Back to Journals » International Journal of Nanomedicine » Volume 12
Nanostructured delivery systems with improved leishmanicidal activity: a critical review
Authors Bruni N, Stella B ![]() , Giraudo L
, Giraudo L ![]() , Della Pepa C, Gastaldi D, Dosio F
, Della Pepa C, Gastaldi D, Dosio F ![]()
Received 24 April 2017
Accepted for publication 12 June 2017
Published 26 July 2017 Volume 2017:12 Pages 5289—5311
DOI https://doi.org/10.2147/IJN.S140363
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Natascia Bruni,1 Barbara Stella,2 Leonardo Giraudo,1 Carlo Della Pepa,2 Daniela Gastaldi,3 Franco Dosio2
1Candioli Pharmaceutical Institute Srl, Beinasco, Italy; 2Department of Drug Science and Technology, University of Turin, Turin, Italy; 3Department of Molecular Biotechnology and Health Sciences, University of Turin, Turin, Italy
Abstract: Leishmaniasis is a vector-borne zoonotic disease caused by protozoan parasites of the genus Leishmania, which are responsible for numerous clinical manifestations, such as cutaneous, visceral, and mucocutaneous leishmaniasis, depending on the site of infection for particular species. These complexities threaten 350 million people in 98 countries worldwide. Amastigotes living within macrophage phagolysosomes are the principal target of antileishmanial treatment, but these are not an easy target as drugs must overcome major structural barriers. Furthermore, limitations on current therapy are related to efficacy, toxicity, and cost, as well as the length of treatment, which can increase parasitic resistance. Nanotechnology has emerged as an attractive alternative as conventional drugs delivered by nanosized carriers have improved bioavailability and reduced toxicity, together with other characteristics that help to relieve the burden of this disease. The significance of using colloidal carriers loaded with active agents derives from the physiological uptake route of intravenous administered nanosystems (the phagocyte system). Nanosystems are thus able to promote a high drug concentration in intracellular mononuclear phagocyte system (MPS)-infected cells. Moreover, the versatility of nanometric drug delivery systems for the deliberate transport of a range of molecules plays a pivotal role in the design of therapeutic strategies against leishmaniasis. This review discusses studies on nanocarriers that have greatly contributed to improving the efficacy of antileishmaniasis drugs, presenting a critical review and some suggestions for improving drug delivery.
Keywords: amphotericin B, drug delivery systems, drug targeting, human leishmaniasis, polymeric nanoparticle
Introduction
Leishmaniasis is a parasitic disease caused by Leishmania spp., transmitted to humans and animals by species of Phlebotomine and Lutzomyia sandflies. The Leishmania parasite has two distinct morphological forms in its life cycle: promastigote and amastigote. The sandfly-transmitted flagellated metacyclic promastigotes rapidly enter into cells of the mononuclear phagocyte system (MPS) as part of the normal phagocytic response. The phagosome formed around the parasites undergoes a process of maturation and remodeling of the membrane and forms a new organelle, the parasitophorous vacuole (PV). Within the PV, the promastigotes differentiate into amastigotes, which multiply until eruption of the MPS cell (over a period of 4–6 days); the infection then spreads further. Having different tropism characteristics, parasites may infect either superficial cells or visceral cells. The parasite’s life cycle is completed when an uninfected sandfly takes a blood meal from the infected host. Because the infection involves several overlapping species and sandfly vectors, the taxonomy, ecology, epidemiology, and pathogenicity of the disease are complex.1,2
Leishmaniasis has numerous clinical manifestations, depending on which it is classified into three types: 1) cutaneous leishmaniasis (CL), the most common; 2) mucocutaneous leishmaniasis (ML), which may disseminate to the mucosa; and 3) visceral leishmaniasis (VL), also known as kala-azar, the most serious form of the disease, which can be fatal if not properly treated and could be disseminated to several organs.3
Leishmania infantum is responsible for VL infections in Latin America and North Africa, while Leishmania donovani is responsible for VL infections in areas of the Indian subcontinent and of East Africa. Although >90% of VL infections are concentrated in India, Brazil, Bangladesh, Nepal, and Sudan,4 a study shows a rapid increase in VL infections worldwide and climate change is expected to cause VL to severely impact Europe in the near future.5
Treatment failure and relapse rates are particularly high in cases of impaired cellular immunity, especially in human immunodeficiency virus (HIV) coinfection.6 HIV and Leishmania infection mutually reinforce one another, and HIV patients are more likely to develop VL (due to reactivation of a dormant infection or clinical manifestation after primary infection).
Current chemotherapeutic treatments are successful, to some extent, and the major targets pursued by associations such as the Drugs for Neglected Diseases initiative (DNDi) and the World Health Organization (WHO) in conjunction with foundations and pharmaceutical companies, may chiefly be summarized as to develop an oral, safe, effective, low-cost, and short-course treatment for VL and to develop novel treatment regimens for patients coinfected with HIV and VL. Furthermore, as for all neglected tropical diseases, additional control mechanisms and tools are necessary, such as drugs, vaccines, reliable diagnostics, vector control agents, and control strategies, to eradicate infection.7
Considering the small number of chemotherapeutic agents or innovative antileishmanial medicines that are available, in parallel with the search for more efficient and less toxic antileishmanial drugs, including the development of a successful vaccine, the push to design stable nanotechnology-based drug delivery systems (DDSs) is likely to be the main strategy in fighting the disease. The use of colloidal carriers loaded with active agents is a clear-cut approach, thanks to the physiological uptake route of intravenous (iv) administered nanosystems (the MPS cells).8 Nanosystems can thus lead to high drug concentrations in the intracellular MPS-infected cells and in the PV; colloidal carriers also protect the drug against in vivo degradation.
This review seeks to give an overview of the current therapeutic protocols and new approaches involved in the search for safer and more active drugs, as well as the strategies employed for choosing appropriate delivery systems to produce the next-generation agents for treating VL. Although the role of nanotechnology in DDSs for leishmaniasis has been reviewed elsewhere,9,10 this review also highlights the limits and issues involved in applying modern techniques and illustrates the most promising results. In particular, the different strategies, such as their potential, cost, feasibility, and limits, were critically evaluated and commented. As in vivo studies are imperative for estimating the clinical feasibility of a DDS, particular emphasis was given to approaches reporting preclinical/clinical data and/or to those showing research progress during recent years.
Although alternative immunotherapeutic strategies are potentially advantageous, these strategies and nanodevices that can be used as promising vaccine carriers were not discussed here.9,11
Current chemotherapy
The first therapeutic options were introduced in the early 1900s for mucocutaneous form, and other treatments have been added over the last decade; however, they are not devoid of limitations relating to efficacy, toxicity, cost, length of treatment, increasing parasitic resistance, and difficulty of administration, making treatment a complex issue. The principal drugs currently available are antimonials, amphotericin B (AmpB), paromomycin (PM), and miltefosine (MF). Table 1, taken from the 2010 WHO report, is presented to give a clear view of treatment regimens and their costs (report of a meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva, March 22–26, 2010). Several recent studies clearly describe the various combination approaches that have been used, in the search for the optimal and most cost-effective strategy, not only to treat patients but also to control infections in countries including Morocco, India, Sudan, and Nepal.12–16
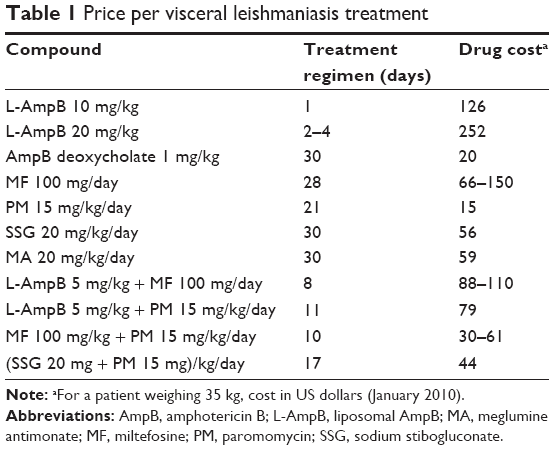 | Table 1 Price per visceral leishmaniasis treatment |
Pentavalent antimonials have been used for the past several decades to treat VL. At present, two types of organoantimony (V) complexes are commercially available: Glucantime® (meglumine antimoniate [MA]) and Pentostam® (sodium stibogluconate [SSG]). These are the standard first-line medicines in most parts of the world (>90% overall cure rate). Initial treatment of VL should be based on a daily injection of 20 mg/kg body weight of Sb(V), and injections are usually given for 28–30 days. The cost for the course of treatment is <60 US dollars (Table 1).
The mechanism of action of these compounds, in one model, is based on the reductive bioconversion of Sb(V) to Sb(III), by the parasite or by the infected host cells, to create an active agent Sb(III)–trypanothione conjugate.17 In a second model, Sb(V) is the active species, which directly exerts activity against Leishmania. Sb(V) is able to act selectively against the topoisomerase of the promastigote rather than against that of the monocyte.18 Sb(V) is also reported to bind ribonucleosides in an environment similar to that of lysosomes.19
Unfortunately for first-line antimonials, the development of resistance is a primary barrier to successful treatment. The resistance mechanism has been found to be multifactorial and is principally due to reduced Sb(III) uptake or/and increased cell efflux/sequestration through an abnormally high level of trypanothione and increased expression of the metalloid-thiol pump.20 Resistance to therapeutics has also been imputed to different efflux pumps or ATP-binding cassette transporters, such as P-glycoprotein and multidrug resistance protein.21
AmpB is a natural antibiotic with a macrolide polyene structure and significant antifungal and antiparasitic activities. Currently, AmpB is widely used to treat systemic Candida albicans and Aspergillus fumigatus infections.22 The mechanism of action of AmpB is based on its binding to the ergosterol in the fungus or parasite rather than to human cholesterol. In general, AmpB binds to ergosterol through a hydrophobic interaction, in a series of events that eventually lead to pore formation in the parasite membrane.23 In clinical trials in India, AmpB formulated with deoxycholate (Fungizone®) was given daily or on alternate days by iv slow infusion at a dose of 0.75–1.0 mg/kg/day for 15–20 doses and was 99% effective.24
To reduce the severe side effects, which include nephrotoxicity and hematotoxicity, different formulations have been developed and are currently available on the market under the trade names Abelcet®, Amphocil®, Amphotec® (lipid complexes), AmBisome® (liposomes), and Ampholip®.
Used in India and Europe, all of these formulations were found to be effective; the liposomal formulation, in which the drug is intercalated in a lipid membrane consisting of hydrogenated soy phosphatidylcholine (PC), cholesterol, and distearoylphosphatidylglycerol, is the most widely used against VL. Given at a dose of 3–4 mg/kg/day, for a total dose of 15–24 mg/kg, administered to various regimens, AmpB liposomal formulation had 90%–98% efficacy in southern Europe.25 Shorter treatments are also effective; in India, a single-course therapy of 10 mg/kg was found to give a 98% cure rate.26 The principal benefit of AmBisome is its reduced toxicity to mammalian cells, allowing higher doses to be administered, rather than any increase in its antileishmanial activity per se. The major drawback of AmBisome is its high cost (126–250 US dollars) vs Fungizone (30 US dollars for a 30-day course of treatment), putting it out of patients’ reach in some countries; it is unclear whether this limitation is applicable to other geographical areas. Nevertheless, WHO and Gilead Sciences have signed agreements to donate hundreds of vials of liposomal AmpB, extending their previous agreement until 2021.195 The new 5-year collaboration, whose estimated value is 20 million US dollars, includes funding that will allow populations affected by VL to benefit from enhanced access to diagnosis and treatment.
In a study investigating the extent to which the side effects of AmpB were reduced by the use of a lipid emulsion, a preformed AmpB lipid emulsion (ABLE) was compared with Fungizone and AmBisome in terms of safety and efficacy.27,28 Indian clinical trials showed that ABLE was a well-tolerated, efficacious, and affordable lipid formulation. In a recent study on a population of 500 patients, the efficacy of single day infusions of ABLE at 15 mg/kg/day was satisfactory, with an initial cure rate of 95.9%, compared to 100% for AmBisome. The proportion of patients with no clinical signs and symptoms of relapse of kala-azar during 6 months follow-up was 85.9% in the ABLE group compared to 98.4% in the AmBisome group.28
Resistance to AmpB has been induced in vitro, and a study has shown it to be related to the increased cell membrane fluidity occurring when ergosterol is modified to its precursor cholesta-5,7,24-trien-3β-ol.29 The modification reduced AmpB’s attachment to sterol-modified membranes. In clinical isolates of L. donovani, a multifactorial response was observed, in which altered membrane composition, ATP-binding cassette transporters, and an upregulated thiol metabolic pathway played roles in conferring AmpB resistance.30
PM (aminosidine) is an aminoglycoside antibiotic, used to treat intestinal infections such as amebiasis and cryptosporidiosis, which was rediscovered as an antileishmanial agent. PM has been shown to be effective in Indian VL: a dose of 15 mg/kg for 21 days administered iv gave a cure rate of 93%–95%. The mechanism of action of PM in Leishmania is not precisely known, but protein synthesis machinery has been proposed as the target. Although there are no known clinical isolates of PM-resistant strains, a resistant L. donovani strain was readily generated experimentally: ribosomal proteins were found to be upregulated.31 PM must be administered by the parenteral route; however, because it undergoes rapid clearance after iv administration, being excreted in the urine upon glomerular filtration, treatment requires multiple dosages. Adverse effects such as nephrotoxicity, ototoxicity, and hepatoxicity have been reported.32
MF (hexadecylphosphocholine) was the first, and still the only, orally available drug for the treatment of VL.33 MF belongs to a class of alkylphosphocholine drugs initially developed in the 1980s to treat solid tumors. Treatment with 150 mg/day per >50 kg body weight for 28 days in immunocompetents gave a cure rate of 94% in India and ~90% in Ethiopia.34 Its antileishmanial activity involves action on several targets; however, no mechanism has been identified definitively: alkyl phospholipid metabolism, glycosylphosphatidylinositol anchor biosynthesis, signal transduction, mitochondrial dysfunction, and immunomodulatory effects might be involved.35 Whatever its indication of use, pharmacovigilance for significant safety issues will remain a priority. Furthermore, MF should not be administered in pregnancy due to its teratogenicity.33
In India, post-kala-azar dermal leishmaniasis (PKDL) is a sequel to VL occurring within a year or up to 32 years after VL has been cured; a study suggests that oral MF for 2–3 months could be considered as a treatment of choice for Indian PKDL.36 MF has a median half-life of ~152 h, which might encourage the development of clinical resistance. Furthermore, being an oral agent, its improper use in endemic countries would increase the risk of resistance and would spread resistant parasites where the prevalence of infection is high.37
The risk of emergence of drug resistance in the field is exacerbated by the relatively easy production of in vitro resistant Leishmania clones, combined with the occurrence of relapses in immunocompetent patients, the presence of HIV/VL coinfections, and high levels of anthroponotic transmission in both Africa and India.38 Combination treatment has the potential advantages not only of shortening the treatment duration (thus increasing compliance) and reducing the overall drug dose (thereby reducing toxic effects and cost) but also of reducing the probability that drug-resistant parasites are selected, and thus prolonging the effective life of available medicines. Several trials of combinations have shown favorable results.39,40 The combination of PM and antimonials produced a higher cure rate in VL patients in a trial in Bihar, than did antimonials alone, in which the lack of response was common.41
Many other compounds are considered to be the second-line drugs for leishmaniasis, including pentamidine and the antifungal azole fluconazole, as well as fexinidazole and oleyl phosphocholine (OlPC), with approval status at different stages. The diguanidine-based compound pentamidine was initially developed as a synthetic analog of insulin. Pentamidine has shown high activity against the acute phase of human African trypanosomiasis;42 it has attracted renewed interest for its use as secondary prophylaxis in HIV-coinfected VL patients.43 The drug is given intramuscularly or, preferably, by iv infusion. However, pentamidine is now no longer recommended as a treatment option for VL. High doses are needed to achieve a cure, and irreversible diabetes has been reported in 4%–12% of cases.44
The primary action of pentamidine against Leishmania is not clearly understood, but the drug’s structural characteristics and experimental results suggest that the active transport system and the mitochondria are the final sites of inhibition. A series of experiments showed that it acts as an inhibitor of polyamine uptake and the arginine transporter,45 while other findings indicate the mitochondrial replication system as possible target (through inhibition of topoisomerase II activity).46 Resistance to pentamidine in Leishmania has been associated with changes in the concentrations of polyamines and arginine within the cell.47
In collaboration with Sanofi, DNDi recently developed fexinidazole, a nitroimidazole derivative DNA synthesis inhibitor, for the oral treatment of human African trypanosomiasis. Fexinidazole has shown potent activity against L. donovani in vitro and in vivo in a VL mouse model.48 A Phase II proof of concept study evaluating the efficacy of fexinidazole for the treatment of primary VL was initiated in November 2013 in Sudan (FEXI VL 001; NCT01980199); this open-label trial was designed to enroll 66 patients. DNDi terminated this trial in September 2015 due to lack of efficacy. However, the combination of fexinidazole and MF is still in trial.196 Other members of the nitroimidazole class of molecules have emerged, and novel products are currently in advanced preclinical evaluations.49,50
Nanostructured anti-Leishmania delivery systems
The use of DDSs stands as a complementary strategy for developing new treatments and combination therapies for VL.9 An appropriate DDS can be used in antileishmaniasis therapy to produce the necessary high drug concentration in the intracellular phagolysosome or PV, where the Leishmania is hosted. The major challenge in using a small drug is the difficulty of enabling it to penetrate inside the macrophages: multiple membranes must be traversed by the therapeutic agent to reach parasitic target. The nonspecificity to macrophages of current treatments leads to their accumulation in normal, healthy tissues, with prevalent toxicity as side effects. Improving the safety/efficacy ratio by delivering a high drug concentration at a controlled rate, using an appropriate targeting strategy, is an attractive goal that has been vigorously pursued.
DDS may also overcome problems relating to the low water solubility of drugs and could protect the active molecule from degradation in biological fluids. Furthermore, alternative routes to parenteral drug delivery (eg, nasal, pulmonary, and topical) that can improve patient compliance are achievable with appropriate DDS. In particular, developing oral formulations of current chemotherapy options is the most attractive prospect, although the lack of treatment supervision may lead to patients failing to complete the full course of treatment. This is a critical issue, as it may facilitate the development of drug resistance in Leishmania. Finally, novel materials that are able to become “smart”, capable of responding to the physiological environment, can also considerably improve drug availability.51
The combination of antileishmanial drugs with nanocarriers is emerging as a promising approach, because the traditional nanocarriers (liposomes and polymer nanoparticles) are readily internalized by macrophages in the liver and spleen, releasing the drug inside the cell and thus leading to a high local concentration, and ultimately killing the protozoa. In this connection, the main strategy in treating Leishmania is to target the drugs directly to macrophages, using appropriate nanosized delivery systems.52 A targeted approach entailing modification of the nanocarrier surface will also increase parasite selectivity.
Another advantage is the possibility of packing multiple payloads in a single carrier, providing a combination therapy that may have a synergist effect (overcoming resistance). However, the sequencing and scale-up of approaches of this type are still very challenging.
The following sections review the recently reported research concerning anti-VL nanotechnology, with special emphasis on the use of liposomes, polymers, polysaccharides, metal nanoparticles, and carbon-based materials loaded with chemotherapeutics currently used in VL treatment (a schematic presentation of different delivery systems is given in Figure 1). Since AmpB is currently the most potent antileishmanial agent, the majority of efforts in nanotechnology have been focused on this drug. For this reason, we pay attention and detail on innovative AmpB delivery systems. Table 2 summarizes the principal characteristics and the results obtained.
 | Figure 1 Schematic representation of different classes of antileishmanial nanometric delivery systems. |
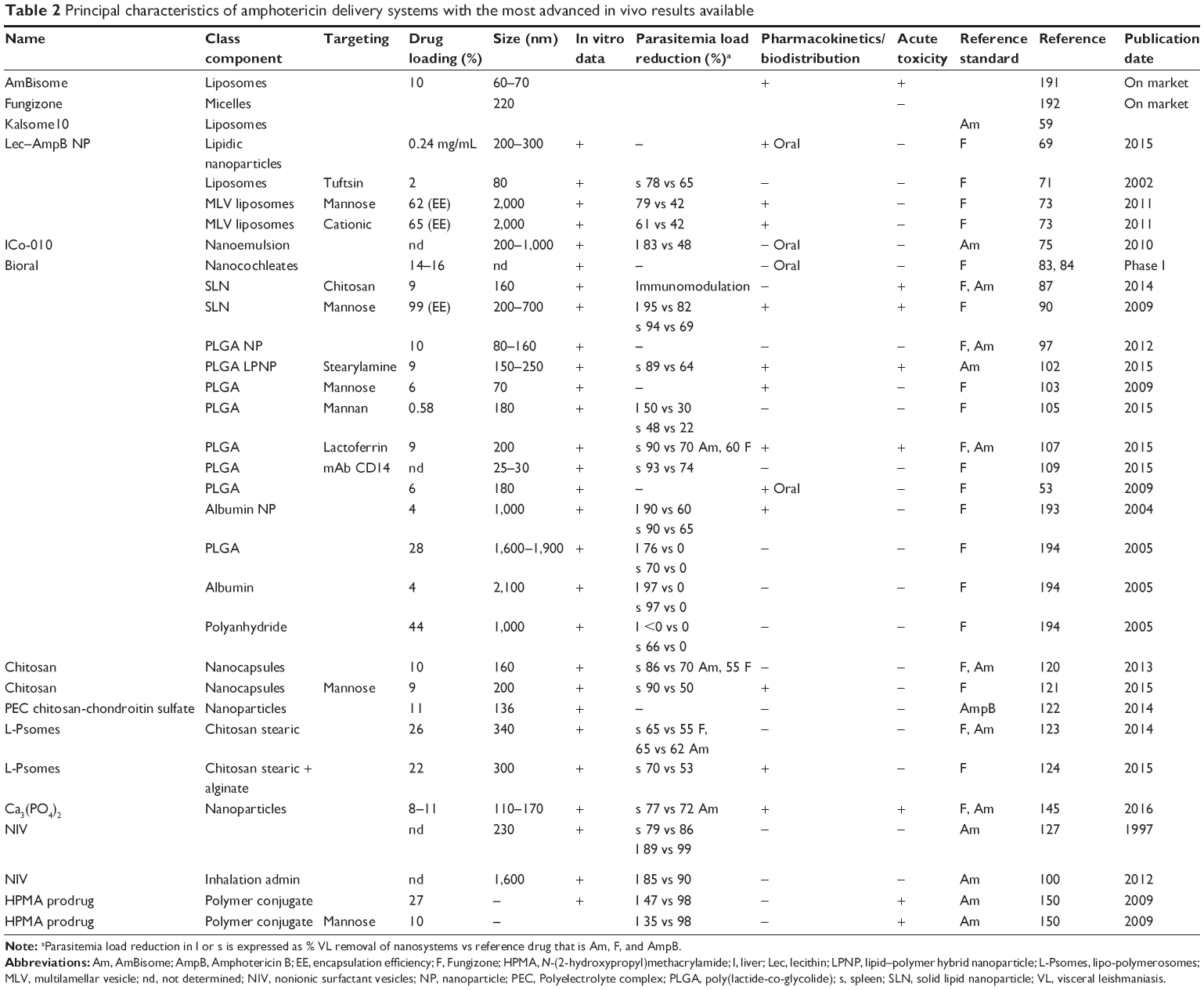 | Table 2 Principal characteristics of amphotericin delivery systems with the most advanced in vivo results available |
In particular, one of the most challenging achievements is the oral administration of AmpB. This is limited because AmpB is absorbed by passive diffusion through the intestinal membrane. However, the process of passive diffusion is dependent on drug’s molecular weight and is efficient for drugs up to molecular weight 500 Da; efficiency then decreases at higher molecular weights. AmpB has a molecular weight of 924.08 Da, and thus, passive diffusion may be expected to be very low.53 Moreover, AmpB is relatively unstable at the acidic pH of the gastrointestinal tract, and upon oral administration might be degraded prior to absorption. In addition, it undergoes extensive P-gp efflux from the enterocytes, which further limits its oral bioavailability. Taken together, these factors lead to very low oral bioavailability of AmpB (~0.3% of the administered dose).
Several nanosystems have also been developed to improve the bioavailability of AmpB (for an exhaustive review refer Torrado et al54), and some of them, related to VL treatment, are reported in the following sections. These delivery systems may have several advantages, as they might 1) enhance AmpB dissolution and also protect it from the acidic gastric environment, 2) increase intestinal permeability using either delivery systems of an appropriate size to enhance lymphatic uptake or carriers able to cross the gastrointestinal barrier, and 3) prolong gastrointestinal transit time through the use of bioadhesive systems. The biodistribution of AmpB is dependent on the type of formulation, and there is a weak correlation between drug plasma and organ levels.55 For this reason, AmpB plasma levels must be considered in combination with target tissue pharmacokinetics. An oral formulation would be of benefit to VL patients, but several issues remain to be resolved.
For completeness, “Novel products with anti-leishmanial activity combined into nanocarriers” section comprises DDS loaded with different products with antileishmaniasis activity.
Currently, all these experimental compounds, as well as the majority of nanostructured systems, have been tested in vitro and in animal (mice or hamsters) models. Thus, a clear comparison with currently available medicines has to be carefully pondered.
Liposomes
Liposomes are small artificial vesicles of spherical shape that can be created from cholesterol and phospholipids. Due to their biocompatibility, stability, ease of scaling up, and ability to carry various molecules as cargo, liposomes are currently used as DDS in a range of different treatments. Most liposomal formulations available on the market target cancers. Information about this application can be find elsewhere.56
The reticuloendothelial system (RES) is the main site of accumulation of liposomes following their systemic administration, and in the RES, liposomes are mainly cleared by resident macrophages. Primary organs associated with the RES include the liver, spleen, kidney, lungs, bone marrow, and lymph nodes.
The best clinical results in VL treatment, as reported earlier, have been achieved with the liposomal formulation AmBisome, which allows to load an high concentration of AmpB (drug loading [DL] =12.5%) (drug weight/[drug + liposomal components] weight) and represents the gold standard for novel delivery systems. Over recent years, lipid complex formulations of AmpB have been proposed,57 showing interesting in vitro activity but less efficient than AmBisome in in vivo evaluations. More recently, liposomal formulations in which cholesterol was replaced by ergosterol, which constitutes 50% molarity of total lipids (Kalsome™ 10; Lifecare Innovations Ltd., Gurugram, India), have been evaluated.58,59 This change followed the in vitro observation that exogenous cholesterol, if added to the culture, enhanced the growth of Leishmania promastigotes; thus, the absence of cholesterol might make the drug more suitable for clearing parasites. Large multilamellar vesicles of ~1,000 nm in diameter were obtained and were kept separately by sonication. In vivo results confirmed Kalsome™ 10 efficacy in a murine VL model without major toxic effects. More recently, a possible cell death mechanism in Kalsome™ 10-treated L. donovani was proposed.60 Interestingly, the component of the liposomal formulation induced apoptotic-like cell death in L. donovani parasites, demonstrating its antileishmanial function.
A number of older anti-Leishmania antimonials, formulated in liposomes, have been investigated, starting in 1977.61 Liposomes with different surface charge (anionic and neutral) were subsequently tested,62 but only recently, a robust effective treatment against infection with SSG-resistant Leishmania parasites in mice has been demonstrated, using cationic PC–stearylamine liposomes. When the charged phospholipids were encapsulated in liposomes, their binding to macrophages was shown to be greatly enhanced.63
To improve liposome stability and enhance blood circulation time, sterically stabilized liposomes have been introduced: the addition of polyethylene glycol (PEG) to the liposome surface slows removal from the blood, improving half-life (eg, Doxil®).64 PEGylated and uncoated liposomes bearing MA in VL-infected dogs were compared;65 the former were more effective, although a mixed formulation of PEGylated and conventional liposomes showed greater activity, than either single formulation, for parasite elimination in the spleen and bone marrow.
Other drugs commonly used as anticancer agents have been encapsulated in sterically stabilized liposomes: camptothecin66 and, more recently, doxorubicin have been investigated to integrate MA treatment, so as to decrease the resistance of Leishmania against currently available drugs.67
From the toxicology standpoint, liposome-mediated delivery leads to greatly increased and prolonged concentrations of antileishmanial drugs in the liver and spleen. This is an advantage in a disease such as VL, where macrophages are the target of the therapy, but the macrophages might already be impaired by the infection, and high tissue accumulation may lead to severe side effects.68
In a study aiming to improve the oral bioavailability of AmpB, the antileishmanial activity of two nanocarriers was compared: soya lecithin (Lec)-based biodegradable nanocarriers and liposomes. The surface of the Lec–AmpB nanoparticles was modified with PEG and Tween 20 to further improve their stability and biological activity. PEG nanoparticles showed more promising antileishmanial activity, possibly because of their better interaction with biological systems and membranes.69
To further increase the specificity of action to the diseased area, liposomes decorated with specific ligands have also been developed. This strategy offers a vast potential for site-specific delivery of drugs to designated cell types or organs, which selectively express or overexpress specific ligands (eg, receptors and cell adhesion molecules) at the site of the disease.70 However, their real therapeutic advantage over nontargeted liposomes is still debatable, with conflicting results being reported; no targeted liposomal product has yet reached the market.
The targeting approach was applied to liposome-encapsulated AmpB, by decorating the particle surface with the naturally occurring macrophage activator tetrapeptide, tuftsin (Thr–Lys–Pro–Arg). Increased in vivo antileishmanial activity vs standard liposomes was observed and appeared to be due both to improved drug tolerance of the liposomal formulation (like that of AmBisome but with a DL of 2%) and to the increased specific uptake of tuftsin by the macrophages.71
C-type lectin receptors (CLRs) are highly conserved pattern recognition receptors with carbohydrate recognition domains that bind sugars (eg, mannose, fucose, and N-acetylglucosamine), present on the surface of many pathogens.72 In particular, the mannose receptor (also called CD206) was the first reported member of a family of four endocytic CLRs receptors; it plays a role in the clearance of endogenous glycoproteins and in pathogen recognition/antigen presentation. The targeting of pattern recognition receptors on macrophages offers the advantage of triggering specific signaling pathways, thus inducing a tailored and robust immune response. Antileishmanial drugs have been encapsulated in mannosylated or fucosylated liposomes to target the corresponding receptors expressed by macrophages; they demonstrated successful treatment of experimental leishmaniasis in a hamster model.73 Furthermore, a biodistribution study clearly showed that uptake of mannosylated liposomes in the liver and spleen was higher than either free AmpB solution or cationic liposomal formulation, indicating that active targeting to the RES occurred.73
Nanoemulsions (NEs) and niosomes
NEs are mixtures of two normally immiscible liquids that are stabilized by using a surfactant (emulsifier). The addition of surfactant is critical for the creation of small droplets (~100 nm) as it decreases the interfacial tension. The emulsifier also plays a role in stabilizing NE through repulsive electrostatic interactions and steric hindrance.74 Lipid AmpB formulations potentially suited to oral administration have been developed based on mono- and diglycerides with or without a lipophilic derivative of vitamin E, D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), as an alternative to the less temperature-stable monoglyceride. The formulation, named ICo-010, retained AmpB in simulated gastric and intestinal fluids and exhibited a significant antileishmanial activity in a VL-infected murine model.75,76 ICo-010, currently being developed by iCo therapeutics, was granted orphan drug status by the FDA, but recent data from the company website do not show any further improvement from preclinical evaluations.197
A NE loaded with doxorubicin (NE–DOX) was further grafted with phosphatidylserine (PS) to enhance the cellular uptake.77 There was significant improvement in activity with PS–NE–DOX on the spleen parasitic burden in Leishmania-infected hamsters, in comparison to uncoated NE or free drug.
Niosomes are vesicles consisting of nonionic surfactants with cholesterol. These are biodegradable, somewhat less toxic and more stable than liposomes, and relatively low in cost, making them an alternative to liposomes. These particular characteristics make niosomes promising candidates for commercial manufacturing.78 In an early study on delivery systems for VL, niosomal SSG was found to be more effective than liposomal vesicular formulations or than free drug, against experimental murine VL.79 More recently, the effect of itraconazole niosomes on the in vitro susceptibility of L. tropica was demonstrated.80 Entrapment efficacy of AmpB into niosomes has been also reported.81
Lipid cochleates
An interesting approach to prepare lipid systems for oral delivery is that of nanocochleates, cigar-shaped nanostructures composed of negatively charged lipid bilayers (usually PS) bridged by a divalent cation, normally calcium.82 Calcium induces dehydration of the interbilayer domains, and consequently, the amount of water in this region is low, allowing better encapsulation efficiency of hydrophobic drugs, which have a high affinity for the hydrophobic interior of the lipid bilayers. AmpB is an ideal model for hydrophobic drugs. Nanocochleates containing AmpB or AmpB deoxycholate were in vitro tested against L. chagasi, and results showed similar activity.83,84 Lipid cochleates containing both AmpB and MF were also produced, and the incorporation stability was estimated.85 Some clinical trials for VL were initiated on the basis of the results obtained with an oral nanocochleate formulation of AmpB (Bioral® Amphotericin B), which reached Phase I development in the USA for the treatment of mycoses, but nowadays, the research appears to be discontinued. However, Matinas BioPharma is developing MAT 2203, comprising nanocochleates of AmpB for oral administration in treating asperigillosis.198
Solid lipid nanoparticles (SLNs)
SLNs are nanospheres made from solid lipids, with size in the range of 50–1,000 nm. They consist of a solid lipid matrix, ie, glycerides, fatty acids, and waxes, stabilized by physiologically compatible emulsifiers, such as phospholipids, bile salts, Tween, polyoxyethylene ethers, and polyvinyl alcohol. The lipids used in their production are solid at room temperature, and most of them have approved status, eg, Generally Recognized as Safe (GRAS), due to their low toxicity.86
Chitosan-coated SLNs loaded with AmpB have been developed for immunoadjuvant chemotherapy of Leishmania infections. The in vitro antileishmanial activity revealed that chitosan-coated AmpB–SLNs were markedly more potent than commercial formulations (AmBisome and Fungizone). Furthermore, cytotoxic evaluation in macrophages and acute toxicity study in mice evidenced a better safety profile of the formulation compared to marketed products.87
More recently, PM was incorporated into SLN made of stearic acid and Tween 80, and both characterization and in vivo data were reported.88,89 As with liposomes, it has been attempted to increase the uptake by macrophages by targeting to mannose receptors. Mannose-coated lipid nanoparticles of AmpB showed higher liver uptake and promising antileishmanial activity.90 Interestingly, SLN can also be used to improve oral administration protocols (AmbiOnp formulation),91 although this has not yet been applied in VL therapy. The loading of AmpB in the nontoxic superaggregated form results in enhancing the oral bioavailability and increasing their safety profile.91
To reduce the side effects that limited the use of MF, delivery in lipid nanoparticles has been proposed.92 MF formulations were prepared using glycerin, cottonseed oil, oleic acid, cholesterol, and the emulsifiers, such as egg PC and Tween 80. A reduction in epithelial irritation in the gastrointestinal tract was observed in an in vivo study performed in Balb/C mice. Compared to the free drug, the formulation reduced in vitro hemolytic action and cytotoxic activity against macrophages and resulted in comparable level of in vitro cytotoxicity against promastigote and amastigote forms of L. chagasi.92
Polymer nanoparticles
Polymer nanoparticles may be of value in treating infectious diseases such as leishmaniasis, since their small size enables them to pass through biological barriers upon parenteral administration, also enhancing cellular uptake and enabling therapeutic agents to be delivered to infected tissues.10 Polymer nanocarriers can be modulated to give them advanced physicochemical properties, such as increased bioavailability, and precisely defined biodegradability, affording release of the encapsulated drug. Polymer system may consist of either a polymeric matrix (nano- or microspheres) or a reservoir system (nano- or microcapsules). Several different synthetic polymers can be used to prepare nanoparticles, including poly(lactic acid) (PLA), poly(glycolic acid) (PGA), poly(lactide-co-glycolide) (PLGA), poly(caprolactone) (PCL), and poly(cyanoacrylate) (PCA).93 However, natural protein polymers, such as albumin and gelatin, and polysaccharides, such as chitosan, alginate, and starch, have also been described as candidates for the delivery of particle-based antileishmanial systems.94 Other polymers, such as PEG and N-(2-hydroxypropyl)methacrylamide (HPMA), have been used predominantly to produce polymer–drug conjugates in the form of macromolecular prodrugs. In this approach, the release mechanism entails splitting the linker between the polymer and the bioactive agent.
Concerning synthetic polymers, the first studies involved the use of PCA and PCL, loading different drugs with interesting results.95 However, in recent years, PLGA has been one of the most attractive polymeric candidates, used to fabricate devices for drug delivery and tissue engineering applications.96 Currently, 15 FDA-approved PLGA-based drug products are available on the US market.199
Different research groups have investigated AmpB-loaded PLGA nanoparticles. In particular, the efficacy of PLGA nanoparticle-encapsulated AmpB was compared to free drug or marketed formulations against intracellular L. infantum amastigotes as well as promastigotes. One nanoparticle formulation was more effective than AmBisome and, at the same time, less cytotoxic and hemotoxic.97 Although very promising, especially as an antifungal agent, no further in vivo clinical applications have since been reported.
AmpB has been entrapped in PLGA nanoparticles to improve its oral bioavailability and to reduce its nephrotoxicity.53 The addition of the stabilizer TPGS permitted the formation of stable nanoparticles of <200 nm in diameter. It is worth remembering that TPGS, reported as a P-gp efflux inhibitor, is currently approved by the FDA for use as an excipient in various nanoparticle formulations and also as a solubilizing and emulsifying agent for poorly soluble compounds to improve their absorption and bioavailability.98 The AmpB–PLGA formulation containing TPGS reduced drug nephrotoxicity on administration by either the oral route or the iv route compared with Fungizone. The relative oral bioavailability of AmpB was found to be about eight times higher when it was formulated in polymer nanoparticles than when it was administered as Fungizone.99 Further study on in vivo activity has only been reported for pulmonary aspergillosis.100
MF loaded in PLGA–PEG nanoparticles also showed a fourfold increase in in vitro activity and in in vivo antileishmanial efficacy and bioavailability vs free drug.101
Lipid–polymer hybrid nanoparticles (LPNPs) loading AmpB with the anionic core composed of PLGA polymer and TPGS surfactant and the shell made of cationic stearylamine lipid have also been prepared.102 Stearylamine was selected for its cationic nature, biocompatibility, benign antiprotozoal activity, and immunopotentiation strength. LPNPs containing an interesting amount of AmpB (DL 9.8%, w/w) were reported. Receptor-mediated LPNP identification ensures considerable uptake into the macrophage cells and RES organs (spleen, liver, and so on), while uptake by the kidneys is very low. Furthermore, the positive charge of LPNPs allows them to bind to negatively charged sialic acid molecules on the macrophage surface, resulting in adsorption-mediated endocytosis. In vivo experiments demonstrated significantly stronger parasite growth inhibition (89.4%) in patients treated with AmpB–LPNPs than in those treated with carrier without stearylamine (63.6% inhibition), whereas 69.3% parasite inhibition was observed with the AmBisome formulation.102
With regard to the active targeting approach, mannose-bearing PLGA nano/microparticles have been designed to further improve macrophage targeting of antileishmanial agents. Mannose-grafted PLGA nanoparticles, with and without a PEG spacer, are reported to target AmpB;103 the presence of PEG spacer resulted in a more efficient inhibition of parasites in comparison to both the one without spacer and the free drug. Furthermore, both drug–polymer interaction and drug miscibility in the polymer affect the drug content of polymer nanoparticles. The PLGA hydrophobic core facilitates the loading of amphiphilic AmpB. The polymer slow precipitation, due to the slow rate of solvent removal, allowed more time for drug molecules to partition into the aqueous phase, leading to low % encapsulation efficiency. However, in the engineered version of polymer nanoparticles, in which mannose was directly linked to PLGA (M–PNPs) or to the edge of PEG (M–PEG–PNPs), an increased entrapment efficiency was observed, probably due to the higher drug–polymer miscibility, permitting the enhanced drug incorporation.103 The significant rise in drug content in M–PNPs and M–PEG–PNPs might be due to the stronger drug–polymer interaction (between the hydroxyl group of mannose and PEG and the amine group of AmpB) and to the high miscibility in the case of amphiphilic AmpB. Another study suggested that the acidic end-group of PLGA is responsible for drug incorporation and that increased chain length may increase the drug–polymer interactions, leading to improved drug incorporation efficiency.104
More recently, carbohydrate (mannose, mannosamine, and mannan)-functionalized PLGA nanospheres loaded with AmpB have been investigated in the treatment of murine VL.105 The uptake of mannan-coated nanoparticles (MNs) by macrophages was elevated. Furthermore, a single injection and a 1-week course of treatment with MN–PLGA AmpB nanoparticles were sufficient to achieve a significant decrease in the visceral organs parasite load, although AmpB loading was only 0.58% (w/w). The successful results were probably due to increased production of cytokines relevant in VL infection, such as INF-gamma, and nitric oxide (NO), which plays a key role in the organism’s defense against parasite infection.105
Polymeric nanosystems are also feasible as carriers for multiple drugs. In a recent study, PLGA nanoparticles loaded with AmpB and doxorubicin and further coated with chitosan as a macrophage targeting agent were developed. Chitosan-coated nanoparticles provide a signal for specialized phagocytes, because chitosan recognition involves multiple macrophage surface receptors inducing the drug internalization into cells. Furthermore, this system demonstrated slow drug release to macrophages, with minimal hemotoxicity and tissue toxicity.106 Currently, no in vivo data on VL infected rodent models are available.
AmpB was recently targeted by PLGA nanoparticles using a coating with glycoprotein lactoferrin (Lcf).107 Lcf is an 80 kDa iron-binding glycoprotein with a single polypeptide chain of ~700 amino acids that provides several potential N-glycosylation sites. These sites of Lcf are recognized by the group of CLRs, ie, mannose receptors, which present as biomarkers on the monocyte/macrophage and dendritic cell surfaces. In vivo tests demonstrated significant in vivo antileishmanial activity of Lcf–PLGA–AmpB, compared with nontargeted and commercial formulations, as well as lower nephrotoxicity, attributed to the slower drug release. This might permit the administration of much higher drug doses, leading to increased efficacy in VL treating. It is worth noting that peptide fragments derived from Lcf, such as lactoferricin, exerted interesting activity against L. donovani promastigotes.108 AmpB encapsulated into PEG–PLGA nanoparticles, further decorated with anti-CD14 antibodies, was recently evaluated for macrophage localization improvement.109 CD14 is mainly present on macrophages surface and, to a lesser extent, on neutrophils and dendritic cells. Interestingly, evaluations on promastigote and amastigote revealed that the inhibition efficiency of nanoparticles was higher than that of free drug.
Polysaccharide polymers
Polysaccharides are ideal candidates for drug delivery and biomedical applications, as they are easily obtained from natural sources. Examples of polysaccharides of plant origin include starch, cellulose, hemicellulose, hyaluronic acids, alginate, and guar gums, whereas polysaccharides originating from animals include chitin and chondroitin sulfate.110 Natural polysaccharides have been evaluated for their ability to act as antileishmanial agents, in some cases based on their use in traditional medicine111 and in others from exploring novel sources such as marine macroalgae, which contain sulfated polysaccharides with antileishmanial activity.112 Polysaccharides with different structures act by different mechanisms in macrophages:113 sulfated heterorhamnan, iota-/nu-carrageenans, and arabinogalactan polysaccharides demonstrated significant macrophage activation and a subsequent inhibition of intracellular amastigotes of L. amazonensis. The antileishmanial effects of the polysaccharides occurred by different mechanisms: for sulfated polysaccharides, this may be an expressive increase in NO production by macrophages, while for arabinogalactan, it may be attributed to stimulation of O2•- and TNF production.113
Polysaccharide polymers have also been used to deliver vanadium complexes, which are reported to be highly active metal-based drugs against a variety of trypanosomatids, including Leishmania species.114 The immunomodulatory effects of the polysaccharides arabinogalactan, galactomannan, and xyloglucan, as well as their oxovanadium (IV/V) complexes, were evaluated on peritoneal macrophages exhibiting significant leishmanicidal activity (~60%–70%). The mechanism of action appears to be due to increasing levels of IL-1b and IL-6 released by macrophages.115 Vanadium complexes of stilbene demonstrated leishmanicidal activity in promastigote forms of L. amazonensis, inducing reactive oxygen species (ROS) production and NO-dependent microbicidal action.116
Biopolymers such as chitosan and sodium alginate are potent macrophages activators, inducing the release of a range of cytokines and cytotoxic agents.117–119 Development of a biocompatible polymers formulation, which enhances immunological responses, might thus provide a new approach to improve leishmaniasis treatment. Naturally occurring polysaccharides, such as chitosan and alginate, have been utilized to deliver antileishmanial agents in nanoparticles. Nanometric AmpB-encapsulated chitosan nanocapsules (CNC–AmpB) have been formulated using a polymeric deposition technique mediated by NE template fabrication.120 The lipid core of the nanocapsule consisted of surfactant, glycerol, soybean oil, and soya lecithin. Enhanced efficacy of CNC–AmpB is likely due to active macrophage targeting by N-acetyl-glucosamine unit of chitosan content of CNC, recognition being mediated by major histocompatibility complex class I and II, Fc receptors, and mannose receptors. The same team improved the targeting capability of chitosan, using mannosylated chitosan (Mnos-CS) copolymer, subsequently formulated in a nanocapsule system (MnosCNc) for the delivery of AmpB.121 Using nanocapsules, DL of 9%–10% (w/w) was obtained. In vivo results showed that more drug was delivered by mannosylated nanocapsules than by CNC or plain AmpB in both liver and spleen.
Polyelectrolyte complexes (PECs) are formed by interactions between two macromolecules bearing oppositely charged groups. Encapsulation of a drug during formation of PECs has shown great promise for preparing drug delivery carriers. For this purpose, the native positively charged polymer chitosan, and chondroitin sulfate, of negative charge was used to load AmpB,122 resulting in a good DL (11%, w/w). In evaluating the antileishmanial activity, it was observed that chitosan itself presented activity and a synergistic interaction between chitosan and chondroitin sulfate also occurred. The activity of AmpB nanoparticles was similar to those with pure AmpB; however, the cytotoxicity of the nanoparticle formulation was approximately one-tenth that of pure AmpB. Neither pure chitosan nor chondroitin sulfate, or the nanoparticles, presented any significant macrophages toxicity.122
Chitosan was also chemically modified into glycol chitosan stearate, an amphiphilic copolymer that forms carriers with a hydrophilic shell and a hydrophobic core comprising stearic acid.123 By increasing the rigidity of the bilayer of so-called lipo-polymerosomes (L-Psomes) using cholesterol, a very large amount of AmpB was loaded (25%), exploiting the hydrophobic interaction with the stearic acid chain. The AmpB–L-Psomes proved to be a biologically safe and stable delivery system, with improved efficacy compared to commercial formulations; they can thus be proposed as an alternative low-cost product. A further study exploited a surface interaction between chitosan and sodium alginate (natural polymer with anionic charge) to stabilize the nanoparticles, maintaining the high AmpB loading (22%).124 The biopolymers sodium alginate and chitosan are potent macrophage activators, inducing the release of different cytokines and cytotoxic agents.118 In vivo study demonstrated that L-Psomes facilitated the AmpB uptake in RES-rich organs and showed that AmpB remains in the tissues for prolonged periods, followed by slower distribution of the drug into the blood stream. Increased tolerance to AmpB was also observed.118
Alginate has also been used as active coating of a liposomal AmpB preparation117 and, more recently, of doxorubicin loaded in an NE (soybean oil with Tween as surfactant and protamine sulfate).125 This treatment strategy is based on the immune response modulation, combined with chemotherapy, to improve efficacy during infection.
Ionic amphiphile biovectors, first reported by Loiseau et al,126 comprised anionic lipids (dipalmitoyl phosphatidyl glycerol) included in a cationic cross-linked maltodextrin matrix, used as reservoir for AmpB. The nanoparticles of 100 nm were loaded with 20% (w/w) AmpB and demonstrated a high stability. The in vitro and in vivo activities remained in the same dose and concentration range as Fungizone and AmBisome, suggesting that their interaction with host cells, their uptake, and intracellular distribution were efficient, although the formulation is slightly less active and safe than AmBisome.126 Although in the early development stage, ionic amphiphile biovectors have not been further improved or described. However, hydroxypropyl-γ-cyclodextrin was used to solubilize AmpB solution (AmpB–dextrin solution) by inclusion; it was then formulated into nonionic surfactant vesicles (NIVs) (consisting of a 3:3:1 molar ratio of mono-n-hexadecylether tetraethylene glycol to cholesterol to dicetylphosphate). The antileishmanial activity of NIV and chitosan-coated NIV was compared with that of commercial AmpB formulations.127 It has been shown that the pulmonary route is not only suitable for lung conditions but can also be used to target the liver: treatment with an NIV formulation of AmpB significantly reduced L. donovani liver parasite burdens in treated mice and was also effective at reducing Aspergillus levels in the lungs.100 Furthermore, a reduced toxicity in L. major-infected mice after several (15) doses indicated that both formulations, as well as the pulmonary route, minimized systemic exposure to AmpB.
Polymeric micelles
Polymeric micelles are nanoscopic core/shell structures made up of amphiphilic block copolymers. Both the intrinsic and the modifiable properties of polymeric micelles make them particularly well suited for drug delivery purposes.128 Among the polymers used for micelle preparation, poloxamers (α-hydro-ω-hydroxypoly[oxyethylene]a poly[oxypropylene]b poly[oxyethylene]a block copolymers) present interesting behavior and have been applied in a wide range of biomedical and pharmaceutical industries. Particularly, poloxamer in aqueous media exhibits micellar structures, which can convert into gel-like structures depending on their length, concentration, and temperature. In the search for new delivery systems to treat tegumentary leishmaniasis, AmpB-containing a polymeric micelle system (namely AmpB/M) was developed using poloxamer P407 (Pluronic® F127) and its in vitro and in vivo potency against L. amazonensis was evaluated.129 When administered by subcutaneous injection in mice, the AmpB/M formulation turns into a semirigid gel on contact with local tissue, creating a reservoir system and keeping the drug in the extracellular space; this allows it to act against parasites at the local infection site. Over time, as the gel matrix is diluted by body fluids and phagocytosis, the drug is gradually released into systemic circulation, enabling its systemic action to be controlled.
A poloxamer P407-based paramomycin-containing micelle nanogel system has been evaluated. The system was tested for in vivo tolerance, ex vivo cytotoxicity on cells, and antileishmanial activity against promastigotes. The results showed the product to possess negligible toxicity and effective antileishmanial activity against L. major and L. infantum promastigotes. It was concluded that the formulation provides controlled, effective, and safe delivery of paramomycin in mice.130
An interesting approach to obtain stable micelles with high DL is based on the self-assembling ability of prodrugs with amphiphilic properties. Regarding pentavalent antimonials, the use of nonionic surfactants as complex agents has been investigated.131 The formation of an amphiphilic antimony (V) complex using N-alkyl-N-methylglucamide (with carbon chain lengths of 8 and 10) produced stable nanodispersions (~115 nm). The most significant advantages of the system were its great solubility in water, its superior bioavailability, and sustained drug release properties. These results were much better than those obtained with a cyclodextrin-based oral formulation of MA proposed by the same group.132 Micelles of the amphiphilic antimony (V) complex may also offer a possibility for solubilizing lipophilic drugs and achieving oral coadministration of anti-VL drugs.
Inorganic compounds as delivery systems
The majority of nanoparticles of pharmaceutical interest are made of organic polymers (biodegradable or not), but inorganic systems, as well as organic–inorganic hybrid materials, are now also receiving considerable attention in the pharmaceutical field. Mesoporous silica nanoparticles, hydroxyapatite nanostructures, metallic nanoparticles such as Fe3O4, gold, and silver nanoparticles, and carbon-based nanostructures (nanotubes, graphene, nano-onions, and horns) exhibit several interesting features and enable different functionalities (eg, diagnostic and therapeutic) to be combined in a single device.133,134
Small clearly defined aggregates of noble metals can be produced with very precise and controlled methods, modulating the reaction conditions used in their preparation. Metals in nanoparticulate form might be of interest in treating leishmaniasis, owing to their ability to produce ROS. In particular, silver nanoparticles135 were found to inhibit the survival of amastigotes in host cells and, in the presence of UV light, the effect was more significant. L. tropica promastigotes lost their shape, and their internal organelles were no longer distinguishable.135
Metal oxide nanoparticles, especially titanium dioxide (TiO2), silver oxide (Ag2O), zinc oxide (ZnO), magnesium oxide (MgO), and, more recently, selenium oxide (SeO2) nanoparticles, have been extensively explored, demonstrating a significant antibacterial activity.136 Among metals, the highest antileishmanial activity was observed for nanoparticles composed of Ag, followed by Au, Ti, Zn, and Mg. Both UV and infrared irradiation increased the antileishmanial properties of all these nanoparticles. However, they were found to possess macrophages cytotoxicity. Selenium nanoparticles demonstrated less cytotoxic effect on uninfected macrophages but have stronger antiamastigote and antipromastigote activities than SeO2.137
Another study focused on the development of six biocompatible nanoparticles of ZnO doped with different concentrations of Cu and their in vitro efficacy against the Leishmania parasite.138 Especially those from ZnCuO to ZnCuO5 have been proposed against leishmaniasis, because of their biocompatibility and enhanced ROS yield: after irradiation, they generated measurable quantities of singlet oxygen. Similarly, with Ag as dopant of the semiconductor, nanoparticles of zinc oxide demonstrated the production of ROS when activated by daylight and were able to kill L. tropica.139
More recently, two approaches were combined, using AmpB adsorbed on preformed spherical biogenic silver nanoparticles surface. AmB–Ag nanoparticles were more potent in inhibiting L. tropica in in vitro tests. Furthermore, the antileishmanial activity of the prepared nanoparticles was enhanced upon irradiation with visible light, which may be attributed to the formation of excess ROS by the released silver ions. The study limitation is the lack of the information about the amount of AmpB loaded.140
Among the inorganic nanomaterials, carbon compounds are attracting interest due to their excellent mechanical, thermal, and optical properties. Drugs may be covalently linked to carbon nanotubes (CNTs) or strongly absorbed by π–π stacking on their surface (eg, doxorubicin). However in its early ages in anti-Leishmania applications, a way of using multiwalled CNT to deliver covalently bound AmpB has been devised.141 Unusually, this product was obtained without using any condensing agent and was thought to be covalently linked via an ether bond instead of an amide bond. The intraperitoneal administration route led to a higher percentage suppression of parasites in the spleen with CNT–AmpB (89.8%) than with AmpB (68.9%). Without any improvement in characterization, the same formulation was orally administered, resulting in antileishmanial activity in hamster spleen tissues.142
Due to their dimensions, similar to those of the inorganic components of calcified tissues, nanosized calcium phosphate materials are expected to have better bioactivity and biocompatibility than conventional materials. Furthermore, calcium phosphate is a cheap drug cargo that is chemically stable, composed of inorganic minerals native to the body, which can be metabolized as a nontoxic degradation product.143,144
The preparation of calcium phosphate nanoparticles loaded with AmpB has been reported.145 The method involved mixing two microemulsions containing, separately, calcium chloride and disodium phosphate, and sodium silicate in the presence of a surfactant. Nanoparticles <200 nm in size, with an interesting AmpB loading of 11%, were obtained. In vivo efficacy of the formulation was assessed in a L. donovani-infected golden hamster model. The in vivo activity of AmpB-loaded nanoparticles was stronger than that of marketed formulations or AmpB suspension, which is the collective outcome of higher internalization by macrophages via clathrin receptor-mediated phagocytosis and enhancement of Th1-biased immune response by nanoparticles, which ultimately targeted macrophages and retarded nonspecific distribution of AmpB. Furthermore, the calcium phosphate nanoformulation was able to release the encapsulated drug in a slow and sustained manner, with preferential accumulation in macrophage-rich organs such as liver and spleen. A toxicity assessment study was also reported.145
Similar approach was also used to investigate the potential oral administration of pentavalent antimonials. Nanoparticle phosphate-based composites containing Sb(V) have been explored in terms of their in vitro activity.146 This system allowed the Sb(V) delivery into the cells in which the parasite is internalized, and the elevation of calcium concentrations inside the vacuoles might increase parasite apoptosis.
Covalently linked drug to polymers
In the nanosystems described earlier, the drug was always adsorbed, loaded, or entrapped in its native form. The covalent conjugation of drugs to an appropriate carrier is another strategy used to deliver drugs to particular target sites. The development of specific carrier–drug conjugates was first proposed for the delivery of small hydrophobic drug molecules to their sites of action147 and has recently reached significant results in anticancer therapy with antibody–drug conjugates. These delivery systems basically comprise a water-soluble polymer, bearing a number of drug molecules covalently linked. The main advantages of this approach are 1) increased water solubility of little soluble or insoluble drugs, therefore enhancing drug bioavailability; 2) protection of drugs from deactivation and relative activity preservation during circulation; and 3) most importantly, specifically drug targeting to the site of action. Unlike other approaches, in this case, the active agent must be considered as a prodrug that generates the activity only after release. For this reason, the role of linker between polymer and drug is of paramount importance, because it defines the mechanism for drug release (pH, enzyme derived, chemical reaction, and so on). Although many are undergoing the final phase of clinical trials and several polymer–protein conjugates have been approved by the FDA for clinical use, to date no polymer–synthetic drug conjugate has been approved.148
Polymer–drug conjugates have been used for targeting macrophages and have already shown potential in antileishmanial chemotherapy. HPMA is undoubtedly the best-known synthetic polymer exploited in macromolecular conjugates. It is water soluble and nonimmunogenic and has undergone clinical trials in the cancer field.149 In a detailed article, the synthesis of HPMA conjugate of AmpB obtained through a degradable GlyPheLeuGly linker is described. The linker was chosen as it is known to be cleaved by cathepsin B, which has been found in the PV.150 Its in vitro activity was comparable to that of free AmpB and Fungizone, and although less active than AmBisome in in vivo experiments (though of a similar order), no signs of toxicity were recorded, even at the maximum used dose of 3 mg/kg AmpB equivalent used. It should be noted that this dose is above the reported 50% lethal dose (LD50) of 2.5 mg/kg for Fungizone. No significant difference in antileishmanial activity was found using a similar but targeted conjugate with mannose.150 Further studies on polymer–drug conjugates in this field have not been reported, and clinical evaluation is thus lacking.
Another interesting natural polymer that can act as delivery platform and targeting scaffold simultaneously is hyaluronic acid.151 Recently, a versatile synthetic method to link pentamidine to hyaluronic acid, affording DL of up to 33% (w/w), was presented. Assayed against the intracellular amastigote form of the parasite, the bioconjugate was more efficient than free drug in the internalization of infected macrophages but, at the same time, less cytotoxic.152
Novel products with antileishmanial activity combined into nanocarriers
The need for alternative treatments, in particular to reduce the duration of clinical treatment, led to a program to screen natural products for potential use in leishmaniasis therapy, as recent reviews report,153–156 and also to the screening of currently available medicines with potential use in VL. In parallel, quantitative structure–activity relationship (QSAR) study has been used to identify new organic compounds with stronger activity.157 Another approach examined the role played by known active molecules in medicinal plants traditionally used against leishmaniasis, as well as in documented plants that have yet to be explored.158–161
Several active agents from different chemical classes are also reported to have in vitro antileishmanial activity. The more promising candidates appear to be the nitroimidazoles, quinoline scaffold-based derivatives such as indolyl quinoline analogs, naphthoquinones, the cytotoxic drug doxorubicin, and also antimicrobial peptides.108,162 A recent review clearly summarizes these results.163 Disulfiram, a drug extensively used to treat alcoholism, has recently been reported to be active at nanomolar concentrations against the intracellular amastigotes of L. donovani.164 Although clinical trials have not yet been carried out to assess the efficacy of disulfiram as monotherapy or in combination to treat different types of leishmaniasis, the implementation of a clinical trial is feasible, since disulfiram is a known and approved drug and, furthermore, is administered orally.
OlPC is a structural analog of MF. The in vitro efficacy of OlPC against the intracellular amastigotes of different Leishmania spp. was similar to the profile of MF, with IC50 values in the low micromolar range, except for L. major and L. braziliensis.165 When tested on a VL hamster model with L. infantum, OlPC was found to be more potent than MF after multiple or single oral dosing. Furthermore, short oral treatment with OlPC improves clinical signs of canine L. infantum leishmaniasis.166 The pharmacokinetic profiles of OlPC, administered either in water or as a liposomal preparation, have also been reported recently.167
Natural agent, namely andrographolide, is a diterpenoid lactone extracted from the leaves of Andrographis paniculata, with strong antiparasitic and antileishmanial activity.168 Further progress was achieved upon studying andrographolide’s behavior regarding resistance, with PLGA nanoparticles loaded with andrographolide and stabilized with vitamin E TPGS.169 Nanoparticles were delivered into macrophage cells infested with sensitive and drug-resistant amastigotes of L. donovani parasites. Antileishmanial activity was found to be significant for the nanoparticles with TPGS at about one-tenth of the free drug dosage and one-third of the nanoparticles without TPGS dosage. When andrographolide was incorporated in TPGS nanoparticles, the selectivity of andrographolide increased eightfold in resistant cells.169
Recently, the same approach was used to deliver oleanolic acid (OA), a naturally occurring pentacyclic triterpene with antileishmanial activity.170 In vivo evaluation indicated that OA suppressed the amastigote burden in the spleen after the fourth dose (67.69%±4.12%), whereas the percentage of suppression was increased to 98.82%±1.92% in the case of OA PLGA nanoformulation.
A further example concerns bisnaphthalimidopropyl (BNIP) derivatives, which exert significant antiproliferative activity on L. infantum.171 Encapsulation of BNIP aminooctane derivative into PLGA nanoparticles resulted in a reduction of aspecific toxicity and in the 80% increase of in vivo efficiency on L. infantum compared to free drug.172 This nanoformulation remained active after oral administration in infected BALB/c mice.
Artemisinin, a potent drug used against Plasmodium falciparum malaria, potentially possesses antileishmanial activity, although bioavailability and stability are low.173 Encapsulating artemisinin into PLGA nanoparticles reduced the free drug macrophage toxicity, increasing leishmanicidal activity.174 Furthermore, NO production may intercede in the antileishmanial effect of the nanoformulations. Unfortunately, the curative effect was not as high as the standard liposomal AmpB formulation.175
Curcumin (diferuloylmethane), the main yellow bioactive component of turmeric, has been shown to have a wide spectrum of biological action, including leishmanicidal activity in vitro.176 Different approaches have been proposed to deliver curcumin. A particular type of prodrug-based approach, named “squalenoylation” has been proposed.177 In this approach, the derivatization of small molecules by conjugation with a squalene moiety enables conjugates to be produced that spontaneously self-assemble into 60–300 nm nanoparticles. Although to date it has mainly been applied in the anticancer field,178 a squalenoyl prodrug of curcumin was recently investigated in promastigote and amastigote forms of L. donovani and showed enhanced activity vs the parent drug.179 Recently, PLGA nanoparticles loaded with curcumin (nanocurcumin) were proposed to support MF in exerting leishmanicidal effect at a subcurative dose through immunomodulation. Nanocurcumin monotherapy or combination therapy with MF did not exhibit any hepato- or nephrotoxicity effect.180
Compound 8-hydroxyquinoline (8-HQN) and its derivates have been experimentally evaluated in different studies as chemotherapeutic agents with interesting antileishmaniasis activity.181,182 Recently, polymeric micelle system was exploited to deliver 8-HQN.183 After daily subcutaneous injections for 15 days in L. infantum-infected mice, the treated group showed a more marked reduction in the parasite load than the control group. The formulation was highly effective in treating the disease, more so than free 8-HQN or AmpB; the highest levels of nitrite were found in the 8-HQN/M-treated mice, together with a significant Th1 immune response.
In a different approach NPC1161, a model 8-aminoquinoline drug with antileishmanial activity was conjugated with HPMA and targeted with N-acetylmannosamine.184
Inorganic carrier such as CNTs was recently used to deliver betulin, a pentacyclictriterpenoid with antileishmanial properties.185 Counting the amastigote cells inside the infected macrophage cells using a microscope, a dose-dependent response was observed with betulin and with CNT-linked betulin, whereas CNT alone did not show any antileishmanial effect. In addition, CNT was seen to downregulate the activity of P-gp efflux, which would help to increase the bioavailability of any drug incorporated into the carrier system and overcome resistance mechanisms. However, toxicity data are still limited, and further effort will be required to evaluate in depth not only the potency but also the in vivo fate of these formulations (eg, biodistribution and elimination from the body).185 A complete toxicological evaluation must also be made before they can be administered safely.
Discussion
At present, only a small number of antileishmanial drugs are available for clinical use: three injectables and the sole orally administrable MF. There is thus a pressing need to develop other antileishmanial drugs as each of the available drugs suffers from various drawbacks (toxicity, repeated dose requirement, and so on).
To reduce dosage, enable improved release, and promote high drug concentrations in the intracellular (macrophage) vesicle where Leishmania is hosted, DDSs may play a crucial role. Colloidal carriers can also protect drugs against extracellular degradation, improving the pharmacokinetic profile of the active agent. The most significant result is the case of AmBisome, which has significantly improved VL treatment, as it provides almost complete eradication of parasite burden, with minor toxic effects. However, the cost of this lipid formulation is a severe deterrent, in the light of the need to provide a reasonably priced therapeutic option to third world countries. Resistance to AmpB has not yet been observed in clinical practice, although it has been reported in in vitro study.29 Thus, the DDS approach had not been yet evaluated on resistant lines.
Since AmpB is currently the most potent antileishmanial agent, the majority of efforts in nanotechnology have been addressed to improving its specificity, reducing its adverse effects, and in some cases, using noninvasive route. For this reason, in this section, we critically evaluate and discuss the role and advantages that may be played by innovative AmpB delivery systems. The principal systems, methods and results, described in the text, are summarized in Table 2. Considering the structure, the majority of the systems comprise polymeric nanoparticles or nanocapsules of size (100–300 nm) greater than that of AmBisome. As far as, the stability of the carrier is concerned, the polymeric systems are much more stable than liposomes, and from the regulatory standpoint, the use of polymers that are already approved by FDA/EMA may reduce the need for toxicological studies, in comparison to other systems. PLA and PLGA are widely used in many FDA-approved drug products.96 DL is an important parameter to predict the strength of an approach. If drug capacity is <4%–5% (w/w) of the carrier material, either the quantity of drug administered will not be sufficient to achieve a pharmacologically active concentration in the relevant body compartment capable of eradicating the parasite burden, or the amount of carrier material required will be too large, leading to toxicity.186 The hydrophobic core of PLGA nanoparticle (NP) and LPNP facilitates the loading of AmpB, which can reach significant levels. Another issue in nanoparticle systems that may lead to lower activity, and potentially to more toxicity, is the rapid release or premature “burst release” of the encapsulated drug. This phenomenon is not frequent in the case of AmpB, because in polymeric NP, only a small fraction of the drug is present in the free, monomeric form, the majority remaining superaggregated and associated with the delivery system.97
The majority of nanostructures achieved interesting ranges of DL (up to 44% in one case). Passive or “natural” targeting, since the MPS cells are considered to be the main target for therapeutic interventions in leishmaniasis, is a mechanism that may be used to enhance drug concentration, reducing aspecific toxicity (hematotoxicity and kidney toxicity). Nevertheless, the active targeting approach (using mannose, stearylamine, mannan, and antibody fragments) appears to slightly increase the uptake in liver and spleen lesions. In particular, the surface charge of LPNPs created by stearylamine enhanced uptake by macrophages, demonstrating interesting immunoadjuvant properties. In LPNP, the lipid component of the formulation also behaves as a targeting agent, while in the majority of formulations, surface decoration was preferred. In the latter case, clearly illustrated by the use of antibodies, difficulties may arise because there is as yet no clear information concerning the “optimum” range of ligand characteristics (ie, valency, avidity, conformation, and packing) on the surface of nanocarriers that results in reproducible target binding and internalization. Manufacturing these products may be challenging; this is clearly of great practical importance, as is shown by the lack of actively targeted liposomes on the market for cancer therapy.
Owing to their complexity, site-directed DDSs will always require full and in-depth characterization before being proposed for industrial development and manufacturing. Especially when novel components (lipids, lipid polysaccharides, and so on) with unknown toxicological properties are added to the formulation, or when too many complex steps are required, industrial interest decreases sharply. Regarding cost-effectiveness, PLGA, a pharmaceutically accepted polymer, including from the regulatory standpoint, is more advantageous than more “sophisticated” systems, although these may be of interest, as is the case of L-Psomes or targeted systems.
Nevertheless, it is important to notice that each study, able to combine novelty and promising in vivo results, should be compared with the best treatment available (AmBisome). This comparison was done only in 44% of the studies reported in Table 2.
Also molecular conjugates, typically prepared to increase drug pharmacokinetic profile, did not significantly improve the AmpB antileishmanial activity.150 A remarkable review by Duncan148 reflects very clearly on the current status of polymer therapeutics, the major issues concerned, and future opportunities.
Conclusion
Bearing in mind the results obtained in some clinical trials where a single-course iv therapy of 10 mg/kg AmBisome was found to give a 98% cure,26 the space for robust and market-oriented innovation seems to be represented mainly by the delivery of AmpB by oral route, but this delivery must be directed in targeted tissues to elicit its pharmacological effect while reducing its nephrotoxicity and infusion-related side effects.
Even if it remained difficult to select and design an appropriate formulation to improve AmpB oral absorption, some novel DDSs have emerged as effective. In particular, lipid cochleates and an SLN formulation (AmbiOnp) appear very promising, not least because they are already involved in industrial development. Both approaches provide high DL-reduced AmpB toxicity and production protocols that avoid expensive compounds or processes. Further improvement could involve a polymer such as chitosan, which combines bioadhesion and absorption-enhancing properties, to prolong contact between the DDS and the mucosa, increasing the amount of AmpB that can be absorbed.
In recent years, material sciences and biomedical sciences have produced numerous new systems and materials with the potential to treat disease. However, two major limitations must be borne in mind. First, as the recent literature concerning DDSs shows, there is a flourishing strain of articles in which increased complexity appears to be seen as an advantage, without any real justification in terms of the balance between complexity and feasibility. Second, complex delivery systems obviously cost more to develop and manufacture than do conventional therapeutic agents. The resulting higher prices may only be acceptable if the performance of these systems is sufficiently increased, thereby reducing the treatment period and overall cost, leading to savings to the health care system. It is becoming ever more imperative to formulate standards for system characterization, and robust means of comparative testing, so as to facilitate close and critical analysis of proposed strategies. Further efforts and investments will still be necessary for this approach to achieve greater clinical applicability in humans.
Meanwhile, with regard to the identification of novel drugs, there can be no doubt that the availability of the complete genome sequence of Leishmania has given the scientific community the possibility for large-scale analysis, which may lead to a better understanding of the parasite’s biology and consequent identification of novel drug targets.187 Furthermore, the use of powerful CRISPR–Cas9 techniques of genome editing is now making it possible to accelerate the characterization of Leishmania genes, so as to develop new drugs, diagnostics, and vaccines.188 From another standpoint, the medicinal chemistry approach continues to improve “old drugs”, suggesting novel AmpB derivatives with high activity and increased safety.189
However, since the goal is to eliminate parasitic infections, a “discovery science” for the development of drugs and diagnostics must be conducted on the global stage, involving, in the local context, scientists, clinicians, regulators, and health ministries in disease-endemic countries. Furthermore, “implementation science” must take place in the local context. To achieve the goal, it is of fundamental importance to involve multidisciplinary teams, including pharmaceutical and medical scientists, policymakers, social scientists, health administrators, and communication scientists, working together in new ways.190
Disclosure
The authors report no conflicts of interest in this work.
References
Bañuls AL, Hide M, Prugnolle F. Leishmania and the leishmaniases: a parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv Parasitol. 2007;64(1–109):455–458. | ||
Séguin O, Descoteaux A. Leishmania, the phagosome, and host responses: the journey of a parasite. Cell Immunol. 2016;309:1–6. | ||
Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366(9496):1561–1577. | ||
Alvar J, Vélez ID, Bern C, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5):e35671. | ||
Lindgren E, Andersson Y, Suk JE, Sudre B, Semenza JC. Public health: monitoring EU emerging infectious disease risk due to climate change. Science. 2012;336(6080):418–419. | ||
Van Griensven J, Carrillo E, López-Vélez R, Lynen L, Moreno J. Leishmaniasis in immunosuppressed individuals. Clin Microbiol Infect. 2014;20(4):286–299. | ||
Hotez PJ, Pecoul B, Rijal S, et al. Eliminating the neglected tropical diseases: translational science and new technologies. PLoS Negl Trop Dis. 2016;10(3):e0003895. | ||
Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle uptake: the phagocyte problem. Nano Today. 2015;10(4):487–510. | ||
Gutiérrez V, Seabra AB, Reguera RM, Khandare J, Calderón M. New approaches from nanomedicine for treating leishmaniasis. Chem Soc Rev. 2016;45(1):152–168. | ||
Asthana S, Gupta PK, Chaurasia M, Dube A, Chourasia MK. Polymeric colloidal particulate systems: intelligent tools for intracellular targeting of antileishmanial cargos. Expert Opin Drug Deliv. 2013;10(12):1633–1651. | ||
Srivastava S, Shankar P, Mishra J, Singh S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasit Vectors. 2016;9(1):277. | ||
Biswas S, Subramanian A, Elmojtaba IM, Chattopadhyay J, Sarkar RR. Optimal combinations of control strategies and cost-effective analysis for visceral leishmaniasis disease transmission. PLoS One. 2017;12(2):e0172465. | ||
Meheus F, Balasegaram M, Olliaro P, et al. Cost-effectiveness analysis of combination therapies for visceral leishmaniasis in the Indian subcontinent. PLoS Negl Trop Dis. 2010;4(9):e818. | ||
Meheus F, Abuzaid AA, Baltussen R, et al. The economic burden of visceral leishmaniasis in Sudan: an assessment of provider and household costs. Am J Trop Med Hyg. 2013;89(6):1146–1153. | ||
Tachfouti N, Najdi A, Alonso S, et al. Cost of pediatric visceral leishmaniasis care in Morocco. PLoS One. 2016;11(6):e0155482. | ||
Uranw S, Meheus F, Baltussen R, Rijal S, Boelaert M. The household costs of visceral leishmaniasis care in south-eastern Nepal. PLoS Negl Trop Dis. 2013;7(2):e2062. | ||
Shaked-Mishan P, Ulrich N, Ephros M, Zilberstein D. Novel intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J Biol Chem. 2001;276(6):3971–3976. | ||
Walker J, Saravia NG. Inhibition of Leishmania donovani promastigote DNA topoisomerase I and human monocyte DNA topoisomerases I and II by antimonial drugs and classical antitopoisomerase agent. J Parasitol. 2004;90(5):1155–1162. | ||
Demicheli C, Frézard F, Lecouvey M, Garnier-Suillerot A. Antimony(V) complex formation with adenine nucleosides in aqueous solution. Biochim Biophys Acta. 2002;1570(3):192–198. | ||
Jeddi F, Piarroux R, Mary C. Antimony resistance in Leishmania, focusing on experimental research. Re Dai Yi Xue Za Zhi. 2011;2011:695382. | ||
Leandro C, Campino L. Leishmaniasis: efflux pumps and chemoresistance. Int J Antimicrob Agents. 2003;22(3):352–357. | ||
Walsh TJ, Goodman JL, Pappas P, et al. Safety, tolerance, and pharmacokinetics of high-dose liposomal amphotericin B (AmBisome) in patients infected with Aspergillus species and other filamentous fungi: maximum tolerated dose study. Antimicrob Agents Chemother. 2001;45(12):3487–3496. | ||
Brajtburg J, Powderly WG, Kobayashi GS, Medoff G. Amphotericin B: current understanding of mechanisms of action. Antimicrob Agents Chemother. 1990;34(2):183–188. | ||
Olliaro PL, Guerin PJ, Gerstl S, Haaskjold AA, Rottingen J-A, Sundar S. Treatment options for visceral leishmaniasis: a systematic review of clinical studies done in India, 1980–2004. Lancet Infect Dis. 2005;5(12):763–774. | ||
Balasegaram M, Ritmeijer K, Lima MA, et al. Liposomal amphotericin B as a treatment for human leishmaniasis. Expert Opin Emerg Drugs. 2012;17(4):493–510. | ||
Sundar S, Chakravarty J, Agarwal D, Rai M, Murray HW. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. N Engl J Med. 2010;362(6):504–512. | ||
Sundar S, Chakravarty J, Agarwal D, Shah A, Agrawal N, Rai M. Safety of a pre-formulated amphotericin B lipid emulsion for the treatment of Indian Kala-azar. Trop Med Int Health. 2008;13(9):1208–1212. | ||
Sundar S, Pandey K, Thakur CP, et al. Efficacy and safety of amphotericin B emulsion versus liposomal formulation in Indian patients with visceral leishmaniasis: a randomized, open-label study. PLoS Negl Trop Dis. 2014;8(9):e3169. | ||
Mbongo N, Loiseau PM, Billion MA, Robert-Gero M. Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob Agents Chemother. 1998;42(2):352–357. | ||
Purkait B, Kumar A, Nandi N, et al. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother. 2012;56(2):1031–1041. | ||
Chawla B, Jhingran A, Panigrahi A, Stuart KD, Madhubala R. Paromomycin affects translation and vesicle-mediated trafficking as revealed by proteomics of paromomycin–susceptible–resistant Leishmania donovani. PLoS One. 2011;6(10):e26660. | ||
Sundar S, Chakravarty J. Paromomycin in the treatment of leishmaniasis. Expert Opin Investig Drugs. 2008;17(5):787–794. | ||
Dorlo TPC, Balasegaram M, Beijnen JH, de Vries PJ. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67(11):2576–2597. | ||
World-Health-Organization. Control of the Leishmaniasis: Report of a Meeting of the WHO Expert Committee (N° 949) on the Control of Leishmaniases. Geneva: World Health Organization; 2010. | ||
Lux H, Heise N, Klenner T, Hart D, Opperdoes FR. Ether-lipid (alkyl-phospholipid) metabolism and the mechanism of action of ether-lipid analogues in Leishmania. Mol Biochem Parasitol. 2000;111(1):1–14. | ||
Sundar S, Sinha P, Jha TK, et al. Oral miltefosine for Indian post-kala-azar dermal leishmaniasis: a randomised trial. Trop Med Int Health. 2013;18(1):96–100. | ||
Srivastava S, Mishra J, Gupta AK, Singh A, Shankar P, Singh S. Laboratory confirmed miltefosine resistant cases of visceral leishmaniasis from India. Parasit Vectors. 2017;10(1):49. | ||
Alvar J, González U, Pinart M, et al. Interventions for visceral leishmaniasis. Cochrane Database Syst Rev. 2010;6. | ||
van Griensven J, Boelaert M. Combination therapy for visceral leishmaniasis. Lancet. 2011;377(9764):443–444. | ||
Sundar S, Sinha PK, Rai M, et al. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet. 2011;377(9764):477–486. | ||
Wasunna M, Njenga S, Balasegaram M, et al. Efficacy and safety of ambisome in combination with sodium stibogluconate or miltefosine and miltefosine monotherapy for African visceral leishmaniasis: phase II randomized trial. PLoS Negl Trop Dis. 2016;10(9):e0004880. | ||
Barrett MP, Boykin DW, Brun R, Tidwell RR. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol. 2007;152(8):1155–1171. | ||
Diro E, Ritmeijer K, Boelaert M, et al. Use of pentamidine as secondary prophylaxis to prevent visceral leishmaniasis relapse in HIV infected patients, the first twelve months of a prospective cohort study. PLoS Negl Trop Dis. 2015;9(10):e0004087. | ||
Georgiadou SP, Stefos A, Spanakos G, et al. Current clinical, laboratory, and treatment outcome characteristics of visceral leishmaniasis: results from a seven-year retrospective study in Greece. Int J Infect Dis. 2015;34:46–50. | ||
Basselin M, Coombs GH, Barrett MP. Putrescine and spermidine transport in Leishmania. Mol Biochem Parasitol. 2000;109(1):37–46. | ||
Singh G, Dey CS. Induction of apoptosis-like cell death by pentamidine and doxorubicin through differential inhibition of topoisomerase II in arsenite-resistant L. donovani. Acta Trop. 2007;103(3):172–185. | ||
Basselin M, Denise H, Coombs GH, Barrett MP. Resistance to pentamidine in Leishmania mexicana involves exclusion of the drug from the mitochondrion. Antimicrob Agents Chemother. 2002;46(12):3731–3738. | ||
Wyllie S, Patterson S, Stojanovski L, et al. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci Transl Med. 2012;4(119):119re1. | ||
Thompson AM, O’Connor PD, Blaser A, et al. Repositioning antitubercular 6-Nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for neglected tropical diseases: structure–activity studies on a preclinical candidate for visceral leishmaniasis. J Med Chem. 2016;59(6):2530–2550. | ||
Gupta S, Yardley V, Vishwakarma P, et al. Nitroimidazo-oxazole compound DNDI-VL-2098: an orally effective preclinical drug candidate for the treatment of visceral leishmaniasis. J Antimicrob Chemother. 2015;70(2):518–527. | ||
Lu Y, Aimetti AA, Langer R, Gu Z. Bioresponsive materials. Nat Rev Mater. 2016;1:16075. | ||
Jain K, Jain NK. Novel therapeutic strategies for treatment of visceral leishmaniasis. Drug Discov Today. 2013;18(23–24):1272–1281. | ||
Italia JL, Yahya MM, Singh D, Ravi Kumar MNV. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous fungizone®. Pharm Res. 2009;26(6):1324–1331. | ||
Torrado JJ, Serrano DR, Uchegbu IF. The oral delivery of amphotericin B. Ther Deliv. 2013;4(1):9–12. | ||
Torrado JJ, Espada R, Ballesteros MP, Torrado-Santiago S. Amphotericin B formulations and drug targeting. J Pharm Sci. 2008;97(7):2405–2425. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Larabi M, Yardley V, Loiseau PM, et al. Toxicity and antileishmanial activity of a new stable lipid suspension of amphotericin B. Antimicrob Agents Chemother. 2003;47(12):3774–3779. | ||
Mishra J, Dey A, Singh N, Somvanshi R, Singh S. Evaluation of toxicity and therapeutic efficacy of a new liposomal formulation of amphotericin B in a mouse model. Indian J Med Res. 2013;137(4):767–776. | ||
Asad M, Bhattacharya P, Banerjee A, Ali N. Therapeutic and immunomodulatory activities of short-course treatment of murine visceral leishmaniasis with KALSOME™10, a new liposomal amphotericin B. BMC Infect Dis. 2015;15(1):188. | ||
Shadab M, Jha B, Asad M, Deepthi M, Kamran M, Ali N. Apoptosis-like cell death in Leishmania donovani treated with KalsomeTM10, a new liposomal amphotericin B. PLoS One. 2017;12(2):e0171306. | ||
New RRC, Chance ML, Thomas SC, Peters W. Antileishmanial activity of antimonials entrapped in liposomes. Nature. 1978;272(5648):55–56. | ||
Carter KC, Dolan TF, Alexander J, Baillie AJ, McColgan C. Visceral leishmaniasis: drug carrier system characteristics and the ability to clear parasites from the liver, spleen and bone marrow in Leishmania donovani infected BALB/c mice. J Pharm Pharmacol. 1989;41(2):87–91. | ||
Roychoudhury J, Sinha R, Ali N. Therapy with sodium stibogluconate in stearylamine-bearing liposomes confers cure against SSG-resistant Leishmania donovani in BALB/c mice. PLoS One. 2011;6(3):e17376. | ||
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. | ||
Azevedo EG, Ribeiro RR, da Silva SM, et al. Mixed formulation of conventional and pegylated liposomes as a novel drug delivery strategy for improved treatment of visceral leishmaniasis. Expert Opin Drug Deliv. 2014;11(10):1551–1560. | ||
Proulx M-E, Désormeaux A, Marquis J-F, Olivier M, Bergeron MG. Treatment of visceral leishmaniasis with sterically stabilized liposomes containing camptothecin. Antimicrob Agents Chemother. 2001;45(9):2623–2627. | ||
Keighobadi M, Fakhar M, Emami S. Hypothesis: the potential application of doxorubicin against cutaneous leishmaniasis. Trop Parasitol. 2015;5(1):69–70. | ||
Alving CR. Delivery of liposome-encapsulated drugs to macrophages. Pharmacol Ther. 1983;22(3):407–424. | ||
Javed I, Hussain SZ, Ullah I, et al. Synthesis, characterization and evaluation of lecithin-based nanocarriers for the enhanced pharmacological and oral pharmacokinetic profile of amphotericin B. J Mater Chem B. 2015;3(42):8359–8365. | ||
Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42(5):439–462. | ||
Agrawal AK, Agrawal A, Pal A, Guru PY, Gupta CM. Superior chemotherapeutic efficacy of Amphotericin B in tuftsin-bearing liposomes against Leishmania donovani infection in hamsters. J Drug Target. 2002;10(1):41–45. | ||
Vázquez-Mendoza A, Carrero JC, Rodriguez-Sosa M. Parasitic infections: a role for C-type lectins receptors. Biomed Res Int. 2013;2013:456352. | ||
Rathore A, Jain A, Gulbake A, et al. Mannosylated liposomes bearing amphotericin B for effective management of visceral leishmaniasis. J Liposome Res. 2011;21(4):333–340. | ||
Gupta A, Eral HB, Hatton TA, Doyle PS. Nanoemulsions: formation, properties and applications. Soft Matter. 2016;12(11):2826–2841. | ||
Wasan EK, Gershkovich P, Zhao J, et al. A novel tropically stable oral amphotericin B formulation (iCo-010) exhibits efficacy against visceral leishmaniasis in a murine model. PLoS Negl Trop Dis. 2010;4(12):1–6. | ||
Hnik P, inventor; iCo Therapeutics Inc., Can.; Laherty, Carol D, assignee. Stable formulations for the oral administration of amphotericin B. US patent WO2016112339A12016. 2016. | ||
Kansal S, Tandon R, Dwivedi P, et al. Development of nanocapsules bearing doxorubicin for macrophage targeting through the phosphatidylserine ligand: a system for intervention in visceral leishmaniasis. J Antimicrob Chemother. 2012;67(11):2650–2660. | ||
Shingitha KP. A review: niosomes a novel tool for drug delivery. Int J Pharm Technol. 2015;6(3):3017–3026. | ||
Baillie AJ, Coombs GH, Dolan TF, Laurie J. Non-ionic surfactant vesicles, niosomes, as a delivery system for the anti-leishmanial drug, sodium stibogluconate. J Pharm Pharmacol. 1986;38(7):502–505. | ||
Khazaeli P, Sharifi I, Talebian E, Heravi G, Moazeni E, Mostafavi M. Anti-leishmanial effect of itraconazole niosome on in vitro susceptibility of Leishmania tropica. Environ Toxicol Pharmacol. 2014;38(1):205–211. | ||
Salerno C, Chiappetta DA, Monteagudo E, Bregni C. Influence of surfactant structure in the encapsulation and stability of amphotericin B in niosomes. Latin Am J Pharm. 2011;30(9):1728–1736. | ||
Zarif L. Drug delivery by lipid cochleates. Methods Enzymol. 2005;391:314–329. | ||
Sesana AM, Monti-Rocha R, Vinhas SA, Morais CG, Dietze R, Lemos EM. In vitro activity of amphotericin B cochleates against Leishmania chagasi. Mem Inst Oswaldo Cruz. 2011;106(2):251–253. | ||
Mannino RJ, inventor; Biodelivery Sciences International, Inc., USA, assignee. Pharmaceutical cochleate compositions comprising amphotericin B for treating leishmaniasis. US patent WO2010091090A12010. 2010. | ||
Pham TT, Barratt G, Michel JP, Loiseau PM, Saint-Pierre-Chazalet M. Interactions of antileishmanial drugs with monolayers of lipids used in the development of amphotericin B-miltefosine-loaded nanocochleates. Colloids Surf B Biointerfaces. 2013;106:224–233. | ||
Battaglia L, Gallarate M. Lipid nanoparticles: state of the art, new preparation methods and challenges in drug delivery. Expert Opin Drug Deliv. 2012;9(5):497–508. | ||
Jain V, Gupta A, Pawar VK, et al. Chitosan-assisted immunotherapy for intervention of experimental leishmaniasis via amphotericin B-loaded solid lipid nanoparticles. Appl Biochem Biotechnol. 2014;174(4):1309–1330. | ||
Heidari-Kharaji M, Taheri T, Doroud D, Habibzadeh S, Rafati S. Solid lipid nanoparticle loaded with paromomycin: in vivo efficacy against Leishmania tropica infection in BALB/c mice model. Appl Microbiol Biotechnol. 2016;100(16):7051–7060. | ||
Heidari-Kharaji M, Taheri T, Doroud D, Habibzadeh S, Badirzadeh A, Rafati S. Enhanced paromomycin efficacy by solid lipid nanoparticle formulation against Leishmania in mice model. Parasite Immunol. 2016;38(10):599–608. | ||
Veerareddy PR, Vobalaboina V, Ali N. Antileishmanial activity, pharmacokinetics and tissue distribution studies of mannose-grafted amphotericin B lipid nanospheres. J Drug Target. 2009;17(2):140–147. | ||
Chaudhari MB, Desai PP, Patel PA, Patravale VB. Solid lipid nanoparticles of amphotericin B (AmbiOnp): in vitro and in vivo assessment towards safe and effective oral treatment module. Drug Deliv Transl Res. 2016;6(4):354–364. | ||
da Gama Bitencourt JJ, Pazin WM, Ito AS, et al. Miltefosine-loaded lipid nanoparticles: improving miltefosine stability and reducing its hemolytic potential toward erythtocytes and its cytotoxic effect on macrophages. Biophys Chem. 2016;217:20–31. | ||
Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 2012;41(7):2545–2561. | ||
Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv Drug Deliv Rev. 2008;60(15):1650–1662. | ||
Couvreur P, Puisieux F. Nano- and microparticles for the delivery of polypeptides and proteins. Adv Drug Deliv Rev. 1993;10(2):141–162. | ||
Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel). 2011;3(3):1377–1397. | ||
Van De Ven H, Paulussen C, Feijens PB, et al. PLGA nanoparticles and nanosuspensions with amphotericin B: potent in vitro and in vivo alternatives to Fungizone and AmBisome. J Control Release. 2012;161(3):795–803. | ||
Zhang Z, Tan S, Feng S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Italia JL, Ravi Kumar MNV, Carter KC. Evaluating the potential of polyester nanoparticles for per oral delivery of amphotericin B in treating visceral leishmaniasis. J Biomed Nanotechnol. 2012;8(4):695–702. | ||
Alsaadi M, Italia JL, Mullen AB, et al. The efficacy of aerosol treatment with non-ionic surfactant vesicles containing amphotericin B in rodent models of leishmaniasis and pulmonary aspergillosis infection. J Control Release. 2012;160(3):685–691. | ||
Kumar R, Sahoo GC, Pandey K, et al. Development of PLGA-PEG encapsulated miltefosine based drug delivery system against visceral leishmaniasis. Mater Sci Eng C Mater Biol Appl. 2016;59:748–753. | ||
Asthana S, Jaiswal AK, Gupta PK, Dube A, Chourasia MK. Th-1 biased immunomodulation and synergistic antileishmanial activity of stable cationic lipid–polymer hybrid nanoparticle: biodistribution and toxicity assessment of encapsulated amphotericin B. Eur J Pharm Biopharm. 2015;89:62–73. | ||
Nahar M, Jain NK. Preparation, characterization and evaluation of targeting potential of amphotericin b-loaded engineered PLGA nanoparticles. Pharm Res. 2009;26(12):2588–2598. | ||
Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336(2):367–375. | ||
Barros D, Lima SAC, Cordeiro-Da-Silva A. Surface functionalization of polymeric nanospheres modulates macrophage activation: relevance in leishmaniasis therapy. Nanomedicine. 2015;10(3):387–403. | ||
Singh PK, Sah P, Meher JG, et al. Macrophage-targeted chitosan anchored PLGA nanoparticles bearing doxorubicin and amphotericin B against visceral leishmaniasis. RSC Adv. 2016;6(75):71705–71718. | ||
Asthana S, Gupta PK, Jaiswal AK, Dube A, Chourasia MK. Targeted chemotherapy of visceral leishmaniasis by lactoferrin-appended amphotericin B-loaded nanoreservoir: in vitro and in vivo studies. Nanomedicine. 2015;10(7):1093–1109. | ||
Silva T, Abengózar MA, Fernandez-Reyes M, et al. Enhanced leishmanicidal activity of cryptopeptide chimeras from the active N1 domain of bovine lactoferrin. Amino Acids. 2012;43(6):2265–2277. | ||
Kumar R, Sahoo GC, Pandey K, Das V, Das P. Study the effects of PLGA-PEG encapsulated Amphotericin B nanoparticle drug delivery system against Leishmania donovani. Drug Deliv. 2015;22(3):383–388. | ||
Pushpamalar J, Veeramachineni AK, Owh C, Loh XJ. Biodegradable polysaccharides for controlled drug delivery. ChemPlusChem. 2016;81(6):504–514. | ||
Sharma G, Kar S, Basu Ball W, Ghosh K, Das PK. The curative effect of fucoidan on visceral leishmaniasis is mediated by activation of MAP kinases through specific protein kinase C isoforms. Cell Mol Immunol. 2014;11(3):263–274. | ||
Lehnhardt Pires C, Rodrigues SD, Bristot D, et al. Evaluation of macroalgae sulfated polysaccharides on the Leishmania (L.) amazonensis promastigote. Mar Drugs. 2013;11(3):934–943. | ||
Kangussu-Marcolino MM, do Rosário MMT, Noseda MD, et al. Acid heteropolysaccharides with potent antileishmanial effects. Int J Biol Macromol. 2015;81:165–170. | ||
Matte C, Marquis JF, Blanchette J, et al. Peroxovanadium-mediated protection against murine leishmaniasis: role of the modulation of nitric oxide. Eur J Immunol. 2000;30(9):2555–2564. | ||
Amaral AED, Petkowicz CLO, Mercê ALR, et al. Leishmanicidal activity of polysaccharides and their oxovanadium(IV/V) complexes. Eur J Med Chem. 2015;90:732–741. | ||
Machado PA, Mota VZ, Cavalli ACL, et al. High selective antileishmanial activity of vanadium complex with stilbene derivative. Acta Trop. 2015;148:120–127. | ||
Singodia D, Khare P, Dube A, Talegaonkar S, Khar RK, Mishra PR. Development and performance evaluation of alginate-capped amphotericin B lipid nanoconstructs against visceral leishmaniasis. J Biomed Nanotechnol. 2011;7(1):123–124. | ||
Yang D, Jones KS. Effect of alginate on innate immune activation of macrophages. J Biomed Mater Res A. 2009;90(2):411–418. | ||
Porporatto C, Bianco ID, Riera CM, Correa SG. Chitosan induces different L-arginine metabolic pathways in resting and inflammatory macrophages. Biochem Biophys Res Commun. 2003;304(2):266–272. | ||
Asthana S, Jaiswal AK, Gupta PK, Pawar VK, Dube A, Chourasia MK. Immunoadjuvant chemotherapy of visceral leishmaniasis in hamsters using amphotericin B-encapsulated nanoemulsion template-based chitosan nanocapsules. Antimicrob Agents Chemother. 2013;57(4):1714–1722. | ||
Asthana S, Gupta PK, Jaiswal AK, Dube A, Chourasia MK. Overexpressed macrophage mannose receptor targeted nanocapsules-mediated cargo delivery approach for eradication of resident parasite: in vitro and in vivo studies. Pharm Res. 2015;32(8):2663–2677. | ||
Ribeiro TG, Chávez-Fumagall MA, Valadares DG, et al. Novel targeting using nanoparticles: an approach to the development of an effective anti-leishmanial drug-delivery system. Int J Nanomedicine. 2014;9(1):877–890. | ||
Gupta PK, Jaiswal AK, Kumar V, et al. Covalent functionalized self-assembled lipo-polymerosome bearing amphotericin B for better management of leishmaniasis and its toxicity evaluation. Mol Pharm. 2014;11(3):951–963. | ||
Gupta PK, Jaiswal AK, Asthana S, et al. Self assembled ionically sodium alginate cross-linked amphotericin B encapsulated glycol chitosan stearate nanoparticles: applicability in better chemotherapy and non-toxic delivery in visceral leishmaniasis. Pharm Res. 2015;32(5):1727–1740. | ||
Kansal S, Tandon R, Verma A, et al. Coating doxorubicin-loaded nanocapsules with alginate enhances therapeutic efficacy against Leishmania in hamsters by inducing Th1-type immune responses. Br J Pharmacol. 2014;171(17):4038–4050. | ||
Loiseau PM, Imbertie L, Bories C, Betbeder D, De Miguel I. Design and antileishmanial activity of amphotericin B-loaded stable Ionic Amphiphile Biovector formulations. Antimicrob Agents Chemother. 2002;46(5):1597–1601. | ||
Mullen AB, Carter KC, Baillie AJ. Comparison of the efficacies of various formulations of amphotericin B against murine visceral leishmaniasis. Antimicrob Agents Chemother. 1997;41(10):2089–2092. | ||
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–131. | ||
Mendonça DVC, Lage LMR, Lage DP, et al. Poloxamer 407 (Pluronic® F127)-based polymeric micelles for amphotericin B: in vitro biological activity, toxicity and in vivo therapeutic efficacy against murine tegumentary leishmaniasis. Exp Parasitol. 2016;169:34–42. | ||
Brugués AP, Naveros BC, Calpena Campmany AC, Pastor PH, Saladrigas RF, Lizandra CR. Developing cutaneous applications of paromomycin entrapped in stimuli-sensitive block copolymer nanogel dispersions. Nanomedicine. 2015;10(2):227–240. | ||
Fernandes FR, Ferreira WA, Campos MA, et al. Amphiphilic antimony(V) complexes for oral treatment of visceral leishmaniasis. Antimicrob Agents Chemother. 2013;57(9):4229–4236. | ||
Ribeiro RR, Ferreira WA, Martins PS, et al. Prolonged absorption of antimony(V) by the oral route from non-inclusion meglumine antimoniate-β-cyclodextrin conjugates. Biopharm Drug Dispos. 2010;31(2–3):109–119. | ||
Ali-Boucetta H, Kostarelos K. Carbon nanotubes in medicine & biology – therapy and diagnostics. Adv Drug Deliv Rev. 2013;65(15):1897–1898. | ||
Baeza A, Colilla M, Vallet-Regí M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin Drug Deliv. 2015;12(2):319–337. | ||
Allahverdiyev AM, Abamor ES, Bagirova M, et al. Antileishmanial effect of silver nanoparticles and their enhanced antiparasitic activity under ultraviolet light. Int J Nanomedicine. 2011;6:2705–2714. | ||
Jebali A, Kazemi B. Nano-based antileishmanial agents: a toxicological study on nanoparticles for future treatment of cutaneous leishmaniasis. Toxicol In Vitro. 2013;27(6):1896–1904. | ||
Soflaei S, Dalimi A, Abdoli A, et al. Anti-leishmanial activities of selenium nanoparticles and selenium dioxide on Leishmania infantum. Comp Clin Pathol. 2014;23(1):15–20. | ||
Nadhman A, Nazir S, Khan MI, et al. Visible-light-responsive ZnCuO nanoparticles: benign photodynamic killers of infectious protozoans. Int J Nanomedicine. 2015;10:6891–6903. | ||
Nadhman A, Nazir S, Ihsanullah Khan M, et al. PEGylated silver doped zinc oxide nanoparticles as novel photosensitizers for photodynamic therapy against Leishmania. Free Radic Biol Med. 2014;77:230–238. | ||
Ahmad A, Wei Y, Syed F, et al. Isatis tinctoria mediated synthesis of amphotericin B-bound silver nanoparticles with enhanced photoinduced antileishmanial activity: a novel green approach. J Photochem Photobiol B. 2016;161:17–24. | ||
Prajapati VK, Awasthi K, Gautam S, et al. Targeted killing of Leishmania donovani in vivo and in vitro with amphotericin β attached to functionalized carbon nanotubes. J Antimicrob Chemother. 2011;66(4):874–879. | ||
Prajapati VK, Awasthi K, Yadav TP, Rai M, Srivastava ON, Sundar S. An oral formulation of amphotericin B attached to functionalized carbon nanotubes is an effective treatment for experimental visceral leishmaniasis. J Infect Dis. 2012;205(2):333–336. | ||
Kester M, Heakal Y, Fox T, et al. Calcium phosphate nanocomposite particles for in vitro imaging and encapsulated chemotherapeutic drug delivery to cancer cells. Nano Lett. 2008;8(12):4116–4121. | ||
Chen F, Zhu Y, Wu J, Huang P, Cui D. Nanostructured calcium phosphates: preparation and their application in biomedicine. Nano Biomed Eng. 2012;4(1):41–49. | ||
Chaurasia M, Singh PK, Jaiswal AK, et al. Bioinspired calcium phosphate nanoparticles featuring as efficient carrier and prompter for macrophage intervention in experimental leishmaniasis. Pharm Res. 2016;33(11):2617–2629. | ||
Alvarenga BM, Melo MN, Frézard F, et al. Nanoparticle phosphate-based composites as vehicles for antimony delivery to macrophages: possible use in leishmaniasis. J Mater Chem B. 2015;3(48):9250–9259. | ||
Singh J, Desai S, Yadav S, Narasimhan B, Kaur H. Polymer drug conjugates: recent advancements in various diseases. Curr Pharm Des. 2016;22(19):2821–2843. | ||
Duncan R. Polymer therapeutics: top 10 selling pharmaceuticals – what next? J Control Release. 2014;190:371–380. | ||
Duncan R, Vicent MJ. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv Drug Deliv Rev. 2010;62(2):272–282. | ||
Nicoletti S, Seifert K, Gilbert IH. N-(2-hydroxypropyl)methacrylamide-amphotericin B (HPMA-AmB) copolymer conjugates as antileishmanial agents. Int J Antimicrob Agents. 2009;33(5):441–448. | ||
Dosio F, Arpicco S, Stella B, Fattal E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv Drug Deliv Rev. 2016;97:204–236. | ||
Micale N, Piperno A, Mahfoudh N, et al. A hyaluronic acid-pentamidine bioconjugate as a macrophage mediated drug targeting delivery system for the treatment of leishmaniasis. RSC Adv. 2015;5(116):95545–95550. | ||
Le Pape P. Development of new antileishmanial drugs – current knowledge and future prospects. J Enzyme Inhib Med Chem. 2008;23(5):708–718. | ||
Mishra BB, Kale RR, Singh RK, Tiwari VK. Alkaloids: future prospective to combat leishmaniasis. Fitoterapia. 2009;80(2):81–90. | ||
Espuelas S, Plano D, Nguewa P, et al. Innovative lead compounds and formulation strategies as newer kinetoplastid therapies. Curr Med Chem. 2012;19(25):4259–4288. | ||
Penã S, Scarone L, Serra G. Macrocycles as potential therapeutic agents in neglected diseases. Future Med Chem. 2015;7(3):355–382. | ||
Castillo-Garit JA, Abad C, Rodríguez-Borges JE, Marrero-Ponce Y, Torrens F. A review of QSAR studies to discover new drug-like compounds actives against leishmaniasis and trypanosomiasis. Curr Top Med Chem. 2012;12(8):852–865. | ||
Tariq A, Adnan M, Amber R, Pan K, Mussarat S, Shinwari ZK. Ethnomedicines and anti-parasitic activities of Pakistani medicinal plants against Plasmodia and Leishmania parasites. Ann Clin Microbiol Antimicrob. 2016;15(1):52. | ||
Polonio T, Efferth T. Leishmaniasis: drug resistance and natural products (review). Int J Mol Med. 2008;22(3):277–286. | ||
Nagle AS, Khare S, Kumar AB, et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem Rev. 2014;114(22):11305–11347. | ||
Sangshetti JN, Kalam Khan FA, Kulkarni AA, Arote R, Patil RH. Antileishmanial drug discovery: comprehensive review of the last 10 years. RSC Adv. 2015;5(41):32376–32415. | ||
Marr AK, McGwire BS, McMaster WR. Modes of action of leishmanicidal antimicrobial peptides. Future Microbiol. 2012;7(9):1047–1059. | ||
Sundar S, Chakravarty J. Investigational drugs for visceral leishmaniasis. Expert Opin Investig Drugs. 2015;24(1):43–59. | ||
Peniche AG, Renslo AR, Melby PC, Travi BL. Antileishmanial activity of disulfiram and thiuram disulfide analogs in an ex vivo model system is selectively enhanced by the addition of divalent metal ions. Antimicrob Agents Chemother. 2015;59(10):6463–6470. | ||
Fortin A, Hendrickx S, Yardley V, Cos P, Jansen H, Maes L. Efficacy and tolerability of oleylphosphocholine (OLPC) in a laboratory model of visceral leishmaniasis. J Antimicrob Chemother. 2012;67(11):2707–2712. | ||
Hernández L, Gálvez R, Montoya A, et al. First study on efficacy and tolerability of a new alkylphosphocholine molecule (oleylphosphocholine – OlPC) in the treatment of canine leishmaniosis due to Leishmania infantum. Parasitol Res. 2014;113(1):157–164. | ||
Fortin A, Dorlo TPC, Hendrickx S, Maes L. Pharmacokinetics and pharmacodynamics of oleylphosphocholine in a hamster model of visceral leishmaniasis. J Antimicrob Chemother. 2016;71(7):1892–1898. | ||
Roy P, Das S, Bera T, Mondol S, Mukherjee A. Andrographolide nanoparticles in leishmaniasis: characterization and in vitro evaluations. Int J Nanomedicine. 2010;5(1):1113–1121. | ||
Mondal S, Roy P, Das S, Halder A, Mukherjee A, Bera T. In Vitro susceptibilities of wild and drug resistant Leishmania donovani amastigote stages to andrographolide nanoparticle: role of vitamin e derivative TPGS for nanoparticle efficacy. PLoS One. 2013;8(12):e81492. | ||
Ghosh S, Kar N, Bera T. Oleanolic acid loaded poly lactic co-glycolic acid-vitamin E TPGS nanoparticles for the treatment of Leishmania donovani infected visceral leishmaniasis. Int J Biol Macromol. 2016;93:961–970. | ||
Tavares J, Ouaissi A, Silva AM, Lin PKT, Roy N, Cordeiro-da-Silva A. Anti-leishmanial activity of the bisnaphthalimidopropyl derivatives. Parasitol Int. 2012;61(2):360–363. | ||
Costa Lima SA, Resende M, Silvestre R, et al. Characterization and evaluation of BNIPDaoct-loaded PLGA nanoparticles for visceral leishmaniasis: in vitro and in vivo studies. Nanomedicine. 2012;7(12):1839–1849. | ||
Yang DM, Liew FY. Effects of qinghaosu (artemisinin) and its derivatives on experimental cutaneous leishmaniasis. Parasitology. 1993;106(1):7–11. | ||
Want MY, Islamuddin M, Chouhan G, Dasgupta AK, Chattopadhyay AP, Afrin F. A new approach for the delivery of artemisinin: formulation, characterization, and ex-vivo antileishmanial studies. J Colloid Interface Sci. 2014;432:258–269. | ||
Want MY, Islamuddin M, Chouhan G, et al. Therapeutic efficacy of artemisinin-loaded nanoparticles in experimental visceral leishmaniasis. Colloids Surf B Biointerfaces. 2015;130:215–221. | ||
Fouladvand M, Barazesh A, Tahmasebi R. Evaluation of in vitro antileishmanial activity of curcumin and its derivatives “Gallium curcumin, Indium curcumin and Diacethyle curcumin”. Eur Rev Med Pharmacol Sci. 2013;17(24):3306–3308. | ||
Couvreur P, Stella B, Harivardhan Reddy L, et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006;6(11):2544–2548. | ||
Maksimenko A, Dosio F, Mougin J, et al. A unique squalenoylated and nonpegylateddoxorubicin nanomedicine with systemiclong-circulating properties and anticancer activity. Proc Natl Acad Sci U S A. 2014;111(2):E217–E226. | ||
Cheikh-Ali Z, Caron J, Cojean S, et al. “Squalenoylcurcumin” nanoassemblies as water-dispersible drug candidates with antileishmanial activity. ChemMedChem. 2015;10(2):411–418. | ||
Tiwari B, Pahuja R, Kumar P, Rath SK, Gupta KC, Goyal N. Nanotized curcumin and miltefosine, a potential combination for treatment of experimental visceral leishmaniasis. Antimicrob Agents Chemother. 2017;61(3):e01169-16. | ||
Paloque L, Verhaeghe P, Casanova M, et al. Discovery of a new antileishmanial hit in 8-nitroquinoline series. Eur J Med Chem. 2012;54:75–86. | ||
Costa Duarte M, dos Reis Lage LM, Lage DP, et al. An effective in vitro and in vivo antileishmanial activity and mechanism of action of 8-hydroxyquinoline against Leishmania species causing visceral and tegumentary leishmaniasis. Vet Parasitol. 2016;217:81–88. | ||
Duarte MC, Lage LMDR, Lage DP, et al. Treatment of murine visceral leishmaniasis using an 8-hydroxyquinoline-containing polymeric micelle system. Parasitol Int. 2016;65(6):728–736. | ||
Nan A, Croft SL, Yardley V, Ghandehari H. Targetable water-soluble polymer-drug conjugates for the treatment of visceral leishmaniasis. J Control Release. 2004;94(1):115–127. | ||
Saudagar P, Dubey VK. Carbon nanotube based betulin formulation shows better efficacy against Leishmania parasite. Parasitol Int. 2014;63(6):772–776. | ||
Crommelin DJA, Florence AT. Towards more effective advanced drug delivery systems. Int J Pharm. 2013;454(1):496–511. | ||
de Menezes JP, Guedes CE, Petersen AL, Fraga DB, Veras PS. Advances in development of new treatment for leishmaniasis. Biomed Res Int. 2015;2015:815023. | ||
Zhang W-W, Lypaczewski P, Matlashewski G. Optimized CRISPR-Cas9 genome editing for Leishmania and its use to target a multigene family, induce chromosomal translocation, and study DNA break repair mechanisms. mSphere. 2017;2(1):e00340-16. | ||
Antillón A, De Vries AH, Espinosa-Caballero M, et al. An amphotericin B derivative equally potent to amphotericin B and with increased safety. PLoS One. 2016;11(9):e0162171. | ||
Olliaro PL, Shamsuzzaman TA, Marasini B, et al. Investments in research and surveillance are needed to go beyond elimination and stop transmission of Leishmania in the Indian subcontinent. PLoS Negl Trop Dis. 2017;11(1):e0005190. | ||
Hiemenz JW, Walsh TJ. Lipid formulations of amphotericin B: recent progress and future directions. Clin Infect Dis. 1996;22(suppl 2):S133–S144. | ||
Thakur CP, Singh RK, Hassan SM, Kumar R, Narain S, Kumar A. Amphotericin B deoxycholate treatment of visceral leishmaniasis with newer modes of administration and precautions: a study of 938 cases. Trans R Soc Trop Med Hyg. 1999;93(3):319–323. | ||
Sánchez-Brunete JA, Dea MA, Rama S, et al. Treatment of experimental visceral leishmaniasis with amphotericin B in stable albumin microspheres. Antimicrob Agents Chemother. 2004;48(9): 3246–3252. | ||
Sánchez-Brunete JA, Dea MA, Rama S, et al. Influence of the vehicle on the properties and efficacy of microparticles containing amphotericin B. J Drug Target. 2005;13(4):225–233. | ||
World Health Organization. Neglected tropical diseases: WHO and Gilead Sciences extend collaboration against visceral leishmaniasis. Available from: http://www.who.int/neglected_diseases/news/WHO_and_Gilead_Sciences_extend_collaboration/en/. | ||
Drugs for Neglected Diseases Initiative. Fexinidazole/Miltefosine combination (VL). Available from: https://www.dndi.org/diseases-projects/portfolio/completed-projects/fexinidazole-vl/. | ||
iCo Therapeutics Inc. Pipeline. Available from: http://www.icotherapeutics.com/pipeline/. | ||
Matinas Biopharma. Learn more about cochleate technology. Available from: http://www.matinasbiopharma.com. | ||
American Pharmaceutical Review. FDA’s regulatory science program for generic PLA/PLGA-based drug products. Available from: http://www.americanpharmaceuticalreview.com/Featured-Articles/188841-FDA-s-Regulatory-Science-Program-for-Generic-PLA-PLGA-Based-Drug-Products/. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.