")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Nanoparticles inhibit cancer cell invasion and enhance antitumor efficiency by targeted drug delivery via cell surface-related GRP78
Authors Zhao L, Li H, Shi Y, Wang G, Liu L, Su C, Su R
Received 24 September 2014
Accepted for publication 9 November 2014
Published 30 December 2014 Volume 2015:10(1) Pages 245—256
DOI https://doi.org/10.2147/IJN.S74868
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Liang Zhao,1,* Hongdan Li,2,* Yijie Shi,1 Guan Wang,2 Liwei Liu,1 Chang Su,3 Rongjian Su2
1School of Pharmacy, Liaoning Medical University, Jinzhou, People’s Republic of China; 2Central Laboratory of Liaoning Medical University, Jinzhou, People’s Republic of China; 3School of Veterinary Medicine, Liaoning Medical University, Jinzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Nanoparticles (NPs) which target specific agents could effectively recognize the target cells and increase the stability of chemical agents by encapsulation. As such, NPs have been widely used in cancer treatment research. Recently, over 90% of treatment failure cases in patients with metastatic cancer were attributed to resistance to chemotherapy. Surface-exposed glucose-regulated protein of 78 kDa (GRP78) is expressed highly on many tumor cell surfaces in many human cancers and is related to the regulation of invasion and metastasis. Herein, we report that NPs conjugated with antibody against GRP78 (mAb GRP78-NPs) inhibit the adhesion, invasion, and metastasis of hepatocellular carcinoma (HCC) and promote drug delivery of 5-fluorouracil into GRP78 high-expressed human hepatocellular carcinoma cells. Our new findings suggest that mAb GRP78-NPs could enhance drug accumulation by effectively transporting NPs into cell surface GRP78-overexpressed human hepatocellular carcinoma cells and then inhibit cell proliferation and viability and induce cell apoptosis by regulating caspase-3. In brief, mAb GRP78-NPs effectively inhibit cancer cell invasion and enhance antitumor efficiency by targeted drug delivery.
Keywords: 5-Fu, apoptosis, HCC, caspase-3
Introduction
Nanoparticles (NPs) have been used as a novel drug and gene delivery system for the targeted therapy of human cancers.1–5 With developments in preparation technique, physical properties, and material synthesis, NPs show many advantages in the biomedical therapy of malignant tumors, including hepatocellular carcinoma. NPs increase the stability of chemical agents by encapsulation, protecting these agents from the extracellular environment and regulating the drug release in a prolonged and controllable manner.6–9 Furthermore, NPs can be engulfed into cells by phagocytosis and actively transfer the cargoes, chemical agents and nucleotides, into cells.
Recently, researchers have found that some specific molecules used as targeting agents, such as carbohydrate,10 folic acid,11,12 transferrin,13,14 and some peptides,15,16 could be modified onto the surface of NPs and achieve active targeting therapy with high specificity, selectivity, and affinity through coupling targeting molecules with their specific receptor expressed on the cell surface.
The glucose-regulated protein 78 (GRP78) is a stress-induced endoplasmic reticulum chaperone.17 Many data have shown that GRP78 is overexpressed on the cell surface in many human cancers such as melanoma, lung cancer, breast cancer, and hepatocellular carcinoma.18–20 Cell surface GRP78 is involved in the regulation of many cells’ biology processes, such as survival,21 proliferation,22 apoptosis,23 invasion, and metastasis.24,25 Blockade of cell surface GRP78 inhibited the proliferation and induced apoptosis in melanoma and prostate cancer.26,27 Inhibition of cell surface GRP78 inhibited the tumor invasion and metastasis in hepatocellular carcinoma and colorectal cancer cell.28–30 Nowadays, cell surface GRP78 is regarded as a potential target for the targeted therapy of many human cancers.31–33
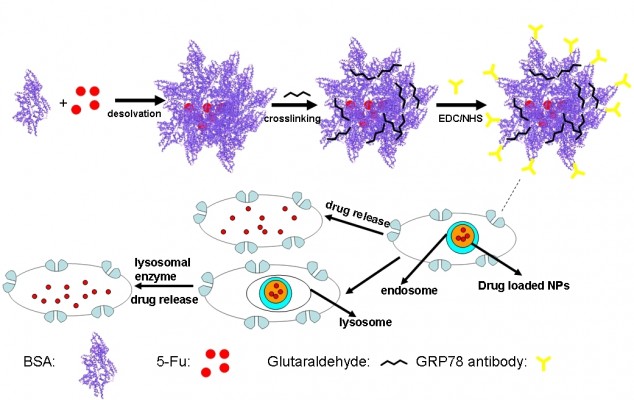
In this paper, we show that bovine serum albumin (BSA) NPs conjugated with the monoclonal antibody against GRP78 (mAb GRP78) could inhibit the adhesion, invasion, and metastasis of hepatocellular carcinoma SMMC-7721, in which GRP78 is overexpressed. The mAb GRP78-NPs combined with GRP78 receptors situated at the surface of cancer cells and were internalized to intracellular compartments to form endosomes. Furthermore, endosomes containing drug-loaded NPs were degraded in cells to release drug, and, alternatively, endosomes fused with intracellular vesicles and were internalized into lysosomes. Then, NPs broke down through degradation activated by secreted lysosomal proteinases and drugs escaped from lysosomes and were transferred into the cytoplasm (Figure 1).34–37 5-fluorouracil (5-Fu) as a model drug was encapsulated into mAb GRP78-NPs for the drug loading and in vitro release studies. Furthermore, the cell uptake, in vitro cytotoxicity, and cellular apoptosis were investigated to prove the mAb GRP78-mediated tumor targeting ability of mAb GRP78-NPs.
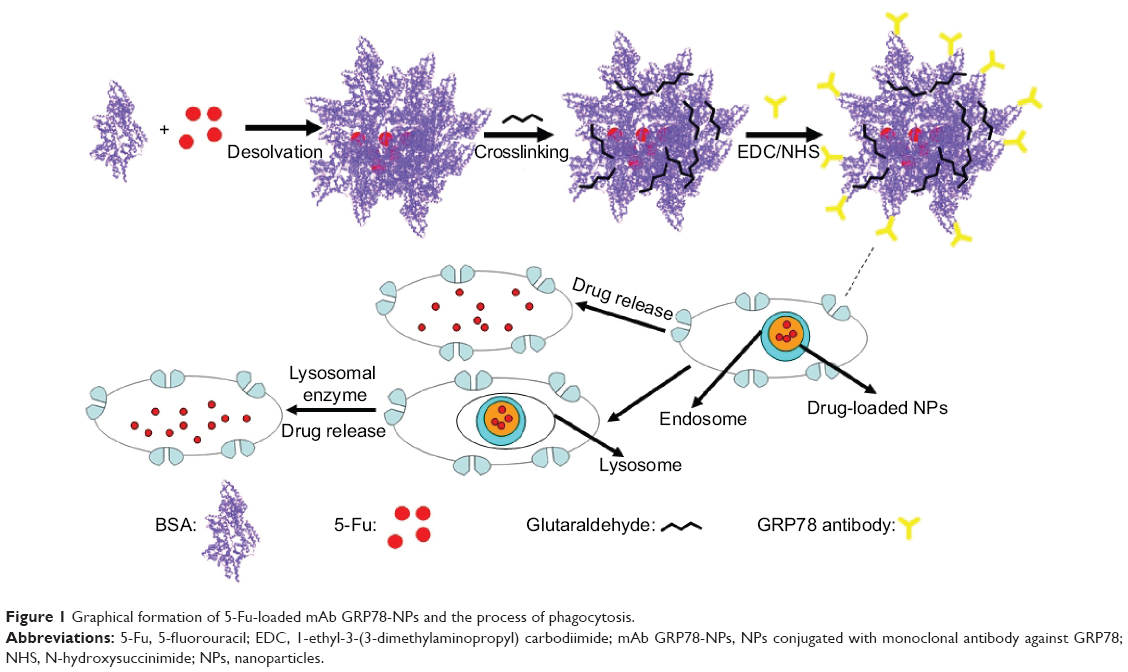 | Figure 1 Graphical formation of 5-Fu-loaded mAb GRP78-NPs and the process of phagocytosis. |
Materials and methods
Chemical agents and antibodies
BSA was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 5-Fu was purchased from Nantong Jinghua Pharmaceutical Co., Ltd (Nantong, People’s Republic of China). N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and acetic acid were obtained from Sigma-Aldrich Co. All other chemicals were of reagent level and were used as received. The antibody against GRP78 and isotype IgG were purchased from Santa Cruz Biotechnology Inc., Dallas, TX, USA.
The preparation of BSA NPs
BSA NPs were prepared by a desolvation method described previously.38 For the conjugation of mAb GRP78 as a targeting ligand or isotype IgG as a nonspecific antibody, the carboxyl unit of NPs was activated by N-hydroxysuccinimide and EDC in pH 7.4 phosphate-buffered saline (PBS) by constant vibration for 4 hours. The mAb GRP78 or isotype IgG were then added into NP suspension and stirred for 6 hours in darkness. Finally, antibody-conjugated NPs were collected and washed three times with deionized water and centrifuged at 16,000 rpm to remove uncoupled residues.
Characterization of BSA NPs
The morphology and shape of the NPs were determined by means of a transmission electron microscope (JEM-1200EX; JEOL, Tokyo, Japan). Particle size and surface charge were measured by dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). Determination of the encapsulation efficiency of 5-Fu in NPs was estimated using a previously reported method.38
Cell culture
Human hepatocellular carcinoma cells SMMC-7721 and PLC were purchased from the Type Culture Collection of Chinese Academy of Sciences (Shanghai, People’s Republic of China). The cells were passaged in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 2 mM glutamine, and 100 μg/mL streptomycin at 37°C, 5% CO2 and propagated every 3–5 days.
Identification of antibody-conjugated NPs with immunofluorescence
BSA NPs conjugated with GRP78 antibody or isotype IgG were extensively washed with PBS and incubated with fluorescein isothiocyanate (FITC)-labeled anti-goat IgG (1:200) for 1 hour. After incubation, to clear away the unbound antibody, PBS was used to rinse NPs three times. Then, NPs were dropped on the microslides and their fluorescence was detected by means of a fluorescent microscope (DMI4000B; Leica Microsystems, Wetzlar, Germany).
Assessment of tumor invasion
Twenty-four-well Transwell chamber inserts (Corning Incorporated, Corning, NY, USA) with 8μM porous polycarbonate filters were coated with 80μg of basement membrane Matrigel (BD Biosciences, San Jose, CA, USA) onto the upper surface and with fibronectin (10μg/mL) onto the lower surface. The cells were counted and dispensed into the upper chambers (2×104 cells/well) and incubated at 37°C for 48hours in the presence of mAb GRP78- or isotype IgG-NPs. Cells were fixed with 3.7% methanal and stained with 1% crystal violet. A cotton swab was used to remove non-invading cells situated on the top of the filter, and the filter was separated from the chamber and mounted on a microscope slide. Invasiveness was determined by counting cells on the lower surface of the filter.
Assessment of cell adhesion
Ninety-six-well culture plates were coated with fibronectin (10μg/mL) overnight at 4°C, washed with pH 7.4 PBS, and then incubated with 1% heat-inactivated BSA for 1hour at 37°C to block the nonspecific binding to FN. Cells (5×103 each well) pretreated with mAb GRP78- or isotype IgG-NPs were suspended in serum-free DMEM and dispensed into each well, incubated at 37°C for 1hour, and washed three times with PBS. Cells adhering to the bottom of the plate were stained with 1% crystal violet, dissolved with dimethyl sulfoxide overnight at room temperature, and examined using a microplate reader (Synergy 2; Biotek, Winooski, VT, USA) at 595nm.
Assessment of drug release
The in vitro drug release from mAb GRP78-NPs was estimated using a method reported previously.39 Briefly, 2 mL of mAb GRP78-NPs and free drug at the same concentration of 5-Fu, respectively, were placed in a dialysis bag with a molecular weight cutoff of 1,000 molecular weight (Mw) and dialyzed against 60 mL of PBS and stirred continuously at 60 rpm at 37°C. After certain time intervals (0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48 hours), the aliquots from the release media were taken out and were replaced with the same volume of fresh PBS. The amount of released 5-Fu was assayed by detecting its absorbance at 256 nm with ultraviolet-visible spectrophotometry. The in vitro drug release assay was performed thrice.
In vitro cellular uptake
In order to further explore the intracellular internalization and distribution of mAb GRP78-NPs, confocal laser scanning microscopy was utilized to locate the positions of NPs in cells. Briefly, FITC as the fluorescent marker was encapsulated for labeling NPs using a previously reported method.40 SMMC-7721 cells and PLC cells were incubated in six-well flat-bottom plates and allowed to adhere overnight. When the cells reached 70%–80% confluence, NPs were added into the medium and incubated with cells. At predetermined intervals, the cellular distributions of FITC-labeled IgG-NPs and mAb GRP78-NPs were observed by confocal laser scanning microscopy.
MTT assay
Cytotoxicity of all the samples was tested on both SMMC-7721 cells and PLC cells by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (CellTiter 96® AQueous One Solution reagent). SMMC-7721 cells and PLC cells in full growth media were seeded in a 96-well plate (1×104 cells/well). Then, cells were treated with 5-Fu-loaded IgG-NPs, 5-Fu-loaded mAb GRP78-NPs and free 5-Fu containing the same amount of 5-Fu for 48 hours at 37°C. The culture medium was removed and 100 μL DMEM was added. Then, added 20 μL MTT with concentration of 5 mg/mL into the 96-well plate and incubated for 4 hours at 37°C. Finally, a combination of MTT and culture medium was removed carefully and 150 μL of dimethyl sulfoxide was added into each well, then the absorbance was measured at 490 nm using a microplate reader (Synery-2; BioTek, Winooski, VT, USA). All experiments were performed thrice.
Annexin V–FITC/propidium iodide staining by flow cytometry
Annexin V–FITC/propidium iodide staining was performed by FCM on the cells treated with the same concentrations of free 5-Fu, 5-Fu-loaded IgG-NPs, and 5-Fu-loaded mAb GRP78-NPs for 48 hours at 37°C, according to the protocol of our previous study.38
Intracellular adenosine triphosphate level assay
In order to evaluate intracellular ATP level of cells treated with different NPs, the luciferin–luciferase-based ATP bioluminescence assay kit was used to determine the change of intracellular ATP level using a previously reported method.40 Seeded SMMC-7721 cells and PLC cells (5×104 cells/well, 96-well plate, for 24 hours) were incubated with 5-Fu-loaded IgG-NPs, 5-Fu-loaded mAb GRP78-NPs, with all free 5-Fu at the same drug concentration for 48 hours. We took ATP level of untreated cells as the control group and the changing rates of intracellular ATP level (CR[%]) were calculated using the following equation:
|
|

Endocytic tracking assay of NPs in cells
SMMC-7721 cells and PLC cells were separately pretreated with cytochalasin D, genistein, and chlorpromazine, and then treated with mAb GRP78-NPs and IgG-NPs to track the uptake pathways. In the experiment, we seeded SMMC-7721 cells and PLC cells in a 96-well plate, 5×103 cells/well, for 24 hours. The cells were separately incubated with 10 μg/mL chlorpromazine (clathrin-mediated uptake inhibitor), 1 μg/mL genistein (inhibition of caveolae-mediated uptake), and 30 μM of cytochalasin D (inhibition of macropinocytosis) for 30 minutes. Quantitative analysis of the internalization of NPs preincubated with all kinds of inhibitors was carried out to compare the intracellular fluorescent intensity between treatment with inhibitors and non-inhibitors.
Western blot assay
After treatment with 5-Fu-loaded IgG-NPs, 5-Fu-loaded mAb GRP78-NPs, and free 5-Fu for 48 hours, cells were collected, washed twice with ice-cold PBS, then lysed in radioimmunoprecipitation assay buffer (1% Nonidet P 40, 1% phenylmethanesulfonyl fluoride, 150 mM NaCl, 1 mM PMSF, 1 mM aprotinin, 10 μg/mL leupeptin, 50 mM Tris-Cl, pH 7.4). The cell lysate was cleared by centrifuging at 12,000 rpm for 25 minutes. Cell lysate containing 50 μg protein was separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and the protein was transferred onto polyvinylidene fluoride membrane. After blocking with 1% BSA, the polyvinylidene fluoride membrane was incubated with the primary antibodies (caspase-3, tubulin) at 4°C overnight, followed by incubation with appropriate secondary antibody for 1 hour, and stained with enhanced chemiluminescence (ECL). The level of the targeted proteins was photographed and analyzed using a UVP gel analysis system (iBox Scientia 600; UVP, LLC., CA, USA).
In-cell Western assay
The cells were harvested and diluted with DMEM containing 10% fetal bovine serum to 75,000 cells/mL, then 200μL of the cell suspension was dispensed per well in a Nunc™ 96-well MicroWell™ plate under sterile conditions. After 24hours, we removed the supernatant and dispensed 100μL of serum-free media (DMEM) per well of the 96-well microplate in 4–6hours. Then, media were removed manually. Cells were immediately fixed with 4% methanal in 1× PBS for 20 minutes at room temperature then washed five times with 1× PBS containing 0.1% Triton X-100 (cell permeabilization) for 5 minutes per wash (to determine the expression of GRP78 in cell surface without permeabilization). The wells were blocked by adding 150μL of Blocking Buffer (Odyssey; LI-COR, Nebraska, USA) to each well in 1.5hours. Then, we removed the blocking buffer from the blocking step and added 50μL of primary antibody (GRP78, 1:100; GAPDH, 1:100), with incubation overnight at 4°C, and washed the plate five times with 1× PBST (PBS with 0.1% Tween® 20) for 5 minutes at room temperature with slight shaking, using a generous amount of buffer. Then, we added 50μL of the secondary antibody (IRDye® 800CW-labeled secondary antibody, 1:100,000) solution to each well, incubated for 60 minutes with mild shaking at room temperature (protecting the plate from light during incubation), and washed the plate five times with 1× PBST for 5 minutes at room temperature with gentle shaking, using abundant buffer. The plates were scanned at 800nm using an Odyssey CLx Infrared Imaging System.
Statistical analysis
The data were analyzed using one-way analysis of variance (ANOVA). A P-value less than 0.05 was considered statistically significant.
Results
The characterization of mAb GRP78-NPs and isotype IgG-NPs
Using immunofluorescence, we observed green fluorescence in mAb GRP78- or isotype IgG-conjugated NPs (Figure 2A and B). However, no fluorescence was observed in unconjugated NPs (Figure 2C and D), suggesting that mAb GRP78 and isotype IgG were successfully conjugated on the surface of BSA NPs. The morphology of NPs was observed using scanning electron microscopy, as shown in Figure 2E and F, and demonstrated that mAb GRP78-NPs were characterized by homogeneous and spherical spheres with a smooth surface and nanometric size. The average size of mAb GRP78-NPs was 224.9 nm, and the zeta potential was -4.91mV. The polydispersity index was 0.023, indicating good monodispersity and uniformity. The average encapsulation efficiency of 5-Fu in mAb GRP78-NPs was 87.6%.
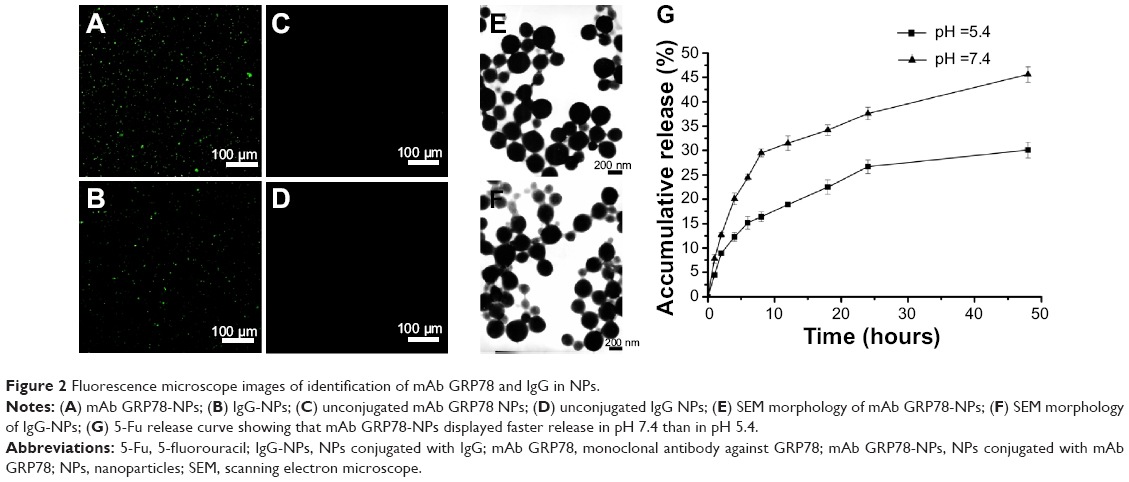 | Figure 2 Fluorescence microscope images of identification of mAb GRP78 and IgG in NPs. |
5-Fu-loaded mAb GRP78-NPs exhibited promising controlled release behavior with sustained release for more than 48 hours. The 5-Fu-loaded mAb GRP78-NPs presented an initial burst release owing to the fast release of drugs attached on the surface of NPs. As the medium solution penetrated into the interior of the NPs, the polymer matrix eroded and drug diffused slowly from the matrix of NPs, thus leading to the constant and smooth drug release. It was clearly observed that NPs displayed a faster release in pH 7.4 than in pH 5.4 (Figure 2G), possibly suggesting that the matrix of BSA protein tended to be compact at pH 5.4 which is closer to the isoelectric points of BSA (pH 4.9), therefore, drug could be difficult to penetrate the interior of NPs and released slowly from NPs. With the increase of pH, the protein molecules began to stretch and became loose, leading to the rapid release of drug.
mAb GRP78-NPs inhibit the invasion and adhesion of human hepatocellular carcinoma cells
Using in-cell Western assay, we detected GRP78 expression on the cell surface of SMMC-7721 and PLC. We found that GRP78 was expressed on the cell surface of SMMC-7721 and PLC cells at different levels. As shown in Figure 3 SMMC-7721 cells showed higher GRP78 expression than PLC cells on the cell surface. We first determined that targeting cell surface GRP78 using mAb GRP78-NPs affected the invasion and metastasis of SMMC-7721 and PLC cells. Using cell adhesion assay, we found that mAb GRP78-NPs inhibited the adhesion of SMMC-7721 and PLC to different degrees. The binding ability of SMMC-7721 cells to substrate was inhibited to 50% as compared to PLC cells, in which it was inhibited to 65%. Using Transwell assay, we found that mAb GRP78-NPs inhibited the invasiveness of SMMC-7721 cells to a greater degree than PLC cells. However, isotype IgG-NPs and BSA NPs did not affect the invasion and metastasis of SMMC-7721 and PLC.
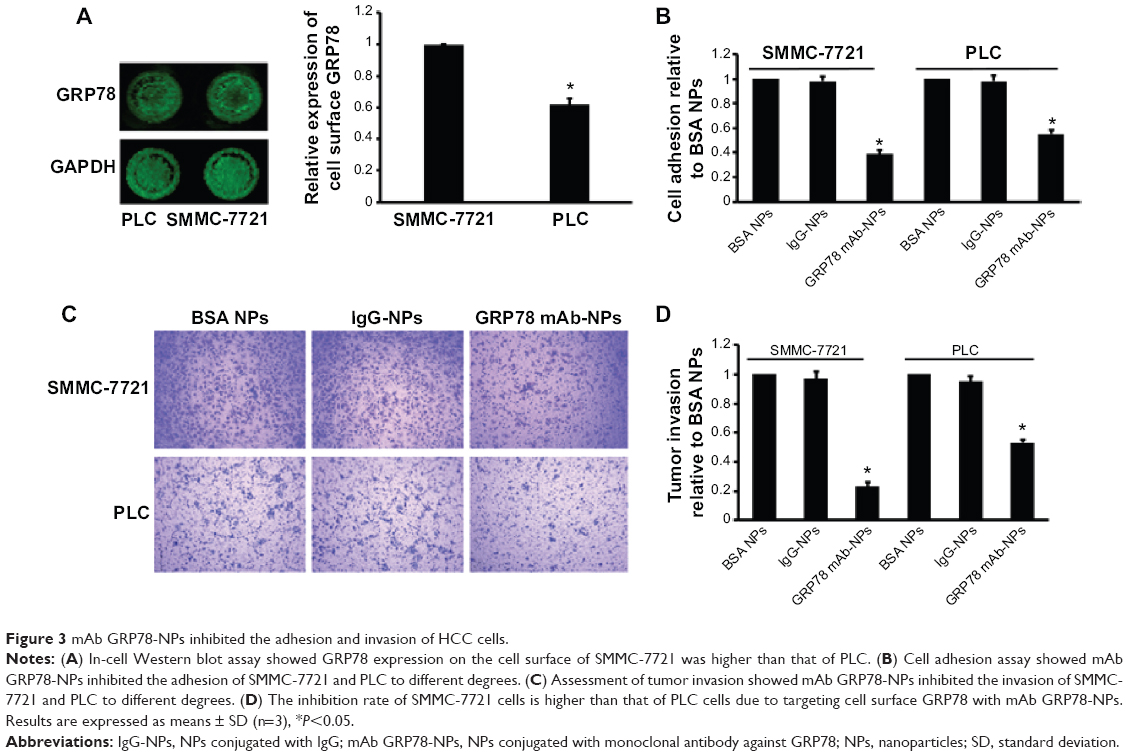 | Figure 3 mAb GRP78-NPs inhibited the adhesion and invasion of HCC cells. |
mAb GRP78-NPs effectively enhance drug accumulation in cell surface GRP78-overexpressed cancer cells mediated through clathrin and macropinocytosis
The cellular uptake of FITC-labeled NPs of SMMC-7721 and PLC cells was performed by confocal laser scanning microscopy, as shown in Figure 4. When the cells were treated with IgG-NPs, the green fluorescence intensity of the cells was weak, indicating that most of the NPs were not taken up by tumor cells and that only a small portion of NPs were internalized into cells by endocytic process. On the contrary, increased fluorescent spots were observed in SMMC-7721 cells with high expression of GRP78 when processed with mAb GRP78-NPs, as compared to cells treated with isotype IgG-NPs. However, the number of fluorescent spots in mAb GRP78-NP-treated PLC cells paralleled that of the cells treated with isotype IgG-NPs. This observation indicates that mAb GRP78-NPs could effectively promote drug accumulation in cancer cells in which GRP78 is highly expressed on the cell surface, suggesting the role of specific binding between mAb-GRP78 and cell surface GRP78.
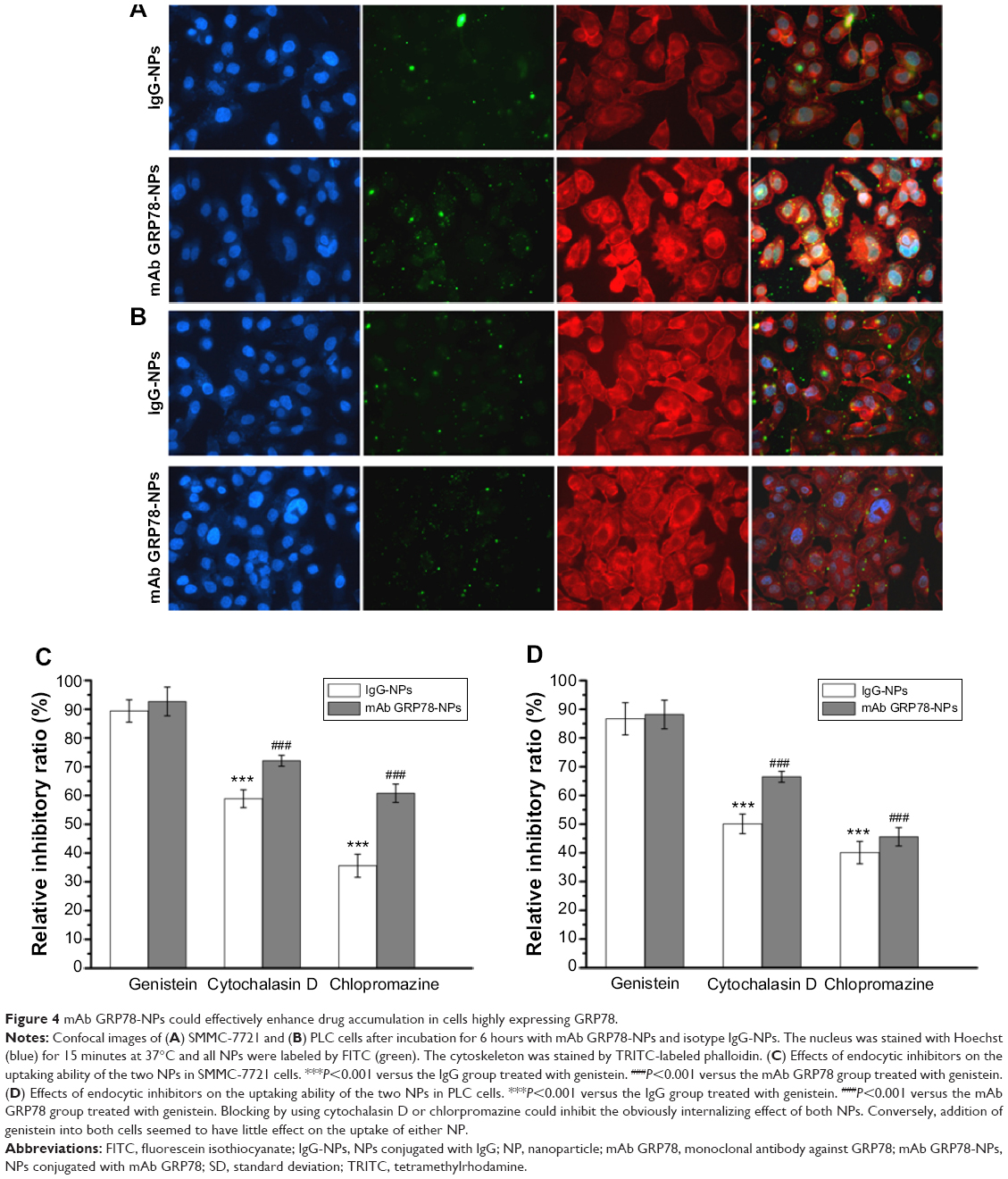 | Figure 4 mAb GRP78-NPs could effectively enhance drug accumulation in cells highly expressing GRP78. |
In order to better ascertain which uptake mechanisms are related to the cellular internalization of NPs, different endocytic inhibitors specific for various endocytic pathways were used. SMMC-7721 cells and PLC cells were preincubated with genistein (1 μg/mL) as an inhibitor to block caveolae-mediated endocytosis and then treated with IgG-NPs and mAb GRP78-NPs. The uptakes of targeted NPs were 89.4% and 92.7% in SMMC-7721 cells and 86.7% and 88.2% in PLC cells treated with IgG-NPs and mAb GRP78-NPs, respectively, compared with that of the cells not disposed with genistein, demonstrating that there was no significant difference in the internalization of both NPs and suggesting a minor role of caveolae-mediated endocytosis. Interestingly, when 30 μM of cytochalasin D (inhibitor of macropinocytosis) was used to treat both cells respectively, NP uptake was inhibited, and a 72.1% in SMMC-7721 and 66.5% in PLC cells for mAb GRP78-NPs was observed in comparison to a larger reduction of intracellular uptake of IgG-NPs (58.9% in SMMC-7721 cells and 50.1% in PLC cells). Remarkably, it was also observed that internalization of both NPs in cells with chlorpromazine treatment (clathrin-mediated endocytosis) was significantly lower than that in untreated cells. Moreover, a 60.8% in SMMC-7721 and 45.6% in PLC cells in mAb GRP78-NPs was observed in comparison with 35.6% in SMMC-7721 and 40.1% reduction in PLC cells of intracellular uptake of IgG-NPs. The results demonstrate that the significant uptake reduction of both NPs in treated cells with chlorpromazine and cytochalasin D proved that the internalization of both NPs into cells could rely on clathrin-mediated endocytosis and macropinocytosis. By contrast, the influence of caveolae-mediated endocytosis on NPs internalization was not significantly different from that of the untreated group.
mAb GRP78-NPs effectively facilitate 5-Fu-induced cell viability decrease in cell surface GRP78-overexpressed cancer cells
The cytotoxic effects of different kinds of naked NPs and 5-Fu-loaded NPs were evaluated by MTT assay using hepatocellular carcinoma cell line SMMC-7721 and PLC cells (Figure 5). Cell viability assay showed that no obvious cytotoxic activities were observed in SMMC-7721 and PLC cells when treated with naked mAb GRP78-NPs, IgG-NPs, and blank NPs (Figure 5A and B). As shown in Figure 5C and D, treatment of both cells with 5-Fu-loaded mAb GRP78-NPs caused an obvious decrease in the cell viability at 48 hours, as compared with that of free 5-Fu and 5-Fu-loaded IgG-NPs. The half-maximal inhibitory concentration value of 5-Fu-loaded mAb GRP78-NP-treated SMMC-7721 cells at 48 hours was 18.4 μg/mL, with values of 20.3 μg/mL for free 5-Fu and 29.4 μg/mL for 5-Fu-loaded IgG-NPs. The IC50 value of 5-Fu-loaded mAb GRP78-NP-treated PLC cells at 48 hours was 22.0 μg/mL, with values of 22.3 μg/mL for free 5-Fu and 30.3 μg/mL for 5-Fu-loaded IgG-NPs. These data suggest that conjugation of mAb GRP78 on NPs could transport more NPs into cancer cells expressing GPR78 at high levels due to the interaction between mAb GRP78 and its receptors.
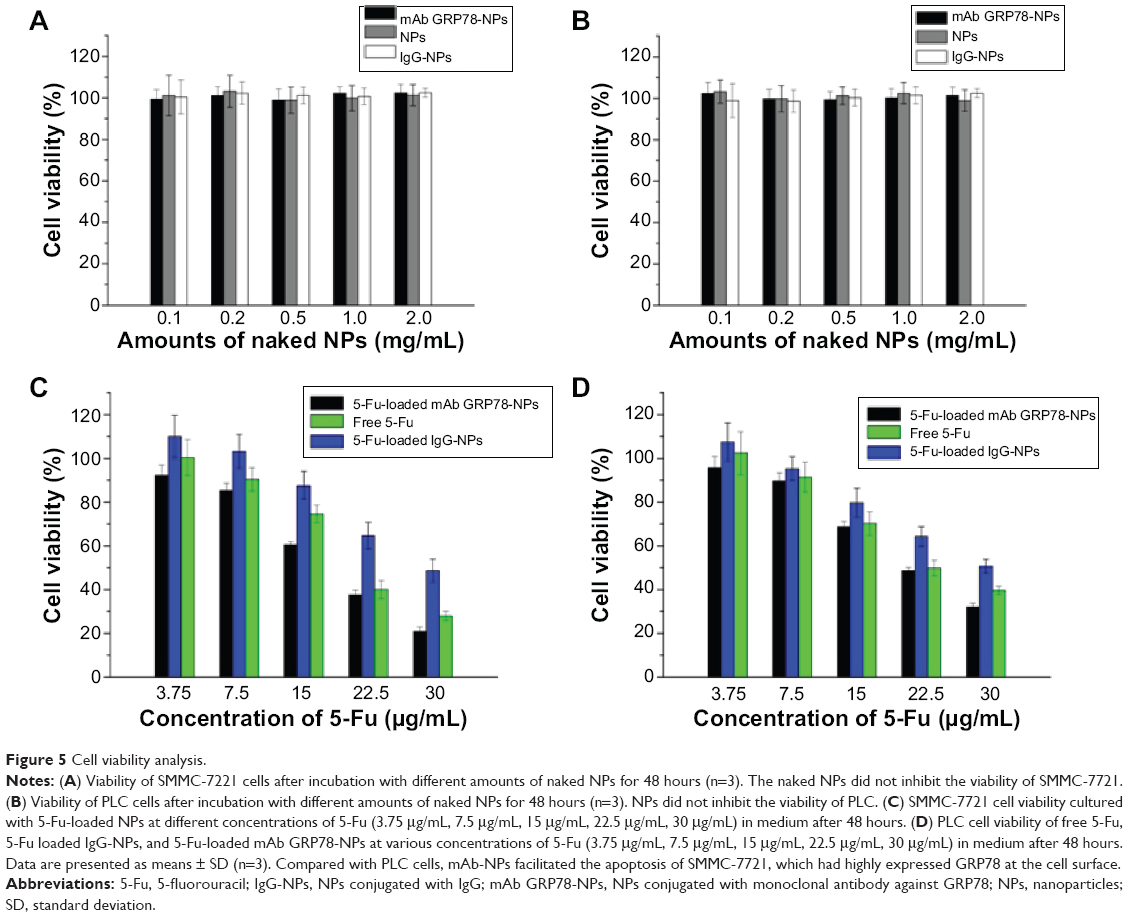 | Figure 5 Cell viability analysis. |
5-Fu-loaded mAb GRP78-NPs significantly enhance 5-Fu-induced apoptosis with the mediation of mAb GRP78-GRP78
As shown in Figure 6A and B, flow cytometry analysis revealed that after 48 h incubation with the treatment of 5-Fu loaded mAb GRP78-NPs, free 5-Fu and 5-Fu loaded IgG-NPs, the ratio of double (annexin V/propidium iodide) positive cells in SMMC-7721 cells was increased to 89.34% for 5-Fu loaded mAb GRP78-NPs, 41.29% for free 5-Fu and 51.05% for 5-Fu loaded IgG-NPs. As for PLC cells, the ratio was increased to 61.35% for 5-Fu loaded mAb GRP78-NPs, 54.17% for free 5-Fu and 38.12% for 5-Fu loaded IgG-NPs. It is worth noting that the ratio of double positive fractions in 5-Fu-loaded mAb GRP78-NP-treated cells was markedly higher than with other treatments. This proves that 5-Fu-loaded NPs with the mediation of mAb GRP78–GRP78 significantly enhanced 5-Fu-induced apoptosis.
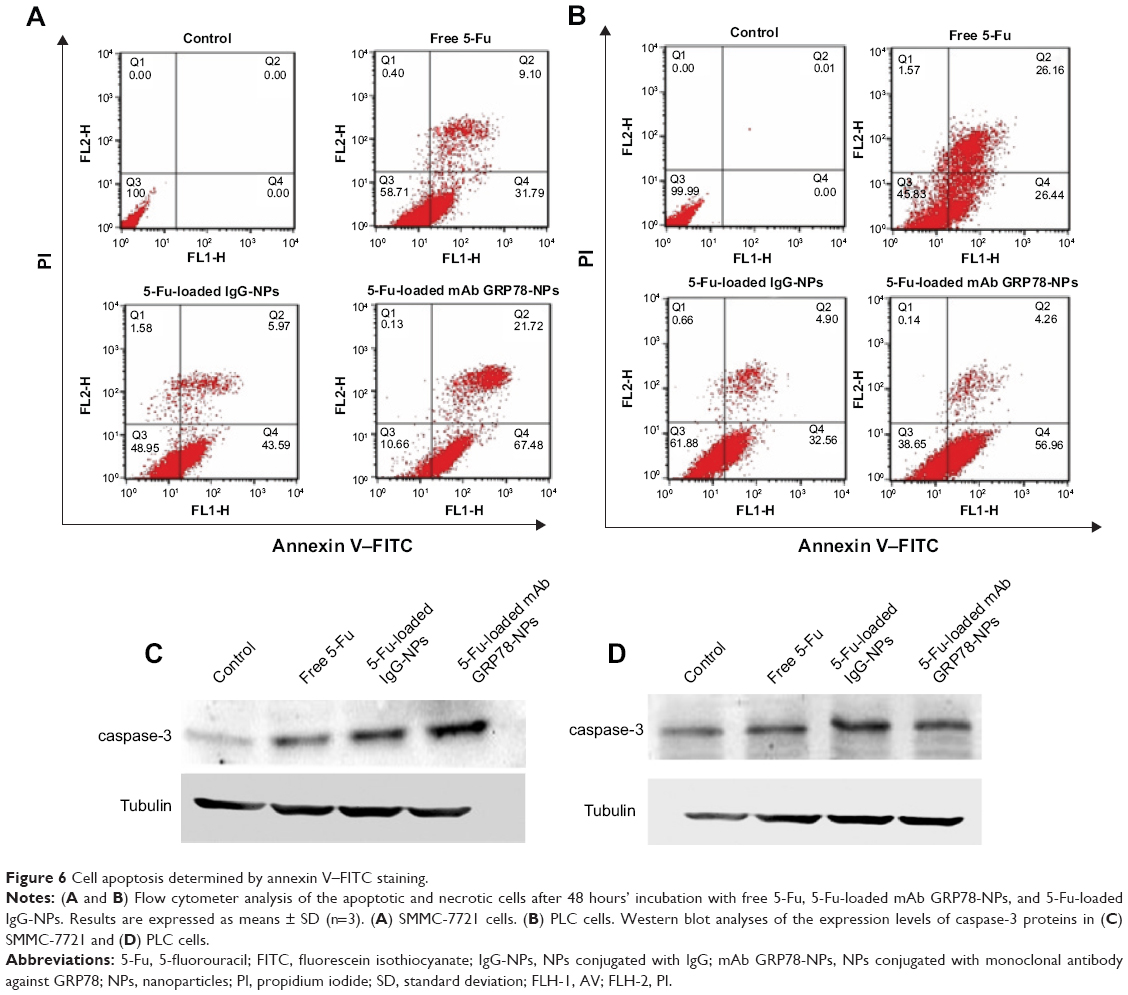 | Figure 6 Cell apoptosis determined by annexin V–FITC staining. |
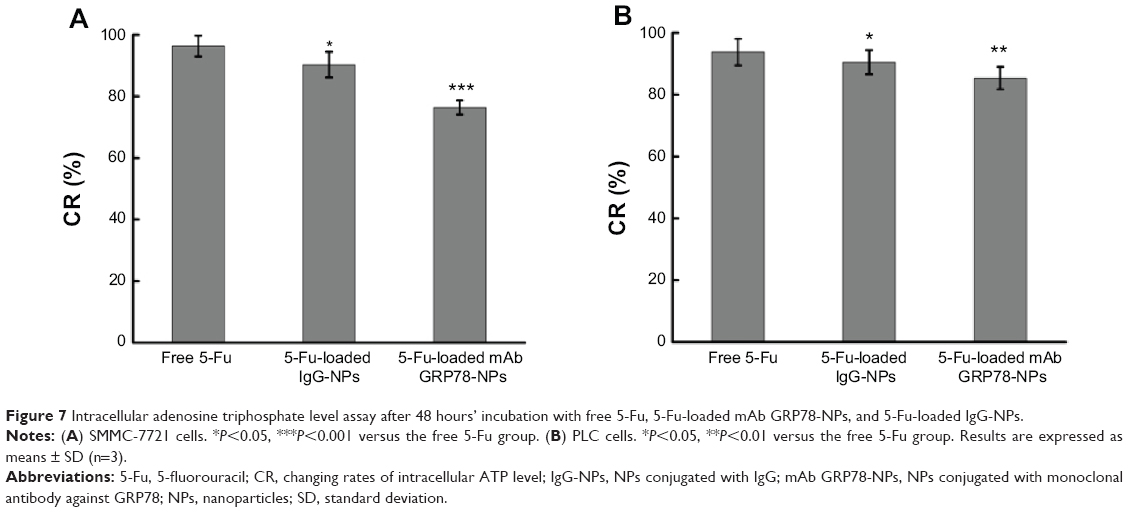 | Figure 7 Intracellular adenosine triphosphate level assay after 48 hours’ incubation with free 5-Fu, 5-Fu-loaded mAb GRP78-NPs, and 5-Fu-loaded IgG-NPs. |
To explore the apoptosis of 5-Fu-loaded NPs, Western blot assay was performed to detect the expression of caspase-3. From Figure 6C and D, we can see that, compared to the treatment group with the free 5-Fu and 5-Fu-loaded IgG-NPs, 5-Fu-loaded mAb GRP78-NPs induced the highest expression of caspase-3 in SMMC-7721 cells. In PLC cells, 5-Fu-loaded mAb GRP78-NPs also promoted the expression of caspase-3. Moreover, the specific mAb GRP78-NPs and IgG-NPs had no significant difference on caspase-3 with in PLC cells (Figure 6D), illustrating that, compared with the free 5-Fu and 5-Fu-loaded IgG-NP groups, when SMMC-7721 cells with a high expression of GRP78 were treated with 5-Fu-loaded mAb GRP78-NPs, a large amount of drug-loaded mAb GRP78-conjugated NPs were accumulated in cells due to the interaction between GRP78 and mAb GRP78, leading to the significant apoptosis and increasing expression of caspase-3, as the main apoptosis-relevant protein, in Western blot. However, due to the insufficient amount of GRP78 at the surface of PLC cells, the internalization of 5-Fu-loaded mAb GRP78-NPs was not greatly enhanced by the mediation of mAb GRP78; therefore, there was no obvious difference in the caspase-3 protein expression between the 5-Fu-loaded mAb GRP78-NP group and the free 5-Fu and 5-Fu-loaded IgG-NP groups.
mAb GRP78-NPs mediate intracellular 5-Fu uptake by decreasing intracellular ATP
Compared with the ATP level of the control group, after 48 hours’ incubation with the free 5-Fu, 5-Fu-loaded IgG-NPs and 5-Fu-loaded mAb GRP78-NPs, the changing rates of intracellular ATP level were decreased to 96.4%, 90.3%, and 76.4% in SMMC-7721 cells and 93.8%, 90.5%, and 85.4% in PLC cells, respectively. It was also found that more drug-loaded mAb GRP78-NPs were internalized into cells and increased the intracellular drug concentration with the mediation of GRP78, thus leading to the significant decrease of intracellular ATP and accelerating the apoptosis of cells by accumulation of drugs in cells.
Discussion
GRP78, an endoplasmic reticulum chaperone, could express on cell surfaces in many human cancers. Cell surface GRP78 is involved in the adjustment of many biological processes. Many researchers have reported that cell surface GRP78 could promote invasion and metastasis of tumor cells and that blockade of cell surface GRP78 could reverse this effect.26,27
Recent investigations10,11,15 have revealed that specific molecules targeting cell surface agents could administrate entry and subsequent cytosolic access of NPs into living cells, for example, carbohydrate, folic acid, transferrin, and some peptides. From Figure 3, we can see that GRP78 is more highly expressed on the cell surface of SMMC-7721 cells than on the cell surface of PLC cells, and many studies23–25 have shown that GRP78 plays an important role in tumor cells’ invasion and metastasis. After investigating the adhesion and invasion abilities of SMMC-7721 and PLC, we found that SMMC-7721 cells are stronger than PLC cells. Moreover, by blocking the expression of GRP78 on cell surface with mAb GRP78-NPs, the invasion and adhesion abilities are correspondingly decreased. Our immunofluorescence results clearly show that mAb GRP78-NPs could effectively adhere to cell surface GRP78 and enhance 5-Fu accumulation in plasma (Figure 3). To examine the effect of mAb GRP78-NPs on cell surface target, we used 5-Fu to induce cell apoptosis, and the results showed that 5-Fu highly decreased the extent of cell viability and highly promoted the extent of apoptosis in cancer cells that have highly expressed cell surface GRP78 (Figure 5 and 6). By Western blot (WB), caspase-3 bands showed that the 5-Fu-induced apoptosis was associated with mAb GRP78-NPs, and intracellular ATP level assay showed mAb GRP78-NPs mediated 5-Fu into cytoplasm decreased intracellular ATP. As such, these data suggest that conjugation of mAb GRP78 on NPs could transport more NPs into cancer cells. To explore how mAb GRP78-NPs introduce 5-Fu into cytosol, cells were treated with cytochalasin D, genistein, and chlorpromazine, and the results showed that mAb GRP78-NP uptake was mediated by clathrin and macropinocytosis (Figure 4).
In summary, our results provide convincing evidence that mAb GRP78-NPs could effectively transport drug from cell surface into cytosol; the drug delivery function of mAb GRP78-NPs may be applied to the development of personalized medicine through antibody-based modeling.
Conclusion
Briefly, we have clarified that the mechanisms of mAb GRP78-NP uptake in tumor cells are strongly dependent on cell surface GRP78 expression level. mAb GRP78-NPs provide a possibility to develop nanocarriers of specific therapeutic action that can reduce the cytotoxicity of normal cells. Because normal cells usually do not express GRP78 on cell surface, mAb GRP78-NPs will not target them and the drug will not release to normal cells. As such, application of mAb GRP78-NPs can decrease chemotherapeutic side effects.
The results of this study indicate the significance of mAb GRP78-NPs in targeting tumor cells and signify the role of NPs that target tumor cell surface molecules in nanomedicine.
Acknowledgments
This work was supported by the Foundation of Liao’ning Educational Committee (number L2014339), Natural Science Foundation of Liaoning Province (number 2013022035), and Grant of Liaoning Medical University (number XZJJ20130104-05).
Disclosure
The authors report no conflicts of interest in this work.
References
Kumar V, Toffoli G, Rizzolio F. Fluorescent carbon nanoparticles in medicine for cancer therapy. ACS Med Chem Lett. 2013;4(11):1012–1013. | ||
Wang C, Ravi S, Garapati US, et al. Multifunctional chitosan magnetic-graphene (CMG) nanoparticles: a theranostic platform for tumor-targeted co-delivery of drugs, genes and MRI contrast agents. J Mater Chem B Mater Biol Med. 2013;1(35):4396–4405. | ||
Xu H, Hou Z, Zhang H, et al. An efficient Trojan delivery of tetrandrine by poly(N-vinylpyrrolidone)-block-poly(ε-caprolactone) (PVP-b-PCL) nanoparticles shows enhanced apoptotic induction of lung cancer cells and inhibition of its migration and invasion. Int J Nanomedicine. 2014;9:231–242. | ||
Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano. 2013;7(11):9571–9584. | ||
Friedman H, Holt AT, Pham W. Lipid nanoparticles as ideal delivery modules for siRNA. Nanomedicine (Lond). 2013;8(12):1910–1911. | ||
Miao Q, Xu D, Wang Z, et al. Amphiphilic hyper-branched co-polymer nanoparticles for the controlled delivery of anti-tumor agents. Biomaterials. 2010;31(28):7364–7375. | ||
Li X, Qin F, Yang L, Mo L, Li L, Hou L. Sulfatide-containing lipid perfluorooctylbromide nanoparticles as paclitaxel vehicles targeting breast carcinoma. Int J Nanomedicine. 2014;9:3971–3985. | ||
Yang J, Huang Y, Gao C, Liu M, Zhang X. Fabrication and evaluation of the novel reduction-sensitive starch nanoparticles for controlled drug release. Colloids Surf B Biointerfaces. 2014;115:368–376. | ||
Ragelle H, Riva R, Vandermeulen G, et al. Chitosan nanoparticles for siRNA delivery: optimizing formulation to increase stability and efficiency. J Control Release. 2014;176:54–63. | ||
Yu Z, Schmaltz RM, Bozeman TC, et al. Selective tumor cell targeting by the disaccharide moiety of bleomycin. J Am Chem Soc. 2013; 135(8):2883–2886. | ||
Sun L, Wei Z, Chen H, et al. Folic acid-functionalized up-conversion nanoparticles: toxicity studies in vivo and in vitro and targeted imaging applications. Nanoscale. 2014;6(15):8878–8883. | ||
Yuan Y, Zhang J, An L, Cao Q, Deng Y, Liang G. Oligomeric nanoparticles functionalized with NIR-emitting CdTe/CdS QDs and folate for tumor-targeted imaging. Biomaterials. 2014;35(27):7881–7886. | ||
Liu T, Kempson I, de Jonge M, Howard DL, Thierry B. Quantitative synchrotron X-ray fluorescence study of the penetration of transferrin-conjugated gold nanoparticles inside model tumour tissues. Nanoscale. 2014;6(16):9774–9782. | ||
Wu LC, Chu LW, Lo LW, Liao YC, Wang YC, Yang CS. Programmable cellular retention of nanoparticles by replacing the synergistic anion of transferrin. ACS Nano. 2013;7(1):365–375. | ||
Shirazi AN, Paquin KL, Howlett NG, Mandal D, Parang K. Cyclic peptide-capped gold nanoparticles for enhanced siRNA delivery. Molecules. 2014;19(9):13319–13331. | ||
Jiang T, Yu X, Carbone EJ, Nelson C, Kan HM, Lo KW. Poly aspartic acid peptide-linked PLGA based nanoscale particles: potential for bone-targeting drug delivery applications. Int J Pharm. 2014;475(1–2) 547–557. | ||
Zhang H, Lv M, Zhao Z, et al. Glucose-regulated protein 78 may play a crucial role in promoting the pulmonary microvascular remodeling in a rat model of hepatopulmonary syndrome. Gene. 2014;545(1):156–162. | ||
Delie F, Petignat P, Cohen M. GRP78-targeted nanotherapy against castrate-resistant prostate cancer cells expressing membrane GRP78. Target Oncol. 2013;8(4):225–230. | ||
Delie F, Petignat P, Cohen M. GRP78 protein expression in ovarian cancer patients and perspectives for a drug-targeting approach. J Oncol. 2012;2012:468615. | ||
Liu Y, Steiniger SC, Kim Y, Kaufmann GF, Felding-Habermann B, Janda KD. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol Pharm. 2007;4(3):435–447. | ||
Li Z, Wang Y, Wu H, Zhang L, Yang P, Li Z. GRP78 enhances the glutamine metabolism to support cell survival from glucose deficiency by modulating the β-catenin signaling. Oncotarget. 2014; 5(14):5369–5380. | ||
Yang L, Yang S, Liu J, et al. Expression of GRP78 predicts taxane-based therapeutic resistance and recurrence of human gastric cancer. Exp Mol Pathol. 2014;96(2):235–241. | ||
Guo YS, Sun Z, Ma J, et al. 17β-Estradiol inhibits ER stress-induced apoptosis through promotion of TFII-I-dependent Grp78 induction in osteoblasts. Lab Invest. 2014;94(8):906–916. | ||
Su R, Li Z, Li H, et al. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer. 2010;10:20. | ||
Zhang D, Tai LK, Wong LL, et al. Proteomic characterization of differentially expressed proteins in breast cancer: expression of hnRNP H1, RKIP and GRP78 is strongly associated with HER-2/neu status. Proteomics Clin Appl. 2008;2(1):99–107. | ||
Misra UK, Pizzo SV. Receptor-recognized α2-macroglobulin binds to cell surface-associated GRP78 and activates mTORC1 and mTORC2 signaling in prostate cancer cells. PLoS One. 2012;7(12):e51735. | ||
Liu R, Li X, Gao W, et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin Cancer Res. 2013;19(24):6802–6811. | ||
Zhang XX, Li HD, Zhao S, et al. The cell surface GRP78 facilitates the invasion of hepatocellular carcinoma cells. Biomed Res Int. 2013;2013:917296. | ||
Li Z, Zhang L, Zhao Y, et al. Cell-surface GRP78 facilitates colorectal cancer cell migration and invasion. Int J Biochem Cell Biol. 2013;45(5):987–994. | ||
Miao YR, Eckhardt BL, Cao Y, et al. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin Cancer Res. 2013;19(8):2107–2116. | ||
Raiter A, Vilkin A, Gingold R, et al. The presence of anti-GRP78 antibodies in the serum of patients with colorectal carcinoma: a potential biomarker for early cancer detection. Int J Biol Markers. Epub 2014 May 1. | ||
Chen M, Zhang Y, Yu VC, Chong YS, Yoshioka T, Ge R. Isthmin targets cell-surface GRP78 and triggers apoptosis via induction of mitochondrial dysfunction. Cell Death Differ. 2014;21(5):797–810. | ||
Kang J, Zhao G, Lin T, et al. A peptide derived from phage display library exhibits anti-tumor activity by targeting GRP78 in gastric cancer multidrug resistance cells. Cancer Lett. 2013;339(2):247–259. | ||
Wang Q, Liu P, Gong T, Sun X, Duan Y, Zhang Z. Uptake mechanism and endosomal fate of drug-phospholipid lipid nanoparticles in subcutaneous and in situ hepatoma. J Biomed Nanotechnol. 2014;10(6):993–1003. | ||
Gao H, Yang Z, Zhang S, Pang Z, Jiang X. Internalization and subcellular fate of aptamer and peptide dual-functioned nanoparticles. J Drug Target. 2014;22(5):450–459. | ||
He X, Li J, An S, Jiang C. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv. 2013;4(12):1499–1510. | ||
Kocbek P, Kralj S, Kreft ME, Kristl J. Targeting intracellular compartments by magnetic polymeric nanoparticles. Eur J Pharm Sci. 2013; 50(1):130–138. | ||
Zhao L, Su R, Cui W, Shi Y, Liu L, Su C. Preparation of biocompatible heat-labile enterotoxin subunit B-bovine serum albumin nanoparticles for improving tumor-targeted drug delivery via heat-labile enterotoxin subunit B mediation. Int J Nanomedicine. 2014;9:2149–2156. | ||
Song H, Su C, Cui W, et al. Folic acid-chitosan conjugated nanoparticles for improving tumor-targeted drug delivery. Biomed Res Int. 2013; 2013:723158. | ||
Su C, Li H, Shi Y, et al. Carboxymethyl-β-cyclodextrin conjugated nanoparticles facilitate therapy for folate receptor-positive tumor with the mediation of folic acid. Int J Pharm. 2014;474(1–2):202–211. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.