Back to Journals » International Journal of Nanomedicine » Volume 20
Nanoparticles with Cell-Penetrating Peptides for Oral Delivery: A Case for Oral Delivery of Insulin
Authors Wang Y, Song W, Wang T, Sheng Y, Xue S, Dang Y, Rasool A, Zhang G
Received 22 March 2025
Accepted for publication 23 June 2025
Published 29 November 2025 Volume 2025:20 Pages 14283—14312
DOI https://doi.org/10.2147/IJN.S529791
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Yunyun Wang,1 Wangdi Song,1 Taiyu Wang,1 Yue Sheng,1 Shengnan Xue,1 Yanyan Dang,1 Aamir Rasool,2 Genlin Zhang1
1School of Chemistry and Chemical Engineering/State Key Laboratory Incubation Base for Green Processing of Chemical Engineering, Shihezi University, Shihezi, 832003, People’s Republic of China; 2Institute of Biochemistry, University of Balochistan, Quetta, 78300, Pakistan
Correspondence: Genlin Zhang, Email [email protected] Aamir Rasool, Email [email protected]
Abstract: The expanding protein-based drug market is facing limitations from invasive delivery methods. These methods can cause discomfort and pose infection risk, particularly for the chronic disease patients such as diabetes requiring insulin with adherence challenges. Oral insulin, though preferred, suffers from < 2% bioavailability, thus, nano-drug delivery system (NDDS) is becoming a highly promising strategy to enhance bioavailability and stability. However, the low expression of receptors and limited uptake capacity remain challenge. The use of cell-penetrating peptides (CPPs) will enhance the permeability of epithelial cells, and combining them with nanoparticles (NPs) can further improve the stability of protein-based drugs in blood circulation and facilitate the development of efficient delivery carriers. This comprehensive review delves into the design, synthesis, classification, challenges, and cellular uptake mechanisms of CPPs-cargo complexes and CPPs-NP nanocarriers for insulin delivery. Furthermore, it provides an in-depth exploration of the challenges and prospects of these innovative approaches.
Keywords: diabetes, cell-penetrating peptides, insulin, nanoparticles, oral delivery
Introduction
The protein-based drug market is growing at an average annual rate of 20%, which is significantly higher than the overall pharmaceutical market’s annual growth of 9%and reached $369.11 billion by 2024.1 And it is projected to reach a value of $1326.16 billion by 2032. Protein-based drugs are primarily delivered through intravenous (IV), intraperitoneal (IP), intramuscular (IM), and subcutaneous (SC) routes. These delivery systems cause physical discomfort, pose a risk of infection, and are costly due to needle-based administration, which often leads to disapproval by patients, particularly those suffering from chronic conditions like diabetes who require regular dosing.1,2 The oral insulin delivery system is a groundbreaking treatment for patients with diabetes and is preferred by those struggling with hyperinsulinemia, discomfort, infections, and adherence issues.3 However, the FDA has approved only a few protein-based drugs for oral delivery because the gastrointestinal tract presents significant physiological barriers. The bioavailability of oral insulin is reduced to less than 2% due to the action of stomach acid and proteases, as well as the impermeable mucus layer of the intestinal epithelium.4,5
The researchers have addressed the above-mentioned obstacles by developing various nanoparticle (NP) platforms for oral insulin delivery. Recently, many nanocarriers, including lipid-based nanocarriers (liposomes, micelles), carbon nanotubes (CNTs), inorganic nanocarriers (quantum dots), gold nanoparticles and polymeric nanoparticles (PNPs, including polysaccharides, proteins, peptides, nucleic acids, metal-organic frameworks (MOFs), porous organic polymers (POPs)) have been reported.6 Although NPs protect encapsulated protein-based drugs from acidic denaturation and enzymatic degradation, several factors still limit their application in oral insulin delivery systems. Firstly, the viscous, semi-permeable mucus layer acts as an initial barrier to the diffusion of NPs toward the surface of the intestinal epithelium.7 Secondly, the tight junctions linking intestinal epithelial cells significantly hinder the uptake of macromolecules via the paracellular route.8,9 Thirdly, macromolecules’ large size and hydrophilic nature also prevent their transcellular passage.8
Biomolecules such as vitamin B12 and transferrin have been observed to cross the mucus layer and intestinal epithelial cells through various carriers and receptor-mediated transport systems.10,11 However, these systems face multiple challenges, including low expression of target receptors and insufficient uptake capacity.12
Recently, cell-penetrating peptides (CPPs), composed of 4–30 amino acids and capable of penetrating cell membranes,13,14 have been employed to facilitate the systemic delivery of biological molecules across the intestinal mucosa and epithelial cells.13 CPPs are encapsulated in NPs or specialized devices to protect them from harsh environments, such as low-pH gastric conditions and intestinal proteases.15,16 Additionally, CPPs can be integrated with various drug carriers to combine their benefits, forming innovative multifunctional drug delivery systems that enhance stability during blood circulation.17–19 These advancements have also greatly facilitated the development of novel cargos, enabling targeted delivery and controlled release of biomolecules. The widespread application of CPPs in developing drug delivery cargos will help overcome the physiological barriers encountered by biomacromolecules, such as protein-based drugs, during oral delivery.20–22
This review summarizes the latest developments in CPPs-NPs as nanocarriers and their use for oral insulin delivery. Specifically, it covers (i) the design, synthesis, and classification of CPPs; (ii) the challenges and mechanisms involved in the cellular uptake of CPPs-cargos; and (iii) a detailed analysis of the advancements in utilizing CPPs-NPs as oral delivery systems for insulin. The aim is to assist researchers in designing and developing CPPs-NPs for drug delivery applications.
Cell-Penetrating Peptides
Definition of CPPs
Cell-penetrating peptides (CPPs), also known as protein transduction domains (PTDs) or membrane transduction peptides (MTPs), are typically composed of 4–30 amino acids23 and can penetrate cell membranes without significant toxicity.17,18 CPPs enter the cell through various mechanisms; therefore, they can enable the intracellular delivery of bioactive substances, which are either covalently or non-covalently bound, such as nucleic acids and low molecular weight drugs.24 In 1988, the first CPPs, known as Tat, were reported.25 In the following years, CPPs have been extensively employed for the treatment of various ailments, including cancer,26–28 muscular dystrophy,29 anti-prion diseases,30 both viral and bacterial infections,31–33 and diabetes.34–39 CPPs can be derived from DNA- and RNA-binding proteins,40 homeoproteins,41 heparin-binding proteins,3 bacterial membrane proteins,42,43 and signal peptides.44 CPPs database (CPPsite 2.0: http://crdd.osdd.net/raghava/CPPsite/) was updated in 2015 and contains 1855 entries, including peptide sequences, the nature of the peptides, chemical modifications, experimental validation techniques, peptide structures, and types of cargo delivered.45
Classification of CPPs
CPPs are generally categorized based on their physicochemical properties, origin, structural conformation and cell targeting (Figure 1).46
 |
Figure 1 Schematic diagram illustrating the types of CPPs. |
Physicochemical Properties of CPPs
CPPs can be categorized into cationic, amphipathic, and hydrophobic CPPs based on their physicochemical properties (Table 1). Cationic CPPs comprise positively charged amino acid residues (Arg & Lys).47,48 Amphiphilic CPPs are made up of both hydrophilic- (Arg & Lys) and hydrophobic amino acid residues (Val, Leu, Ile, and Ala).49 Amphiphilic CPPs are classified into primary amphipathic-, secondary amphipathic- and Pro-rich CPPs.50,51 Primary-amphipathic CPPs inherently possess hydrophilic and hydrophobic amino acid residues distributed across their molecular surfaces. This dual nature allows them to interact effectively with aqueous environments and lipid-rich cell membranes.49 Secondary-amphipathic CPPs are conformationally dynamic and exhibit amphipathic properties only upon adopting a specific secondary structure, such as an α-helix or β-sheet.52 Another interesting class of amphiphilic peptides are Pro-rich CPPs, for which various families with different sequences and structures have been reported, but all contain a proline-pyrrolidine template. Pro has unusual properties among the 20 genetically encoded amino acids due to the rigidity imparted by the pyrrolidine ring. Additionally, since the α-amino group lacks hydrogen, it cannot provide hydrogen bonds (in the peptide structure) to stabilize α-helices or β-sheets. Unlike other amino acids that are found almost exclusively in the trans structure of a polypeptide, proline can be found in the cis structure of a polypeptide.49 Hydrophobic CPPs contain hydrophilic amino acid residues (Leu, Ile, Val, Phe, and Ala) and few hydrophilic amino acid residues.53
 |
Table 1 The Name, Sequences, Physicrochemical Properties, Origin, Confirmation and Cell Targeting of CPPs |
Origin of CPPs
CPPs are also categorized into three types based on their sources: protein-derived, chimeric, and synthetic CPPs. Protein-derived CPPs originate from naturally occurring proteins, which are inherently equipped to penetrate cell membranes.94 Tat CPPs (transcription activator of the human immunodeficiency virus) is one of the earliest and well-studied examples of protein-derived CPPs.45 Tat CPPs interacts with proteoglycans on the cell surface through its positively charged amino acid residues. It efficiently delivers drugs into the cell without requiring or saturating the specific ligand-receptor. Tat CPPs lacks selectivity, which limits its use in synthesizing systems needed for targeted oral delivery.55,56 Chimeric CPPs are synthesized by combining two distinct peptides of different origins. For instance, the transportan (TPGWTLNSAGYLLGKINLKAKISIL) is produced by fusing a segment of the hormone galanin and wasp venom mastoparan.95 Synthetic CPPs are designed and synthesized artificially; therefore, their features, such as stability in blood circulation, resistance to endolysosomal breakdown, enhanced cellular uptake, and pH sensitivity, can be tuned.19 MAP-amphipathic model peptide (KLALKLALKALKAALKLA), a synthetic CPPs, is a highly amphiphilic α-helical structure that has been several times used to deliver multiple bioactive molecules into the cell through endocytosis.66 Glutamate (Glu) and aspartate (Asp) function as pH sensors due to their inherent trait of losing organic protons depending on the environmental pH.96 His, with its imidazole group’s pKa of 6.5, is a buffering agent in physiological conditions and demonstrates a proton-sponge effect, which facilitates the molecule’s escape from endolysosomes. Additionally, novel synthetic CPPs can be engineered with customized features using the appropriate proportion of Glu, Asp, and His residues.48
Structural Conformations of CPPs
CPPs are classified into two groups based on their structural conformation: linear and cyclic CPPs (Figure 2). Linear CPPs have disadvantages such as poor stability, endosomal entrapment, toxicity, and suboptimal cell penetration, whereas cyclic CPPs can avoid these limitations. Therefore, cyclic CPPs demonstrate increased cell permeability, higher resistance to proteolysis, effective endosomal escape, a higher affinity for target receptors, and nuclear-targeting properties. The amphipathic cyclic CPPs are composed of positively charged amino acid residues (His, Arg, and Lys) and hydrophobic amino acid residues (Trp, Phe, and Val), which enable them to interact efficiently with and penetrate cell membranes. Positive and negatively charged amino acid residues on the ring of cyclic CPPs are important factors affecting cellular uptake and their use for drug delivery.89
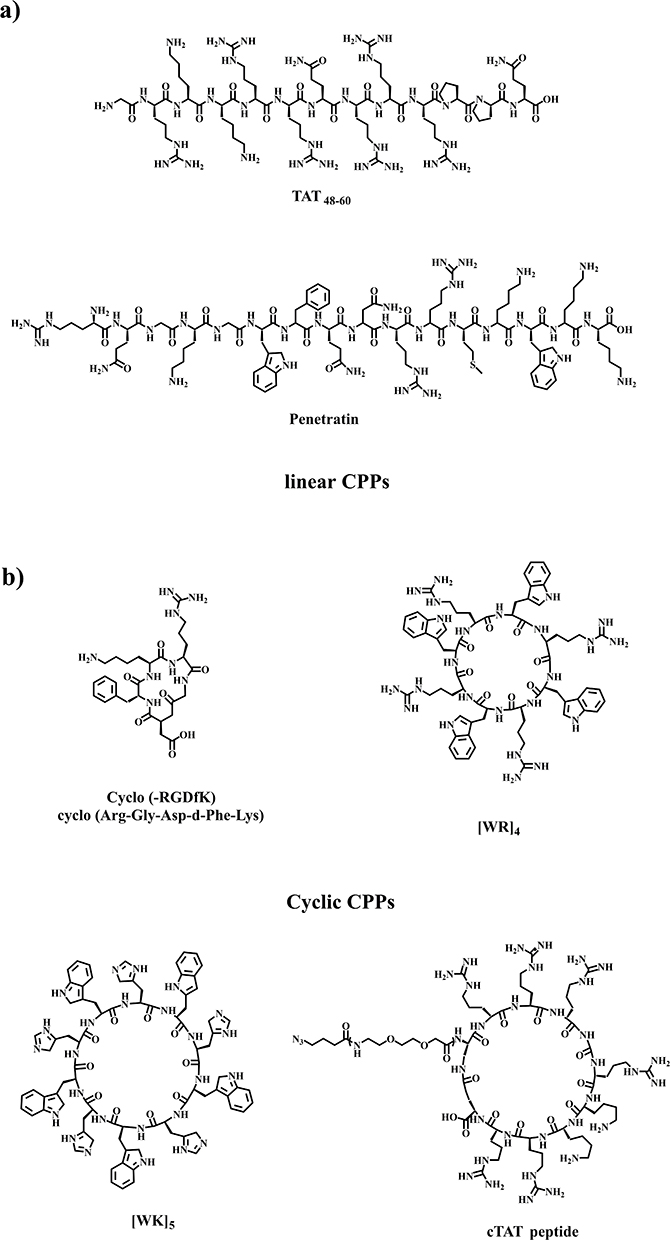 |
Figure 2 The chemical structures of (a) linear and (b) cyclic CPPs.55,58,86–89 |
Cell Targeting by CPPs
CPPs are categorized into two types based on their targeting specificity: tissue-specific CPPs, which selectively target particular cells, and non-tissue-specific CPPs, which target cells indiscriminately to deliver the drugs.97 Non-tissue-specific CPPs are categorized into three types based on their structural characteristics: cationic peptides, hydrophobic peptides, and amphipathic peptides.98 Cationic peptides comprise 6–12 residues of positively charged amino acids such as TAT-PTD,35,54 Tat-HA257 and penetratin,58 etc. The positive charge of these peptides facilitates cellular uptake by promoting interactions with the negatively charged cellular membranes.99 Hydrophobic peptides, rich in hydrophobic amino acids, are derived from the leader sequences of secreted growth factors or cytokines. Due to their hydrophobic nature, these peptides easily traverse lipid membranes.85 Amphipathic peptides are formed by fusing hydrophobic sequences with nuclear localization signals (NLSs), creating a structure that allows them to effectively penetrate cell membranes and deliver their cargo to the nucleus.100 However, non-tissue-specific CPPs will appear off-target in vivo. Tissue-specific CPPs could address this issue. Tissue-specific CPPs are identified through extensive screening of large peptide phage display libraries. This approach enables researchers to isolate the peptides that exhibit high affinity for specific tissue types.101 These peptides have widespread applications in both diagnostic and therapeutic fields. For instance, they facilitate the delivery of various compounds, including fluorescent or radioactive molecules, for imaging purpose. Moreover, these peptides can transport therapeutic peptides and proteins. Additionally, they significantly enhance the uptake of nucleic acids such as DNA, RNA, and siRNA and enable the delivery of viral particles. The broad range of applications underscores the importance of CPPs in advancing therapeutic strategies and enhancing the effectiveness of treatment modalities.102
Designing of CPPs
The following parameters must be considered when designing CPPs: guanidinium groups, hydrophobic residues, structure, and targeted performance.
Guanidinium Group
Due to its unique chemical properties, the guanidinium group is essential for designing CPPs because it significantly enhances membrane permeability and cellular uptake. The guanidinium group of the Arg side chain demonstrates a stronger affinity for hydrophobic anions than Lys, which is why arginine-rich peptides form strong bonds with the lipid bilayer.103 This interaction helps to reinforce transient pores formation and allows the peptides to embed themselves within the bilayer. The distance between the guanidine group and the peptide backbone influences its cellular uptake capability. As the flexibility of the backbone or side chain increases, the guanidine group interacts more closely with the cell surface, thereby neutralizing the negatively charged regions of the cell membrane more effectively.104,105
Cell-permeable polydisulfides (CPDs) are derived from a guanidine-rich polyarginine (pArg) peptide backbone that is substituted with disulfide polymers.106 These CPDs demonstrate improved cell penetration capability by employing dynamic covalent disulfide exchange reactions that occur on the cell surface with thiol compounds.107,108
Hydrophobic Residues
Hydrophobic amino acid residues play a crucial role in the insertion and penetration of CPPs into the lipid bilayer of the cell membrane.109 A proposed mechanism for CPPs penetration suggests the formation of a hydrophobic counter-ion around the guanidinium-rich backbone of CPPs.110 This counter-ion formation is initiated through hydrogen-bonding interactions between arginine residues in the CPPs and components of the cell membrane. The resulting complex then facilitates the crossing of the CPPs across the cell membrane. However, the binding strength of counter-ions in Lys-rich CPPs is lower than that of the guanidinium moieties of arginine, highlighting the critical role of the guanidinium group in this process.111 The self-activating characteristics of these peptides are associated with the formation of counter-ion complexes by the hydrophobic segments of CPPs. Consequently, while hydrophobicity warrants further investigation, exploring the functional contributions of individual aliphatic and aromatic groups is imperative.105 Hydrophobicity can be achieved through the incorporation of either aliphatic or aromatic moieties.112 The process of lipidation entails attaching hydrocarbon chains of varying lengths to the N-termini of well-characterized CPPs.113 This alteration is crucial for improving the capability of these peptides to penetrate cellular membranes. In addition, alkylation is recognized as a conventional technique to bolster the internalization of peptides by reinforcing their hydrophobic interactions with cellular membranes. This approach utilizes the fundamental characteristics of alkyl groups to enhance the interaction between the peptide and the lipid elements of the membrane, consequently increasing the efficiency of cellular uptake. Through these strategies, the bioavailability of cell-penetrating peptides can be significantly improved, which is essential for their effectiveness in therapeutic applications.114 The inclusion of aromatic amino acid residues, like Trp, Phe, and Tyr, can also improve hydrophobicity. In addition to boosting hydrophobicity, aromatic groups can engage in π-π stacking with membrane proteins that feature aromatic residues. This interaction can help promote and stabilize the binding of CPPs to the membrane, thus assisting in their translocation and drug delivery.51
Secondary Structure
The secondary structure of CPPs (eg, α-helices and β-sheets) also significantly influences their ability to penetrate the cells. The influence of secondary structures on the translocation capability of CPPs is determined through two key factors: the strength of the interaction between CPPs and cell membranes, and their capacity to fold in the presence of these membranes.51 Weak non-covalent interactions, such as electrostatic interactions, hydrogen bonds, hydrophobic effects, and van der Waals forces, primarily influence the internalization of CPPs. Therefore, understanding the role of these non-covalent forces is essential to unveil the driving factors that support the ability of CPPs to penetrate cellular membranes. Furthermore, this knowledge will facilitate the design of novel CPPs with improved cell-penetrating capabilities, as well as more effective drug delivery cargos.111 Yamashita et al115 investigated the effect of the secondary structure of CPPs on cell penetration activity. They developed a cationic dAA (α,α-disubstituted amino acid), ApiC2Gu, as an Arg mimic and replaced the hydrophobic Aib residues in peptide A (FAM-β-Ala-(L-Arg-L-Arg-Aib)3-NH2) with cationic ApiC2Gu residues. The cationic peptide B (FAM-β-Ala-(LArg-L-Arg-Aib)3C2Gu-NH2) also formed a stable helical structure and exhibited greater cell permeability than nona-arginine (R9).20 The results showed that peptides capable of altering their secondary structure in response to environmental conditions may demonstrate superior cell penetration capability compared to CPPs with a fixed helical secondary structure.
Rigidity is gaining recognition as an important concept in the design of CPPs, as it can influence the interaction of CPPs with cell membranes and their overall efficiency. In the case of linear CPPs, the efficiency of internalization is affected by multiple factors: sequence length, quantity of arginine residues, and positioning of arginine residues.116 The comparison of free Arg residues in linear CPPs with Arg residues in cyclic CPPs revealed that cyclization enhances the membrane interaction by facilitating improved charge distribution and greater penetration efficacy. Previously, the insertion of a spacer, such as Gly, into pArg CPPs was shown to enhance the cell permeability. This improvement is attributed to the increased distance between guanidinium groups, which resultantly reduces the steric hindrance and allows more effective interactions with the negatively charged components of cell membrane.104 Traditional CPPs derived from natural L-amino acids are sensitive to proteolytic enzymes, which presents a significant obstacle to their use in vivo for drug delivery. Consequently, the chirality of the amino acids plays a fundamental role in the development and optimization of these CPPs.
Hence, the stability of the primary structure can be improved through the incorporation of non-natural amino acids and their chiral isomers, especially D-stereoisomers, which enable these CPPs to withstand proteolytic degradation. This characteristic allows them to remain stable and functional in the biological environment. However, the exact mechanism of how chirality of amino acid residues plays a role in enhancing the stability and cellular penetration of CPPs remains elusive, but it is evident that the strategic use of D-stereoisomers significantly enhances the performance of CPPs in cellular applications.51
Targeted Performance
The researcher design targeting CPPs through employing three different strategies: (i) the development of novel CPPs that function as targeting ligands; (ii) the attachment of CPPs to specific targets, including small molecules, peptides, and proteins; and (iii) the modulation of CPPs absorption through stimuli-responsive signals, such as UV light, ultrasound, temperature variations, enzymatic activity, and pH levels.105 Pep-1 was designed from native CPPs by incorporating fusion sequences, which include a hydrophobic segment composed of a patch of five tryptophan residues, a positively charged segment of Lys residues, and a Pro-rich spacer region. The hydrophobic segment enhances cellular penetration, the positively charged segment improves nuclear traversal capability, and the Pro-rich region increases the flexibility of the Pep-1 CPPs.117 Fusing RGD (arginine-glycine-aspartic acid; target integrin receptors) or homing peptides (specific for tissue markers, cells, or pathological conditions, eg, tumors or inflamed tissues) improves the specificity and affinity of CPPs.118,119 The pH-responsive CPPs are engineered by substituting Lys with His residues. These CPPs target selectins that are overexpressed due to the acidic microenvironment produced in inflamed tissues or tumor sites.120 Additionally, thermoresponsive polymers are coupled with CPPs to enable them to detect variations in the temperature of the cellular microenvironment. As a consequence, this enhances their capability for cell penetration and drug delivery.121
Synthesis of CPPs
Generally, CPPs are synthesized through two methods: microbial synthesis and chemical synthesis.122 The synthesis of CPPs by microorganisms mainly involves enzymatic hydrolysis, genetic engineering, and fermentation (Figure 3a). Enzymatic hydrolysis uses enzymes to degrade plant or animal proteins into small peptides. However, this process fails to meet industrial production levels due to its low yield, significant investment, long cycle time, and the severe pollution associated with it.123 The production of CPPs through genetic engineering offers several advantages, including solid expression orientation, safety and health benefits, a comprehensive source of raw materials, and low cost. However, the production of CPPs through genetic engineering also encounters setbacks, including difficulties in achieving industrial-scale production levels due to issues with efficient expression, separation challenges, and low yield. The fermentation method involves the use of simple nutrients by microbial organisms to produce peptides, such as ɛ-polylysine (ɛ-PL), γ-polyglutamate (γ-PGA), and cyanobacterial peptide.124 Microbial synthesis is only helpful for synthesizing peptides with natural amino acid residues125 and is typically employed for the production of longer peptides (> 40 amino acids). However, due to their low yield and the long synthesis time required, CPPs are rarely synthesized on an industrial scale using microorganisms.126
 |
Figure 3 CPPs synthesis methods. (a) microbial synthesis; (b) chemical synthesis. |
Nowadays, chemical synthesis is the standard technique for peptide production because it is fast, allows the incorporation of non-standard amino acid residues, and poses minimal risk of endotoxin contamination.126 Three approaches are commonly used for the chemical synthesis of polypeptides: solid-phase peptide synthesis (SPPS), solution-phase coupling, and ring-opening polymerization (ROP) of α-amino acid N-carboxy anhydrides (NCAs) (Figure 3b).127 The SPPS is recognized as the benchmark method for synthesizing peptides, and its efficiency has significantly improved since Merrifield’s pioneering synthesis. The linker, which bears reactive functional groups (either inherent to the resin or added at the outset of synthesis), secures the first amino acid to the resin. This linker is designed to facilitate the release of the complete peptide from the resin once the synthesis is completed. In SPPS, peptides are constructed on the resin from the C-terminus to the N-terminus using side chain-protected amino acids, which prevent side reactions during the coupling process.128 SPPS is commonly employed to synthesize short peptides consisting of fewer than 50 residues.125
The solution-phase coupling technique utilizes the short peptide sequence as the starter to produce larger quantities of CPPs. Therefore, this method is always used to synthesize CPPs containing repetitive short sequences (ca. 3 to 10 residues in length).129 Nevertheless, this method also faces challenges in synthesizing initial oligopeptides, the cyclization processes, and the low molecular weight of the resulting products.127 The ROP of NCAs represents a fundamental and widely embraced methodology for synthesizing cyclic- and linear polypeptides.130 The NCAs are synthesized through two methods: (i) the Leuchs method, which involves the reaction between N-alkyloxycarbonylamino acids and halogenating agents like thionyl chloride, and (ii) the Fuchs-Farthing method, where α-amino acids react with phosgene (COCl2).130 These methods are essential for creating a robust framework for synthesizing NCAs, which subsequently undergo polymerization initiated by a diverse array of nucleophiles and bases, including amines and metal alkoxides. There are two predominant mechanisms: the activated monomer (AMM) pathway and the normal amine (NAM) pathway through which NCAs are aggregated.131 The AMM mechanism uses bases such as tertiary amines and alkoxides to initiate the polymerization reaction. These bases abstract a proton from the nitrogen of the NCA (designated as 3-N) and form an NCA anion. This deprotonation process allows the NCA anion to initiate the polymerization by attacking the carbonyl group of another NCA; as a consequence, carbon dioxide (CO2) is released with the formation of a new anion that continues the polymerization reaction.125 However, it is essential to note that polymerizations based on the AMM are usually uncontrolled and less desirable for synthesizing well-defined cyclic peptides due to their inherent unpredictability. In contrast, the NAM mechanism is typically used for NCA polymerizations initiated by nonionic initiators with at least one mobile hydrogen atom, such as primary and secondary amines, alcohols, and water. The NAM mechanism offers advantages by providing control over the molecular weight of the resulting polymer and ensuring the fidelity of end groups, which is crucial for synthesizing high-quality cyclic peptides.132 Traditional NCA polymerization is typically a slow process, often lasts 2–3 days or more, and is sensitive to moisture, frequently necessitating using anhydrous solvents and handling within a glovebox. Recently, Wu et al130 introduced a superfast NCA ring-opening polymerization method initiated by lithium hexamethyldisilazane (LiHMDS), conducted in an open vessel outside glovebox environments.
Clinical Challenges of CPPs-Cargo
CPPs maintain the integrity of cell membranes and are therefore regarded as both highly effective and safe in contrast to the traditional methods of sonoporation, microinjection, electroporation, and bead loading.133–135 Furthermore, other key aspects that must be evaluated for clinical use of CPPs include therapeutic effectiveness, the practicality of large-scale production, physical and chemical characteristics, formulation, administration routes, stability, toxicity, and pharmacokinetics (PK).136 CPPs are rapidly degraded by proteases in systemic circulation and subjected to intracellular digestion by lysosomes and proteasomes, significantly limiting their plasma half-life and stability. The rapid barrier penetration and cellular uptake of CPPs result in the clearance of CPP conjugates from the bloodstream within minutes, thereby reducing protease-mediated degradation in circulation. However, this characteristic complicates pharmacokinetic analysis through plasma concentration measurements of these conjugates. The short half-life is a primary factor diminishing the in vivo efficacy of CPP-cargo complexes.17 To address these limitations, peptide stabilization strategies have been developed, including: (i) D-amino acid and unnatural amino acid substitutions,137 (ii) N-terminal or C-terminal modifications,138 (iii) fatty acid conjugation,139 (iv)formulation optimization140 and (v) cyclizing.141 Additionally, the coupling of CPPs with therapeutic cargo peptides to generate fusion peptides is often not cost-effective for large-scale synthesis. Given the limited development of peptide-based therapeutics for human diseases, drug design strategies must be rigorously evaluated by integrating scientific and economic considerations. CPP conjugates may accumulate in non-target cells/tissues, particularly the liver and kidneys, posing potential toxicity risks.142 Consequently, comprehensive assessment of the pharmacokinetics, biodistribution, and hepatotoxicity/nephrotoxicity of CPP conjugates is essential to mitigate toxicity. Localized administration rather than systemic delivery can minimize systemic toxicity. Encouragingly, CPP-based therapies have demonstrated preliminary safety in Phase I clinical trials. However, immunogenicity concerns persist for non-human-derived or synthetic CPPs, underscoring the preference for human-derived CPPs in clinical applications.143 While novel CPP-based candidates continue to enter clinical trials with recent advancements. Ja-Hyun Koo et al discussed this in detail.136 However, no FDA-approved drugs incorporating CPPs are currently available due to limitations in human trial validation. Importantly, in the clinical application of CPPs, it is imperative to rigorously uphold patient autonomy through informed consent, ensure transparency in trial design, prioritize safety-first pharmacovigilance, and enforce stringent screening of raw materials to mitigate environmental contamination risks.
Cellular Uptake Mechanisms of CPPs
A defining characteristic of CPPs lies in their capacity to perturb cellular membranes, thereby enabling subsequent intracellular transduction. The efficiency of cellular transduction and active targeting (extending beyond peptide-specific interactions) is constrained and modulated by molecular size and polarity. Small, nonpolar molecules traverse cell membranes via simple diffusion, a process characterized by rapid, concentration-dependent, and energy-independent kinetics. Recent studies highlight that loosened packing of lipid bilayers has been recognized as a pivotal factor governing CPP-mediated membrane translocation. Strategic augmentation of hydrophobic residues within the peptide backbone facilitates penetration through the lipophilic core of the membrane. A proposed mechanism suggests that CPPs may mediate transduction by inducing membrane curvature. Furthermore, enzymatic degradation in extracellular environments (eg, proteases in the gastrointestinal tract) compromises CPPs stability prior to cellular uptake. Beyond intrinsic CPPs properties, membrane composition critically influences translocation efficacy. Notably, specific lipids such as bis (monoacylglycero) phosphate (BMP), enriched in late endosomes, are identified as key mediators of endosomal escape-a rate-limiting step for intracellular delivery.144–146 The precise molecular mechanism governing the entry of CPPs into cells has yet to be fully explained, although many investigators have proposed mechanisms through which CPPs penetrate the plasma membrane.105 The mechanistic models have unveiled several factors which impact the cellular penetration of CPPs: i) the type of cell, ii) the mass and structure of the cargo, iii) the binding method, iv) the concentration of CPPs, and v) the duration and temperature of incubation. The primary mechanisms proposed for CPPs internalization include endocytic routes and direct penetration. (Figure 4).147
 |
Figure 4 Cellular internalization/uptake mechanisms of CPPs. Endocytosis mechanisms mainly include macropinocytosis, clathrin-/cavedin-mediated endocytosis, and clathrin-/cavedin-independent endocytosis. Direct penetration mainly includes the inverted micelles, carpet-like model, membrane thinning, transient pore formation, and induction of multilamellarity alongside the stalk-pore hypothesis. |
Endocytic Routes
Most CPPs enter cells via endocytic pathways, which comprise two key steps: endocytosis and subsequent endosomal escape.148 Endocytosis is the process by which intracellular and extracellular substances are internalized through the invagination of the plasma membrane, forming vesicles that enclose the ingested material.149 Endocytosis is an energy-dependent process that occurs in all cells. The CPPs are translocated into cells primarily through endocytosis rather than direct translocation due to the rapid turnover of the plasma membrane.150 Endocytosis consists of various pathways, including macropinocytosis, clathrin-/cavedin-mediated endocytosis, and clathrin-/cavedin-independent endocytosis (Figure 4).151 All of these endocytic pathways are inhibited under energy-depleting conditions, such as low temperature (4°C) or with sodium azide (NaN3) treatment.152 Macropinocytosis is a unique cellular process that does not initiate through the typical mechanism of membrane invagination; instead, it arises from increased membrane ruffling and cellular activation.125 This form of membrane dynamics leads to protrusions that do not encapsulate a ligand-coated particle but fold back to integrate into the plasma membrane, forming structures referred to as macropinosomes.153 A large body of experimental evidence has indicated the crucial role of macropinocytosis in the cellular penetration of different CPPs.154,155 The mechanism by which the M918 peptide translocates across the cell membrane appears to involve both macropinocytosis and clathrin-mediated endocytosis (CME). Similarly, the TP10 peptide penetrates the cell membrane primarily via CME.156 This suggests that cellular uptake mechanisms are complex and multifaceted. In addition to macropinocytosis, various endocytic processes, such as CME, also play a vital role in the uptake of CPPs by neural cells. The Tat peptide and related transporters rely on caveolin-mediated endocytosis.157,158 Furthermore, the cellular uptake of Tat was partially inhibited by a blocking agent, indicating that caveolin-mediated endocytosis is not solely responsible for its uptake, as other pathways also work in conjunction. A comprehensive study revealed that Tat and pArg utilize three endocytic pathways for cellular translocation: clathrin-mediated endocytosis, caveolin-mediated endocytosis, and micropinocytosis. However, the involvement of these pathways in uptake varies across different CPPs, highlighting the complexity and specificity of their cellular entry mechanisms.159 J.P. Richard proposed that the clathrin-dependent pathway might be involved in the cellular uptake of CPPs and demonstrated that TAT uptake in HeLa cells was reduced by 50% following chlorpromazine-mediated inhibition.160 It is widely accepted that CPPs bind to the plasma membrane before being internalized via any of the aforementioned endocytic pathways.161 The electrostatic interaction between CPPs and glycosaminoglycans is not merely a passive cellular internalization process. In fact, it prompts endocytosis by inducing the aggregation of membrane proteins. The heparan sulfate proteoglycans have been implicated in playing a crucial role in the cellular internalization of CPPs. However, the precise role of proteoglycans in the context is still ambiguous.162,163 Recently, it has been reported that scavenger receptors play a crucial role in the endocytic-mediated internalization of PepFect CPPs, indicating that internalization also depends on these receptors.164 After endocytic internalization, CPPs and CPPs-cargo complexes must escape from endosomes to avoid lysosomal degradation, reach their target site, and exert their biological activity. Endosomal escape is a major limiting factor for the efficient intracellular delivery of functional macromolecules.49 Several reports have shown that the formation of a pH gradient, increased vesicle concentration, and the attraction of CPPs can cause membrane stiffening and rupture, contributing to the escape of CPPs from endosomes.165,166 The endocytic pathway is ideal for delivering drugs to the area of interest. However, the number of NPs that can enter cells is limited by factors such as endocytosis, endosomal maturation, and exocytosis. Studies have shown that NPs must be smaller than 100 nm to efficiently penetrate the cells through vesicle-based transport systems. This transport mechanism is particularly effective for the transcytosis of macromolecules across endothelial cells.167
Direct Penetration
Although nerve-dependent endocytosis is considered the main pathway for the cellular internalization of various CPPs, increasing evidence indicates that some CPPs are internalized through endocytosis-independent pathways. Certain CPPs, for example, are internalized at low temperatures, which may explain why they avoid energy-dependent endocytosis.168–172 Direct penetration is a straightforward, single-step process that occurs independently of energy. In this mechanism, positively charged CPPs interact with negatively charged components of the cell membrane, such as the phospholipid bilayer and heparan sulfate.173 It has been shown that Arg-CPPs translocate through the plasma membrane by the direct penetration mechanism.167–171 The direct translocation of the Arg-rich CPPs is thought to be facilitated by their strong interactions with the lipid head groups of the plasma membrane. Specifically, the positively charged Arg residues bind to negatively charged lipids (eg, phosphatidylserine (PS) and phosphatidylglycerol (PG)). Additionally, the guanidine group forms hydrogen bonds with phosphate, carboxylic acid, or sulfate groups, leading to structural changes in the membrane.174,175 Direct penetration of CPPs can even occur in the presence of cryogenic and endocytic inhibitors. CPPs can penetrate cells via five distinct direct penetration mechanisms: (i) the formation of inverted micelles, (ii) the carpet-like model, (iii) the model of membrane thinning, (iv) transient pore formation, and (v) the induction of multilamellarity alongside the stalk-pore hypothesis (Figure 4).176,177 The first pathway involves the formation of inverted micelles, where CPPs aggregate and insert themselves into the lipid bilayer of the membrane. The carpet-like model demonstrates that CPPs adhere to the membrane surface and disrupt the lipid organizational arrangement, consequently allowing them to penetrate the membrane. Additionally, the membrane thinning model proposes that CPPs rely on reducing the membrane thickness, which subsequently creates favourable conditions for their cellular internalization. The transient pore formation model relies on CPPs forming temporary openings in the cell membrane by interacting with lipid molecules. The induction of multilamellar and the stalk-pore hypothesis highlight how CPPs can promote the formation of multiple lipid layers and transient structures that facilitate their uptake into the cell. These diverse internalization pathways highlight the versatile applications of CPPs in cellular drug delivery, functioning without the need for energy input.
Strategies for CPPs Conjugation to Cargo
CPPs and cargos bind through covalent binding or physical mixtures Recently, the ConjuPepDB database was launched, and it contains information on over 1600 drug molecules conjugated with CPPs, their biomedical applications, and the specific chemical conjugation procedures employed.178
Covalent Binding
Numerous types of covalent bonds exist, including peptide bonds,179 disulfide bonds,180,181 sulfanyl bonds,182 maleimide bonds,183 imine bonds,184 and triazole bonds.184 The attachment of CPPs to cargo through covalent binding enhances the stability, improves cellular uptake, endosomal escape and allows precise control over positional selectivity.185 Morishita et al found that the D-R9-insulin conjugate promoted intestinal insulin absorption and demonstrated an improved hypoglycaemic effect after pulmonary administration in diabetic rats.186 Noriyasu et al noticed an improvement in the intestinal absorption of insulin due to the covalent attachment of D-R8 with insulin. They also observed that the binding ratio between insulin and D-R8 plays a significant role in enhancing intestinal absorption.20 However, covalent conjugation can alter the CPPs or cargo molecule’s natural conformation and functional groups, which may affect their biological activity. And this method faces the challenge of synthesis complexity and inflexibility for dynamic adaptation.187–189 The active fragment of parathyroid hormone (PTH1-34), conjugated with the N-terminal, demonstrated superior biological activity compared to the C-terminal conjugated (VP22-PTH (1–34) of ((CD40 + CD86) / (MHC II + CD11C) and relative SEAP activity increased by 5% and 0.3 compared to PTH (1–34) -VP22, respectively). The coadministration approach proved to be more effective for delivering PTH 1–34 across the Caco-2 monolayer.188 In such cases, non-covalent conjugation strategies are more appropriate.
Physical Mixture
The CPPs-cargo complexes are formed through intermolecular interactions, including hydrophobic interactions, electrostatic forces, hydrogen bonds, and others.77 This method can preserve cargo bioactivity, simplify preparation, and enable dynamic ratio adjustment, as demonstrated by R8-insulin mixtures enhancing intestinal absorption. Kamei et al discovered that the simultaneous administration of insulin and R8, including VEC and RRL helices, following physical mixing, notably enhanced insulin absorption in the ileum of rats. Among various CPPs, the L-CPPs demonstrated the highest efficacy for intestinal absorption of insulin.190 Non-covalent complexes also shield cargo from enzymatic degradation, prolonging serum half-life. Despite limitations in stability under physiological conditions and lower loading efficiency, non-covalent strategies hold significant clinical potential for delivering sensitive therapeutics (eg, nucleic acids, proteins) due to their adaptability, stimulus-responsive release, and reduced immunogenicity. Future advancements may integrate nanocarriers or targeted ligands to address stability and toxicity challenges, positioning non-covalent systems as promising tools for oral biologics, combination therapies, and precision medicine. For instance, negatively charged oligonucleotides interact with positively charged CPPs through electrostatic and hydrophobic forces. In this strategy, CPPs shield the bioactive conjugate, thereby protecting it from degradation by proteases or nucleases, which, in turn, prolong the serum half-life of the cargo.49 Table 2 summarizes the results of more CPPs combined with cargo.
 |
Table 2 List of the Reported Conjugates of CPPs and Cargos |
CPPs and PNPs for Oral Delivery of Insulin
Recently, many nanocarriers, including lipid-based nanocarriers (liposomes, micelles), carbon nanotubes (CNTs), inorganic nanocarriers (quantum dots), gold nanoparticles and polymeric nanoparticles (PNPs) have been reported.6 However, lipid-based nanocarriers (liposomes, micelles) exhibit limitations including immunogenicity, stability challenges, and scalability issues. For instance, polyethylene glycol (PEG) coatings on liposomes may induce anti-PEG antibodies, leading to accelerated blood clearance (ABC phenomenon) or hypersensitivity reactions, compromising long-term efficacy. Repeated dosing of lipid formulations can also trigger complement activation, resulting in drug leakage and toxicity. Furthermore, phospholipid bilayers in solid lipid nanoparticles (SLNs) are prone to drug leakage during storage, while large-scale production via microfluidic techniques remains inefficient. Lipid carriers may also interfere with lipid metabolism by binding to lipoproteins (eg, LDL), potentially exacerbating atherosclerosis.196,197 CNTs are generally limited by poor biodegradability and challenges in systemic clearance.198 Inorganic nanocarriers (quantum dots) and gold nanoparticles, face biocompatibility and toxicity concerns. Quantum dots containing heavy metals (eg, cadmium) pose long-term toxicity risks, while gold nanoparticles often require complex surface modifications to achieve targeting, increasing costs.199,200 The PNPs are oral drug delivery systems which are typically range between 100 to 500 nm. They are classified into two types: natural polymers and synthetic polymers.201 PNPs can improve insulin loading capacity, gastrointestinal stability, higher bioavailability, low toxicity and preparation controllability.202–205 It is especially suitable for the treatment of diseases that require precise delivery and long-term efficacy. However, these systems face multiple challenges, including low expression of target receptors and insufficient uptake capacity.12 CPPs have been widely used in the delivery of protein drugs due to their cell membrane, penetrating and designed targeting function, and good biocompatibility.17–20 The intestinal permeability of insulin is influenced by factors such as the chain length, amphipathic nature, hydrophobic characteristics, and basic properties of CPPs. The attachment of CPPs with macromolecules enhances the permeability of membranes for the associated cargoes.189 Tat-CPPs not only efficiently penetrate the cell membrane but also enhances the translocation of 100 times larger macromolecular drugs. Tat (YGRKKRRQRRR) interacts non-covalently with proteins to mediate their delivery into cells.206,207 In 2005, the first CPPs employed as a peptide vector for insulin transport through the intestinal epithelium was TAT-PTD.34 Since then, additional CPPs have been utilized as potential carriers for intestinal transepithelial and transmucosal delivery of biopharmaceuticals in both in vitro and in vivo studies. Mariko et al found that oligoarginine is likely to be a powerful tool for overcoming insulin’s low permeability through the epithelial cell membrane.186 According to Kristensen et al. Arg residues within the peptide are essential for facilitating insulin transport across the epithelium.188 This is because Arg is the most polar of all the proteinogenic amino acids. The cationic Arg residue is translocated through a nonpolar lipid bilayer, and more acid residues form stronger hydrogen bonds. This forms a salt bridge between the cationic amino acids and the acidic residues.208 As stated by Patel et al209 the covalent linkage of D-R9 to insulin plays a crucial role in its translocation mechanism across the alveolar epithelium in rats. According to Patel et al, the covalent attachment of D-R9 to insulin is essential for the translocation process across the alveolar epithelium in rats. The D-R9-insulin conjugate demonstrated an enhanced hypoglycaemic effect after pulmonary administration in diabetic rats and also promoted the intestinal absorption of insulin.186 Kamei et al demonstrated that the binding rate of insulin to D-R8 is a critical factor in D-R8’s ability to enhance intestinal insulin absorption.20 The initial group demonstrated that CPPs facilitated the oral absorption of a peptide through a physical mixing formulation without the use of PNPs or devices. Nielsen et al observed that, in diluted gastrointestinal fluid, D-penetration exhibited greater stability than L-penetration. By interacting non-covalently with insulin, D-penetration extends its half-life from 25 minutes to 91 minutes. Nonetheless, the effectiveness of this approach was only moderate (PA = 1.2%) compared to the subcutaneous delivery of insulin.210 Bioavailability, stability, selectivity, and in vivo efficacy are enhanced by incorporating CPPs into or coupling with nanomaterials.6,211
Natural Polymers
Natural polymers, such as polysaccharides, proteins, peptides, and nucleic acids, offer numerous advantages for encapsulating and delivering active drug components into the cell. These benefits include biodegradability, biocompatibility, nontoxicity, and non-immunogenic properties.212,213 Biocompatible and biodegradable, natural polymer nanoparticle carriers are the most promising materials for developing innovative oral insulin delivery systems.214
Natural Polysaccharide
Natural polysaccharides primarily include dextran, chitosan (CS), pectin, alginate (ALG), hyaluronic acid (HA), starch, and others.
CS is a cationic polysaccharide copolymer composed of glucosamine and N-acetylglucosamine linked by β (1→4) bonds.215 The structure of CS features numerous active amine and hydroxyl groups. Consequently, chemical modification of CS, along with further modification of its derivatives, can yield new compounds with desired characteristics.216 CS improves the intestinal permeability of biological macromolecules by enhancing adhesiveness and temporarily opening tight junctions, ensuring good biocompatibility and safety for drug delivery applications.217 Guo et al218 developed a delivery system (Tat-CS-NPs) consisting of NPs incorporated with Tat and amphiphilic chitosan derivatives (aCS). The resulting delivery system demonstrated a significant increase in the colonic absorption of the medication. The study on a diabetic rat model revealed that Tat-CS-NPs produced an insulin-lowering effect that was 6.89 times greater than PVA-NPs and 1.79 times greater than CS-NPs, indicating that Tat enhanced penetration efficiency. The nanocarriers, developed by coupling chitosan (CS) with a novel cell-penetrating peptide (SAR6EW), demonstrated embedding and drug loading rates for insulin of 75.36% and 7.58%, respectively. The oral administration of SAR6EW/CS/insulin NPs produced a greater hypoglycaemic effect in diabetic rats compared to CS/insulin NPs without inducing NP-mediated toxicity. The findings indicated that chitosan nanocarriers mediated by SAR6EW effectively deliver insulin orally. Furthermore, this delivery method shows promise for the oral administration of other protein-based medications.219 Liu et al220 employed CRT-modified trimethylchitosan (TMC) NPs for the oral delivery of insulin. Their results showed that CRT-NPs exhibited improved cellular uptake, superior transport across the Caco-2 monolayer, and enhanced absorption via villi, particularly when compared to transferrin receptor-targeted modified NPs (HAI NPs). Furthermore, in vivo investigations confirmed that the hypoglycemic effect and absorption of insulin (INS) were significantly greater in diabetic rats treated with CRT peptide-modified NPs compared to those receiving HAI NPs targeting the transferrin receptor (TfR). These findings suggest that CRT peptides represent a promising alternative for the oral delivery of peptides and proteins.
Modified starch derivatives, such as carboxymethyl short-chain amylose and cyclodextrin (CD), are extensively utilized in drug delivery due to their low toxicity, excellent biocompatibility, and unique biodegradability. CD are cyclic oligosaccharides widely regarded as valuable pharmaceutical excipients.221 β-cyclodextrin (β-CD) is a ring-shaped oligosaccharide featuring a hydrophobic cavity that interacts with specific surface residues of insulin through hydrophobic interactions.222,223 The R8-CM-β-CD conjugate enhanced the intestinal absorption of insulin through various endocytosis mechanisms, inhibited the function of P-glycoprotein (P-gp) efflux pumps, and demonstrated an extraordinary hypoglycemic effect. These outcomes highlight R8-CM-β-CD as a promising candidate for an effective drug delivery system.224 Nanocomplexes loaded with insulin were prepared through self-assembly using CPPs or their derivatives modified with bis-β-cyclodextrin (P-bis-CD), enhancing stability and delivery efficiency. In diabetic rats, the intestinal administration of P-bis-CD nanocomplexes demonstrated a significant hypoglycemic effect that persisted for 6 h. The bioavailability of EN NC and P-bis-CDNC was measured at 3.5% and 10.6%, respectively. These findings indicate that P-bis-CD effectively enhances the epithelial penetration of insulin.12
Alginate, a polyanionic polysaccharide, is widely used in various fields, including medicine and food. Its reliability and biocompatibility make it highly suitable for numerous applications in medical materials.225 In a study, Li et al194 developed R8-modified insulin-SA NPs (INS-SA/R8 NPs) to enhance the oral delivery of insulin. These NPs significantly improved insulin absorption, increased intestinal permeability and enhanced the uptake of villi. Notably, INS-SA/R8 NPs also stimulated the production of endogenous nitric oxide (NO), a naturally occurring signalling molecule that enhances insulin absorption at specific concentrations. When administered orally, INS-SA/R8 NPs exhibited superior hypoglycemic effects and biocompatibility compared to the oral administration of INS-SA NPs in diabetic rats.
Natural Proteins
Proteins are essential natural biomaterials for developing innovative nano-vehicles due to their exceptional properties, including biodiversity, biodegradability, low immunogenicity, non-toxicity, and biocompatibility.226,227 Protein-based nano-vehicles, with their well-defined primary structure, can be customized through pre- or post-functionalization. This unique characteristic allows various drugs, components, and carriers to be combined with the hydrophobic or hydrophilic regions of proteins using a range of reagents. In addition to serving as insulin delivery systems, protein NPs can also provide energy to individuals with diabetes.228,229 Liu et al230 utilized conventional transferrin receptor-targeting ligand (HAIYPRH)-modified NPs (HAI-NPs) for the oral delivery of insulin. The HAI-NPs demonstrated good hypoglycaemic efficacy and insulin absorption. However, while their hypoglycemic efficacy and insulin absorption were significantly improved, they remained lower than those of CRT peptide-modified NPs.
Synthetic Polymers
Synthetic polymers (SPs) offer several advantages over natural polymers and other materials, particularly due to their ability to be tailor-made for specific pathological needs and patient requirements.229 Examples of synthetic polymers (SPs) include poly(lactic-co-glycolic acid) (PLGA), hydrogels, silica, porous coordination polymers such as metal-organic frameworks (MOFs), and porous organic polymers (POPs), among others.230 Synthetic polymers have been shown to enhance bioavailability and slow the release of insulin in oral insulin delivery systems.
PLGA
PLGA is a widely recognized biomaterial used in drug delivery and approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA).231–233 It is metabolized into lactide and glycolic acid in the body, which are further converted into carbon dioxide and water via the Krebs cycle. This indicates that PLGA possesses a low toxicity profile for living organisms. Importantly, as a copolymer composed of two monomers (lactide and glycolate), PLGA’s degradation rates and drug release profiles can be regulated by modifying the ratio of its monomers and the molecular weights of lactide and glycolate.3 Consequently, PLGA is regarded as one of the most essential carriers in modern biomedical research, enabling targeted and extended drug delivery to address various health issues, including diabetes and cancer.234–237
In 2013, Yan et al238 developed Tat-mediated PLGA NPs for delivering insulin to the brain. Their findings indicated that CPPs have the potential to facilitate the transport of macromolecules to cerebral tissue. In 2013, Liu et al239 introduced a method using CPPs-functionalized PLGA-NPs to enhance the oral delivery of insulin. Their results showed that the use of L-R8-modified NPs increased the relative bioavailability of insulin by 3.2 times, while D-R8-modified NPs increased it by 4.4 times. Corresponding improvements in hypoglycemic effects were observed at 2.5 times and 3.7 times, respectively. In 2016, J. Sheng and associates59 developed an insulin-LMWP conjugate, which was subsequently loaded onto mucoadhesive PLGA NPs coated with N-trimethyl chitosan chloride, resulting in the formation of insulin-LMWP NPs. These findings indicated that the pharmacological availability of oral insulin was 17.98 ± 5.61% compared to subcutaneously administered insulin, representing an improvement twice as significant as that observed with MNPs containing native insulin. In 2022, Yang et al240 developed PLGA-NPs (Pep/Gal NPs) modified with pH-responsive stretchable CPPs and a liver-targeting fraction (Gal). They demonstrated effective intestinal absorption and significant intrahepatic deposition of insulin. The highest pharmacological availability (PA) of Pep/Gal-PNPs was 10.1%. Notable, Pep/Gal PNPs supported glucose homeostasis for effective diabetes management by increasing hepatic glycogen production by 7.2-folds.
Hydrogel
Hydrogel systems consist of two or more components, including a stable three-dimensional polymer network and water molecules that fill the spaces between macromolecules. These hydrogels can absorb and retain substantial amounts of water (typically between 70% and 99%); therefore, they are ideal for formulating hydrophilic and sensitive drugs in semi-solid delivery systems.241 The water content influences drug diffusion through the polymer network in the hydrogel. The diffusion coefficient is determined by the number and size of the pores, while water absorption depends on the type of bonds.242 The hydration of hydrophilic groups in the polymer chain triggers the swelling phenomenon. As these polar groups hydrate, the network expands, exposing hydrophobic groups that can interact with water molecules. This process allows the polymer network to absorb additional water, driven by osmotic forces that push it toward a state of infinite dilution. This additional expansion is limited by covalent cross-links, which establish equilibrium. Free water refers to excess water absorbed after the saturation of ionic, polar, and hydrophobic groups. The “free water” occupies the voids within the polymeric network.243 Fukuoka et al38 investigated the effectiveness of combining P(MAA-g-EG) hydrogel with oligoarginine R6 to enhance intestinal insulin absorption. Their findings suggest that P(MAA-g-EG) hydrogel carriers provide protection and enable the controlled release of drugs, presenting a promising approach for oral insulin delivery.
Silica
Silicon NPs (SNs) are widely used as inorganic drug carriers. Mesoporous silica NPs (MSNs) (Mobil Composition of Matter (MCM), Santa Barbara Amorphous (SBA), and Mesocellular Foam (MCF)) have been extensively studied in medical applications such as diagnosis, engineering, and treatment. Among various drug delivery systems, SN-based oral delivery systems have been extensively studied and demonstrate significant potential for the delivery of oral therapeutic proteins and peptides (TPPs) delivery, particularly insulin.244,245
In 2021, Rao et al synthesized porous SNs and modified them with poly (pyridyl disulfide ethylene phosphate/sulfobetaine) polymers, resulting in the formation of P(PyEP-g-SBm)n-AmPSiNPs NPs. The insulin-loaded P(PyEP-g-SB)-AmPSiNPs demonstrated enhanced stability and biocompatibility in-vitro. However, their hypoglycemic effect and bioavailability were suboptimal in vivo, possibly due to the mucus layer acting as a barrier to the nanoparticle’s passing through the intestinal epithelium.244 The same year, Zhang et al246 engineered mesoporous SNs with modification groups (MSN-NH2@COOH/CPPs5) that effectively traversed the mucus layer and passed through the intestinal epithelium by mimicking the surface of viruses. These MSN-NH2@COOH/CPPs5-NPs significantly improved the apical-to-basal transcytosis, primarily through caveolae-mediated endocytosis. Interestingly, the transepithelial transport rate of MSN-NH2@COOH/CPPs5 across the Caco-2 cell monolayer was 2.4-fold higher than that of MSN@NH2 and 2.0-fold higher compared to MSN-NH2@COOH. The bioavailability of insulin encapsulated in the MSN-NH2@COOH/CPPs5 NPs was 2.1 times higher than insulin administered directly into the jejunum. Additionally, these modified NPs demonstrated no significant toxicity in preliminary in vitro and in vivo experiments. This work demonstrates the efficient delivery of peptide or protein drugs by overcoming dual barriers, such as the intestinal mucus layer and epithelium.
Others
Many synthetic polymers, such as carrier peptides and silica-alginate NPs, can orally deliver insulin but have been less extensively studied. Diedrichsen et al247 studied the effect of the physical mixing of carrier peptides with insulin on intestinal absorption. Carrier peptides are novel and promising excipients for oral delivery of therapeutic peptides. The study demonstrated that insulin’s transepithelial permeability was influenced by the extent of complex formation with the insulin-carrier peptide and depended on the stereochemistry of petromax. However, it was not affected by penetration or shuffle. He et al248 suggested that encapsulating CPPs-insulin conjugates into silica-alginate-NPs enhances the intestinal shelf life of insulin. The presence of alginate within the NPs highlights its adhesive role in facilitating transport to the intestinal mucosa. Upon accumulation at the mucosal surface, the strong cell-penetrating capabilities of CPPs enable the rapid passage of released CPPs-insulin conjugates across the epithelial cell layer, directly delivering insulin into the blood circulation. Additionally, the swift internalization of CPPs-mediated NPs significantly reduces the time insulin spends in the luminal cavity, thereby minimizing its degradation by endogenous proteases. In 2020, Abdelhamid et al249 introduced a method for loading simple oligonucleotides (ONs) and achieving efficient release through a synergistic approach. This method combines the adaptable features of ZIF-8 NPs as chemically tunable nanocarriers with those of CPPs, specifically PepFects (PF), functioning as a capping system. Their findings revealed that the use of ZIF-8 and its composites enhances cell transfection and improves the cellular uptake of ONs while maintaining high biocompatibility. Thus, combining CPPs with POPs may significantly enhance drug loading and enable efficient delivery. Surprisingly, the sublingual insulin drops developed by the University of British Columbia (UBC) team, incorporating fish-derived cell-penetrating peptides, enable rapid insulin absorption via sublingual capillaries, demonstrating hypoglycemic efficacy comparable to injections. This technology has been validated in murine models and has entered the commercial licensing phase.250
In summary, CPPs play a vital role as transporters for nanodrugs and demonstrate promising outcomes in the treatment of diabetes. Further, significant research findings are outlined in Table 3.
 |
Table 3 Application of CPPs in the Oral Insulin Delivery |
Conclusion and Perspective
In recent years, numerous nanoparticle systems intended for the oral administration of insulin have been developed. However, the efficacy of novel drug delivery systems (NDDS) is often constrained by the low expression of receptors on enterocytes and their limited absorption capacity. Incorporating or coupling CPPs with nanoparticles (NPs) can enhance bioavailability, stability, selectivity, and in vivo efficacy. Despite being the safe and effective carrier for macromolecular delivery, CPPs have not received significant attention for the oral delivery of insulin. Currently, porous organic polymers (POPs), including hyper-cross-linked polymers (HCPs), metal-organic frameworks (MOFs), and covalent organic frameworks (COFs), are widely utilized in oral insulin delivery systems. Their unique pore architecture, adjustable pore dimensions, customizable structures, and modifiable surface properties make them particularly suitable for this application. POPs can enhance bioavailability and enable controlled insulin release, making them an excellent candidate for oral insulin delivery systems. In the future, the development of multimodal delivery systems (POPs-CPPs nanocomposite carriers) and stimuli-responsive CPPs engineering (eg, pH/enzyme-sensitive peptide motifs) provides a promising strategy to overcome mucosal barriers, achieve enhanced cellular internalization, and optimize future oral insulin delivery platforms. The implementation of CPPs-mediated intranasal and pulmonary delivery systems could establish a complementary therapeutic regimen to oral administration, effectively mitigating dose-dependent complications associated with single-route delivery. Furthermore, CPPs-enabled co-delivery of insulin with metabolic regulators (eg, GLP-1 analogues) facilitates synergistic glycemic control through insulin-GLP-1 combination therapy. To accelerate clinical translation, it is imperative to forge interdisciplinary collaborations between academic institutions and pharmaceutical enterprises, ultimately enabling pain-free and therapeutically efficient treatment modalities for diabetic patients.
Abbreviations
IV, Intravenous; IP, Intraperitoneal; IM, Intramuscular; SC, Subcutaneous; NDDS, Nano-drug delivery systems; CPPs, Cell-penetrating peptides; NPs, Nanoparticles; PTDs, Protein transduction domains; MTPs, Membrane transduction peptides; NLSs, Nuclear localization signals; CPDs, Cell-permeable polydisulfides; SPPS, Solid-phase peptide synthesis; ROP, Ring-opening polymerization; NCAs, N-carboxy anhydrides; AMM, Activated monomer; NAM, Normal amine; LiHMDS Lithium hexamethyldisilazane; PK, Pharmacokinetics; CME, Clathrin-mediated endocytosis; PS, Phosphatidylserine; PG, Phosphatidylglycerol; CS, Chitosan; ALG, Alginate; HA, Hyaluronic acid; TMC, Trimethylchitosan; INS, Insulin; TfR, Transferrin receptor; CD, Cyclodextrin; gp, P-glycoprotein; SPs, Synthetic polymers; PLGA, Poly(lactic-co-glycolic acid); PA, Pharmacological availability; MOFs, Metal-organic frameworks; POPs, Porous organic polymers; FDA, Food and Drug Administration; EMA, European Medicines Agency; SNs, Silicon nanoparticles; MSNs, Mesoporous silica nanoparticles; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous; MCF, Mesocellular Foam; TPPs, Therapeutic proteins and peptides; OAs, Oligonucleotides; PF, PepFects; HCPs, Hyper-cross-linked polymers; COFs, Covalent organic frameworks; ɛ-PL, ɛ-polylysine; γ-PGA, γ-polyglutamate; pArg, Polyarginine; Arg, Arginine; Lys, Lysine; Val, Valine; Leu, Leucine; Ile, Isoleucine; Ala, Alanine; Pro, Proline; Phe, Phenylalanine; Glu, Glutamate; Asp, Aspartate; His, Histidine; Trp, Tryptophan; Tyr, Tycrosine; Gly, Glycine.
Data Sharing Statement
No data was used for the research described in the article.
Acknowledgments
Acknowledge Prof. Genlin Zhang for their fruitful discussion and suggestions during the preparation of this manuscript. We gratefully acknowledge the School of Chemistry and Chemical Engineering/State Key Laboratory Incubation Base for Green Processing of Chemical Engineering (Shihezi University) for providing the facilities to carry out this work. We also appreciate the support of Xinjiang Synthetic Biological Industry Innovation Research Institute.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the International Journal of Nanomedicineto which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Corps Science and Technology Program Project (2022ZD066).
Disclosure
The authors declare that they have no competing interests.
References
1. Wu J, Roesger S, Jones N, Hu C-MJ, Li SD. Cell-penetrating peptides for transmucosal delivery of proteins. J Control Release. 2024;366:864–878. doi:10.1016/j.jconrel.2024.01.038
2. Shi M, McHugh KJ. Strategies for overcoming protein and peptide instability in biodegradable drug delivery systems. Adv Drug Deliv Rev. 2023;199:114904. doi:10.1016/j.addr.2023.114904
3. Pang H, Huang X, Xu ZP, Chen C, Han FY. Progress in oral insulin delivery by PLGA nanoparticles for the management of diabetes. Drug Discov Today. 2023;28:14. doi:10.1016/j.drudis.2022.103393
4. Xiao Y, Tang Z, Wang J, et al. Oral insulin delivery platforms: strategies to address the biological barriers. Angew Chem Int Edit. 2020;59:19787–19795. doi:10.1002/anie.202011449
5. Drucker DJ. Advances in oral peptide therapeutics. Nat Rev. 2020;19:277–289. doi:10.1038/s41573-019-0053-0
6. Wang YY, Li H, Rasool A, Wang HB, Manzoor R, Zhang GL. Polymeric nanoparticles (PNPs) for oral delivery of insulin. J Nanobiotechnol. 2024;22(1). doi:10.1186/s12951-023-02253-y
7. Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61:75–85. doi:10.1016/j.addr.2008.09.008
8. Chen MC, Sonaje K, Chen KJ, Sung HW. A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials. 2011;32:9826–9838. doi:10.1016/j.biomaterials.2011.08.087
9. Salama NN, Eddington ND, Fasano A. Tight junction modulation and its relationship to drug delivery. Drug Deliv Rev. 2006;58:15–28. doi:10.1016/j.addr.2006.01.003
10. Chalasani KB, Russell-Jones GJ, Jain AK, et al. Effective oral delivery of insulin in animal models using vitamin B12-coated dextran nanoparticles. J Control Release. 2007;122:141–150. doi:10.1016/j.jconrel.2007.05.019
11. Zou JJ, Wei G, Xiong C, et al. Efficient oral insulin delivery enabled by transferrin-coated acid-resistant metal-organic framework nanoparticles. Sci Adv. 2022;8:eabm4677. doi:10.1126/sciadv.abm4677
12. Zhu X, Shan W, Zhang P, et al. Penetratin derivative-based nanocomplexes for enhanced intestinal insulin delivery. Mol Pharm. 2014;11:317–328. doi:10.1021/mp400493b
13. Khafagy ES, Morishita M. Oral biodrug delivery using cell-penetrating peptide. Adv Drug Deliv Rev. 2012;64:531–539. doi:10.1016/j.addr.2011.12.014
14. Raucher D, Ryu JS. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med. 2015;21(9):560–570. doi:10.1016/j.molmed.2015.06.005
15. Swiecicki JM, Pisa MD, Lippi F, et al. Unsaturated acyl chains dramatically enhanced cellular uptake by direct translocation of a minimalist oligo-arginine lipopeptide. Chem Commun. 2015;51:14656–14659. doi:10.1039/C5CC06116D
16. Douat C, Aisenbrey C, Antunes S, et al. A cell-penetrating foldamer with a bioreducible linkage for intracellular delivery of DNA. Angew Chem Int Edit. 2015;54:11133–11137. doi:10.1002/ange.201504884
17. Xie J, Bi Y, Zhang H, et al. Cell-penetrating peptides in diagnosis and treatment of human diseases: from preclinical research to clinical application. Front Pharmacol. 2020;11:6979. doi:10.3389/fphar.2020.00697
18. Eudes F, Chugh A. Cell-penetrating peptides. Plant Signal Behav. 2008;3:1246. doi:10.4161/psb.3.8.5696
19. Zhang D, Wang J, Xu D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J Control Release. 2016;229:130–139. doi:10.1016/j.jconrel.2016.03.020
20. Kamei N, Morishita M, Takayama K. Importance of intermolecular interaction on the improvement of intestinal therapeutic peptide/protein absorption using cell-penetrating peptides. J Control Release. 2009;136:179–186. doi:10.1016/j.jconrel.2009.02.015
21. Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60:580–597. doi:10.1016/j.addr.2007.10.005
22. Copolovici DM, Langel K, Eriste E, Langel U. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano. 2014;8:1972–1994. doi:10.1021/nn4057269
23. Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharma Res. 2007;24:1977–1992. doi:10.1007/s11095-007-9303-7
24. Dowaidar M. Uptake pathways of cell-penetrating peptides in the context of drug delivery gene therapy, and vaccine development. Cell Signal. 2024;117:111116. doi:10.1016/j.cellsig.2024.111116
25. Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi:10.1016/0092-8674(88)90263-2
26. Ghaemi B, Tanwar S, Singh A, et al. Cell-penetrating and enzyme-responsive peptides for targeted cancer therapy: role of arginine residue length on cell penetration and in vivo systemic toxicity. ACS Appl Mater Interfaces. 2024;16:11159–11171. doi:10.1021/acsami.3c14908
27. Ueda Y, Wei FY, Hide T, et al. Induction of autophagic cell death of glioma-initiating cells by cell-penetrating D-isomer peptides consisting of Pas and the p53 C-terminus. Biomaterials. 2012;33:9061–9069. doi:10.1016/j.biomaterials.2012.09.003
28. Li Y, Zheng X, Cao Z, et al. Self-assembled peptide (CADY-1) improved the clinical application of doxorubicin. Int J Pharmaceut. 2012;434:209–214. doi:10.1016/j.ijpharm.2012.06.003
29. Jirka SMG, Hoen PACT, Parillas VD, et al. Cyclic peptides to improve delivery and exon skipping of antisense oligonucleotides in a mouse model for duchenne muscular dystrophy. Mol Ther. 2018;26:132–147. doi:10.1016/j.ymthe.2017.10.004
30. Mukundan V, Maksoudian C, Vogel M, et al. Cytotoxicity of prion protein-derived cell-penetrating peptides is independent of amyloid formation. Biophys J. 2016;110:37A–37A. doi:10.1016/j.bpj.2015.11.269
31. Buccini DF, Cardoso MH, Franco OL. Antimicrobial peptides and cell-penetrating peptides for treating intracellular bacterial infections. Front Cell Infect Mi. 2021;10:612931. doi:10.3389/fcimb.2020.612931
32. Huang X, Li G. Antimicrobial peptides and cell-penetrating peptides: non-antibiotic membrane-targeting strategies against bacterial infections. Infect Drug Resist. 2023;16:1203–1219. doi:10.2147/IDR.S396566
33. Zhao M, Tan X, Liu ZQ, et al. Engineered phage with cell-penetrating peptides for intracellular bacterial infections. mSystems. 2023;8. doi:10.1128/msystems.00646-23
34. Liang JF, Yang VC. Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem Biophys Res Commun. 2005;335:734–738. doi:10.1016/j.bbrc.2005.07.142
35. Khafagy ES, Iwamae R, Kamei N, Takeda-Morishita M. Region-dependent role of cell-penetrating peptides in insulin absorption across the rat small intestinal membrane. AAPS J. 2015;17:1427–1437. doi:10.1208/s12248-015-9804-y
36. Zhu S, Chen S, Gao Y, et al. Enhanced oral bioavailability of insulin using PLGA nanoparticles co-modified with cell-penetrating peptides and Engrailed secretion peptide (Sec). Drug Deliv. 2016;23:1980–1991. doi:10.3109/10717544.2015.1043472
37. Kamei N, Shigei C, Hasegawa R, Takeda-Morishita M. Exploration of the key factors for optimizing the in vivo oral delivery of insulin by using a noncovalent strategy with cell-penetrating peptides. Biol Pharm Bull. 2018;41:239–246. doi:10.1248/bpb.b17-00798
38. Fukuoka Y, Khafagy ES, Goto T, et al. Combination strategy with complexation hydrogels and cell-penetrating peptides for oral delivery of insulin. Biol Pharm Bull. 2018;41:811–814. doi:10.1248/bpb.b17-00951
39. Xu Y, Zhang X, Wang N, et al. Cell-penetrating peptide enhanced insulin buccal absorption. Int J Pharmaceut. 2020;584:119469. doi:10.1016/j.ijpharm.2020.119469
40. Futaki S, Nakase I, Tadokoro A, Takeuchi T, Jones AT. Arginine-rich peptides and their internalization mechanisms. Biochem Soc Trans. 2007;35:784–787. doi:10.1042/bst0350784
41. Balayssac S, Burlina F, Convert O, et al. Comparison of penetratin and other homeodomain-derived cell-penetrating peptides: interaction in a membrane-mimicking environment and cellular uptake efficiency. Biochemistry. 2006;45:1408–1420. doi:10.1021/bi0518390
42. Takechi-Haraya Y, Saito H. Current understanding of physicochemical mechanisms for cell membrane penetration of arginine-rich cell penetrating peptides: role of glycosaminoglycan interactions. Curr Protein Pept Sc. 2018;19:623–630. doi:10.2174/1389203719666180112100747
43. Lu S, Tager LA, Chitale S, Riley LW. A cell-penetrating peptide derived from mammalian cell uptake protein of Mycobacterium tuberculosis. Analy Biochem. 2006;353:7–14. doi:10.1016/j.ab.2006.01.044
44. Gori A, Lodigiani G, Colombarolli SG, et al. Cell penetrating peptides: classification, mechanisms, methods of study, and applications. ChemMedChem. 2023:18. doi:10.1002/cmdc.202300236.
45. Agrawal P, Bhalla S, Usmani SS, et al. CPPsite 2.0: a repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016;44:D1098–D1103. doi:10.1093/nar/gkv1266
46. Derakhshankhah H, Jafari S. Cell penetrating peptides: a concise review with emphasis on biomedical applications. Biomed Pharmacother. 2018;108:1090–1096. doi:10.1016/j.biopha.2018.09.097
47. Green M, Ishino M, Loewenstein PM. Mutational analysis of HIV-1 Tat minimal domain peptides: identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell. 1989;58:215–223. doi:10.1016/0092-8674(89)90417-0
48. He H, Ye J, Liu E, et al. Low molecular weight protamine (LMWP): a nontoxic protamine substitute and an effective cell-penetrating peptide. J Control Release. 2014;193:63–73. doi:10.1016/j.jconrel.2014.05.056
49. Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38:406–424. doi:10.1016/j.tips.2017.01.003
50. Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17:850–860. doi:10.1016/j.drudis.2012.03.002
51. Kalafatovic D, Giralt E. Cell-penetrating peptides: design strategies beyond primary structure and amphipathicity. Molecules. 2017;22:1929. doi:10.3390/molecules22111929
52. Radis-Baptista G, Campelo IS, Morlighem J-ERL, Melo LM, Freitas VJF. Cell-penetrating peptides (CPPs): from delivery of nucleic acids and antigens to transduction of engineered nucleases for application in transgenesis. J Biotechnol. 2017;252:15–26. doi:10.1016/j.jbiotec.2017.05.002
53. Marks JR, Placone J, Hristova K, et al. Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J Am Chem Soc. 2011;133:8995–9004. doi:10.1021/ja2017416
54. Chauhan A, Tikoo A, Kapur AK, Singh M. The taming of the cell penetrating domain ofthe HIV Tat: myths and realities. J Control Release. 2007;117:148–162. doi:10.1016/j.jconrel.2006.10.031
55. Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi:10.1074/jbc.272.25.16010
56. Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Dis Markers. 1990;8:34–35. doi:10.1016/0092-8674(88)90262-0
57. Liu Y, Kim YJ, Ji M, et al. Enhancing gene delivery of adeno-associated viruses by cell-permeable peptides. Mol Ther-Meth Clin D. 2014;1:12. doi:10.1038/mtm.2013.12
58. Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. doi:10.1016/s0021-9258(17)34080-2
59. Sheng J, He H, Han L, et al. Enhancing insulin oral absorption by using mucoadhesive nanoparticles loaded with LMWP-linked insulin conjugates. J Control Release. 2016;233:181–190. doi:10.1016/j.jconrel.2016.05.015
60. Kim D, Lee Y, Dreher TW, Cho TJ. Empty turnip yellow mosaic virus capsids as delivery vehicles to mammalian cells. Virus Res. 2018;252:13–21. doi:10.1016/j.virusres.2018.05.004
61. Futaki S, Suzuki T, Ohashi W, et al. Arginine-rich peptides-an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi:10.1074/jbc.m007540200
62. De Coupade C, Fittipaldi A, Chagnas V, et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J. 2005;390:407–418. doi:10.1042/bj20050401
63. Pooga M, Soomets U, Hällbrink M, et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol. 1998;16:857–861. doi:10.1038/nbt0998-857
64. Oehlke J, Krause E, Wiesner B, et al. Extensive cellular-uptake into endothelial cells of an amphipathic beta-sheet forming peptide. FEBS Lett. 1997;415:196–199. doi:10.1016/s0014-5793(97)01123-x
65. Elliott G, Ohare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi:10.1016/s0092-8674(00)81843-7
66. Oehlke J, Scheller A, Wiesner B, et al. Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. BBA-Biomembranes. 1998;1414:127–139. doi:10.1016/s0005-2736(98)00161-8
67. Lundberg P, Magzoub M, Lindberg M, et al. Cell membrane translocation of the N-terminal (1-28) part of the prion protein. Biochem Biophys Res Commun. 2002;299:85–90. doi:10.1016/s0006-291x(02)02595-0
68. Elmquist A, Hansen M, Langel U. Structure-activity relationship study of the cell-penetrating peptide pVEC. J Pept Sci. 2004;10:188. doi:10.1016/j.bbamem.2006.05.013
69. Magzoub M, Sandgren S, Lundberg P, et al. N-terminal peptides from unprocessed prion proteins enter cells by macropinocytosis. Biochem Biophys Res Commun. 2006;348:379–385. doi:10.1016/j.bbrc.2006.07.065
70. Lindgren M, Rosenthal-Aizman K, Saar K, et al. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem Pharmacol. 2006;71:416–425. doi:10.1016/j.bcp.2005.10.048
71. Lundberg P, El-Andaloussi S, Sutlu T, et al. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21:2664–2671. doi:10.1096/fj.06-6502com
72. El-Andaloussi S, Johansson HJ, Holm T, Langel U. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol Ther. 2007;15:1820–1826. doi:10.1038/sj.mt.6300255
73. Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100:201–217. doi:10.1042/bc20070116
74. Johansson HJ, El-Andaloussi S, Holm T, et al. Characterization of a novel cytotoxic cell-penetrating peptide derived from p14ARF protein. Mol Ther. 2008;16:115–123. doi:10.1038/sj.mt.6300346
75. Pujals S, Giralt E. Proline-rich, amphipathic cell-penetrating peptides. Adv Drug Deliv Rev. 2008;60:473–484. doi:10.1016/j.addr.2007.09.012
76. Crombez L, Aldrian-Herrada G, Konate K, et al. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol Ther. 2009;17:95–103. doi:10.1038/mt.2008.215
77. Khafagy ES, Morishita M, Takayama K. The role of intermolecular interactions with penetratin and its analogue on the enhancement of absorption of nasal therapeutic peptides. Int J Pharmaceut. 2010;388:209–212. doi:10.1016/j.ijpharm.2009.12.060
78. Yamada T, Christov K, Shilkaitis A, et al. p28, a first in class peptide inhibitor of cop1 binding to p53. Br J Cancer. 2013;108:2495–2504. doi:10.1038/bjc.2013.266
79. Bae H-D, Lee K. On employing a translationally controlled tumor protein-derived protein transduction domain analog for transmucosal delivery of drugs. J Control Release. 2013;170:358–364. doi:10.1016/j.jconrel.2013.06.010
80. Bae HD, Lee J, Jun KY, Kwon Y, Lee K. Modification of translationally controlled tumor protein-derived protein transduction domain for improved intranasal delivery of insulin. Drug Deliv. 2018;25:1025–1032. doi:10.1080/10717544.2018.1464081
81. Bae HD, Kim M, Lee J, Lee K. Modified translationally controlled tumor protein-derived protein transduction domain enhances nasal delivery of exendin-4 as shown with insulin. Drug Deliv. 2018;25:1579–1584. doi:10.1080/10717544.2018.1491653
82. Rhee M, Davis P, et al. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J Biol Chem. 2006;281:1233–1240. doi:10.1074/jbc.m509813200
83. Gao CS, Mao SL, Ditzel HJ, et al. A cell-penetrating peptide from a novel pVII-pIX phage-displayed random peptide library. Bioorgan Med Chem. 2002;10:4057–4065. doi:10.1016/s0968-0896(02)00340-1
84. Nakayama F, Yasuda T, Umeda S, et al. Fibroblast growth factor-12 (FGF12) translocation into intestinal epithelial cells is dependent on a novel cell-penetrating peptide domain: involvement of internalization in the in vivo role of exogenous FGF12. J Biol Chem. 2011;286:25823–25834. doi:10.1074/jbc.m110.198267
85. Sochacka M, Karelus R, Opalinski L, et al. FGF12 is a novel component of the nucleolar NOLC1/TCOF1 ribosome biogenesis complex. Cell Commun Signal. 2022;20:182. doi:10.1186/s12964-022-01000-4
86. Li D, Su T, Ma L, et al. Dual-acidity-labile polysaccharide-di-drugs conjugate for targeted cancer chemotherapy. Eur J Med Chem. 2020;199:112367. doi:10.1016/j.ejmech.2020.112367
87. Mandal D, Nasrolahi Shirazi A, Parang K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew Chem Int Edit. 2011;50(41):9633–9637. doi:10.1002/anie.201102572
88. Nasrolahi Shirazi A, Tiwari RK, Oh D, et al. Surface decorated gold nanoparticles by linear and cyclic peptides as molecular transporters. Mol Pharm. 2013;10(8):3137–3151. doi:10.1021/mp400199e
89. Park SE, Sajid MI, Parang K, Tiwari RK. Cyclic cell-penetrating peptides as efficient intracellular drug delivery tools. Mol Pharm. 2019;16:3727–3743. doi:10.1021/acs.molpharmaceut.9b00633
90. Oh D, Sun J, Nasrolahi Shirazi A, et al. Antibacterial activities of amphiphilic cyclic cell-penetrating peptides against multidrugresistant pathogens. Mol Pharm. 2014;11(10):3528–3536. doi:10.1021/mp5003027
91. Welch JJ, Swanekamp RJ, King C, Dean DA, Nilsson BL. Functional delivery of sirna by disulfide-constrained cyclic amphipathic peptides. ACS Med Chem Lett. 2016;7(6):584–589. doi:10.1021/acsmedchemlett.6b00031
92. Wallbrecher R, Depre L, Verdurmen WPR, et al. Exploration of the design principles of a cell-penetrating bicylic peptide scaffold. Bioconjugate Chem. 2014;25(5):955–964. doi:10.1021/bc500107f
93. Ichimizu S, Watanabe H, Maeda H, et al. Design and tuning of a cell-penetrating albumin derivative as a versatile nanovehicle for intracellular drug delivery. J Control Release. 2018;277:23–34. doi:10.1016/j.jconrel.2018.02.037
94. Zorko M, Jones S, Langel U. Cell-penetrating peptides in protein mimicry and cancer therapeutics. Adv Drug Deliv Rev. 2022;180:114044. doi:10.1016/j.addr.2021.114044
95. Pooga M, Hällbrink M, Zorko M, Langel Ü. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi:10.1096/fsb2fasebj.12.1.67
96. Tashima T. Intelligent substance delivery into cells using cell-penetrating peptides. Bioorg Med Chem Lett. 2017;27:121–130. doi:10.1016/j.bmcl.2016.11.083
97. Zorko M, Langel U. Studies of cell-penetrating peptides by biophysical methods. Q Rev Biophys. 2022;55:e3. doi:10.1017/s0033583522000026
98. Zahid M, Robbins PD. Cell-type specific penetrating peptides: therapeutic promises and challenges. Molecules. 2015;20:13055–13070. doi:10.3390/molecules200713055
99. Nam SH, Park J, Koo H. Recent advances in selective and targeted drug/gene delivery systems using cell-penetrating peptides. Arch Pharm Res. 2023;46:18–34. doi:10.1007/s12272-022-01425-y
100. Tkachenko AG, Xie H, Coleman D, et al. Multifunctional gold nanoparticle-peptide complexes for nuclear targeting. J Am Chem Soc. 2003;125:4700–4701. doi:10.1021/ja0296935
101. Zahid M, Robbins PD. Identification and characterization of tissue-specific protein transduction domains using peptide phage display. Methods Mol Biol. 2011;683:277–289. doi:10.1007/978-1-60761-919-2_20
102. Cerrato CP, Langeln U. An update on cell-penetrating peptides with intracellular organelle targeting. Expert Opin Drug Deliv. 2022;19:133–146. doi:10.1080/17425247.2022.2034784
103. Tesei G, Vazdar M, Jensen MR, et al. Self-association of a highly charged arginine-rich cell-penetrating peptide. Proc Natl Acad Sci U S A. 2017;114:11428–11433. doi:10.1073/pnas.1712078114
104. Rothbard JB, Kreider E, Vandeusen CL, et al. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J Med Chem. 2002;45:3612–3618. doi:10.1021/jm0105676
105. Kang Z, Ding G, Meng Z, Meng Q. The rational design of cell-penetrating peptides for application in delivery systems. Peptides. 2019;121:170149. doi:10.1016/j.peptides.2019.170149
106. Bruelisauer L, Kathriner N, Prenrecaj M, et al. Tracking the bioreduction of disulfide-containing cationic dendrimers. Angew Chem Int Edit. 2012;51:12454–12458. doi:10.1002/ange.201207070
107. Morelli P, Martin-Benlloch X, Tessier R, et al. Ethynyl benziodoxolones: functional terminators for cell-penetrating poly(disulfide)s. Polym Chem. 2016;7:3465–3470. doi:10.1039/c6py00562d
108. Nhan NTT, Maidana DE, Yamada KH. Ocular delivery of therapeutic agents by cell-penetrating peptides. Cells. 2023;12:1071. doi:10.3390/cells12071071
109. Takayama K, Nakase I, Michiue H, et al. Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas). J Control Release. 2009;138:128–133. doi:10.1016/j.jconrel.2009.05.019
110. Som A, Reuter A, Tew GN. Protein transduction domain mimics: the role of aromatic functionality. Angew Chem Int Edit. 2012;51:980–983. doi:10.1002/anie.201104624
111. Frederix PWJM, Scott GG, Abul-Haija YM, et al. Exploring the sequence space for (tri-) peptide self-assembly to design and discover new hydrogels. Nat Chem. 2015;7:30–37. doi:10.1038/nchem.2122
112. Salonen LM, Ellermann M, Diederich F. Aromatic rings in chemical and biological recognition: energetics and structures. Angew Chem Int Edit. 2011;50:4808–4842. doi:10.1002/anie.201007560
113. Hennig A, Gabriel GJ, Tew GN, Matile S. Stimuli-responsive polyguanidino-oxanorbornene membrane transporters as multicomponent sensors in complex matrices. J Am Chem Soc. 2008;130:10338–10344. doi:10.1021/ja802587j
114. Prata CAH, Zhang X-X, Luo D, et al. Lipophilic peptides for gene delivery. Bioconjugate Chem. 2008;19:418–420. doi:10.1021/bc700451b
115. Yamashita H, Kato T, Oba M, et al. Development of a cell-penetrating peptide that exhibits responsive changes in its secondary structure in the cellular environment. Sci Rep. 2016;6:33033. doi:10.1038/srep33003
116. Kawaguchi Y, Takeuchi T, Kuwata K, et al. Syndecan-4 is a receptor for clathrin-mediated endocytosis of arginine-rich cell-penetrating peptides. Bioconjugate Chem. 2016;27:1119–1130. doi:10.1021/acs.bioconjchem.6b00082
117. Morris MC, Depollier J, Mery J, et al. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19:1173–1176. doi:10.1038/nbt1201-1173
118. Regberg J, Srimanee A, Langel U. Applications of cell-penetrating peptides for tumor targeting and future cancer therapies. Pharmaceuticals. 2012;5:991–1007. doi:10.3390/ph5090991
119. Martin I, Teixido M, Giralt E. Building cell selectivity into CPPs-mediated strategies. Pharmaceuticals. 2010;3:1456–1490. doi:10.3390/ph3051456
120. Zhang W, Song J, Zhang B, et al. Design of acid-activated cell penetrating peptide for delivery of active molecules into cancer cells. Bioconjugate Chem. 2011;22:1410–1415. doi:10.1021/bc200138d
121. Kim C, Lee Y, Kim JS, et al. Thermally triggered cellular uptake of quantum dots immobilized with poly(n-isopropylacrylamide) and cell penetrating peptide. Langmuir. 2010;26:14965–14969. doi:10.1021/la102632m
122. Song Z, Han Z, Lv S, et al. Synthetic polypeptides: from polymer design to supramolecular assembly and biomedical application. Chem Soc Rev. 2017;46:6570–6599. doi:10.1039/c7cs00460e
123. Wang T, Li H, Liang C, et al. Purification and characterization of a novel antioxidant Phelligridin LA produced by Inonotus baumii. J Chem Technol Biotechnol. 2020;95:2483–2494. doi:10.1002/jctb.6430
124. Zenin V, Tsedilin A, Yurkova M. Siniavin A, Fedorov A. Thermostable chaperone-based polypeptide biosynthesis: enfuvirtide model product quality and protocol-related impurities. PLoS One. 2023;18:e0286752. doi:10.1371/journal.pone.0286752
125. Deng C, Wu J, Cheng R, et al. Functional polypeptide and hybrid materials: precision synthesis via α-amino acid N-carboxyanhydride polymerization and emerging biomedical applications. Prog Polym Sci. 2014;39:330–364. doi:10.1016/j.progpolymsci.2013.10.008
126. Collins JM, Singh SK, White TA, et al. Total wash elimination for solid phase peptide synthesis. Nat Commun. 2023;14:8168. doi:10.1038/s41467-023-44074-5
127. Liu Y, Li D, Ding J, Chen X. Controlled synthesis of polypeptides. Chinese Chem l Let. 2020;31:3001–3014. doi:10.1016/j.cclet.2020.04.029
128. Varnava KG, Sarojini V. Making solid-phase peptide synthesis greener: a review of the literature. Chem-Asian J. 2019;14:1088–1097. doi:10.1002/asia.201801807
129. Lee YS. Gram-scale preparation of C-terminal-modified enkephalin analogues by typical liquid-phase peptide synthesis. Curr Protoc Protein Sci. 2019;98:e97–e97. doi:10.1002/cpps.97
130. Wu Y, Chen K, Wang J, et al. Recent advances and future developments in the preparation of polypeptides via N-carboxyanhydride (NCA) ring-opening polymerization. J Am Chem Soc. 2024;146:24189–24208. doi:10.1021/jacs.4c05382
131. Huang J, Heise A. Stimuli responsive synthetic polypeptides derived from N-carboxyanhydride (NCA) polymerisation. Chem Soc Rev. 2013;42:7373–7390. doi:10.1039/c3cs60063g
132. Yan J, Liu K, Li W, et al. Thermoresponsive dendronized polypeptides showing switchable recognition to catechols. Macromolecules. 2016;49:510–517. doi:10.1021/acs.macromol.5b02259
133. Gebbing M, Bergmann T, Schulz E, Ehrhardt A. Gene therapeutic approaches to inhibit hepatitis B virus replication. World J Hepatol. 2015;7:150–164. doi:10.4254/wjh.v7.i2.150
134. Trabulo S, Cardoso AL, Cardoso AMS, et al. Cell-penetrating peptides as nucleic acid delivery systems: from biophysics to biological applications. Curr Pharm Des. 2013;19:2895–2923. doi:10.2174/1381612811319160006
135. Stewart MP, Langer R, Jensen KF. Intracellular delivery by membrane disruption: mechanisms, strategies, and concepts. Chem Rev. 2018;118:7409–7531. doi:10.1021/acs.chemrev.7b00678
136. Koo JH, Kim GR, Nam KH, et al. Unleashing cell-penetrating peptide applications for immunotherapy. Trends Mol Med. 2022;28:482–496. doi:10.1016/j.molmed.2022.03.010
137. Lu J, Xu H, Xia J, et al. D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front Microbiol. 2020;11:563030. doi:10.3389/fmicb.2020.563030
138. Yao JF, Yang H, Zhao YZ, Xue M. Metabolism of peptide drugs and strategies to improve their metabolic stability. Curr Drug Metab. 2018;19(11):892–901. doi:10.2174/1389200219666180628171531
139. Lehto T, Vasconcelos L, Margus H, et al. Saturated fatty acid analogues of cell-penetrating peptide pepfect14: role of fatty acid modification in complexation and delivery of splice-correcting oligonucleotides. Bioconjug Chem. 2017;28(3):782–792. doi:10.1021/acs.bioconjchem.6b00680
140. Paernaste L, Arukuusk P, Langel K, Tenson T, Langel U. The formation of nanoparticles between small interfering rna and amphipathic cell-penetrating peptides. Mol Ther. 2017;7:1–10. doi:10.1016/j.omtn.2017.02.003
141. Gentilucci L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr Pharm Des. 2010;16(28):3185–3203. doi:10.2174/138161210793292555
142. Amantana A, Moulton HM, Cate ML, et al. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18(4):1325–1331. doi:10.1021/bc070060v
143. Habault J, Poyet JL. Recent advances in cell penetrating peptide-based anticancer therapies. Molecules. 2019;24(5):1052301. doi:10.3390/molecules24050927
144. Xu J, Wang F, Ye L, et al. Penetrating peptides: applications in drug delivery. J Drug Deliv Sci Tec. 2023;84:104475. doi:10.1016/j.jddst.2023.104475
145. Murayama T, Masuda T, Afonin S, et al. Loosening of lipid packing promotes oligoarginine entry into cells. Angew Chem Int Ed Engl. 2017;129(26):7644. doi:10.1002/anie.201703578
146. Zhang X, Lei T, Du H. Prospect of cell penetrating peptides in stem cell tracking. Stem Cell Res Ther. 2021;12(1):457. doi:10.1186/s13287-021-02522-3
147. Gagat M, Zielinska W, Grzanka A. Cell-penetrating peptides and their utility in genome function modifications. Int J Mol Med. 2017;40:1615–1623. doi:10.3892/ijmm.2017.3172
148. Ruseska I, Zimmer A. Internalization mechanisms of cell-penetrating peptides. Beilstein J Nanotechnol. 2020;11:101–123. doi:10.3762/bjnano.11.10
149. Cheng X, Chen K, Dong B, et al. Dynamin-dependent vesicle twist at the final stage of clathrin-mediated endocytosis. Nat Cell Biol. 2021;23:859–869. doi:10.1038/s41556-021-00713-x
150. Mahmoudi A, Jalili A, Aghaee-Bakhtiari SH, et al. MicroRNA delivery by arginine-rich cell-penetrating peptides: an investigation on expression and the cellular uptake mechanisms. Colloids Surf. Physicochem Eng Asp. 2024;700. doi:10.1016/j.colsurfa.2024.134749
151. Salloum G, Bresnick AR, Backer JM. Macropinocytosis: mechanisms and regulation. Biochem J. 2023;480:335–362. doi:10.1042/bcj20210584
152. Kim GC, Cheon DH, Lee Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim Biophys Acta Proteins Proteom. 2021;1869:140604. doi:10.1016/j.bbapap.2021.140604
153. Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi:10.1038/nature01451
154. Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102:247–253. doi:10.1016/j.jconrel.2004.10.018
155. Nakase I, Noguchi K, Aoki A, et al. Arginine-rich cell-penetrating peptide-modified extracellular vesicles for active macropinocytosis induction and efficient intracellular delivery. Sci Rep. 2017;7:1991. doi:10.1038/s41598-017-02014-6
156. Eiriksdottir E, Mager I, Lehto T, et al. Cellular internalization kinetics of (luciferin-) cell-penetrating peptide conjugates. Bioconjugate Chem. 2010;21:1662–1672. doi:10.1021/bc100174y
157. Fittipaldi A, Ferrari A, Zoppé M, et al. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278:34141–34149. doi:10.1074/jbc.m303045200
158. Ferrari A, Pellegrini V, Arcangeli C, et al. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther. 2003;8:284–294. doi:10.1016/s1525-0016(03)00122-9
159. Maeger I, Langel K, Lehto T, et al. The role of endocytosis on the uptake kinetics of luciferin-conjugated cell-penetrating peptides. BBA-Biomembranes. 2012;1818:502–511. doi:10.1016/j.bbamem.2011.11.020
160. Richard JP, Melikov K, Brooks H, et al. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–15306. doi:10.1074/jbc.m401604200
161. Nakase I, Tadokoro A, Kawabata N, et al. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 2007;46:492–501. doi:10.1021/bi0612824
162. Letoha T, Keller-Pinter A, Kusz E, et al. Cell-penetrating peptide exploited syndecans. BBA-Biomembranes. 2010;1798:2258–2265. doi:10.1016/j.bbamem.2010.01.022
163. Subrizi A, Tuominen E, Bunker A, et al. Tat(48-60) peptide amino acid sequence is not unique in its cell penetrating properties and cell-surface glycosaminoglycans inhibit its cellular uptake. J Control Release. 2012;158:277–285. doi:10.1016/j.jconrel.2011.11.007
164. Ezzat K, Helmfors H, Tudoran O, et al. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012;26:1172–1180. doi:10.1096/fj.11-191536
165. Borrelli A, Tornesello AL, Tornesello ML, Buonaguro FM. Cell Penetrating peptides as molecular carriers for anti-cancer agents. Molecules. 2018;23:295. doi:10.3390/molecules23020295
166. Xu J, Khan AR, Fu M, et al. Cell-penetrating peptide: a means of breaking through the physiological barriers of different tissues and organs. J Control Release. 2019;309:106–124. doi:10.1016/j.jconrel.2019.07.020
167. Rennick JJ, Johnston APR, Parton RG. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat Nanotechnol. 2021;16:266–276. doi:10.1038/s41565-021-00858-8
168. Hirose H, Takeuchi T, Osakada H, et al. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol Ther. 2012;20:984–993. doi:10.1038/mt.2011.313
169. Kosuge M, Takeuchi T, Nakase I, et al. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjugate Chem. 2008;19:656–664. doi:10.1021/bc700289w
170. Ter-Avetisyan G, Tuennemann G, Nowak D, et al. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284:3370–3378. doi:10.1074/jbc.m805550200
171. Wallbrecher R, Ackels T, Olea RA, et al. Membrane permeation of arginine-rich cell-penetrating peptides independent of transmembrane potential as a function of lipid composition and membrane fluidity. J Control Release. 2017;256:68–78. doi:10.1016/j.jconrel.2017.04.013
172. Jiao CY, Delaroche D, Burlina F, et al. Translocation and endocytosis for cell-penetrating peptide internalization. J Biol Chem. 2009;284:33957–33965. doi:10.1074/jbc.m109.056309
173. Szabo I, Yousef MA, Soltesz D, et al. Redesigning of cell-penetrating peptides to improve their efficacy as a drug delivery system. Pharmaceutics. 2022;14:907. doi:10.3390/pharmaceutics14050907
174. Rothbard JB, Jessop TC, Lewis RS, et al. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–9507. doi:10.1021/ja0482536
175. Li L, Vorobyov I, Allen TW. The different interactions of lysine and arginine side chains with lipid membranes. J Phys Chem B. 2013;117:11906–11920. doi:10.1021/jp405418y
176. Pirhaghi M, Mamashli F, Moosavi-Movahedi F, et al. Cell-penetrating peptides: promising therapeutics and drug-delivery systems for neurodegenerative diseases. Mol Pharm. 2024;21:2097–2117. doi:10.1021/acs.molpharmaceut.3c01167
177. Zakany F, Mándity IM, Varga Z, et al. Effect of the lipid landscape on the efficacy of cell-penetrating peptides. Cells. 2023;12:1700. doi:10.3390/cells12131700
178. Balogh B, Ivánczi M, Nizami B, et al. ConjuPepDB: a database of peptide-drug conjugates. Nucleic Acids Res. 2021;49:D1102–D1112. doi:10.1093/nar/gkaa950
179. Bode SA, Timmermans SBPE, Eising S, et al. Click to enter: activation of oligo-arginine cell-penetrating peptides by bioorthogonal tetrazine ligations. Chem Sci. 2019;10:701–705. doi:10.1039/c8sc04394a
180. Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357–1369. doi:10.1016/j.addr.2012.09.039
181. Turner JJ, Arzumanov AA, Gait MJ. Synthesis, cellular uptake and HIV-1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucleic Acids Res. 2005;33:27–42. doi:10.1093/nar/gki142
182. Kardani K, Milani A, Shabani SH, et al. Cell penetrating peptides: the potent multi-cargo intracellular carriers. Expert Opinion Drug Deliv. 2019;16:1227–1258. doi:10.1080/17425247.2019.1676720
183. Gayraud F, Klussmann M, Neundorf I. Recent advances and trends in chemical CPPs-drug conjugation techniques. Molecules. 2021;26:1591. doi:10.3390/molecules26061591
184. Shabanpoor F, McClorey G, Saleh AF, et al. Bi-specific splice-switching PMO oligonucleotides conjugated via a single peptide active in a mouse model of duchenne muscular dystrophy. Nucleic Acids Res. 2015;43:29–39. doi:10.1093/nar/gku1256
185. Roth L, Agemy L, Kotamraju VR, et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31:3754–3763. doi:10.1038/onc.2011.537
186. Morishita M, Kamei N, Ehara J, et al. A novel approach using functional peptides for efficient intestinal absorption of insulin. J Control Release. 2007;118:177–184. doi:10.1016/j.jconrel.2006.12.022
187. Falanga A, Lombardi L, Galdiero E, et al. The world of cell penetrating: the future of medical applications. Future Med Chem. 2020;12(15):1431–1446. doi:10.4155/fmc-2020-0140
188. Kristensen M, de Groot AM, Berthelsen J, et al. Conjugation of cell-penetrating peptides to parathyroid hormone affects its structure, potency, and transepithelial permeation. Bioconjugate Chem. 2015;26(3):477–488. doi:10.1021/bc5005763
189. Maeng J, Lee K. Systemic and brain delivery of antidiabetic peptides through nasal administration using cell-penetrating peptides. Front Pharmacol. 2022;13:1068495. doi:10.3389/fphar.2022.1068495
190. Kamei N, Morishita M, Eda Y, et al. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J Control Release. 2008;132:21–25. doi:10.1016/j.jconrel.2008.08.001
191. Lin T, Liu E, He H, et al. Nose-to-brain delivery of macromolecules mediated by cell-penetrating peptides. Acta Pharm Sin B. 2016;6:352–358. doi:10.1016/j.apsb.2016.04.001
192. El-Sayed NS, Shirazi AN, Sajid MI, et al. Synthesis and antiproliferative activities of conjugates of paclitaxel and camptothecin with a cyclic cell-penetrating peptide. Molecules. 2019;4:1427. doi:10.3390/molecules24071427
193. Uhl P, Bajraktari-Sylejmani G, Witzigmann D, et al. A nanocarrier approach for oral peptide delivery: evaluation of cell-penetrating-peptide-modified liposomal formulations in dogs. Adv Ther. 2023;6. doi:10.1002/adtp.202300021
194. Li M, Sun Y, Ma C, et al. Design and investigation of penetrating mechanism of octaarginine-modified alginate nanoparticles for improving intestinal insulin delivery. J Pharm Sci. 2021;110:268–279. doi:10.1016/j.xphs.2020.07.004
195. Kamei N, Takeda-Morishita M. Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J Control Release. 2015;197:105–110. doi:10.1016/j.jconrel.2014.11.004
196. Ban Y, Chu Y, Pan F, et al. Lipid-based nanocarriers enabled oral delivery of oleanolic acid derivative DKS26 for diabetes management. Adv Healthc Mater. 2023;12(16). doi:10.1002/adhm.202300639
197. Chauhan A, Chand P, Parashar T, et al. Lipid-based nanoparticles: strategy for targeted cancer therapy. BIO Integr. 2025;6(6). doi:10.15212/bioi-2024-0107
198. Sireesha M, Babu VJ, Ramakrishna S. Functionalized carbon nanotubes in bio-world: applications, limitations and future directions. Mat Sci Eng B-Adv. 2017;223:43–63. doi:10.1016/j.mseb.2017.06.002
199. Wu J, Bai X, Yan L, et al. Selective regulation of macrophage lipid metabolism via nanomaterials’ surface chemistry. Nat Commun. 2024;15(1):8349. doi:10.1038/s41467-024-52609-7
200. Deepak G, Anirudh S, Khushboo D, Sunita N. Targeted drug delivery using graphene quantum dots: approaches, limitations and future perspectives. ECS Trans. 2022;107:16081. doi:10.1149/10701.16081ecst
201. Lohcharoenkal W, Wang L, Chen YC, Rojanasakul Y. Protein nanoparticles as drug delivery carriers for cancer therapy. Biomed Res Int. 2014;2014:1–12. doi:10.1155/2014/180549
202. Zaiki Y, Lim LY, Wong TW. Critical material designs for mucus- and mucosa-penetrating oral insulin nanoparticle development. Int Mater Rev. 2023;68:121–139. doi:10.1080/09506608.2022.2040293
203. Cao J, Li X, Tian H. Metal-organic framework (MOF)-based drug delivery. Curr Med Chem. 2020;2:5949–5969. doi:10.2174/0929867326666190618152518
204. Zhao S, Huang C, Yue X, et al. Application advance of electrosprayed micro/nanoparticles based on natural or synthetic polymers for drug delivery system. Mater Design. 2022;220:110850. doi:10.1016/j.matdes.2022.110850
205. Gedawy A, Martinez J, Al-Salami H, et al. Oral insulin delivery: existing barriers and current counter-strategies. J Pharm Pharmacol. 2018;70:197–213. doi:10.1111/jphp.12852
206. Chen YP, Chen CT, Hung Y, et al. A new strategy for intracellular delivery of enzyme using mesoporous silica nanoparticles: superoxide dismutase. J Am Chem Soc. 2013;135:1516–1523. doi:10.1021/ja3105208
207. Liu J, Zhao Y, Guo Q, et al. TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials. 2012;33:6155–6161. doi:10.1016/j.biomaterials.2012.05.035
208. Calabretta LO, Thomas VM, Raines RT. Canavanine versus arginine: prospects for cell-penetrating peptides. Tetrahedron Lett. 2022;99:153848. doi:10.1016/j.tetlet.2022.153848
209. Patel EN, Wang J, Kim KJ, et al. Conjugation with cationic cell-penetrating peptide increases pulmonary absorption of insulin. Mol Pharm. 2009;6:492–503. doi:10.1021/mp800174g
210. Nielsen EJB, Yoshida S, Kamei N, et al. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J Control Release. 2014;189:19–24. doi:10.1016/j.jconrel.2014.06.022
211. Silva S, Almeida AJ, Vale N. Combination of cell-penetrating peptides with nanoparticles for therapeutic application: a review. Biomolecules. 2019;9:22. doi:10.3390/biom9010022
212. Chowdhury A, Kunjiappan S, Panneerselvam T, et al. Nanotechnology and nanocarrier-based approaches on treatment of degenerative diseases. Int Nano Lett. 2017;7:91–122. doi:10.1007/s40089-017-0208-0
213. Meneguin AB, Silvestre ALP, Sposito L, et al. The role of polysaccharides from natural resources to design oral insulin micro- and nanoparticles intended for the treatment of Diabetes mellitus: a review. Carbohyd Polym. 2021;256:117504. doi:10.1016/j.carbpol.2020.117504
214. Hu Q, Lu Y, Luo Y. Recent advances in dextran-based drug delivery systems: from fabrication strategies to applications. Carbohyd Polym. 2021;264:117999. doi:10.1016/j.carbpol.2021.117999
215. Denkbas EB, Odabasi M. Chitosan microspheres and sponges: preparation and characterization. J Appl Polym Sci. 2000;76:1637–1643. doi:10.1002/(sici)1097-4628(20000613)76:11<1637::aid-app4>3.0.co;2-q
216. Chen J, Huang L, Lai H, et al. Methotrexate-loaded PEGylated Chitosan nanoparticles: synthesis, characterization, and in vitro and in vivo antitumoral activity. Mol Pharm. 2014;11:2213–2223. doi:10.1021/mp400269z
217. Sonaje K, Chuang EY, Lin KJ, et al. Opening of epithelial tight junctions and enhancement of paracellular permeation by chitosan: microscopic, ultrastructural, and computed-tomographic observations. Mol Pharm. 2012;9:1271–1279. doi:10.1021/mp200572t
218. Guo F, Zhang M, Gao Y, et al. Modified nanoparticles with cell-penetrating peptide and amphipathic chitosan derivative for enhanced oral colon absorption of insulin: preparation and evaluation. Drug Deliv. 2016;23:2003–2014. doi:10.3109/10717544.2015.1048489
219. Li L, Yang L, Li M, Zhang L. A cell-penetrating peptide mediated chitosan nanocarriers forimproving intestinal insulin delivery. Carbohyd Polym. 2017;174:182–189. doi:10.1016/j.carbpol.2017.06.061
220. Liu M, Wu L, Shan W, et al. Iron-mimic peptide converts transferrin from foe to friend for orally targeting insulin delivery. J Mater Chem B. 2018;6:593–601. doi:10.1039/c7tb02450a
221. Ponce Cevallos PA, Buera MP, Elizalde BE. Encapsulation of cinnamon and thyme essential oils components (cinnamaldehyde and thymol) in β-cyclodextrin: effect of interactions with water on complex stability. J Food Eng. 2010;99:70–75. doi:10.1016/j.jfoodeng.2010.01.039
222. Lovatt A, Cooper A, Camilleri P. Energetics of cyclodextrin-induced dissociation of insulin. Eur Biophysics J Biophy. 1996;24:354–357. doi:10.1007/bf00180377
223. Irie T, Uekama K. Cyclodextrins in peptide and protein delivery. Adv Drug Deliv Rev. 1999;36:101–123. doi:10.1016/s0169-409x(98)00057-x
224. Yang L, Li M, Sun Y, et al. A cell-penetrating peptide conjugated carboxymethyl-β-cyclodextrin to improve intestinal absorption of insulin. Int J Biol Macromol. 2018;111:685–695. doi:10.1016/j.ijbiomac.2018.01.077
225. Bhattacharyya A, Mukhopadhyay P, Pramanik N, et al. Effect of polyethylene glycol on bis(2-hydroxyethyl) terephthalate-based polyurethane/alginate pH-sensitive blend for oral protein delivery. Adv Polym Tech. 2016;35:21525. doi:10.1002/adv.21525
226. Varanko A, Saha S, Chilkoti A. Recent trends in protein and peptide-based biomaterials for advanced drug delivery. Adv Drug Deliv Rev. 2020;156:133–187. doi:10.1016/j.addr.2020.08.008
227. Ferroni C, Varchi G. Keratin-based nanoparticles as drug delivery carriers. Appl Sci-Basel. 2021;11:9417. doi:10.3390/app11209417
228. Mahobia S, Bajpai J, Bajpai AK. Soya protein as possible potential nanocarriers for in-vitro oral delivery of insulin in simulated gastric fluids (SGFs). Int J Polym Mater. 2018;67:340–350. doi:10.1080/00914037.2017.1327435
229. Rizvi SAA, Saleh AM. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm J. 2018;26:64–70. doi:10.1016/j.jsps.2017.10.012
230. Han L, Peng Y, Ma J, et al. Construction of hypercrosslinked polymers with styrene-based copolymer precursor for adsorption of rare earth elements. Sep Purif Technol. 2022;285:120378. doi:10.1016/j.seppur.2021.120378
231. Allavena P, Palmioli A, Avigni R, et al. PLGA based nanoparticles for the monocyte-mediated anti-tumor drug delivery system. J Biomed Nanotechnol. 2020;16:212–223. doi:10.1166/jbn.2020.2881
232. Angkawinitwong U, Courtenay AJ, Rodgers AM, et al. A novel transdermal protein delivery strategy via electrohydrodynamic coating of PLGA microparticles onto microneedles. ACS Appl Mater Inter. 2020;12:12478–12488. doi:10.1021/acsami.9b22425
233. Mondal J, Khuda-Bukhsh AR. Cisplatin and farnesol co-encapsulated PLGA nano-particles demonstrate enhanced anti-cancer potential against hepatocellular carcinoma cells in vitro. Mol Biol Rep. 2020;47:3615–3628. doi:10.1007/s11033-020-05455-x
234. Wang W, Yu C, Zhang F, et al. Improved oral delivery of insulin by PLGA nanoparticles coated with 5β-cholanic acid conjugated glycol chitosan. Biomed Mater. 2021;16:064163. doi:10.1088/1748-605x/ac2a8c
235. Asal HA, Shoueir KR, El-Hagrasy MA, et al. Controlled synthesis of in-situ gold nanoparticles onto chitosan functionalized PLGA nanoparticles for oral insulin delivery. Int J Biol Macromol. 2022;209:2188–2196. doi:10.1016/j.ijbiomac.2022.04.200
236. Xu B, Jiang G, Yu W, et al. Preparation of poly(lactic-co-glycolic acid) and chitosan composite nanocarriers via electrostatic self assembly for oral delivery of insulin. Mat Sci Eng C-Mater. 2017;78:420–428. doi:10.1016/j.msec.2017.04.113
237. Feltrin F, Agner T, Sayer C, et al. Curcumin encapsulation in functional PLGA nanoparticles: a promising strategy for cancer therapies. Adv Colloid Interfac. 2022;300:102582. doi:10.1016/j.cis.2021.102582
238. Yan L, Wang H, Jiang Y, et al. Cell-penetrating peptide-modified PLGA nanoparticles for enhanced nose-to-brain macromolecular delivery. Macromol Res. 2013;21:435–441. doi:10.1007/s13233-013-1029-2
239. Liu X, Liu C, Zhang W, et al. Oligoarginine-modified biodegradable nanoparticles improve the intestinal absorption of insulin. Int J Pharmaceut. 2013;448:159–167. doi:10.1016/j.ijpharm.2013.03.033
240. Yang T, Wang A, Nie D, et al. Ligand-switchable nanoparticles resembling viral surface for sequential drug delivery and improved oral insulin therapy. Nat Commun. 2022;13:6649. doi:10.1038/s41467-022-34357-8
241. Zielinska A, Eder P, Rannier L, et al. Hydrogels for modified-release drug delivery systems. Curr Pharm Des. 2022;28:609–618. doi:10.2174/1381612828666211230114755
242. Batista RA, Espitia PJP, Vergne DMC, et al. Development and evaluation of superabsorbent hydrogels based on natural polymers. Polymers. 2020;12:2173. doi:10.3390/polym12102173
243. Martins JP, Figueiredo P, Wang S, et al. Neonatal Fc receptor-targeted lignin-encapsulated porous silicon nanoparticles for enhanced cellular interactions and insulin permeation across the intestinal epithelium. Bioact Mater. 2022;9:299–315. doi:10.1016/j.bioactmat.2021.08.007
244. Rao R, Liu X, Li Y, et al. Bioinspired zwitterionic polyphosphoester modified porous silicon nanoparticles for efficient oral insulin delivery. Biomater Sci. 2021;9:685–699. doi:10.1039/d0bm01772h
245. Tan X, Yin N, Liu Z, et al. Hydrophilic and electroneutral nanoparticles to overcome mucus trapping and enhance oral delivery of insulin. Mol Pharm. 2020;17:3177–3191. doi:10.1021/acs.molpharmaceut.0c00223
246. Zhang Y, Xiong M, Ni X, et al. Virus-mimicking mesoporous silica nanoparticles with an electrically neutral and hydrophilic surface to improve the oral absorption of insulin by breaking through dual barriers of the mucus layer and the intestinal epithelium. ACS Appl Mater Interfaces. 2021;13:18077–18088. doi:10.1021/acsami.1c00580
247. Diedrichsen RG, Tuelung PS, Fodera V, Nielsen HM. Stereochemistry and intermolecular interactions influence carrier peptide-mediated insulin delivery. Mol Pharm. 2023;20:1202–1212. doi:10.1021/acs.molpharmaceut.2c00883
248. He H, Ye J, Sheng J, et al. Overcoming oral insulin delivery barriers: application of cell penetrating peptide and silica-based nanoporous composites. Front Chem Sci Eng. 2013;7:9–19. doi:10.1007/s11705-013-1306-9
249. Abdelhamid HN, Dowaidar M, Hallbrink M, et al. Gene delivery using cell penetrating peptides-zeolitic imidazolate frameworks. Micropor Mesopor Mat. 2020;300:110173. doi:10.1016/j.micromeso.2020.110173
250. Wu J, Jones N, Hohenwarter L, et al. Systemic delivery of proteins using novel peptides via the sublingual route. J Control Release. 2024;368:290–302. doi:10.1016/j.jconrel.2024.02.042
251. Chen Z, Han S, Yang X, et al. Overcoming multiple absorption barrier for insulin oral delivery using multifunctional nanoparticles based on chitosan derivatives and hyaluronic acid. Int J Nanomed. 2020;15:4877–4898. doi:10.2147/ijn.s251627
252. Zhang L, Song L, Zhang C, Ren Y. Improving intestinal insulin absorption efficiency through coadministration of cell-penetrating peptide and hydroxypropyl-β-cyclodextrin. Carbohyd Polym. 2012;87:1822–1827. doi:10.1016/j.carbpol.2011.10.002
253. Daimon Y, Kamei N, Kawakami K, et al. Dependence of intestinal absorption profile of insulin on carrier morphology composed of β-cyclodextrin-grafted chitosan. Mol Pharm. 2016;13:4034–4042. doi:10.1021/acs.molpharmaceut.6b00561
254. Guo F, Ouyang T, Peng T, et al. Enhanced oral absorption of insulin using colon-specific nanoparticles co-modified with amphiphilic chitosan derivatives and cell-penetrating peptides. Biomater Sci. 2019;7:1493–1506. doi:10.1039/c8bm01485j
255. Shrestha N, Araújo F, Shahbazi MA, et al. Thiolation and cell-penetrating peptide surface functionalization of porous silicon nanoparticles for oral delivery of insulin. Adv Funct Mater. 2016;26:3405–3416. doi:10.1002/adfm.201505252
256. He H, Sheng J, David AE, et al. The use of low molecular weight protamine chemical chimera to enhance monomeric insulin intestinal absorption. Biomaterials. 2013;34:7733–7743. doi:10.1016/j.biomaterials.2013.06.047
257. Lundquist P, Khodus G, Niu Z, et al. Barriers to the intestinal absorption of four insulin-loaded arginine-rich nanoparticles in human and rat. ACS Nano. 2022;16:14210–14229. doi:10.1021/acsnano.2c04330
258. Niub Z, Samaridou E, Jaumain E, et al. PEG-PGA enveloped octaarginine-peptide nanocomplexes: an oral peptide delivery strategy. J Control Release. 2018;276:125–139. doi:10.1016/j.jconrel.2018.03.004
259. Rehmani S, McLaughlin CM, Eltaher HM, et al. Orally-delivered insulin-peptide nanocomplexes enhance transcytosis from cellular depots and improve diabetic blood glucose control. J Control Release. 2023;360:93–109. doi:10.1016/j.jconrel.2023.06.006
260. Wu Y, Chen K, Wang J, et al. Open-vessel polymerization of N-carboxyanhydride (NCA) for polypeptide synthesis. Nat Protoc. 2024. doi:10.1038/s41596-024-01062-3
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Cross-Sectional Retrospective Study Assessing Potentially Inappropriate Medications for Elderly Diabetic Patients in a Tertiary Care Hospital in Saudi Arabia
Fadil HA, Alrehaili ZS, Alharbi KM, Almuzaini AF, Alharbi RH, Alharbi HS, Elbadawy HM, Alahmadi YM
Risk Management and Healthcare Policy 2024, 17:3227-3238
Published Date: 19 December 2024
Level of Satisfaction Among Patients Using Insulin Administered by Pen vs Vial/Syringe. An Observational Prospective Study
Valladales-Restrepo LF, Delgado-Araujo AC, Oyuela-Gutiérrez MC, Ospina-Arzuaga HD, Machado-Alba JE
Patient Preference and Adherence 2025, 19:65-74
Published Date: 8 January 2025