Back to Journals » International Journal of Nanomedicine » Volume 16
Nanoparticle-Mediated Targeted Drug Delivery to Remodel Tumor Microenvironment for Cancer Therapy
Authors Tang L, Mei Y ![]() , Shen Y, He S, Xiao Q, Yin Y, Xu Y, Shao J, Wang W
, Shen Y, He S, Xiao Q, Yin Y, Xu Y, Shao J, Wang W ![]() , Cai Z
, Cai Z
Received 31 May 2021
Accepted for publication 14 August 2021
Published 25 August 2021 Volume 2021:16 Pages 5811—5829
DOI https://doi.org/10.2147/IJN.S321416
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Lu Tang,1,2,* Yijun Mei,1,2,* Yan Shen,1,2,* Shun He,1,2 Qiaqia Xiao,1,2 Yue Yin,1,2 Yonggang Xu,3 Jie Shao,3 Wei Wang,1,2 Zihao Cai3
1State Key Laboratory of Natural Medicines, Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China; 2NMPA Key Laboratory for Research and Evaluation of Pharmaceutical Preparations and Excipients, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China; 3Department of General Surgery, Huashan Hospital, Fudan University, Shanghai, 200040, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Wang
State Key Laboratory of Natural Medicines, Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China
Tel +86-25-83271102
Email [email protected]
Zihao Cai
Department of General Surgery, Huashan Hospital, Fudan University, No. 12 Middle Urumqi Road, Shanghai, 200040, People’s Republic of China
Tel +86-21-66895079
Email [email protected]
Abstract: Advanced research has revealed the crucial role of tumor microenvironment (TME) in tumorigenesis. TME consists of a complicated network with a variety of cell types including endothelial cells, pericytes, immune cells, cancer-associated fibroblasts (CAFs), cancer stem cells (CSCs) as well as the extracellular matrix (ECM). The TME-constituting cells interact with the cancerous cells through plenty of signaling mechanisms and pathways in a dynamical way, participating in tumor initiation, progression, metastasis, and response to therapies. Hence, TME is becoming an attractive therapeutic target in cancer treatment, exhibiting potential research interest and clinical benefits. Presently, the novel nanotechnology applied in TME regulation has made huge progress. The nanoparticles (NPs) can be designed as demand to precisely target TME components and to inhibit tumor progression through TME modulation. Moreover, nanotechnology-mediated drug delivery possesses many advantages including prolonged circulation time, enhanced bioavailability and decreased toxicity over traditional therapeutic modality. In this review, update information on TME remodeling through NPs-based targeted drug delivery strategies for anticancer therapy is summarized.
Keywords: tumor microenvironment, nanotherapeutics, cancer treatment, targeted delivery, regulation strategy
Graphical Abstract:

Introduction
Cancer is a genetic disorder that represents an intricate disease condition accompanied with uncontrolled cell growth and the capability to escape the natural mechanism of cell death.1 Cancer has also been regarded as an evolutionary process with constant and dynamic interactions between cancer cells and the tumor microenvironment (TME).2 TME encompasses all the non-cancerous cells in the tumor, including surrounding immune cells, endothelial cells, bone marrow-derived inflammatory cells and cancer-associated fibroblasts (CAFs), cancer stem cells (CSCs) as well as its non-cellular components such as the extracellular matrix (ECM), signaling molecules and soluble products like cytokines, growth factors, chemokines, and extracellular vesicles (EVs).3 Interactions between various cells create a TME that influences cancer development and progression.4 For instance, the diversity of ECM components endows cell types with distinct properties to regulate various cellular processes. Cancer-associated fibroblasts (CAFs), the most abundant cells of the tumor stroma, can produce and secrete a variety of growth factors, cytokines and chemokines, playing a vital role in enhancing the tumor phenotype and promoting tumor cell proliferation. Various kinds of immune cells constitute a complicated network that affects the therapeutic outcome of cancer immunotherapy by the immunosuppressive TME (Figure 1). Besides, heterogeneity of TME limits the effect of drug penetration to reach deep tumor tissues in lethal concentration, which greatly decreases the therapeutic efficacy of anticancer drugs.5 Furthermore, different from normal tissue, TME is featured with vascular abnormalities, high interstitial pressure (HIP), hypoxia, acidity and altered metabolic states, which all promotes tumorigenesis.
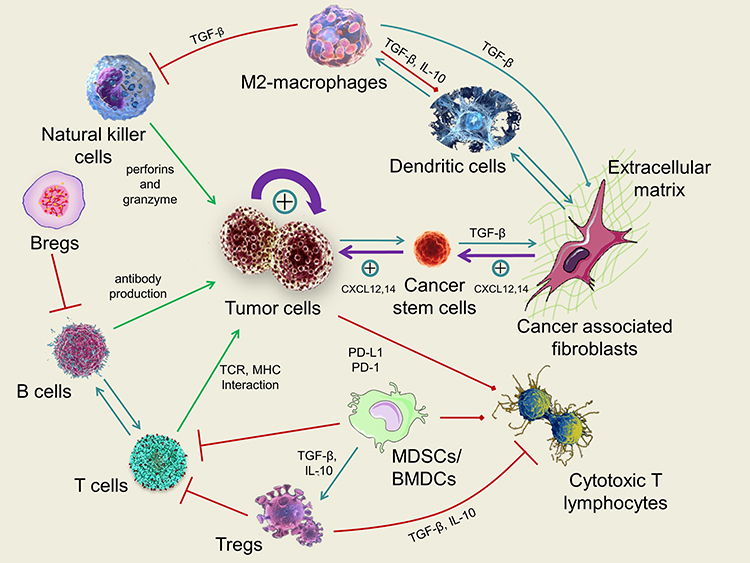 |
Figure 1 Schematic illustration of the most important mechanisms and interactions within the tumor microenvironment (TME). Green arrows indicate the antitumor effect of immune system, red arrows indicate the inhibitions of antitumor effect of immune system, turquoise arrows indicate other interactions within the TME and purple arrows indicate the strong growing effect of tumor cells. TME is composed of an intricate system involving diverse cell types including immune cells, fibroblasts, cancer stem cells as well as the extracellular matrix (ECM). The TME-forming cells dynamically interact with the cancerous cells through various signaling mechanisms and pathways, playing crucial roles in tumor initiation, progression, metastasis, and response to therapies. |
With the growing understanding of TME’s fundamental role in tumor development, many efforts have been made to aim at TME constituents or its signaling pathways. During the past decades, nanotechnology has drawn increasingly attention in cancer treatment. Nanoparticles (NPs) are ideal carriers for many therapeutic agents due to plenty of advantages, such as variable particle shape and size, localized drug delivery ability, enhanced bioavailability, reduced systemic toxicity, and improved pharmacokinetics through their tunable physicochemical properties.6–10 Therefore, regulating TME through NPs-mediated targeted drug delivery might open a new horizon for anticancer therapy. This review focuses on the recent achievements of TME remodeling by nanotechnology, various strategies aiming at regulating TME elements will also be highlighted.
Nanoparticles Designed to Remodel Tumor Microenvironment
Recent decades have witnessed the rapid development of nano-biotechnology, especially the applications of NPs in cancer therapy. Nano-based delivery systems hold an advantage over conventional chemotherapy in that they can transport therapeutic agents preferentially to malignant tumors due to the enhanced permeability and retention (EPR) effect, preventing healthy tissues from dose-limiting side effects.11–13 TME is a complex microenvironment composed of a heterogeneity nature of tumors, which causes several insurmountable barriers and greatly hampers the medical performance in cancer therapy. For instance, the abnormal vasculature in tumor sites contributes to ineffective targeting in some cases; the tumor hypoxia can dramatically blunt the response of tumor cells towards chemotherapeutics; the ECM greatly inhibits the drug penetration into deep tumor tissue; some stromal cells like CAFs and immune cells can secret multiple molecules, cytokine and factors, protecting them from cytotoxic agents and immune clearance.14 Therefore, it is urgent to explore novel therapeutic strategies to address these problems as well as regulate the TME.
To date, a great variety of NPs have been engineered to target and remodel the TME for effective cancer therapy. Organic NPs such as liposomes, micelles, high-density lipoprotein (rHDL), dendrimers, polymeric NPs and nano-emulsions possess high drug-delivery efficiency with less toxicity. Inorganic NPs like gold NPs, magnetic NPs, quantum dots, silica NPs and carbon nanotubes (CNTs) can reach specific sites of action through functionalization.15,16 These NPs can effectively target the cells and components such as CSCs, CAFs, exosomes, ECM within the TME, and could remodel TME through aiming at anti-angiogenesis and immunoregulatory (Figure 2). Additionally, all of these NPs can carry therapeutic agents to target cancerous tissues depend on their unique physical and chemical properties. More excitingly, it has been reported that NPs can alter the complex TME by delivering modulators or receiving external physical signals, overcoming the aforementioned physiological barriers. Besides, nanoplatform based on small-molecule prodrug nanoassembly is an alternative approach that has been developed recently for TME-responsive drug delivery.17,18 For instance, Wang et al designed a self-assembling nanoplatform based on highly toxic cabazitaxel reversibly ligated via a disulfide bioactivatable linker, which could achieve TME-responsive drug release due to the high concentration of glutathione (GSH) in TME.19 Altogether, nanoplatform-based targeted drug delivery strategies are emerging treatment modalities to improve drug potency and efficacy through reprogramming the TME in anticancer therapy.
 |
Figure 2 Schematic illustration of different strategies used to target the tumor microenvironment through various kinds of NPs. These NPs can remodel the TME by either targeting its inside elements and cells such as extracellular matrix (ECM), cancer stem cells (CSCs), cancer-associated fibroblasts (CAFs) and exosomes, or through immunoregulatory and anti-angiogenesis. |
Nanoparticles Remodel Tumor Microenvironment Through Targeting the Extracellular Matrix
Extracellular matrix (ECM) is usually defined as the non-cellular part of three-dimensional macromolecular network that offers multifunctional support for its cellular components.20 The diversity of ECM proteins endows cell types with distinct biochemical and biophysical properties, which regulates various cellular processes. Besides, ECM serves not only as an intercellular filling but also plays a vital role in TME constitution. Fundamentally, ECM is made up of a large variety of macromolecules including collagen, proteoglycans, laminin, and fibronectin. Several studies have demonstrated that ECM was the first barrier in tumor cells’ migration and its dysregulation causes tumor progression.21 Hence, understanding the interplay between tumor cells and ECM as well as revealing promising therapeutic strategies through ECM remodeling will provide more possibilities for anticancer drug delivery.
Drug Delivery Systems That Modulate Collagen
Collagen serves as the basic structure of ECM and is the most abundant protein with 28 unique subtypes discovered in human tissue. Because tumor cells are often embedded in a dense ECM made up of collagen, proteoglycans and glycoproteins, the enhanced matrix density of these substances may limit the efficacy of anticancer therapeutic agents. In addition, structural complexity of ECM hinder the penetration of NPs and influence NPs’ distribution throughout the interstitium.3,5 Therefore, reduction of collagen density is a potential approach to enhance NP penetration and accumulation in tumor tissue.
In recent years, topical administration of proteolytic enzymes such as collagenase (Col) or bromelain before nanotherapy has been reported to significantly promote the penetration of therapeutic drugs into tumor tissues. Collagenases are the endopeptidases that are able to digest native collagen in the triple helix region. Yao et al constructed a nano-enzyme capsule by combining collagenase nanocapsules (Col-nc) with heavy-chain ferritin (HFn) nanocages to encapsulate the anticancer drug doxorubicin (DOX), which could facilitate tumor penetration of the NPs by hydrolyzing collagen from the ECM in 4T1 breast cancer model. Col-nc could protect the enzyme activity before reaching the action sites, after which the released proteolytic enzyme digested collagen, decreasing the density of ECM. In vivo and in vitro studies showed that collagen was effectively degraded by Col-nc/HFn (DOX) and the penetration of NPs was increased in the solid tumor site.22
Besides protein-based method for ECM degradation, administration of certain exogenous substances can also decompose ECM components. Dong et al designed a protein-free strategy to deplete collagen based on the activation of endogenous matrix metalloproteinases (MMPs) by nitric oxide (NO).23 MMPs belong to zinc-dependent endopeptidases family that are produced and secreted by tumor cells and stromal cells like CAFs.24 MMPs can degrade most components of the ECM, and their functions are modulated by tissue inhibitors of metalloproteinases (TIMPs). Because of this, many strategies have been used to improve NP penetration through MMPs activation. NO was one of the substances that was reported to activate MMPs and thus break down matrix proteins to deplete collagen, facilitating the penetration of NPs.25 Dong’s group developed Mesoporous silica NPs (MSN) loaded with DOX and a NO donor (S-nitrosothiol) to create DN@MSN. In vitro and in vivo results demonstrated that administration of DN@MSN led to promoted tumor penetration of NPs, contributing to remarkably enhanced antitumor effect without overt toxicity in 4T1 breast cancer model.23
Drug Delivery Systems That Degrade Hyaluronic Acid
Hyaluronic acid (HA) is another component comprising the major structure of the ECM. HA is a large, negatively charged, unbranched polymer of acetyl glucosamine and D-glucuronic acid that plays a key role in determining the compressive properties of most biological tissues.26 HA is overexpressed in the tumor, contributing to a gel-like ECM that promotes the inhibition of external fluid penetration and drug accumulation, increasing the interstitial fluid pressures (IFP) as well.20 The combinational effect of tensile resistance results from collagen and compression compliance due to HA makes the typical characteristics of ECM. Therefore, HA is another ideal target for anticancer drug delivery and many efforts have been made to increase chemotherapeutics penetration through HA degradation.
Hyaluronidase (HAase) is a family of enzymes that is able to degrade HA and promote the tissue permeability to fluids in a concentration-dependent manner. Chen et al prepared HAase-embedded drug delivery system (NPs-EPI/HAase) that could enhance tumor penetration through regulating the TME. NPs-EPI/HAase was able to effectively degraded the HA and promoted deeper penetration and better accumulation of NPs into HepG2 tumors, resulting in enhanced tumor growth inhibition.27 Another approach is to inhibit and prevent HA synthesis and formation. 4-Methylumbelliferone (4-MU) is a derivative of coumarin that was firstly reported to suppress the synthesis of HA in cultured human skin fibroblasts in 1995.28 Thereafter, many researches revealed that 4-MU could prevent HA formation through reducing the substrate used for HA synthesis in different tumors. A study by Kohli et al developed the 4-MU pro-drug and DOX incorporated liposome, through which the tumor volume was found to decrease significantly after injecting with these formulations. In addition, the improvement of survival rate was identified in comparison with DOXIL® alone in the orthotopic murine tumor.29 In summary, degradation of HA through HAase or synthesis inhibition are two effective strategies to enhance NPs penetration into tumor tissues.
Nanoparticles Remodel Tumor Microenvironment Through Targeting Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) are the most abundant cells of the tumor stroma and they are closely relevant to tumor cell growth through controlling the surrounding TME.30 CAFs can produce or secrete many growth factors including transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), as well as cytokines, chemokines, and ECM proteins such as MMPs.31 Therefore, CAFs play a key role in enhancing the tumor phenotype, promoting tumor cell proliferation, neoangiogenesis, inflammation, and remodeling the ECM. Meanwhile, research has indicated that CAFs can not only contribute to inefficient delivery of nanotherapeutics but also result in drug resistance in cancer treatment.32 Hence, modulation of CAFs is a potential method to remodel the TME for effective anticancer therapy. Currently, several nanotechnology-mediated strategies have been developed to focus on CAFs-based therapies. For example, the disruption of CAFs can directly cut the links between tumor cells and CAFs. Targeting CAFs can take advantage of CAFs as a therapeutic target to increase anticancer efficacy of drugs.
Reprogramming of CAFs to an inactive state or reprogramming of CAFs’ phenotype instead of directly eliminating them also represents as a promising alternative approach.33
For instance, Chen et al designed a versatile tumor stroma-targeted nanovehicle (FH-SSL-Nav) to specifically eliminate CAFs in Hep G2 tumor bearing nude mice model.34 FH-SSL-Nav could break the stromal biological interaction in TME and partly reversed the accompanied drug resistance, contributing to indirect tumor inhibition. In addition, Hu et al prepared a hydrogel loaded with a peptide derivative (C16-GNNQQNYKD-OH) and losartan.35 Losartan was reported as a CAFs-disrupting agent that can destroy stroma barrier.36 This prepared hydrogel was able to exist at the tumor site in a stable condition for more than 9 days after intratumoral injection and losartan exhibited a sustained-release behavior to result in obvious destruction of CAFs in orthotopic 4T1 tumor-bearing mice model. As nano-based drug delivery system has evolved as one of the most potential therapeutic strategies for anticancer treatment, many achievements have been accomplished to explore CAFs as potential targets for cancer therapy. Zhen’s group designed a nanoconjugate called Z@FRT-scFv based on photo-immunotherapy (nano-PIT) approach to selectively eliminate CAFs without causing systemic toxicity.37 They utilized ferritin nanocages to transport a photosensitizer ZnF16Pc to CAFs in breast cancer model. The targeted delivery of photosensitizer to CAFs was realized due to the surface conjugation of an anti-FAP single chain variable fragment (scFv) antibody. Upon photo-irradiation, the nano-PIT could effectively eradicate CAFs in tumors without causing much damage to healthy tissues because of the localized property of this treatment method and selective homing ability to CAFs. In vitro and in vivo results indicated that the elimination of CAFs was accompanied with the destruction of ECM and the reduction of CXCL12 secretion, both resulting in a remarkably CD8+ T cell infiltration enhancement, which eventually led to efficient cancer cell death in 4T1 tumor model. Figure 3 is a schematic illustration of the working mechanism of this nanoconjugate to remodel TME through CAFs targeting.
 |
Figure 3 Schematic illustration of the working mechanism of Z@FRT-scFv based on photo-immunotherapy. After systemic injection, Z@FRT-scFv could home to CAFs in tumors followed by photoirradiation and selectively kill CAFs. This treatment could cause destruction of ECM and lead to reduced secretion of CXCL12, which promoted infiltration of CD8+ T cells that mediated cancer cell death. |
Nanoparticles Remodel Tumor Microenvironment Through Targeting Cancer Stem Cells
Cancer stem cells (CSCs) are a subpopulation of cancer cells defined by their function properties with the self-renew ability and possess the differentiation capability into a great diversity of cell types within the tumor mass.38 Increased evidence indicates that CSCs contribute a lot to regulating TME and are responsible for tumor initiation, progress, metastasis, relapse and poor prognosis.39 Moreover, CSCs are involved in the development of drug resistance. Many researches demonstrated that CSCs are able to withstand conventional therapies such as chemotherapy and radiotherapy via various mechanisms including enhanced drug transporter expression, maintenance of a slow dividing state and rapid DNA repair ability.40 Additionally, due to their incredible survival and tumorigenic abilities, CSCs are able to survive in circulation and possess the ability to seed new tumors in second generation.41,42 In brief, some cancer cells within primary tumor become apoptotic cells after treatment with chemotherapy or radiotherapy. Nevertheless, a group of CSCs can survive even under such treatment because of the resistance of CSCs to the classical therapeutic approaches. Thus, cancer cells can relapse quickly due to the activation of CSCs in the regressive tumor microenvironment, further forming the secondary tumor (Figure 4). Therefore, targeting CSCs within the TME will provide more possibilities for anticancer therapy.
 |
Figure 4 Schematic illustration of the classical therapeutic approaches against tumor growth. In the heterogeneous differentiation model, CSCs can survive under treatment like chemotherapy and radiotherapy while most cancer cells are killed. CSCs show potential resistance to radiation and chemotherapy, which can cause tumor relapse and form the secondary tumor. |
NPs-mediated drug delivery system exhibits excellent performance in targeted drug delivery. Binding to exceptive receptors that are overexpressed or anchoring special signaling pathways are two major strategies for NPs-based targeted delivery. CSCs are found to overexpress CD44 receptors that can be regarded as targets for specific binding.43 Rao et al developed a kind of chemotherapy agent-loaded polymer NPs, which conjugated chitosan on their surface.44 Due to the high affinity between chitosan and CD44 receptors, NPs delivered to the TME were able to targeted CD44 overexpressing CSCs, resulting in increased therapeutic efficacy in mammary tumor spheroids model. Moreover, riboflavin, a fluorescent vitamin, is accumulated in CSCs by ABCG2-coated intracellular vesicles located within cytoplasm, indicating that NPs decorated with riboflavin are potential to target CSCs due to the recognition.45 Li et al constructed a hypoxia/pH-responsive three-pronged NP linked with riboflavin on its surface.46 These NP encapsulates three drugs: irinotecan, cyclopamine, and erlotinib, which can kill undifferentiated CSCs, differentiated MCF-7 cells, and vascular niches in the TME, respectively. Additionally, irinotecan conjugated with riboflavin could accumulate in CSCs and resulted in a remarkable anticancer effect. Furthermore, another investigation by Hembram et al revealed an alternative targeting mechanism due to the extensive DNA repair capacity of CSCs.47 The hybrid NPs of silver and gold with quinacrine inhibited the DNA repair pathway through binding to the DNA of CSCs, contributing to apparent damage of DNA and death of CSCs. In addition, the research by Azmi et al demonstrated CSCs’ targeted therapy for pancreatic ductal adenocarcinoma (PDAC) in both preclinical and phase 1b study.48 Selinexor is a selective inhibitor of nuclear export compounds (SINE), which can activate tumor-suppressor protein to inhibit tumor cell growth. Therefore, they combined the SINE compounds with gemcitabine (GEM) and NPs albumin-bound (nab)-paclitaxel to achieve synergistic effects. According to the results in female tumor-bearing ICR-SCID mice model, the synergistic treatment led to suppression of PDAC cancer cells and disintegration of CSCs. Encouragingly, in the clinical trial based on nine PDAC patients, an outstanding and durable response was identified in one of the responders with progression-free survival of 16 months and overall survival of 22 months. Therefore, preclinical and ongoing clinical study lent support to the application of selinexor-GEM-nab-paclitaxel as an effective therapy for metastatic PDAC.
Nanoparticles Remodel Tumor Microenvironment Through Anti-Angiogenesis
Blood vessels are very critical part of our bodies, and they are responsible for various organism activities including nutrient supply, metabolic waste disposal, endocrine function and communication between tissues. The microarchitecture of normal vasculature is hierarchically coordinated with mature vessels that are evenly distributed to enable adequate oxygen perfusion and nutrients to each cell in the body (Figure 5A). However, the tumor vasculature is characterized by disordered vessel labyrinth with inconsistent diameter and uneven shape, in addition, the accompanying lymphatic vessels are also dilated, leaky and discontinuous, which all diminish their ability to deliver nutrients and remove waste products.49 Besides, tumor vessels are more permeable than normal vessels, and the increased vessel permeability causes the accumulation of vascular contents and enhanced IFP. In addition, tumor cells can produce a large amount of hydrogen ions, lactate and pyruvate through glycolysis, thus creating an acidic microenvironment (Figure 5B). Hence, these abnormalities create a complicated TME that is hypoxic and acid with high IFP, promoting tumor development, immunosuppression and drug resistance. In fact, as tumors will not grow beyond a diameter of 2 mm unless they are well vascularized and they often depend on angiogenesis to grow and metastasize, therefore, anti-angiogenesis has become as a potential cancer treatment strategy.50
 |
Figure 5 Schematic illustration of the comparison between (A) normal vasculature and (B) tumor vasculature. The normal vascular networks are hierarchically organized with mature vessels. However, the tumor vasculature is immature, tortuous and hyperpermeable which leads to a complicated TME characterized by heterogeneity, acid, hypoxia and high interstitial fluid pressure, promoting tumor cell migration and facilitating tumor progression. |
Several compounds have been approved by FDA for anti-angiogenesis therapy in clinic, such as bevacizumab, aflibercept and sorafenib, which are aimed to combine with VEGF or VEGFR to inhibit angiogenesis.51 However, the working mechanism of small molecule anti-vascular agents is comprehensive, which might lead to inevitable system toxicity and myelosuppression. Thus, targeted drug delivery through NPs can overcome these defects by targeting VEGF directly or indirectly, increasing therapeutic efficacy and meanwhile reducing systemic toxicity. For example, compared with anti-vascular drugs, silencing VEGF gene by siRNA loaded NPs tends to be more specific and more powerful to inhibit VEGF expression. Ding et al designed and constructed polyethylenimine (PEI)-modified single-walled carbon nanotubes (SWNTs) conjugated with candesartan, which could transport VEGF-siRNA in a synergistic for the targeted therapy of angiogenesis.52 Candesartan is an Ang-II receptor blocker with a strong affinity for the angiotensin II type-1 receptor (AT1R) and SWNTs possess ultrahigh surface area with the excellent capability to penetrate through the cell membrane. Except for the target effect, candesartan is also able to reduce the expression of VEGF in many tumor types. According to the in vivo results, the co-delivery system resulted in great antitumor efficacy in PANC-1 tumor xenograft nude mice model through the synergistic anti-angiogenesis effect.
Integrin has also been reported to regulate cellular activities during angiogenesis. Integrin ανβ3 and α5β1 inhibitors can cause vascular endothelial cell apoptosis in tumor, which will cause an antitumor effect.53 The classical cRGD peptide (cyclic Arg-Gly-Asp) can specifically bind with integrin ανβ3 and α5β1, and thus could be utilized as targeting ligand in NPs-based delivery system. In our previous study, Wang et al designed a targeted drug delivery system based on multi-walled carbon nanotubes (MWNTs) for anti-angiogenesis in lung cancer treatment.54 iRGD peptide and candesartan were connected to PEI-modified MWNTs, and the functionalized MWNT was assembled with plasmid angiotensin II type 2 receptor (pAT2) to form iRGD-PEI-MWNT-CD/pAT2 complexes. Similar as SWNTs, MWNTs as drug carrier could also penetrate through both cancer and angiogenic endothelium cell membrane to deliver candesartan and pAT2 into cytoplasm. As the in vivo and in vitro experiments indicated, this nanocomplexes exhibited remarkable tumor growth suppression effect in A549 xenograft nude mice through the efficient inhibition of angiogenesis by pAT2 and candesartan. Up till now, many efforts have been made to design a simultaneous co-delivery system of gene drugs and chemotherapy drugs for effective angiogenesis inhibitory to kill tumor cells. For example, Wang et al utilized the characteristics of TME to design a sequential delivery system to carry VEGF-siRNA (siVEGF) and paclitaxel (PTX) via a multi-functional polymer micelle (PTX/siVEGF-CPPs/TMPM).55 Due to the acid environment of TME, this co-delivery system could not only transport both siVEGF and PTX to the specific action sites but also release the payload independently with a controllable profile, which eventually destroyed abnormal blood vessels around the tumor sites, inhibited neo-vasculature inside the tumor parenchyma, induced the apoptosis of tumor cells, and effectively suppressed the tumor growth in MCF-7 tumor-bearing nude mice.
Nanoparticles Remodel Tumor Microenvironment Through Targeting Exosomes
During the past decades, the cancer metabolism has received wide attention. From the discovery by Warburg in 1956, cancer cells were found to consume more glucose than normal cells and the glycolysis rate in them is much higher than oxidative phosphorylation even under adequate oxygen condition.56 Therefore, the metabolic reprogramming ability of cancer cells is one of their hallmarks which distinguishes them from normal cells. Remarkably, extracellular vesicles (EVs) that carried miRNA can silence the glycolytic enzyme pyruvate kinase and are capable of reprogramming the metabolism of cells within the TME efficiently.57 Although EVs were initially regarded as vehicle to transfer waste of cells, they were reported to play a significant role in cellular interactions. EVs derived from cancer cells have been demonstrated to involve in cancer dissemination and participate in cancer metastasis.58 Generally, EVs include exosomes and microvesicles, which are different in composition, size, and biological mechanism of production. Exosomes are 30–100 nm membrane-coated vesicles that can be synthesized in the endosomal pathway of both normal and tumor cells. Accumulated evidences indicate that exosomes are able to transfer biologically functional molecules to the recipient cells, promoting tumor development within TME. Because of this, exosomes are regarded as key signaling mediators in regulating the TME and targeting tumor-derived exosomes (TDEs) is of great importance in anticancer therapy.
TDEs play a significant role in the interaction and function of TME, resulting in tumor metastasis due to their ability to carry numerous cargoes. In addition, many recent studies have shown that TDEs have a distinct content compared with normal cells, which represents a rich source of biomarkers and targets for cancer therapy based on exosomes.59 Lu et al demonstrated that TDEs could mediate communication among cells and remodel the TME by transport of miRNA.60 Firstly, they transfected nasopharyngeal carcinoma (NPC) cells with lentivirus that overexpresses miR-9, an inhibitor of tumor progression and a biomarker of NPC metastasis. Subsequently, TDEs were secreted from NPC cells, actioned as transports of miR-9 to endothelial cells. At last, functional analysis revealed that due to the miR-9 carried by TDEs, endothelial tube formation and tumor angiogenesis were inhibited by suppressing the target gene, eventually contributing to inhibition of NPC progression.
Apart from endothelial cells, TDEs can also communicate with other type of cells like immune cells within the TME. Rao et al revealed that TDEs could provoke a strong dendritic cells-mediated immune response due to a series of antigens associated with hepatocellular carcinoma (HCC) within them. Then, the TME was remodeled, resulting in a significant increased number of T lymphocytes and interferon-γ level in TME.61 Moreover, NPs-based drug delivery system is an alternative approach as non-viral vector in targeting TDEs. Amiji’s Group synthesized a nucleic acid construct delivery system by conjugating HA with PEI and PEG, forming HA-PEI/HA-PEG NPs through self-assembly.62 This NPs-mediated delivery system, encapsulating the plasmid DNA that expresses microRNA-155 and microRNA-125b2, transfected pancreatic cancer cells successfully. TDEs from transfected pancreatic cancer cells showed significant higher microRNA-155 and microRNA-125b2 expression compared with controls, which led to alteration of macrophages state from M2 to M1 phenotype, further promoting differential communication and apoptosis of tumor cells. The results demonstrated enhanced tumor suppression in vivo by reprogramming the TME through TDEs.
Exosome has been considered as a novel target in NPs-based drug delivery for cancer therapy. Cancer cells can secrete exosomes that carry various compounds to promote tumor development. A number of preclinical studies have clearly demonstrated the significant role of exosomes in TME, which also indicates their translational potential in clinic. Therefore, targeting exosomes through NPs-based strategy is a promising approach to regulate TME, and more attention should be paid to in clinical research to develop the treatment strategy based on exosomes.
Nanoparticles Remodel Tumor Microenvironment Through Immunoregulatory
As is shown in Figure 6, immune cells are the most abundant cell types in the TME and are closely relevant to patient’s prognosis. Recently, various types of cancer immunotherapy including immune checkpoint therapy, cancer vaccines therapy, monoclonal antibodies (mAbs) therapy, and adoptive cancer therapy, are emerging.63 However, many limitations that severely circumvent the antitumor effect of immunotherapy still exist. For instance, due to the low immunogenicity of tumor tissue and the large quantity of inhibitory factors in the TME, the effect of immunotherapy can be obviously attenuated. Besides, immunotherapy’s treatment effect varies depending on tumor types, and only a small number of patients show obvious therapeutic efficacy upon immunotherapy. In addition, severe side effects might be aroused unavoidably during systemic immunotherapy. Thus, it is necessary to seek a method to solve these aforementioned obstacles of cancer immunotherapy. As nanotechnology has long been employed to overcome the physiology barrier inside the TME, recently, this strategy has also been utilized to remodel the TME for robust immunotherapeutic outcomes.
 |
Figure 6 Schematic illustration of tumor immune microenvironment. TME possesses highly immunosuppressive properties due to poor immunogenicity of tumor cells, immunosuppressive factors, and the suppression of Teff functions. The application of NPs can be an effective method to regulate TME from immunosuppressive to immunostimulatory. Large amount of tumor-associated antigens can be released after the immunological cell death (ICD) of tumor cells, which can effectively induce the maturation of DCs. Then, the proliferation and activation of immunostimulatory cells like DCs and CD8+ T cells will be triggered to produce an outstanding tumoricidal effect. Meanwhile, the amount of immunosuppressive factors in TME can be decreased obviously. Red plus indicates immunostimulatory factors and green minus indicates immunosuppressive factors in TME. |
As shown in Table 1, NPs-mediated drug delivery has been applied to target various types of immunocytes, namely dendritic cells (DCs), cytotoxic T lymphocytes (CTLs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages with M2 phenotype (M2 TAMs), and regulatory T cells (Tregs), to remodel TME during antitumor therapy. DCs and CTLs are both important immunostimulatory cells, and the application of nanodrug delivery system (DDS) with active targeting ability can further enhance their immunostimulatory abilities in TME. In contrast, MDSCs, M2 TAMs, and Tregs exhibit immunosuppressive properties. Thus, it is essential to circumvent their immunosuppressive effects through nanotechnology in cancer therapy.
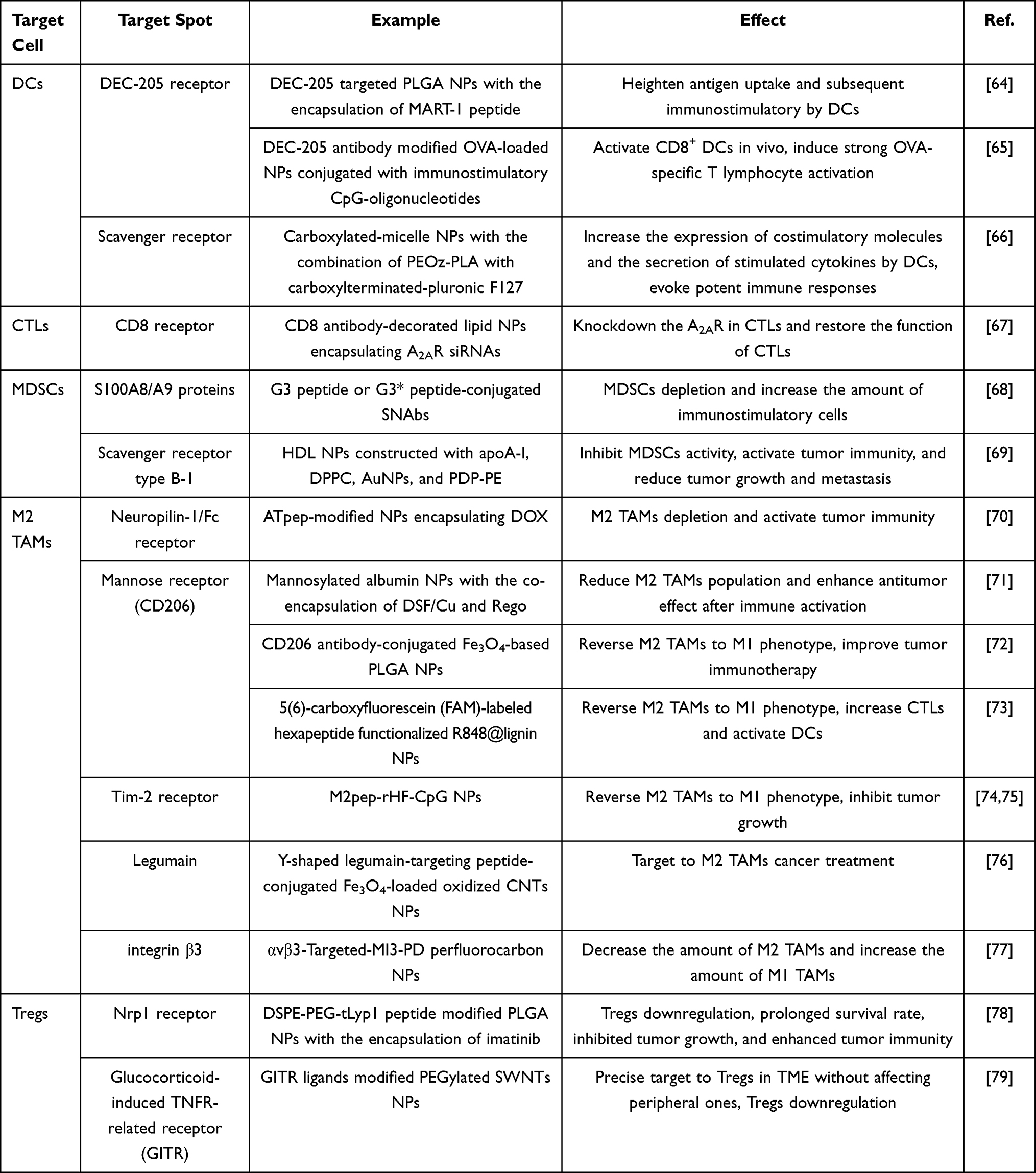 |
Table 1 Summary of NPs-Mediated Immunoregulatory Based on DCs, CTLs, MDSCs, M2 TAMs, and Tregs in TME |
Immunostimulatory Cells
Immunostimulatory cells, such as DCs, T lymphocytes, natural killer cells (NK cells), tumor-associated macrophages with M1 phenotype (M1 TAMs), and tumor-associated neutrophils with N1 phenotype (N1 TANs), often play crucial roles in cancer immunotherapy. DCs are one of the most powerful antigen presenting cells (APCs) and the maturation of DCs represents the activation of tumor immunity. With the existence of matured DCs, not only NK cells and M1 TAMs can be recruited to TME but T lymphocytes can also be activated and keep proliferating in TME for tumor immunotherapy. Unfortunately, TME usually shows high immunosuppression property, which leads to inadequacy or anergy of immunostimulatory cells as well as weakens the tumor immunity. Therefore, in order to ensure the efficacy of tumor immunotherapy, altering the TME from immunosuppression to immunostimulation is of great importance.
Dendritic Cells
DCs, which have been regarded as the most powerful antigen presenting cells for many years, were first discovered by Steinman in 1973.80 DCs distribute in both peripheral tissues and immune organs (like thymus, bone marrow, spleen, and lymph gland), playing important roles in specific immune response. There are abundant amount of antigen-presenting molecules (MHC-I and MHC-II), costimulatory factors (CD80, CD86, CD40, and CD40L) and adhesion factors (ICAM-1, ICAM-2, ICAM-3, LFA-1, and LFA-3) exist on the surface of DCs.81 Immature DCs possess strong antigen phagocytosis capacity, while mature DCs express high levels of costimulatory factors and adhesion factors. The immune response mechanisms of DCs in cancer immunotherapy can be concluded as the following five parts: 1) recognition of tumor antigen molecules; 2) activation of DCs and subsequent recruitment of nonspecific cells (such as macrophages and NK cells); 3) uptake of tumor antigens by DCs and subsequent presentation to T lymphocytes; 4) proliferation and activation of specific T lymphocytes; 5) migration of antigen-specific T lymphocytes to tumor site.82 Thus, DCs play critical roles during cancer immunotherapy process.
Specific targeting to DCs through nanotechnology in vivo has been proved as a promising way to enhance the immunostimulatory effect evoked by DCs in TME. DEC-205 is a member of the C-type lectin macrophages mannose receptor family. DEC-205 is predominantly expressed on DCs in the T cell area of lymph nodes, occupying vital positions in the cross-presentation of antigen.83 Saluja et al designed a type of biotinylated DEC-205 antibody-modified PLGA NPs with the encapsulation of MART-1 peptide.64 MART-1 peptide is a well-characterized immune-dominant epitope for human leukocyte antigen that can be used for the development of immunotherapeutic strategies.84 After the co-culture of the aforementioned NPs with immature DCs in vitro, large number of DCs became mature through the active targeting effect of DEC-205 antibody and the stimulation effect of MART-1 peptide. Subsequently, more efficient stimulation of CTLs can be observed in contrast to the NPs without DEC-205 active targeting ability. Therefore, stronger immunostimulatory effect could be achieved.
Scavenger receptor (SR) is another type of surface receptor displayed on DCs, which can be applied for targeted drug delivery. Li et al constructed a type of mixed micelle NPs through the combination of amphiphilic diblock copolymer poly (2-ethyl-2-oxazoline)-poly-(
Cytotoxic T Lymphocytes
CTLs, which can be produced through the maturation of naive CD8+ T lymphocytes, are one of the main effector cells during tumor immunotherapy.85 Due to the restriction and specificity of MHC-I on CTLs to tumor associated antigen (TAA), tumor cells can be specifically killed by CTLs without affecting any normal cells. In addition, the production of perforin and other cytotoxin after the activation of CTLs further ensures the effect of cancer immunotherapy. Adenosine is a compound formed through the conjugation between adenine and D-ribose. It can accumulate in TME and contribute to the inhibition of CTLs function via the A2A receptor (A2AR).86 In order to rescue CTLs function without engendering side effects, Newton et al reported an anti-CD8 antibody decorated lipid NPs with the encapsulation of A2AR siRNAs.67 The siRNA-loaded NPs precisely targeted to CTLs in vitro, and the A2AR in CTLs was effectively knocked down through the released A2AR siRNAs from the NPs. Expectedly, the function of CTLs restored and the effect of cancer immunotherapy was ensured.
Other Immunostimulatory Cells
Except for DCs and CTLs, there are many other immunostimulatory cells, such as T helper (Th) cells, NK cells, M1 TAMs and tumor-associated neutrophils with N1 phenotype (N1 TANs). All of them exhibit great importance to cancer immunotherapy.
Th cells are a group of immunocytes with immunostimulatory effect and is able to express CD4 molecular on their surface. Th cells can be divided into several subgroups, namely Th1, Th2, and Th17.87 Various types of cytokines can be produced from Th1, while the cytokines secreted from Th2 promote humoral immune response. In addition, Th17 may also play a vital role in cancer immunotherapy.88
NK cells belong to innate immune cells that can be regarded as the first line of defense during cancer immunotherapy. Cytotoxic effect can be directly mediated by NK cells without pre-sensitization. After the activation of NK cells, perforin, granzyme, and TNF-α can be released for antitumor therapy.89
Tumor-associated macrophages (TAMs) are the most abundant immunocytes in tumors, which can be further classified into M1 (immunostimulatory property) and M2 (immunosuppressive property) phenotype.90 M1 TAMs are a group of immune cells distributed in the TME of solid tumors. Similar to NK cells, M1 TAMs can also be classified as innate immune cells and possess board antitumor activity. Different tumoricidal factors, such as superoxide, NO, and tumor necrosis factor (TNF), can be released after the activation of M1 TAMs. In addition, antigens can also be presented by M1 TAMs, which further promotes the activation of tumor immunity.91
N1 TANs are another important group of immunostimulatory cells with short-living property and a mature phenotype in TME.92 N1 TANs possess outstanding tumoricidal ability and immunostimulatory property (such as the activation of CTLs) with the existence of IFN, the absence of TGF-β, or the increased production of TNF. Besides, N1 TANs also exhibit advantage of forming immune memory in TME.93
Although these aforementioned immunostimulatory functions can be produced by immunocytes, many existing problems remain unsolved. For example, for NK cells, their functions are always downregulated with the development of cancer and the cytotoxic effect caused by NK cells is subsequently compromised.94 Besides, both TAMs and TANs with antitumor phenotype may convert into pro-tumor phenotype under the promotion of tumor and immunosuppressive cytokines like TGF-β, and eventually leading to tumor growth, metastasis, and immunosuppressive TME.95 In addition, the clinical researches on nano-based targeting strategy to immunostimulatory cells are inadequate. Thus, in order to ensure the therapeutic efficacy of cancer immunotherapy, more researches need to be investigated to target these immunostimulatory cells through the application of NPs.
Immunosuppresive Cells
In contrast to immunostimulatory cells, immunosuppressive cells infiltrated in TME severely hamper tumor immunity, which usually leads to tumor progression, metastasis and relapse. MDSCs, M2 TAMs, and Tregs are three main types of immunosuppressive cells in TME, which can deactivate the immunostimulatory cells and compromise the effect of tumor immunotherapy accordingly. Therefore, depletion, downregulation or phenotype reversion of immunosuppressive cells through nanotechnology can enhance the immunogenicity of tumor tissue, and thus circumventing the immunosuppressive effect in TME.
Myeloid-Derived Suppressor Cells
MDSCs are a type of immunosuppressive cells derived from bone marrow progenitor cells and immature bone marrow cells (IMCs).96 IMCs can differentiate into mature granulocytes, macrophages, DCs, and other mature bone marrow cells under normal physiological conditions. While under tumor or other pathological conditions, IMCs cannot differentiate into mature bone marrow cells due to the influence of cytokines, and thus designated as MDSCs.97 MDSCs can be divided into monocytes (M-MDSCs) and polymorphonuclear cells (PMN-MDSCs), which both play important roles in immunosuppressive TME. With the existence of various inflammatory factors like IL-6 and IL-1β, MDSCs can be recruited to TME, and subsequently promote tumor growth, invasion and metastasis through the inactivation of immune cells.
In order to circumvent the immunosuppressive TME caused by MDSCs, Liu et al constructed an entirely synthetic, multivalent, Janus nano-therapeutic platform called synthetic nanoparticle antibodies (SNAbs), which is a type of MDSC-targeting NPs for the effective depletion of MDSCs in TME.68 In contrast to other cell types in TME, MDSCs express tenfold higher cell surface S100A8/A9 proteins, and either G3 peptide or G3* peptide conjugated in the NPs shows excellent binding affinity to S100A8/A9 proteins. In vitro and in vivo experiments based on 4T1 mouse breast cancer model demonstrated that this NPs significantly reduced the viability and the total amount of MDSCs. Besides, the percentage of various immunostimulatory cells, such as T lymphocytes and DCs, increased accordingly in TME due to the reduction of MDSCs.
Tumor-Associated Macrophages with M2 Phenotype
M2 TAMs are another type of macrophage phenotype in TME that highly express CD206, IL-10 and ARG1.98 Different from M1 phenotype, M2 TAMs mainly exert immunosuppressive functions and contribute to tumor progression and metastasis. With the existence of M2 TAMs in TME, the expression of VEGF can be obviously promoted, leading to tumor angiogenesis and tumorigenesis. Besides, M2 TAMs upregulate various soluble protein molecules (such as MMP-1 and MMP-7) in TME, further promoting tumor angiogenesis and enhancing tumor metastasis.99 In addition, the M2 TAMs in tumor tissue also lead to the anergy of T lymphocytes, forming immunosuppressive TME and inhibit tumoricidal effect.100 Thus, it is essential to decrease the amount of M2 TAMs infiltrating in TME during antitumor therapy.
Peng et al designed a multifunctional peptide (ATpep)-modified NPs with the targeting ability to M2TAMs, achieving excellent antitumor effect during treatment.70 After the i.v. administration to 4T1 tumor-bearing mice, this NPs can be activated in TME and phagocytosed by M2 TAMs through neuropilin-1/Fc receptor pathways. Eventually, the M2 TAMs in TME can be effectively depleted. Mannose receptor (MR), also known as CD206, is another receptor overexpressed by M2 TAMs.101 Therefore, MR is also a promising target spot for precisely delivering NPs to M2 TAMs. Zhao et al constructed mannosylated albumin NPs with the co-encapsulation of disulfiram/copper complex (DSF/Cu) and regorafenib (Rego).71 In vitro and in vivo experiments based on a drug-resistant colon cancer mouse model showed that this type of NPs was precisely delivered to M2 TAMs in TME, reducing M2 macrophage population after the release of DSF/Cu and Rego. Thus, the antitumor effect was further enhanced. Except for the direct targeting of NPs to M2 TAMs, the phenotype conversion of TAMs from M2 to M1 can also be achieved through the delivery of NPs to TME. For example, Shi et al designed a lipoprodrug through the encapsulation of DHA-tethered cabazitaxel prodrug.90 After i.v. administration, the lipoprodrug eventually internalized into the tumor tissue and promoted the phenotype conversion of TAMs from M2 to M1, revealing the TME reprogramming ability of this lipoprodrug.
Regulatory T Cells
Tregs are a subpopulation of CD4+ T lymphocytes with immunosuppressive function. The mechanism of the production of immunosuppressive TME caused by Tregs is complicated. For instance, various immunosuppressive cytokines (such as IL-10 and TGF-β) can be produced by Tregs, which weakens the effect of tumor immunity.102 Besides, Tregs can inhibit the activation of CD4+ and CD8+ T lymphocytes, and compromise the cytotoxic effect of NK cells and NKT cells. In addition, antigen presentation can also be inhibited by Tregs through the negative regulation of NF-κB pathway in DCs, which influences the activation of DCs and the following immune activation.103
tLyp1 peptide possesses high affinity to Nrp1 receptor expressed on Tregs.104 In order to prevent Tregs from exerting immunosuppressive effect, Ou et al developed a tLyp1 peptide-modified NPs with excellent targeting ability to Tregs in TME.78 In vitro and in vivo experiments based on melanoma mouse model showed that the Imatinib loaded in NPs was precisely delivered to Tregs, downregulating the Tregs in TME through inhibiting the phosphorylation of STAT3 and STAT5. Thus, reduced Tregs in TME, prolonged survival rate, inhibited tumor growth, and enhanced tumor immunity were all observed during antitumor therapy. Glucocorticoid-induced TNFR-related receptor (GITR) is another receptor abundantly expressed on the Tregs in TME.105 Sacchetti et al modified PEGylated SWNTs with GITR ligands, constructing a type of NPs with Tregs-targeting ability.79 After precise delivery, these NPs could be internalized by Tregs in TME based on B16 mouse breast cancer model through receptor-mediated endocytosis and then transported into cytoplasm and nucleus. Besides, the expression of GITR on peripheral Tregs is much lower than the ones in TME.79 Thus, the active targeting property based on GITR applied in this research exhibited low toxicity and side effect during antitumor treatment. In conclusion, these NPs show promising prospects after the encapsulation of the agents acting on Tregs.
Other Immunosuppressive Cells
As mentioned earlier, MDSCs, M2 TAMs and Tregs are three common types of immunosuppressive cells that lead to immunosuppressive TME and subsequent tumor growth, metastasis and relapse. In addition, regulatory B cells (Bregs) and tumor-associated neutrophils with N2 phenotype (N2 TANs) are two newly discovered immunocytes with various immunosuppressive functions. Bregs can be activated through the interaction between CD40 on Bregs and CD40L on tumor cells.106 After activation, IL-10 can be produced by Bregs, leading to the anergy of CD8+ T lymphocytes and decrease the IFN-γ level produced by CD8+ T lymphocytes and NK cells. And ultimately, the tumor immunity can be obviously suppressed. N2 TANs are a type of TANs that can cause tumor growth and metastasis through the production of various tumor growth promoting factor like CXCL2 and IL-8.107 The existence of G-CSF, IL-35, and TGF-β in TME can induce neutrophils to polarize into N2 phenotype, which leads to N2 TANs’ infiltration into tumor tissue, thus causing immunosuppressive effect.108 In addition, with the existence of IFN-γ and TNF-α, the phenotype of N2 TANs can change to N1 that possesses strong tumoricidal function and is available for antitumor treatment.109
Although the application of nano-based targeting strategy to these novel immunosuppressive cells is still under investigation at the present. More intelligent NPs will be definitely discovered to accurately target these immunosuppressive cells for cancer immunotherapy in the future. Moreover, the clinical researches on NPs-mediated active targeted delivery to various immunosuppressive cells are also rare. Therefore, it is necessary to pay more attention to the clinical trials on nano-based delivery strategy to immunosuppressive cells.
Conclusion
Cancer remains a worldwide threat to human health and is a multifactorial disorder that requires multichannel therapy. Although great efforts have been made in the delivery of therapeutic agents to solid tumors over the past few decades, the clinical outcome for cancer therapy remains limited. The TME is a complex ecology that contains extracellular matrix, stromal cells and immune cells. This local environment not only plays a vital role during tumor initiation, progression, and metastasis but also significantly influences the therapeutic response for cancer treatment. Hence, it is essential to modify the TME to overcome these physiological barriers. As nanotechnology has gained much attention due to its inherent advantages and remodeling, the TME through targeting its primary elements can effectively address the obstacles during anticancer therapy. Therefore, many strategies based on nanotechnology have been widely investigated to target the TME components such as ECM, CAFs, CSCs, exosomes and to modify this complicated microenvironment through immunoregulatory and anti-angiogenesis. In addition, these versatile nanotools are able to efficiently transport chemotherapeutics along with multiple targeting ligands to specific target within the TME, enhancing therapeutic efficacy with minimal adverse effects. Besides, owing to their unique physiochemical properties, various NPs are available to aim at the TME elements to enhance drug exposure and penetration to tumor tissue, opening a new era for cancer therapy.
Of course, there are still some challenges or issues that should be paid attention to during nano-based therapies to remodel TME. For instance, although NPs used for TME remodeling have shown attractive potentials in preclinical research, numerous difficulties remain to be unsolved in their clinical translation. Firstly, the safety of nanomaterials restricts their successful application in clinic and requires these nanomaterials to be preferably biodegradable without causing biological aggregation and resistance. Secondly, the controllable industrial production of NPs also restricts their successful clinical translation because some NPs require complex synthesis and purification processes, which make it difficult to reproduce products with the same quality at large-scale preparation. Thirdly, considering the heterogeneity of TME and the diversity of tumor structures, the therapeutic responses of distinct tumors to the same therapy often vary. Therefore, there are individual differences in the therapeutic outcome of nanomedicine. Fourthly, the regulation strategies of TME that may change its structure in part or through immunoregulation have potential risks to promote of tumor metastasis, so the long-term effects of TME regulation strategy should be further investigated. For future prospective, more clinically relevant in vivo and in vitro models should be established to sufficiently simulate the complicated interactions between NPs and the TME. More innovative and effective nanomaterial-based drug delivery systems should be explored and developed to accurately target multiple TEM components at the same time.
Altogether, we are confident that with the rapid development of nanomedicine, these aforementioned obstacles will be overcome one day and more intelligent nanoplatforms will be constructed and designed for precise TME targeting. This review provides a summary of the recent advance of TME regulation through NPs mediated drug delivery and various strategies aiming at targeting the TME components based on nanotechnology are also outlined. We hope that nano-based strategies will contribute a bit to accelerate the progress of current cancer therapy.
Funding
This work was funded by the National Nature Science Foundation of China (Nos. 31872756 and 32071387), National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2016ZX09101031), Six Talent Peaks Project in Jiangsu Province (JY-079) and “Double First-Class” Construction Technology Innovation Team Project of China Pharmaceutical University (Nos. CPU2018GY23 and CPU2018GY26).
Disclosure
The authors declare no conflict of interest in this work.
References
1. Thakkar S, Sharma D, Kalia K, Tekade RK. Tumor microenvironment targeted nanotherapeutics for cancer therapy and diagnosis: a review. Acta Biomater. 2020;101:43–68. doi:10.1016/j.actbio.2019.09.009
2. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2020;1:107753.
3. Arneth B. Tumor Microenvironment. Medicina. 2019;56(1):15. doi:10.3390/medicina56010015
4. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591–5596. doi:10.1242/jcs.116392
5. Muntimadugu E, Kommineni N, Khan W. Exploring the potential of nanotherapeutics in targeting tumor microenvironment for cancer therapy. Pharmacol Res. 2017;126:109–122. doi:10.1016/j.phrs.2017.05.010
6. Zhang J, Wang N, Li Q, Zhou Y, Luan Y. A two-pronged photodynamic nanodrug to prevent metastasis of basal-like breast cancer. Chem Commun (Camb). 2021;57(18):2305–2308. doi:10.1039/D0CC08162K
7. Zhang M, Qin X, Xu W, et al. Engineering of a dual-modal phototherapeutic nanoplatform for single NIR laser-triggered tumor therapy. J Colloid Interface Sci. 2021;594:493–501. doi:10.1016/j.jcis.2021.03.050
8. Tian H, Zhang M, Jin G, Jiang Y, Luan Y. Cu-MOF chemodynamic nanoplatform via modulating glutathione and H2O2 in tumor microenvironment for amplified cancer therapy. J Colloid Interface Sci. 2021;587:358–366. doi:10.1016/j.jcis.2020.12.028
9. Ren X, Wang N, Zhou Y, et al. An injectable hydrogel using an immunomodulating gelator for amplified tumor immunotherapy by blocking the arginase pathway. Acta Biomater. 2021;124:179–190. doi:10.1016/j.actbio.2021.01.041
10. Zhou Y, Ren X, Hou Z, Wang N, Jiang Y, Luan Y. Engineering a photosensitizer nanoplatform for amplified photodynamic immunotherapy via tumor microenvironment modulation. Nanoscale Horiz. 2021;6(2):120–131. doi:10.1039/D0NH00480D
11. Lin G, Chen S, Mi P. Nanoparticles targeting and remodeling tumor microenvironment for cancer theranostics. J Biomed Nanotechnol. 2018;14(7):1189–1207. doi:10.1166/jbn.2018.2546
12. Miao L, Huang L. Exploring the tumor microenvironment with nanoparticles. Cancer Treat Res. 2015;166:193–226.
13. Mei Y, Tang L, Xiao Q, et al. Reconstituted high density lipoprotein (rHDL), a versatile drug delivery nanoplatform for tumor targeted therapy. J Mater Chem B. 2021;9(3):612–633. doi:10.1039/D0TB02139C
14. Anchordoquy TJ, Barenholz Y, Boraschi D, et al. Mechanisms and barriers in cancer nanomedicine: addressing challenges, looking for solutions. ACS Nano. 2017;11(1):12–18. doi:10.1021/acsnano.6b08244
15. Sailor MJ, Park JH. Hybrid nanoparticles for detection and treatment of cancer. Adv Mater. 2012;24(28):3779–3802. doi:10.1002/adma.201200653
16. Ashfaq UA, Riaz M, Yasmeen E, Yousaf MZ. Recent advances in nanoparticle-based targeted drug-delivery systems against cancer and role of tumor microenvironment. Crit Rev Ther Drug Carrier Syst. 2017;34(4):317–353. doi:10.1615/CritRevTherDrugCarrierSyst.2017017845
17. Nguyen A, Bottger R, Li SD. Recent trends in bioresponsive linker technologies of prodrug-based self-assembling nanomaterials. Biomaterials. 2021;275:120955. doi:10.1016/j.biomaterials.2021.120955
18. Xie H, Zhu H, Zhou K, et al. Target-oriented delivery of self-assembled immunosuppressant cocktails prolongs allogeneic orthotopic liver transplant survival. J Control Release. 2020;328:237–250. doi:10.1016/j.jconrel.2020.08.043
19. Zhou L, Xie H, Chen X, et al. Dimerization-induced self-assembly of a redox-responsive prodrug into nanoparticles for improved therapeutic index. Acta Biomater. 2020;113:464–477. doi:10.1016/j.actbio.2020.07.007
20. Walker C, Mojares E, Del Rio Hernandez A. Role of extracellular matrix in development and cancer progression. Int J Mol Sci. 2018;19(10):3028. doi:10.3390/ijms19103028
21. Harisi R, Jeney A. Extracellular matrix as target for antitumor therapy. Onco Targets Ther. 2015;8:1387–1398.
22. Yao H, Guo X, Zhou H, et al. Mild acid-responsive “nanoenzyme capsule” remodeling of the tumor microenvironment to increase tumor penetration. ACS Appl Mater Interfaces. 2020;12(18):20214–20227. doi:10.1021/acsami.0c03022
23. Dong X, Liu HJ, Feng HY, et al. Enhanced drug delivery by nanoscale integration of a nitric oxide donor to induce tumor collagen depletion. Nano Lett. 2019;19(2):997–1008. doi:10.1021/acs.nanolett.8b04236
24. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. doi:10.1126/science.1067100
25. Liu P, Wang Y, Liu Y, Tan F, Li J, Li N. S-nitrosothiols loaded mini-sized Au@silica nanorod elicits collagen depletion and mitochondrial damage in solid tumor treatment. Theranostics. 2020;10(15):6774–6789. doi:10.7150/thno.42661
26. Haider T, Sandha KK, Soni V, Gupta PN. Recent advances in tumor microenvironment associated therapeutic strategies and evaluation models. Mater Sci Eng C Mater Biol Appl. 2020;116:111229.
27. Chen E, Han S, Song B, et al. Mechanism investigation of hyaluronidase-combined multistage nanoparticles for solid tumor penetration and antitumor effect. Int J Nanomedicine. 2020;15:6311–6324. doi:10.2147/IJN.S257164
28. Nakamura T, Takagaki K, Shibata S, Tanaka K, Higuchi T, Endo M. Hyaluronic-acid-deficient extracellular matrix induced by addition of 4-methylumbelliferone to the medium of cultured human skin fibroblasts. Biochem Biophys Res Commun. 1995;208(2):470–475. doi:10.1006/bbrc.1995.1362
29. Kohli AG, Kivimae S, Tiffany MR, Szoka FC. Improving the distribution of Doxil(R) in the tumor matrix by depletion of tumor hyaluronan. J Control Release. 2014;191:105–114. doi:10.1016/j.jconrel.2014.05.019
30. Truffi M, Mazzucchelli S, Bonizzi A, et al. Nano-strategies to target breast cancer-associated fibroblasts: rearranging the tumor microenvironment to achieve antitumor efficacy. Int J Mol Sci. 2019;20(6):1263. doi:10.3390/ijms20061263
31. Luo H, Tu G, Liu Z, Liu M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. 2015;361(2):155–163. doi:10.1016/j.canlet.2015.02.018
32. Miao L, Wang Y, Lin CM, et al. Nanoparticle modulation of the tumor microenvironment enhances therapeutic efficacy of cisplatin. J Control Release. 2015;217:27–41. doi:10.1016/j.jconrel.2015.08.027
33. Liu Q, Chen F, Hou L, et al. Nanocarrier-Mediated chemo-immunotherapy arrested cancer progression and induced tumor dormancy in desmoplastic melanoma. ACS Nano. 2018;12(8):7812–7825. doi:10.1021/acsnano.8b01890
34. Chen B, Dai W, Mei D, et al. Comprehensively priming the tumor microenvironment by cancer-associated fibroblast-targeted liposomes for combined therapy with cancer cell-targeted chemotherapeutic drug delivery system. J Control Release. 2016;241:68–80. doi:10.1016/j.jconrel.2016.09.014
35. Hu C, Liu X, Ran W, et al. Regulating cancer associated fibroblasts with losartan-loaded injectable peptide hydrogel to potentiate chemotherapy in inhibiting growth and lung metastasis of triple negative breast cancer. Biomaterials. 2017;144:60–72. doi:10.1016/j.biomaterials.2017.08.009
36. Patel K, Doddapaneni R, Chowdhury N, Boakye CH, Behl G, Singh M. Tumor stromal disrupting agent enhances the anticancer efficacy of docetaxel loaded PEGylated liposomes in lung cancer. Nanomedicine. 2016;11(11):1377–1392. doi:10.2217/nnm.16.37
37. Zhen Z, Tang W, Wang M, et al. Protein Nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic t cell infiltration and tumor control. Nano Lett. 2017;17(2):862–869. doi:10.1021/acs.nanolett.6b04150
38. Cao Z, Weygant N, Chandrakesan P, Houchen CW, Peng J, Qu D. Tuft and Cancer Stem Cell Marker DCLK1: a new target to enhance anti-tumor immunity in the tumor microenvironment. Cancers. 2020;12(12):3801. doi:10.3390/cancers12123801
39. Das M, Law S. Role of tumor microenvironment in cancer stem cell chemoresistance and recurrence. Int J Biochem Cell Biol. 2018;103:115–124. doi:10.1016/j.biocel.2018.08.011
40. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi:10.1038/nature05236
41. Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14(3):306–321. doi:10.1016/j.stem.2014.02.002
42. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298–306. doi:10.1038/nature17038
43. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253–1261. doi:10.1056/NEJMra061808
44. Rao W, Wang H, Han J, et al. Chitosan-decorated doxorubicin-encapsulated nanoparticle targets and eliminates tumor reinitiating cancer stem-like cells. ACS Nano. 2015;9(6):5725–5740. doi:10.1021/nn506928p
45. Miranda-Lorenzo I, Dorado J, Lonardo E, et al. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nat Methods. 2014;11(11):1161–1169. doi:10.1038/nmeth.3112
46. Li Y, Zhang R, Lu Z, et al. Microenvironment-responsive three-pronged approach breaking traditional chemotherapy to target cancer stem cells for synergistic inoperable large tumor therapy. Small. 2016;12(40):5516–5523. doi:10.1002/smll.201601932
47. Hembram KC, Dash SR, Das B, et al. Quinacrine based gold hybrid nanoparticles caused apoptosis through modulating replication fork in oral cancer stem cells. Mol Pharm. 2020;17(7):2463–2472. doi:10.1021/acs.molpharmaceut.0c00197
48. Azmi AS, Khan HY, Muqbil I, et al. Preclinical assessment with clinical validation of selinexor with gemcitabine and nab-paclitaxel for the treatment of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2020;26(6):1338–1348. doi:10.1158/1078-0432.CCR-19-1728
49. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev. 2011;37(1):63–74. doi:10.1016/j.ctrv.2010.05.001
50. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. doi:10.1056/NEJM197111182852108
51. Goel S, Duda DG, Xu L, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi:10.1152/physrev.00038.2010
52. Ding X, Su Y, Wang C, et al. Synergistic Suppression of tumor angiogenesis by the co-delivering of vascular endothelial growth factor targeted siRNA and candesartan mediated by functionalized carbon nanovectors. ACS Appl Mater Interfaces. 2017;9(28):23353–23369. doi:10.1021/acsami.7b04971
53. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi:10.1038/nrc2748
54. Su Y, Hu Y, Wang Y, et al. A precision-guided MWNT mediated reawakening the sunk synergy in RAS for anti-angiogenesis lung cancer therapy. Biomaterials. 2017;139:75–90. doi:10.1016/j.biomaterials.2017.05.046
55. Yang Y, Meng Y, Ye J, et al. Sequential delivery of VEGF siRNA and paclitaxel for PVN destruction, anti-angiogenesis, and tumor cell apoptosis procedurally via a multi-functional polymer micelle. J Control Release. 2018;287:103–120. doi:10.1016/j.jconrel.2018.08.028
56. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi:10.1126/science.123.3191.309
57. Fong MY, Zhou W, Liu L, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17(2):183–194. doi:10.1038/ncb3094
58. Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. doi:10.1016/j.cell.2016.01.043
59. Meng W, Hao Y, He C, Li L, Zhu G. Exosome-orchestrated hypoxic tumor microenvironment. Mol Cancer. 2019;18(1):57. doi:10.1186/s12943-019-0982-6
60. Lu J, Liu QH, Wang F, et al. Exosomal miR-9 inhibits angiogenesis by targeting MDK and regulating PDK/AKT pathway in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2018;37(1):147. doi:10.1186/s13046-018-0814-3
61. Rao Q, Zuo B, Lu Z, et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64(2):456–472. doi:10.1002/hep.28549
62. Su MJ, Aldawsari H, Amiji M. Pancreatic cancer cell exosome-mediated macrophage reprogramming and the role of microRNAs 155 and 125b2 transfection using nanoparticle delivery systems. Sci Rep. 2016;6:30110. doi:10.1038/srep30110
63. Zhang HM, Chen JB. Current status and future directions of cancer immunotherapy. J Cancer. 2018;9(10):1773–1781. doi:10.7150/jca.24577
64. Saluja SS, Hanlon DJ, Sharp FA, et al. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. Int J Nanomedicine. 2014;9:5231–5246.
65. Shen LM, Krauthauser S, Fischer K, et al. Vaccination with trifunctional nanoparticles that address CD8(+) dendritic cells inhibits growth of established melanoma. Nanomedicine. 2016;11(20):2647–2662. doi:10.2217/nnm-2016-0174
66. Li C, Zhang X, Chen Q, et al. Synthetic polymeric mixed micelles targeting lymph nodes trigger enhanced cellular and humoral immune responses. ACS Appl Mater Interfaces. 2018;10(3):2874–2889. doi:10.1021/acsami.7b14004
67. Newton H, Arnold MJ, Chimote AA, Conforti L. Selective knockdown of A(2A)R in CD8(+) T cells using CD8-targeting nanoliposomes. J Immunol. 2018;200:1.
68. Liu J, Toy R, Vantucci C, et al. Bifunctional janus particles as multivalent synthetic nanoparticle antibodies (SNAbs) for selective depletion of target cells. Nano Lett. 2021;21(1):875–886. doi:10.1021/acs.nanolett.0c04833
69. Plebanek MP, Bhaumik D, Bryce PJ, Thaxton CS. Scavenger receptor type B1 and lipoprotein nanoparticle inhibit myeloid-derived suppressor cells. Mol Cancer Ther. 2018;17(3):686–697. doi:10.1158/1535-7163.MCT-17-0981
70. Peng H, Wang JH, Guo F, et al. Legumain protease-activated tuftsin-functionalized nanoparticles for dual-targeting TAMs and cancer chemotherapy. Colloids Surf B Biointerfaces. 2021;197:111442. doi:10.1016/j.colsurfb.2020.111442
71. Zhao PF, Yin WM, Wu AH, et al. Dual-targeting to cancer cells and M2 macrophages via biomimetic delivery of mannosylated albumin nanoparticles for drug-resistant cancer therapy. Adv Funct Mater. 2017;27(44):15.
72. Zhou Y, Que KT, Tang HM, Zhang P, Fu QM, Liu ZJ. Anti-CD206 antibody-conjugated Fe3O4-based PLGA nanoparticles selectively promote tumor-associated macrophages to polarize to the pro-inflammatory subtype. Oncol Lett. 2020;20(6):298. doi:10.3892/ol.2020.12161
73. Figueiredo P, Lepland A, Scodeller P, et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomater. 2020;S1742(20):30561–30564.
74. Todorich B, Zhang X, Slagle-Webb B, Seaman WE, Connor JR. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J Neurochem. 2008;107(6):1495–1505. doi:10.1111/j.1471-4159.2008.05678.x
75. Shan H, Dou W, Zhang Y, Qi M. Targeted ferritin nanoparticle encapsulating CpG oligodeoxynucleotides induces tumor-associated macrophage M2 phenotype polarization into M1 phenotype and inhibits tumor growth. Nanoscale. 2020;12(43):22268–22280. doi:10.1039/D0NR04520A
76. Yan L, Gao Y, Pierce R, Dai L, Kim J, Zhang M. Development of Y-shaped peptide for constructing nanoparticle systems targeting tumor-associated macrophages in vitro and in vivo. Mater Res Express. 2014;1:2. doi:10.1088/2053-1591/1/2/025007
77. Esser AK, Ross MH, Fontana F, et al. Nanotherapy delivery of c-myc inhibitor targets protumor macrophages and preserves antitumor macrophages in breast cancer. Theranostics. 2020;10(17):7510–7526. doi:10.7150/thno.44523
78. Ou W, Thapa RK, Jiang L, et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J Control Release. 2018;281:84–96. doi:10.1016/j.jconrel.2018.05.018
79. Sacchetti C, Rapini N, Magrini A, et al. In vivo targeting of intratumor regulatory T cells using PEG-modified single-walled carbon nanotubes. Bioconjug Chem. 2013;24(6):852–858. doi:10.1021/bc400070q
80. Bashyam H. Ralph Steinman: dendritic cells bring home the Lasker. J Exp Med. 2007;204(10):2245–2248. doi:10.1084/jem.20071995
81. Wu J, Zhang H, Zheng Y, et al. The Long Noncoding RNA MALAT1 Induces Tolerogenic Dendritic Cells and Regulatory T Cells via miR155/Dendritic Cell-Specific Intercellular Adhesion Molecule-3 Grabbing Nonintegrin/IL10 Axis. Front Immunol. 2018;9:1847. doi:10.3389/fimmu.2018.01847
82. Kochman S, Bernard J. Antitumour immune response and cancer vaccination: the critical role of dendritic cells. Curr Med Res Opin. 1999;15(4):321–326. doi:10.1185/03007999909116504
83. Bonifaz LC, Bonnyay DP, Charalambous A, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–824. doi:10.1084/jem.20032220
84. Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180(1):347–352. doi:10.1084/jem.180.1.347
85. van Stipdonk MJB, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2(5):423–429. doi:10.1038/87730
86. Yu F, Zhu C, Xie Q, Wang Y. Adenosine A2A Receptor Antagonists for Cancer Immunotherapy. J Med Chem. 2020;63(21):12196–12212. doi:10.1021/acs.jmedchem.0c00237
87. Xu X, Wang R, Su Q, et al. Expression of Th1- Th2- and Th17-associated cytokines in laryngeal carcinoma. Oncol Lett. 2016;12(3):1941–1948. doi:10.3892/ol.2016.4854
88. Asadzadeh Z, Mohammadi H, Safarzadeh E, et al. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. 2017;322:15–25. doi:10.1016/j.cellimm.2017.10.015
89. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):671–688. doi:10.1038/s41577-018-0061-z
90. Shi L, Wang Y, Wang Q, et al. Transforming a toxic drug into an efficacious nanomedicine using a lipoprodrug strategy for the treatment of patient-derived melanoma xenografts. J Control Release. 2020;324:289–302. doi:10.1016/j.jconrel.2020.05.025
91. Moradi-Chaleshtori M, Bandehpour M, Heidari N, Mohammadi-Yeganeh S, Hashemi SM. Exosome-mediated miR-33 transfer induces M1 polarization in mouse macrophages and exerts antitumor effect in 4T1 breast cancer cell line. Int Immunopharmacol. 2021;90:107198. doi:10.1016/j.intimp.2020.107198
92. Huang H, Zhang HJ, Onuma AE, Tsung A. Neutrophil elastase and neutrophil extracellular traps in the tumor microenvironment. Adv Exp Med Biol. 2020;1263:13–23.
93. Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol. 2019;9:16. doi:10.3389/fonc.2019.01146
94. Chretien AS, Le Roy A, Vey N, et al. Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front Immunol. 2014;5:122. doi:10.3389/fimmu.2014.00122
95. Bi J, Zeng X, Zhao L, et al. miR-181a induces macrophage polarized to M2 henotype and promotes M2 macrophage-mediated tumor cell metastasis by targeting KLF6 and C/EBP alpha. Mol Ther Nucleic Acids. 2016;5(9):e368. doi:10.1038/mtna.2016.71
96. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77. doi:10.1158/0008-5472.CAN-09-2587
97. Groth C, Hu X, Weber R, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16–25. doi:10.1038/s41416-018-0333-1
98. Cucak H, Grunnet LG, Rosendahl A. Accumulation of M1-like macrophages in type 2 diabetic islets is followed by a systemic shift in macrophage polarization. J Leukoc Biol. 2014;95(1):149–160. doi:10.1189/jlb.0213075
99. Choi JW, Kwon MJ, Kim IH, Kim YM, Lee MK, Nam TJ. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int J Mol Med. 2016;38(2):666–674. doi:10.3892/ijmm.2016.2656
100. Kim KJ, Wen XY, Yang HK, Kim WH, Kang GH. Prognostic implication of M2 macrophages are determined by the proportional balance of tumor associated macrophages and tumor infiltrating lymphocytes in microsatellite-unstable gastric carcinoma. PLoS One. 2015;10(12):e0144192. doi:10.1371/journal.pone.0144192
101. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi:10.1016/j.cell.2006.01.007
102. Shen Y, Wei Y, Wang Z, et al. TGF-beta regulates hepatocellular carcinoma progression by inducing Treg cell polarization. Cell Physiol Biochem. 2015;35(4):1623–1632. doi:10.1159/000373976
103. Vander Lugt B, Riddell J, Khan AA, et al. Transcriptional determinants of tolerogenic and immunogenic states during dendritic cell maturation. J Cell Biol. 2017;216(3):779–792. doi:10.1083/jcb.201512012
104. Roth L, Agemy L, Kotamraju VR, et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31(33):3754–3763. doi:10.1038/onc.2011.537
105. Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol. 2013;43(9):2421–2429. doi:10.1002/eji.201343451
106. Mizoguchi A, Bhan AK. A case for regulatory B cells. J Immunol. 2006;176(2):705–710. doi:10.4049/jimmunol.176.2.705
107. Ohms M, Moeller S, Laskay T. An attempt to polarize human neutrophils toward N1 and N2 phenotypes in vitro. Front Immunol. 2020;2:11.
108. Zou JM, Qin J, Li YC, et al. IL-35 induces N2 phenotype of neutrophils to promote tumor growth. Oncotarget. 2017;8(20):33501–33514. doi:10.18632/oncotarget.16819
109. Ohms M, Moller S, Laskay T. An attempt to polarize human neutrophils toward N1 and N2 phenotypes in vitro. Front Immunol. 2020;11:12. doi:10.3389/fimmu.2020.00532
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.