Saper CB. Restoration: potential for compensatory changes in numbers of neurons in adult human brain: restoration. Ann Neurol. 2013;74(6):762–764. doi:10.1002/ana.24039
30. Quinnell T, Wozniak D. Unmet needs of patients with narcolepsy: perspectives on emerging treatment options. Nat Sci Sleep. 2015;7:51–61. doi:10.2147/NSS.S56077
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.