")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Nanoparticle delivery of an AKT/PDK1 inhibitor improves the therapeutic effect in pancreatic cancer
Authors Lucero-Acuña A, Jeffery J, Abril E, Nagle R, Guzman R, Pagel M, Meuillet E
Received 28 May 2014
Accepted for publication 24 July 2014
Published 3 December 2014 Volume 2014:9(1) Pages 5653—5665
DOI https://doi.org/10.2147/IJN.S68511
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Armando Lucero-Acuña,1 Justin J Jeffery,2 Edward R Abril,3,4 Raymond B Nagle,3,4 Roberto Guzman,1 Mark D Pagel,2,3,5,6 Emmanuelle J Meuillet3,7,8
1Department of Chemical and Environmental Engineering, University of Arizona, 2Department of Biomedical Engineering, University of Arizona, 3University of Arizona Cancer Center, 4Department of Pathology, University of Arizona, 5Department of Chemistry and Biochemistry, University of Arizona, 6Department of Medical Imaging, University of Arizona, 7Department of Molecular and Cell Biology, University of Arizona, 8Department of Nutritional Sciences, University of Arizona, Tucson, AZ, USA
Abstract: The K-ras mutation in pancreatic cancer can inhibit drug delivery and increase drug resistance. This is exemplified by the therapeutic effect of PH-427, a small molecule inhibitor of AKT/PDK1, which has shown a good therapeutic effect against a BxPC3 pancreatic cancer model that has K-ras, but has a poor therapeutic effect against a MiaPaCa-2 pancreatic cancer model with mutant K-ras. To increase the therapeutic effect of PH-427 against the MiaPaCa-2 pancreatic cancer model with mutant K-ras, we encapsulated PH-427 into poly(lactic-co-glycolic acid) nanoparticles (PNP) to form drug-loaded PH-427-PNP. PH-427 showed a biphasic release from PH-427-PNP over 30 days during studies in sodium phosphate buffer, and in vitro studies revealed that the PNP was rapidly internalized into MiaPaCa-2 tumor cells, suggesting that PNP can improve PH-427 delivery into cells harboring mutant K-ras. In vivo studies of an orthotopic MiaPaCa-2 pancreatic cancer model showed reduced tumor load with PH-427-PNP as compared with treatment using PH-427 alone or with no treatment. Ex vivo studies confirmed the in vivo results, suggesting that PNP can improve drug delivery to pancreatic cancer harboring mutant K-ras.
Keywords: nanoparticles, pancreatic cancer, AKT, bioluminescence imaging, drug delivery
Introduction
Drug delivery is a particularly confounding problem in the treatment of pancreatic cancer (PCA).1–3 This type of cancer can form an extensive desmoplasia caused by tumor-stroma interactions, resulting in a dense extracellular matrix surrounding the tumor that contributes to inefficient drug delivery. The K-ras gene mutation is a common molecular biomarker of PCA that promotes tumor-stroma interactions and desmoplasia.4 Mutant K-ras upregulates Hedgehog signaling, RAC1, and STAT3, that can each stimulate the formation of fibroinflammatory stroma.5–7 Mutant K-ras potentiates the effects of inhibition of transforming growth factor-beta (TGF-β) or INK4 m/ARF deficiency, that each lead to formation of an extensive extracellular matrix.8,9 Mutant K-ras is correlated with the recruitment of myeloid cells to the stroma, and the appearance of lipidic deposits at the tumor-stroma interface.10,11 Therefore, drug delivery to pancreatic tumors harboring the K-ras mutation may be particularly challenging.
Our previous research exemplifies the difficulty in treating PCA that has a K-ras mutation. We have developed PH-427 as a novel inhibitor of AKT/PDK112,13 that is activated in PCA.14,15 When PH-427 prevents activation of AKT at the plasma membrane, AKT cannot initiate an important cell survival signaling pathway, leading to death of pancreatic tumor cells. We have previously shown that PH-427 is highly efficient in treating a BxPC3 xenograft model that has wild-type K-ras, but is poorly efficient in a MiaPaCa-2 subcutaneous xenograft model with mutant K-ras. 12,13 These previous results suggest that PCAs with mutant K-ras require a higher dose or longer drug exposure to PH-427 to overcome the protective stromal layer surrounding the pancreatic tumor. Therefore, methods that improve drug delivery or retention may potentially improve treatment of PCA with mutant K-ras.
Greater drug delivery may be especially required to treat PCA harboring the K-ras mutation, because a hallmark of the K-ras mutation in PCA is enhanced drug resistance.16–19 For example, our in vitro studies have shown that PH-427 inhibits AKT activity at low μM concentrations in BxPC3 PCA cell lines, whereas MiaPaCa-2 PCA cell lines were more resistant to PH-427 with half maximal inhibitory concentrations (IC50 values) above 100 μM.12,13 In addition, PH-427 is a hydrophobic drug that is insoluble in aqueous medium. This property obviates intravenous injection of PH-427, and therefore the drug can only be delivered via intraperitoneal injection. However, intravenous injection can often provide faster drug delivery to a tumor, and can also result in a greater amount of drug delivered to the tumor. Therefore, methods to improve delivery of PH-427 to PCA harboring the K-ras mutation would seem to be required for effective therapy.
Polymeric nanoparticles have the potential to successfully address problems related to drug delivery and retention. Approved by the US Food and Drug Administration, poly(lactic-co-glycolic acid) (PLGA) is a polymer used in a host of therapeutic applications, and is arguably one of the most successfully used biodegradable polymers in nanomedicine.20 PLGA undergoes hydrolysis in the body to produce monomeric lactic acid and glycolic acid, which are further biodegraded to carbon dioxide and water.21,22 PLGA nanoparticles have been prepared by several methods, including solvent emulsion-evaporation,21,23 solvent emulsification-diffusion,24,25 and nanoprecipitation,26,27 which provides several routes for loading drugs based on the drug’s physicochemical properties. These properties may be tuned to improve the average nanoparticle size, size distribution, drug loading capacity, and drug release rate for specific drug delivery applications. Furthermore, the hydrophilicity of PLGA can be used to mask the hydrophobicity of PH-427, thereby allowing drug delivery via intravenous injection.
We hypothesized that encapsulating PH-427 into PLGA nanoparticles (PNP) to form PH-427-PNP would improve the delivery and therapeutic effect of this treatment in a PCA tumor model of MiaPaCa-2 harboring mutant K-ras. We performed a drug release study over a period of 50 days to evaluate the stability of PH-427-PNP. We also investigated the cytotoxic effects of PH-427-PNP compared with the drug alone in in vitro studies with BxPC3 and MiaPaCa-2 PCA cell lines. Finally, we conducted in vivo imaging studies with an orthotopic MiaPaCa-2 tumor model, followed by ex vivo studies to complement the imaging results, to evaluate the potential improvement offered by PH-427-PNP relative to PH-427 alone. Together, these studies represent a useful multidisciplinary approach for investigating improvements in the treatment of PCA with a PNP-encapsulated chemotherapy.
Materials and methods
Materials
Rhodamine-6G, poly(vinyl alcohol) with an average molecular weight of about 31,000 amu and 86.7%–88.7 mol% hydrolysis, and PLGA with 50:50 carboxyl-terminated end groups, a molecular weight of 7,000–17,000 amu, and an inherent viscosity of 0.16–0.24 dL/g were obtained from a commercial vendor (Sigma-Aldrich Inc., Milwaukee, WI, USA). Dichloromethane was purchased from another commercial vendor (Fisher Scientific Inc., Fair Lawn, NJ, USA). PH-427 was synthesized as previously described.12
Preparation of PLGA nanoparticles
PH-427-PNP and Rhodamine-6G in PNP (Rhodamine-PNP) were prepared with PLGA using a single emulsification method followed by solvent evaporation (Figure 1).21,23 Briefly, 50 mg of PLGA and 2.5 mg of PH-427 or 2.5 mg of Rhodamine-6G were dissolved in 5 mL of dichloromethane. Next, 10 mL of aqueous 5% poly(vinyl alcohol) was added to the organic phase. The mixture was emulsified in an ice bath with an XL2020 ultrasonicator (Misonix Inc., Farmingdale, NY, USA) operating at 55 W power output for 1 minute. The organic solvent was removed with a rotary evaporator operated at 38°C and low agitation velocity for 1 hour. Next, a 10-minute centrifugation cycle at 850 rcf was employed to remove agglomerations, using an IEC Centra-4B centrifuge (International Equipment Inc., Nashville, TN, USA). The solution was washed by three centrifugation cycles at 4,900 rcf for 50 minutes. During the first two centrifugation cycles, the supernatant was removed and PH-427-PNP or Rhodamine-PNP was resuspended in 10 mL of distilled water. On the final centrifugation cycle, the nanoparticles were resuspended in 0.3 mL of 10 mM sodium phosphate buffer with a pH of 7.4.
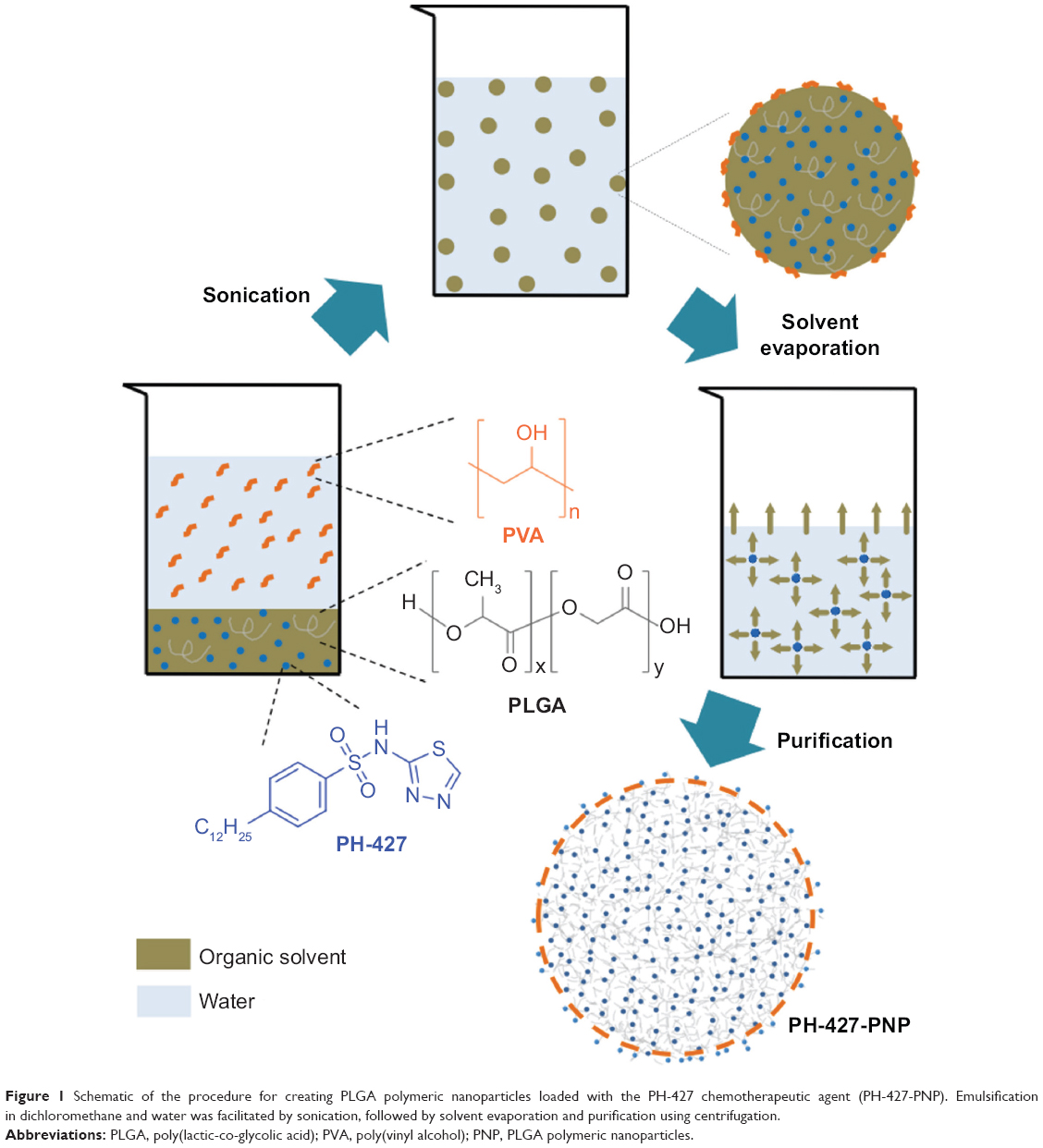 | Figure 1 Schematic of the procedure for creating PLGA polymeric nanoparticles loaded with the PH-427 chemotherapeutic agent (PH-427-PNP). Emulsification in dichloromethane and water was facilitated by sonication, followed by solvent evaporation and purification using centrifugation. |
Characterization of nanoparticles
Nanoparticle sizes were measured with a Zetasizer Nano ZEN3600 particle size analyzer (Malvern Instruments, Westborough, MA, USA). The refraction index used in the experiments was 1.33 and water was used as the dispersant. The average of ten measurements was used for each size analysis. The morphological characteristics of PH-427-PNP were observed using an S-4800 field emission scanning electron microscope (Hitachi Corporation, Tokyo, Japan). The samples were prepared by immobilization onto carbon-coated 400-mesh copper grids (Ted Pella Inc., Redding, CA, USA).
The encapsulation efficiency was determined as the mass ratio of entrapped PH-427 in PNP to the theoretical maximum loading, which was taken to be the point where all the supplied PH-427 was encapsulated in the PNP.28 The solution containing the PH-427-PNP was centrifuged and the supernatant removed. Next, the PH-427-PNP in the solid residue after centrifugation was dissolved in 1 M sodium hydroxide, and hydrochloric acid was used to equilibrate the pH of the solution at 7.4. The drug content was analyzed by spectrophotometry with absorbance at 270 nm using a UV-1800 spectrophotometer (Shimadzu Corporation Ltd., Kyoto, Japan). The drug loading was determined as the mass ratio of drug entrapped in the PNP to the mass of PH-427-PNP recovered.29
Drug release studies
The release rate of drug from PH-427-PNP was determined using dialysis under sink conditions.25,30,31 PH-427-PNP dispersed in 2.5 mL of 10 mM sodium phosphate buffer at pH 7.4 was placed into a dialysis membrane with a 12,000–14,000 molecular weight cut-off (Spectrum Laboratories Inc., Rancho Dominguez, CA, USA), and incubated in 30 mL of 10 mM sodium phosphate buffer at pH 7.4 and 37°C. At time intervals ranging from 0 to 50 days, 1 mL samples was withdrawn from the incubation medium and analyzed for content of PH-427 by spectrophotometry with absorbance at 270 nm using a UV-1800 spectrophotometer. Each volume withdrawn was replenished with 1 mL of 10 mM sodium phosphate buffer at pH 7.4.
Cell culture
Human BxPC3 and MiaPaCa-2 PCA cells were obtained from a qualified source (American Type Culture Collection, Rockville, MD, USA). Cells were maintained in bulk culture in Roswell Park Memorial Institute 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 4.5 g/L glucose, 100 U/mL penicillin, and 100 μg/mL streptomycin in a 5% carbon dioxide atmosphere. Cells were passaged using 0.25% trypsin and 0.02% ethylenediamine tetraacetic acid. Cells were confirmed to be mycoplasma-free with an enzyme-linked immunosorbent assay kit (Roche-Boehringer Mannheim, Indianapolis, IN, USA). MiaPaCa-2 PCA cells were transgenetically modified to carry the luciferase gene (MiaPaCa-2-LucE cells) by the Experimental Mouse Shared Service of the University of Arizona Cancer Center, and were maintained as described above. To develop this cell line, the MiaPaCa-2 cells were transfected with a luciferase plasmid and a stable clone was selected.32 This cell line was grown for 12 weeks in culture and luminescence was validated every 2 weeks to ensure that the cell line was stably transfected.
Fluorescence microscopy
Sterile coverslips were placed into 6-well plates and seeded with MiaPaCa-2 cells at 1,000 cells/well in the presence of complete medium. Cells were stained by adding Rhodamine-6G-loaded PNP to the wells for 1 hour and 24 hours. Rhodamine-6G was selected for this study because previous reports demonstrated that PNP labeled with this fluorophore cause cell staining throughout the cytosol that facilitates interpretation, and the staining can be attributed to Rhodamine-6G within the PNP and not due to free fluorophore.33,34 Coverslips were then washed in sodium phosphate buffer, air-dried, mounted using ProLong® Gold Antifade Reagent with diamidino-2-phenylindole (DAPI; Invitrogen, Grand Island, NY, USA), and stored at 4°C. Cells were visualized using excitation and emission wavelengths of 460 nm and 540 nm, respectively, at 40× using an Olympus Bx-50 microscope equipped with an LCD camera (Olympus Corporation, Central Valley, PA, USA).
Cell viability assays
A standard 96-well microcytoxicity assay was performed as previously described.12,13 Wells were seeded with 4,000 BxPC3 or MiaPaCa-2 cells for drug studies lasting 1 or 5 days, and with 2,000 cells for the drug study lasting 7 days. PH-427 or PH-427-PNP were added directly to the medium at various concentrations ranging from 1 to 150 μM. After the single treatment, the percentages of cell survival were determined by spectrophotometric determination of the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) each day for 7 consecutive days. Dimethylsulfoxide was used as the solvent for dissolution of the PH-427 drug, because this drug is too hydrophobic for dissolution in aqueous medium. An equivalent amount of dimethylsulfoxide was tested with a separate set of cells to ensure that cell viability was not affected by this solvent.
In vivo animal studies and bioluminescence imaging
All animal procedures took place at the Experimental Mouse Shared Service and the Cancer Imaging Shared Service of the University of Arizona Cancer Center. These procedures were in accordance with institutional guidelines and with approval from the Institutional Animal Care and Use Committee of the University of Arizona. Housing and care of the animals was provided by the Animal Resource Center of the Arizona Health Sciences Center. Nine female severe combined immunodeficient mice aged 4–5 weeks were utilized to create an orthotopic xenograft model of PCA. This model was developed by exposing the pancreas via a single abdominal incision using sterile surgical technique, directly injecting approximately one million cells into the exposed pancreas, and suturing the incision. The nine mice were injected with MiaPaCa-2-LucE cells for bioluminescence imaging (BLI) studies. The average weight of each mouse at the beginning of the experiments was 16 g. Mice were scanned with BLI prior to treatment to confirm the presence of viable pancreatic tumors. PH-427-PNP treatments at 10 mg/kg of PH-427 were injected intravenously into four mice, and PH-427 at 10 mg/kg was injected intraperitoneally into two mice, once per week for 4 weeks. The remaining three mice were not treated. PH-427 was injected intraperitoneally because this drug is too hydrophobic to be administered intravenously. Mice were then monitored with BLI at the end of treatment. At the end of the study, the mice were sacrificed, sections of the pancreatic tumor(s) were paraffin-embedded, and the number, size, and location of metastases were recorded.
Bioluminescence images were acquired using an AMI1000 scanner (Spectral Instruments Imaging Inc., Tucson, AZ, USA) and AMIView software for acquisition and analysis.35 All scans were acquired with a 30-second exposure time, 8-fold binning, a 1.2 f/stop, and a 25 cm field of view. All mice were injected intraperitoneally with 100 mg/kg firefly luciferin 10 minutes before image acquisition. Four minutes after injection, the mice were anesthetized with 4% isoflurane gas at 1 L/minute oxygen flow and sedation was maintained during imaging with 1.5% isoflurane gas at 0.5 L/min oxygen flow. Mice were oriented in a supine position in the scanner. Regions of interest were carefully placed around all bioluminescent signals while minimizing the inclusion of scatter signal. Radiance units were used for relative comparison of the sum of photons between scans.
Immunohistochemistry
Tissues were harvested, fixed in 10% neutral buffered formalin for 24 hours, processed, and embedded in paraffin. Routine hematoxylin and eosin stains were performed on sections of tissue cut with a width of 3 microns from the formalin-fixed, paraffin-embedded blocks. Immunohistochemistry of total AKT was performed using rabbit monoclonal antibodies (#4685S; Cell Signaling Technology, Danvers, MA, USA), with human prostate cancer tissues used as the positive tissue control. Immunochemistry of Ki67 was performed with a rabbit polyclonal antibody (#NCL-Ki67p; Leica Biosystems GmbH, Nussloch, Germany), using human tonsil tissue as a positive control. Immunohistochemistry of HMGA2 was performed using a rabbit polyclonal antibody (#5269S; Cell Signaling Technology), using human colon tissue as a positive control. Tissue sections were stained with a Discovery XT Automated Immunostainer (Ventana Medical Systems Inc., Tucson, AZ, USA; VMSI) using Ventana Medical Systems Inc.-validated reagents for deparaffinization, cell conditioningvia antigen retrieval with a borate-ethylenediamine tetraacetic acid buffer, primary antibody staining, detection and amplification using a biotinylated-streptavidin-horseradish peroxidase and diaminobenzidine system, and hematoxylin counterstaining. Following staining with the instrument, the slides were dehydrated through graded alcohols to xylene and coverslipped with mounting medium.
Images were captured using a BX50 microscope equipped with a DP72 camera and CellSens® imaging software (Olympus Corporation). All images were standardized for light intensity and white balance. Microphotographs were taken with a 40× objective for all treatment groups stained with the following stained with antibodies directed against total AKT, HMGA2, phosphor-S6, and Ki-67. An experienced clinical pathologist performed the immunohistochemistry analyses. Potentially inflammatory cells and other reactive background cells were excluded from the analysis. Analysis of Ki-67 was expressed as percent proliferation, while total AKT and HMGA2 were scored using pathology long scores. A long score (LS) is calculated by multiplying the staining intensity ranging from 1+ to 4+ (I) by the percentage of positively stained cells ranging from 1% to 100% (P), with the formula: LS = P × I. The LS minimum and maximum were 1 and 300, respectively.
The mean and standard deviation of dynamic light scattering measurements was determined using the software for the Zetasizer Nano ZEN3600 particle size analyzer. The means and standard deviations of IC50 values, in vivo bioluminescence imaging results, and ex vivo immunohistochemistry results were determined with Excel version 14.4.1 (Microsoft Corporation, Redmond, WA, USA). Statistically significant differences between measurements were determined using the Student’s t-test with Excel version 14.4.1.
Results
Preparation and release studies of polymeric PLGA nanoparticles
The PNP was synthesized with 1% w/w PH-427 following a single nanoemulsion method to create PH-427-PNP, resulting in an 18.8% encapsulation efficiency (Figure 1). Both PNP had a smooth surface as depicted by scanning electron microscopy (Figure 2A). Dynamic light scattering spectra confirmed the average diameter of PNP to be 165±2.2 nm with a polydispersity index of 0.063±0.001, while the average diameter of PH-427-PNP was 274±0.9 nm with a polydispersity index of 0.39±0.027 (Figure 2B).
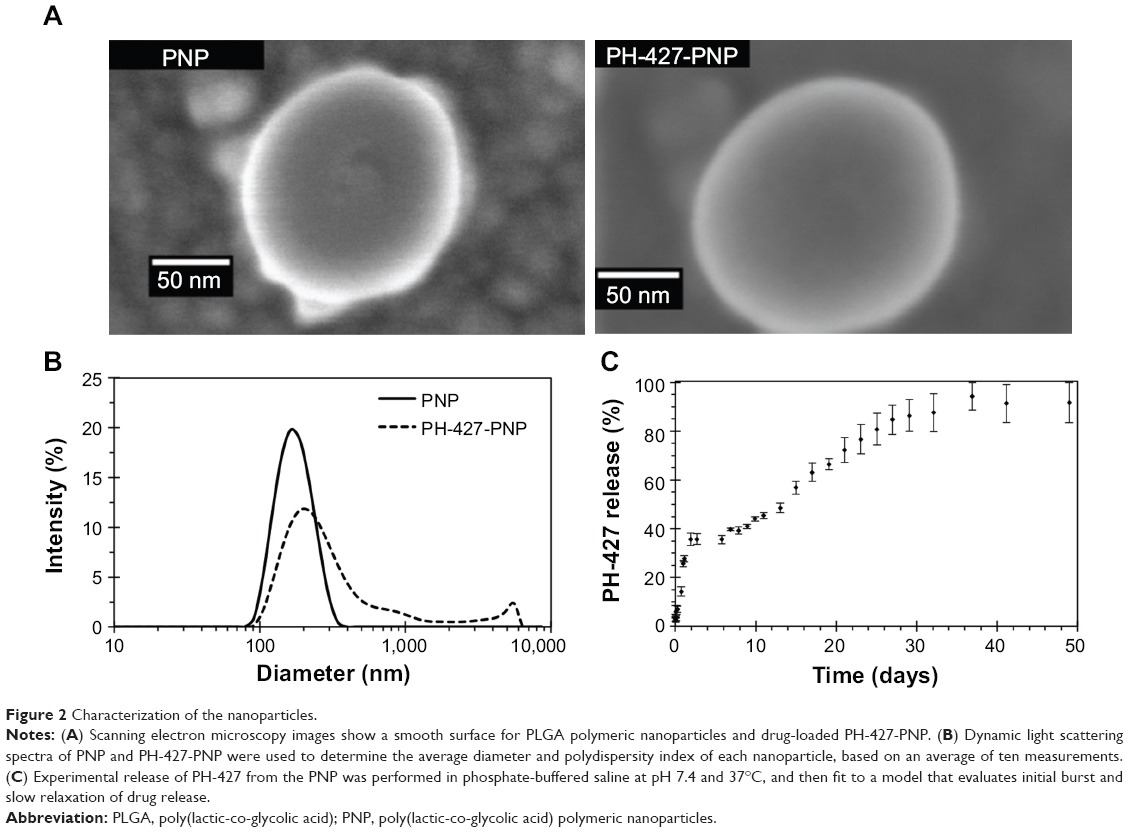 | Figure 2 Characterization of the nanoparticles. |
The release curve of PH-427 from the PH-427-PNP at 37°C showed that 26.2% of the drug was released within 1 day, followed by slow release of an additional 13.5% of drug by day 7, followed by a moderate drug release of an additional 47.1% by day 30; finally, the remaining 13.2% of drug was released by 50 days (Figure 2C). The release curve showed biphasic behavior relating to a relatively rapid initial release followed by a moderately slow second release stage due to particle relaxation or polymer degradation.
In vitro studies
Using fluorescence microscopy, MiaPaCa-2 cells treated with Rhodamine-PNP demonstrated cellular uptake of PNP within 1 hour, as demonstrated by Rhodamine fluorescence surrounding and within the cell nuclei that was localized with DAPI fluorescence (Figure 3). Cells retained fluorescence after 24 hours of incubation with Rhodamine-PNP. These results demonstrated the cellular uptake of the PNP, and suggested that an intracellular environment surrounded the PNP during the in vitro studies, which differed from the purely aqueous environment used to test the release of PH-427 during chemical solution studies.
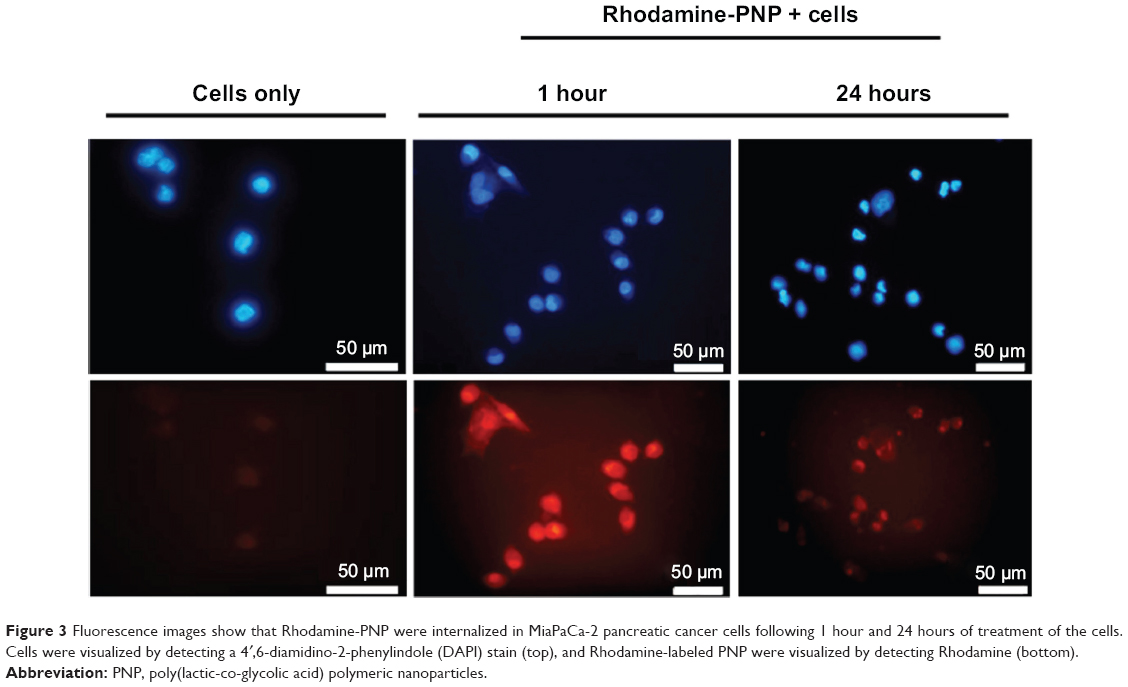 | Figure 3 Fluorescence images show that Rhodamine-PNP were internalized in MiaPaCa-2 pancreatic cancer cells following 1 hour and 24 hours of treatment of the cells. Cells were visualized by detecting a 4′,6-diamidino-2-phenylindole (DAPI) stain (top), and Rhodamine-labeled PNP were visualized by detecting Rhodamine (bottom). |
PCA cell viabilities following treatment with PH-427 or PH-427-PNP were evaluated with BxPC3 and MiaPaCa-2 PCA cell lines (Figure 4A and B). PH-427 caused greater cytotoxicity in BxPC3 PCA with wild-type K-ras relative to MiaPaCa-2 PCA with mutant K-ras (Figure 4A and B), which agreed with our previous results.12,13 The average IC50 value for PH-427 against BxPC3 and MiaPaCa-2 PCA was 46.5±2.5 μM and 93.8±2.7 μM, respectively, with a statistically significant difference (P<0.001).
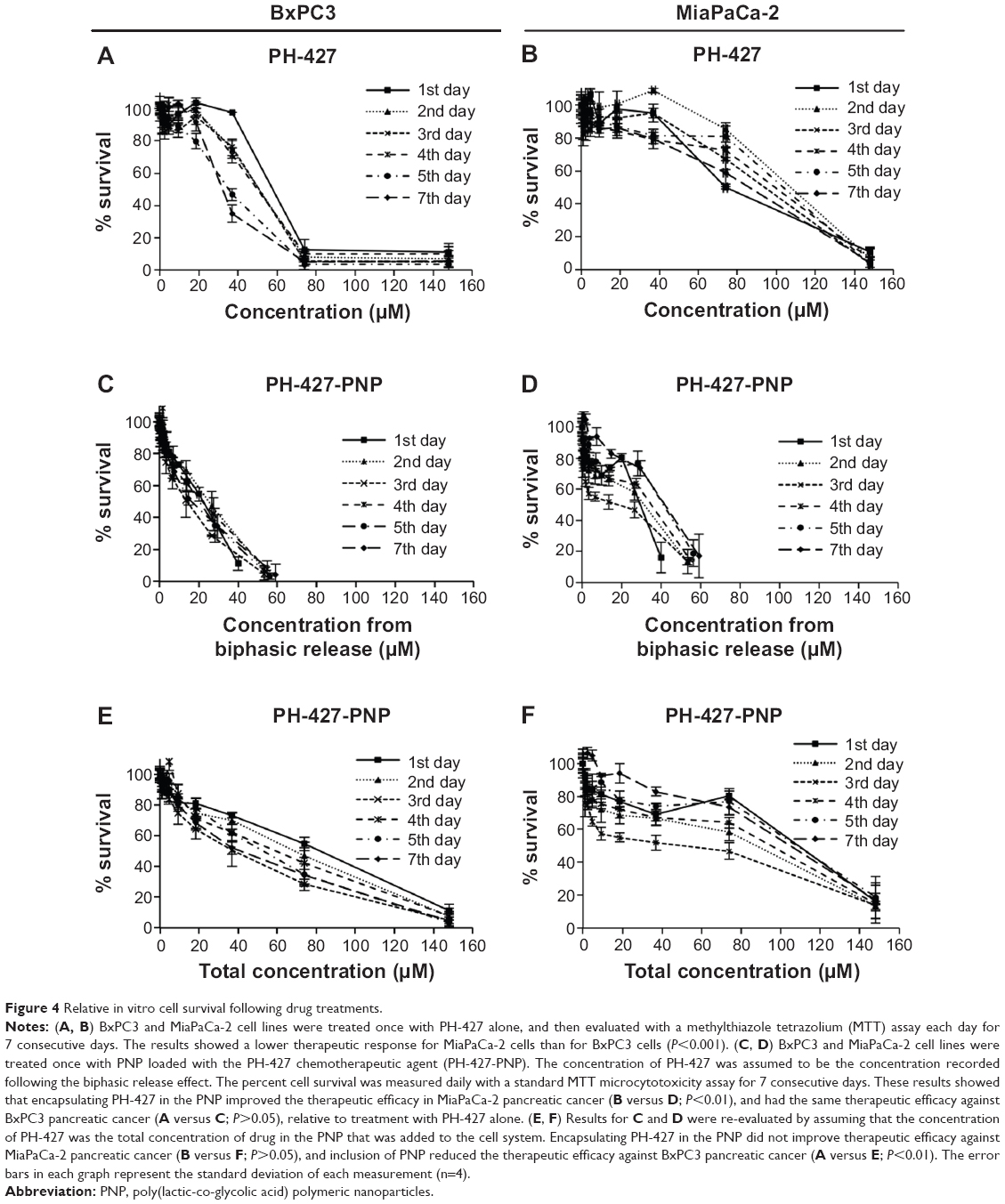 | Figure 4 Relative in vitro cell survival following drug treatments. |
To initially compare the therapeutic efficacy of PH-427-PNP relative to PH-427, we assumed that the biphasic release effect is identical in solution and in vitro. Our assumption was based on attributing the rapid initial “burst” of the biphasic release to drug adsorbed onto the surface of the nanoparticles, which may be similar in solution and in vitro.36 Therefore, we used the released concentration of PH-427 measured each day in solution to evaluate the therapeutic efficacy in vitro (Figure 4C and D). These results showed that encapsulating PH-427 in PNP improved the therapeutic efficacy acting against MiaPaCa-2 PCA (Figure 4B versus Figure 4D; P<0.01), and had the same therapeutic efficacy acting against BxPC3 PCA (Figure 4A versus Figure 4C; P>0.05), relative to treatment with PH-427 alone.
We then investigated an alternative assumption that the PH-427-PNP rapidly delivered its entire contents to the cytosol soon after cell internalization. We used the total concentration of PH-427 in the PNP that was added to the cell samples to evaluate the therapeutic efficacy in vitro (Figure 4E and F). These results showed that encapsulating PH-427 in the PNP did not improve the therapeutic efficacy against MiaPaCa-2 PCA (Figure 4B versus Figure 4F; P>0.05), and inclusion of PNP reduced the therapeutic efficacy acting against BxPC3 PCA (Figure 4A versus Figure 4E; P<0.01). Our assumption of rapid intracellular drug release was based on rapid degradation by enzymes and an acidic environment that PH-427-PNP may experience during endocytosis or pinocytosis, which may accelerate the release of all of the PH-427 from the PNP.37 In addition, the in vitro studies of MiaPaCa-2 PCA cells treated with PH-427-PNP did not show consistently greater cytotoxicity between days 1 and 7 (Figure 4D), which did not match the consistent release of an additional 13.5% of PH-427 from the PNP between days 1 and 7 (Figure 4C), suggesting that the biphasic release effect was not present during in vitro studies.
In vivo studies
We evaluated the antitumor effects of PH-427 and PH-427-PNP in a MiaPaCa-2-LucE orthotopic PCA model using BLI (Figures 5 and 6). The amplitude of the BLI signal is a semiquantitative indication of the pancreatic tumor load. These imaging results were confirmed by ex vivo necropsy analysis (Figure 6). Control mice with no treatment showed high average BLI signal amplitude from the pancreas, indicating a high pancreatic tumor load. Treatment with PH-427 also showed high average BLI signal amplitude, indicating that the drug had no effect on pancreatic tumor load. This result matched our previous studies with PH-427 in the treatment of the orthotopic tumor model of MiaPaCa-2 without the luciferase gene.13 Treatment with PH-427-PNP showed low average BLI signal amplitude, indicating low tumor load and a significant therapeutic effect by the drug-loaded nanoparticles in this tumor model (P<0.001). The standard deviations represented by error bars in Figure 5B were approximately 10% for each group. This indicated that the BLI study was performed with high fidelity, which is critical for interpreting results from this semiquantitative imaging technique. No change in body weight was observed after treatment with PH-427 alone or with PH-427-PNP, and there were no visual signs of toxicity upon gross examination after each treatment. This evidence indicates that PH-427 and PH-427-PNP were not toxic at the dose levels administered during these studies.
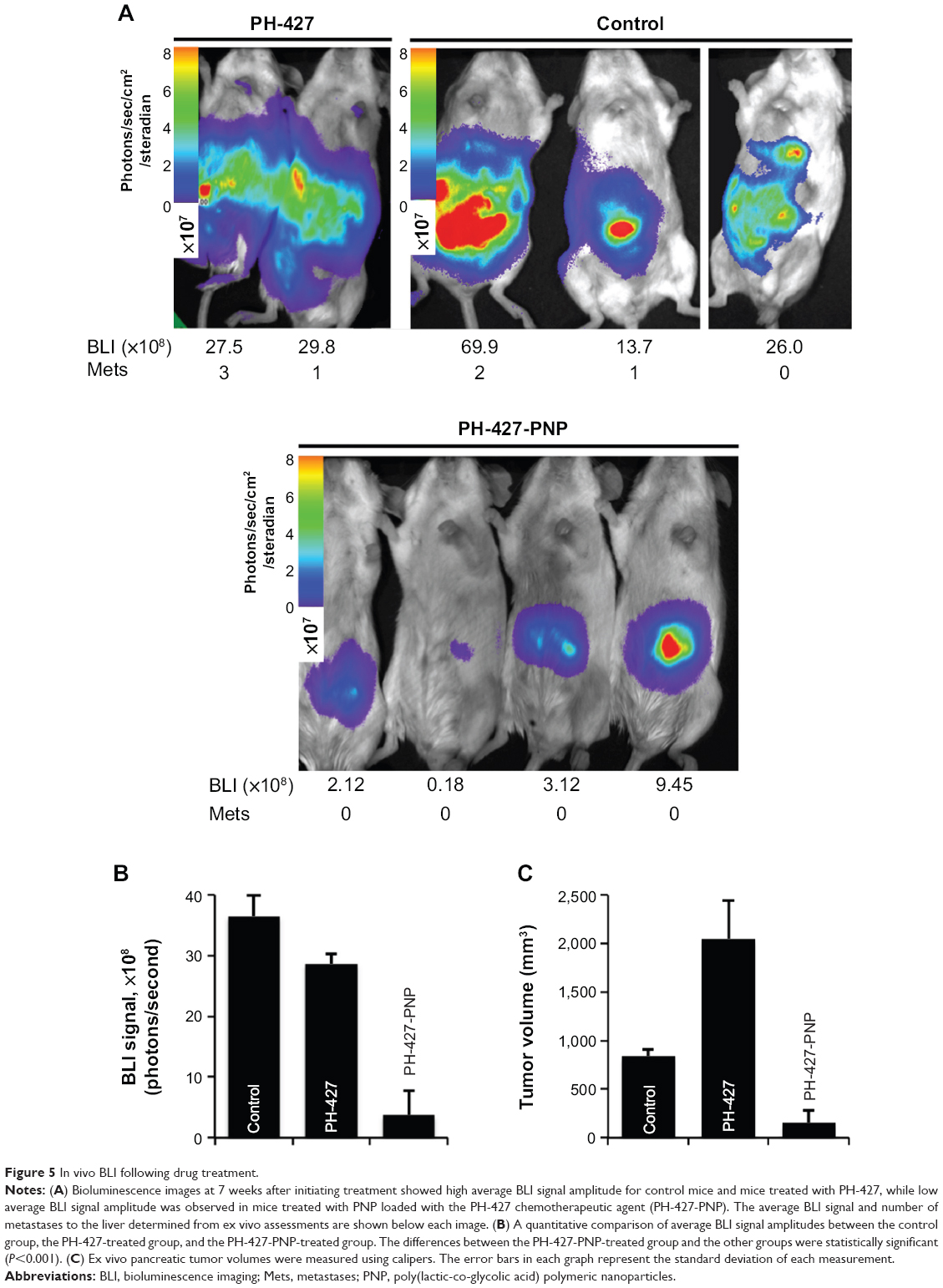 | Figure 5 In vivo BLI following drug treatment. |
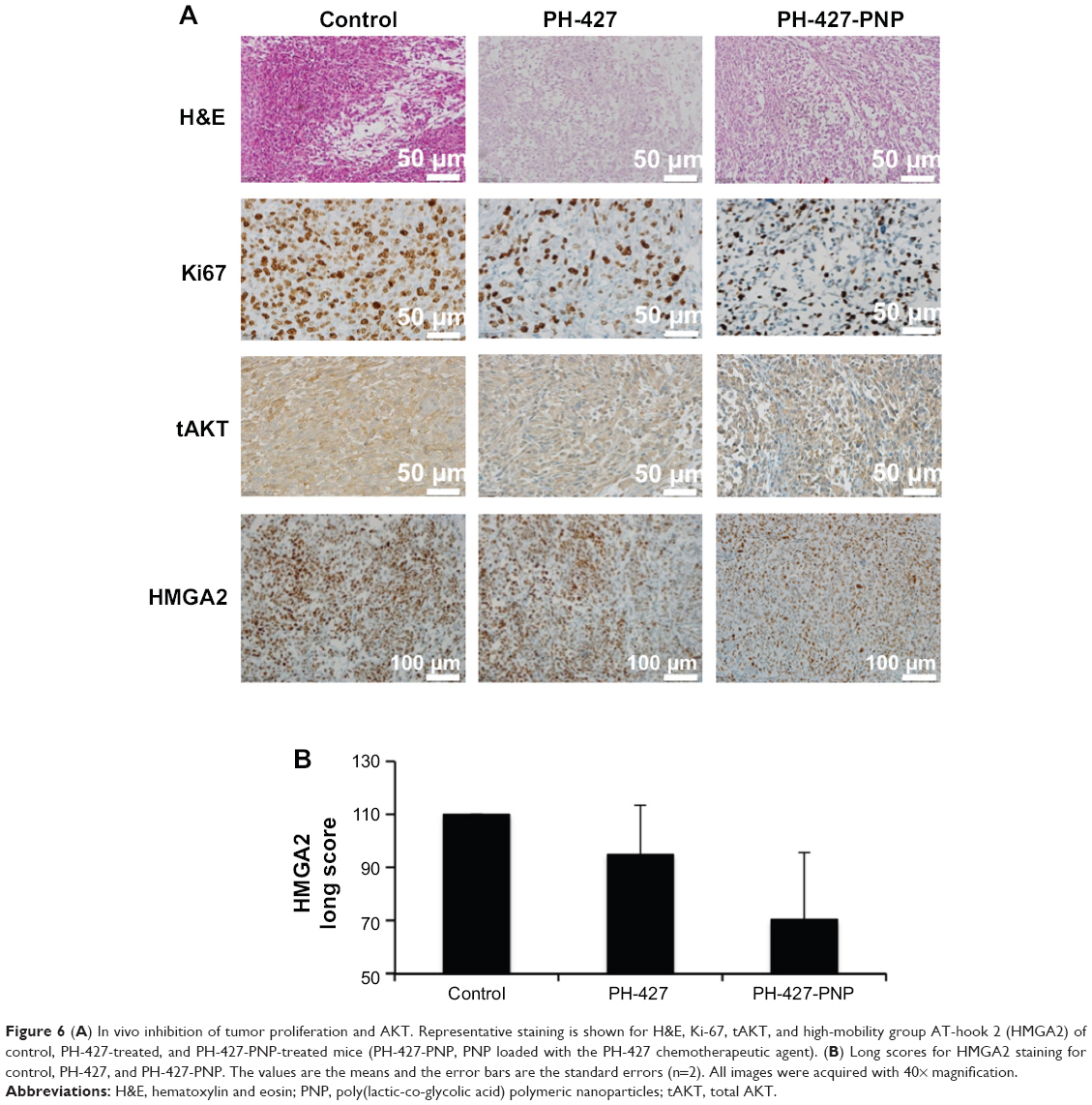 | Figure 6 (A) In vivo inhibition of tumor proliferation and AKT. Representative staining is shown for H&E, Ki-67, tAKT, and high-mobility group AT-hook 2 (HMGA2) of control, PH-427-treated, and PH-427-PNP-treated mice (PH-427-PNP, PNP loaded with the PH-427 chemotherapeutic agent). (B) Long scores for HMGA2 staining for control, PH-427, and PH-427-PNP. The values are the means and the error bars are the standard errors (n=2). All images were acquired with 40× magnification. |
Ex vivo studies
At the end of the study, the mice were sacrificed, their pancreatic tumors were excised, tumor volumes were measured, and metastases to the liver were counted. Following treatment with PH-427-PNP, pancreatic tumor volumes were significantly lower as compared with control or treatment with PH-427 alone (Figure 5C). These results agreed with the BLI study (Figure 5B).
Metastatic tumor lesions were identified in all PH-427-treated mice and in two of the three control mice that did not receive treatment, but no liver tumor metastases were identified in any mice treated with PH-427-PNP. These results were qualitatively correlated with the tumor loads assessed with BLI, providing more evidence that PH-427-PNP improves the therapeutic effect on pancreatic tumor load that indirectly results in reduced metastases. In addition, PNP are known to passively accumulate in liver tissues, so that PH-427-PNP may also directly affect the early-stage metastases in liver tissue.38 Therefore, the combination of BLI and assessments of metastatic liver lesions shows that the PNP drug nanocarrier provided multiple therapeutic benefits.
Immunohistochemistry was performed with these pancreatic tumors using the antibody biomarker Ki-67. Analysis of the treatment groups stained with the tissue proliferation marker Ki-67 demonstrated that the proliferative index was significantly lower for the PH-427-PNP treatment group than for the PH-427 treatment group, and both of these groups had a lower proliferative index than the control group. More specifically, the proliferative indices were 45%, 50%, and 95% for the PH-427-PNP, PH-427, and control groups, respectively. This decrease in tumor proliferation is strongly indicative of a reduction in tumor regeneration by these treatments, and validated an improved therapeutic effect with PH-427-PNP.
Treatment with PH-427-PNP was verified to cause direct target inhibition and downstream target inhibition as evidenced by immunohistochemistry. Staining for total AKT showed a decrease in both staining intensity and number of stained cells for the groups treated with PH-427 and PH-427-PNP compared with the control. LS for total AKT staining control, PH-427 treatment, and PH-427-PNP treatment groups were calculated at 102±34.2 (standard error [SE], n=4), 82.5±17.5 (SE, n=2), and 70±10 (SE, n=3), respectively. Of particular interest, we also stained for HMGA2, a driver of metastasis.39,40 Analyses of the stained tissues demonstrated a reduction in HMGA2 staining in the PH-427-PNP treatment group as compared with PH-427 alone or control (Figure 6B).
Discussion
Our current in vitro and in vivo studies with PH-427 alone confirmed our previous results for the therapeutic efficacy of this drug.13 The BxPC3 cell line and the tumor xenograft model are sensitive to PH-427 while the MiaPaCa-2 cell line and xenograft model are resistant to PH-427. We have attributed this difference between BxPC3 and MiaPaCa-2 PCAs to the difference in their K-ras status, based on extensive evidence for the role of mutant K-ras in PCA, PCA drug resistance, and our previous work regarding profiling of the tumor types responsive or resistant to PH-427.13 These results drove our interest in investigating PNP as a method for improving PH-427 efficacy against PCA with mutant K-ras.
Our in vitro studies with PH-427-PNP were interpreted with and without the biphasic release effect observed in solution. These two interpretations emphasize that the physicochemical properties of a drug-loaded PNP must be evaluated in solution to facilitate an understanding of drug release, and yet the cellular environment and cell uptake mechanisms must also be considered during in vitro studies. Most importantly, the combination of these interpretations showed that PH-427-PNP either improved the therapeutic effect or was not detrimental to the treatment of MiaPaCa-2 PCa, and either reduced the therapeutic effect or was not detrimental to the treatment of BxPC3 PCA. Therefore, these results demonstrate that drug delivery with PNP may be beneficial when treating drug-resistant PCA with mutant K-ras, but may not be beneficial for PCA with wild-type K-ras.
The in vivo and ex vivo results also demonstrated that PNP provides benefit when treating PCA harboring mutant K-ras that can inhibit drug delivery. These studies tested only two to four mice in each treatment group (Figure 5). Even with a limited number of mice, a statistically significant difference in tumor load was detected with bioluminescence between the group of mice treated with PH-427-PNP relative to the PH-427-treated and PH-427-nontreated groups of mice. Future studies should include PNP without drug as an additional control study to ensure that the therapeutic effect is attributed to the delivered drug.
The in vivo results demonstrated that PH-427-PNP could be delivered intravenously, while delivery of PH-427 was limited to intraperitoneal delivery. This additional benefit of masking the hydrophobicity of PH-427 with the hydrophilic PLGA polymer may contribute to the improved therapeutic effect of PH-427-PNP relative to the drug alone. Therefore, these results add to the evidence that intravenous delivery of drug-loaded nanoparticles has merits relative to intraperitoneal delivery.
PNP have been used in one other study of PCA. For this in vivo study, PNP were augmented with a poly(ethylene glycol) coating to prolong circulation.41 However, this study only tested the drug-loaded nanoparticle against a single model of PCA. Based on the promising results of our current study, a poly(ethylene glycol)-coated PNP should be tested against multiple PCA models to ensure that the coating is sensitive to the K-ras status of the PCA.
Other nanoparticles have been used to improve drug delivery and retention in PCA. Examples include liposomes,42 proteins,43 inorganic nanoparticles consisting of silica, gold, iron, or semiconductors,44 and nanoparticles with targeting moieties.45 These nanoparticles have been used to improve the therapeutic effects of cisplatin,42 paclitaxel,43 chemoradiation therapy,46 and gene therapies.47 Almost all of these previous studies have only tested the drug-loaded nanoparticle against one type of in vivo PCA model. Our results indicate that drug-loaded nanoparticles should be tested against multiple pancreatic tumor models, such as models that have wild-type K-ras and mutant K-ras genotypes, to investigate phenotypes that have different drug delivery and retention characteristics.
In conclusion, encapsulating PH-427 in PNP improved the therapeutic effect of PH-427 against an in vivo MiaPaCa-2 PCA model harboring the K-ras mutation. These results were confirmed by ex vivo histopathology studies. Furthermore, in vitro studies demonstrated that the PNP rapidly internalized into MiaPaCa-2 PCA cells. Our study also showed that care should be taken when interpreting results of drug release in solution and in vitro. The combination of these results indicate that investigations of drug-loaded nanoparticles for treating PCA should test multiple PCA cell types and tumor models to ensure that different phenotypes of drug delivery and retention are interrogated.
Acknowledgments
This work was supported by the National Cancer Institute through NIH (National Institutes of Health) award CA139503, and the Hirshberg Foundation for Pancreatic Cancer Research to EJM. The Tissue Acquisition and Cellular/Molecular Analysis Shared Service is supported by a University of Arizona Cancer Center Support Grant (NIH CA023074). AL-A was funded by a CONACYT fellowship. The authors thank Mr Joseph Kobes and Ms Dina Hingorani for helpful discussions and assistance during preparation of this manuscript.
Disclosures
EJM has financial interests in PHusis Therapeutics Inc., (Houston, TX, USA). All other authors report that they have no commercial associations, current and within the past 5 years, that might pose a potential, perceived, or real conflict of interest. These include grants, patent licensing arrangements, consultancies, stock or other equity ownership, advisory board memberships, or payments for conducting or publicizing the study.
References
Kollmannsberger C, Peters HD, Fink U. Chemotherapy in advanced pancreatic adenocarcinoma. Cancer Treat Rev. 1998;24:133–156. | ||
Dimou A, Syrigos KN, Saif MW. Overcoming the stromal barrier: technologies to optimize drug delivery in pancreatic cancer. Ther Adv Med Oncol. 2012;4:271–279. | ||
Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–1197. | ||
Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. | ||
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. | ||
Heid I, Lubeseder-Martellato C, Sipos B, et al. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology. 2011;141:719–730. | ||
Corcoran RB, Contino G, Deshpande V, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71: 5020–5029. | ||
Ijichi H. Genetically-engineered mouse models for pancreatic cancer. World J Clin Oncol. 2011;2:195–202. | ||
Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2002;17:3112–3126. | ||
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic cancer. Cancer Cell. 2012;21:836–847. | ||
Grippo PJ, Fitchev PS, Bentrem DJ, et al. Concurrent PEDF deficiency and Kras mutation induce invasive pancreatic cancer and adipose-rich stroma in mice. Gut. 2012;61:1454–1464. | ||
Moses SA, Ali MA, Zuohe S, et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res. 2009;69:5073–5081. | ||
Meuillet EJ, Zuohe S, Lemos R, et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol Cancer Ther. 2010;9:706–717. | ||
Fahy BN, Schlieman M, Virudachalam S, Bold RJ. AKT inhibition is associated with chemosensitization in the pancreatic cancer cell line MIA-PaCa-2. Br J Cancer. 2003;89:391–397. | ||
Mortenson MM, Galante JM, Schlieman MG, Bold RJ. AKT: a novel target in pancreatic cancer therapy. Cancer Ther. 2004;2:227–238. | ||
Wei F, Liu Y, Bellail AC, et al. K-ras mutation-mediated IGF-1-induced feedback ERK activation contributes to the rapalog resistance in pancreatic ductal adenocarcinomas. Cancer Lett. 2012;322: 58–69. | ||
Jing J, Greshock J, Holbrook JD, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11:720–729. | ||
Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17:2744–2756. | ||
Kelber JA, Reno T, Kaushal S, et al. Kras induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res. 2012;72:2554–2564. | ||
Parveen S, Sahoo SK. Polymeric nanoparticles for cancer therapy. J Drug Target. 2008;16:108–123. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010; 75:1–18. | ||
Cascone MG, Zhu Z, Borselli F, Lazzeri L. Poly(vinyl alcohol) hydrogels as hydrophilic matrices for the release of lipophilic drugs loaded in PLGA nanoparticles. J Mater Sci Mater Med. 2002;13:29–32. | ||
Murakami H, Kobayashi M, Takeuchi H, Kawashima Y. Further application of a modified spontaneous emulsification solvent diffusion method to various types of PLGA and PLA polymers for preparation of nanoparticles. Powder Technol. 2000;107:137–143. | ||
Sahana DK, Mittal G, Bhardwaj V, Kumar MN. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J Pharm Sci. 2008;97:1530–1542. | ||
Chorny M, Fishbein I, Danenberg HD, Golomb G. Lipophilic drug loaded nanospheres prepared by nanoprecipitation: effect of formulation variables on size, drug recovery and release kinetics. J Control Release. 2002;83:389–400. | ||
Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008; 105:17356–17361. | ||
Park J, Fong PM, Lu J, et al. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine. 2009;5:410–418. | ||
Lei T, Srinivasan S, Tang Y, et al. Comparing cellular uptake and cytotoxicity of targeted drug carriers in cancer cell lines with different drug resistance mechanisms. Nanomedicine. 2011;7:324–332. | ||
Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release. 2002;79:123–135. | ||
Liang C, Yang Y, Ling Y, Huang Y, Li T, Li X. Improved therapeutic effect of folate-decorated PLGA-PEG nanoparticles for endometrial carcinoma. Bioorgan Med Chem. 2011;19:4057–4066. | ||
Wendt MK, Molter J, Flask CA, Schiemann WP. In vivo dual substrate bioluminescent imaging. J Vis Exp. 2011;56:3245. | ||
Bentancourt T, Shah K, Brannon-Peppas L. Rhodamine-loaded poly(lactic-co-glycolic acid) nanoparticles for investigation of in vitro interactions with breast cancer cells. J Mater Sci Mater Med. 2009;20:387–395. | ||
Cartiera MS, Johnson KM, Rajendran V, Caplan MJ, Saltzman WM. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials. 2009;30:2790–2798. | ||
Tuli R, Surmak A, Reyes J, Hacker-Prietz A, Armour M, Leubner A. Development of a novel preclinical pancreatic cancer research model: bioluminescence image-guided focal irradiation and tumor monitoring of orthotopic xenografts. Transl Oncol. 2012;5:77–84. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010; 75:1–18. | ||
Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev. 2007;59:718–728. | ||
Watanabe S, Ueda Y, Akaboshi S, Hino Y, Sekita Y, Nakao M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am J Pathol. 2009;174:854–868. | ||
Morishita A, Zaidi MR, Mitoro A, et al. HMGA2 is a driver of tumor metastasis. Mol Cell Pathol. 2013;73:4289–4299. | ||
Pamujula S, Hazari S, Bolden G, et al. Preparation and in-vitro/in-vivo evaluation of surface-modified poly(lactide-co-glycolide) fluorescent nanoparticles. J Pharm Pharmacol. 2010;62:422–429. | ||
Ding H, Yong KT, Roy I, et al. Bioconjugated PLGA-4-arm-PEG branched polymeric nanoparticles as novel tumor targeting carriers. Nanotechnology. 2011;22:165101. | ||
Teni B. Clinical overview on lipoplatin: a successful liposomal formulation of cisplatin. Expert Opin Investig Drugs. 2009;18:1197–1218. | ||
Casaluce F, Sgambato A, Maione P, Ciardiello F, Gridelli C. Emerging mitotic inhibitors for non-small cell carcinoma. Expert Opin Emerg Drugs. 2013;18:97–107. | ||
Yang F, Jin C, Subedi SL, et al. Emerging inorganic nanomaterials for pancreatic cancer diagnosis and treatment. Cancer Treat Rev. 2012;38: 566–579. | ||
Yu X, Zhang Y, Chen C, Yao Q, Li M. Targeted drug delivery in pancreatic cancer. Biochim Biophys Acta. 2010;1805:97–104. | ||
Werner ME, Foote MB, Wang AZ. Chemoradiotherapy of human tumors: novel approaches from nanomedicine. Curr Pharm Des. 2012; 18:2830–2837. | ||
Xu J, Jin C, Hao S-J, Luo G-P, Fu D-L. Pancreatic cancer: gene therapy approaches and gene delivery systems. Expert Opin Biol Ther. 2010; 10:73–88. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.