")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect
Authors Nehoff H, Parayath NN , Domanovitch L, Taurin S, Greish K
Received 31 January 2014
Accepted for publication 3 April 2014
Published 22 May 2014 Volume 2014:9(1) Pages 2539—2555
DOI https://doi.org/10.2147/IJN.S47129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Hayley Nehoff,1 Neha N Parayath,1 Laura Domanovitch,1 Sebastien Taurin,1 Khaled Greish1,2
1Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand; 2Department of Oncology, Faculty of Medicine, Suez Canal University, Egypt
Abstract: The growing research interest in nanomedicine for the treatment of cancer and inflammatory-related pathologies is yielding encouraging results. Unfortunately, enthusiasm is tempered by the limited specificity of the enhanced permeability and retention effect. Factors such as lack of cellular specificity, low vascular density, and early release of active agents prior to reaching their target contribute to the limitations of the enhanced permeability and retention effect. However, improved nanomedicine designs are creating opportunities to overcome these problems. In this review, we present examples of the advances made in this field and endeavor to highlight the potential of these emerging technologies to improve targeting of nanomedicine to specific pathological cells and tissues.
Keywords: nanomedicine, permeability and retention effect, tissue targeting, cancer treatment, inflammation
Introduction
The use of nanotechnology for medical applications encompassing drug delivery and diagnostics has attracted increasing interest due to the ability of nanosized (macromolecular) formulations to favorably alter drug pharmacokinetics. These macromolecules have a different biodistribution profile, a prolonged plasma half-life, and reduced metabolism of the encapsulated drug.1 One of the most commonly cited reasons, however, for the use of nanomedicine is the ability of macromolecular compounds to exploit the enhanced permeability and retention (EPR) effect to specifically target tumor (Figure 1) and inflamed tissues.1
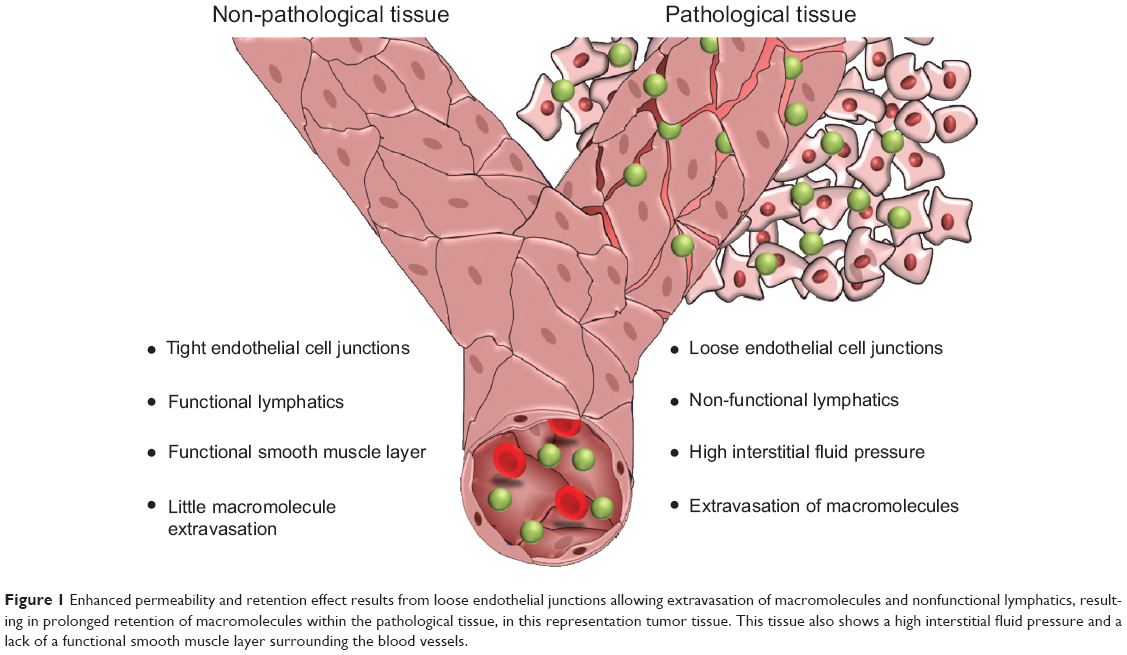 | Figure 1 Enhanced permeability and retention effect results from loose endothelial junctions allowing extravasation of macromolecules and nonfunctional lymphatics, resulting in prolonged retention of macromolecules within the pathological tissue, in this representation tumor tissue. This tissue also shows a high interstitial fluid pressure and a lack of a functional smooth muscle layer surrounding the blood vessels. |
The enhanced permeability observed in the vasculature of tissues undergoing certain pathologies, such as cancer or inflammation, is a result of deregulated angiogenesis and/or increased expression and activation of vascular permeability factors. The imbalance in expression and activation of proangiogenic and antiangiogenic molecules results in a discontinuous endothelial layer, where fenestrations between the endothelial cells may range from 300 nm to 4,700 nm in size.2 In addition to the formation of this porous vasculature, impaired lymphatic drainage may also occur in tumor tissue due to dysfunctional lymphangiogenesis and compression of the lymphatic vessels by growing numbers of cancer cells. As a result of this insufficient lymphatic drainage, constructs present in the interstitial fluid of tumors are retained for longer than in normal tissues with functional lymphatics.3–6
Exploitation of the EPR effect via use of nanoconstructs has been shown to consistently increase the fraction of the injected drug dose that reaches the tumor tissue.7 Furthermore, the absence of large fenestrations in nonpathological tissues prevents the extravasation and deposition of macromolecular constructs in healthy tissue and thereby reduces the level of systemic side effects. The two notable exceptions to this rule are the liver and spleen, because both of these organs commonly have large fenestrations.7 As a result of reduced nonspecific extravasation and circulating free drug, adverse effects are greatly reduced with the use of nanomedicine. This effect was clearly demonstrated with a reduction in doxorubicin-induced cardiotoxicity following treatment with Doxil®, a liposomal formulation of doxorubicin.8 However, an increased incidence of palmar–plantar erythrodysesthesia has been observed to occur as a consequence of use of this construct.9
Despite the ability of the EPR effect to improve the accumulation of macromolecules in pathological tissue, there remains the potential for improvement of tissue targeting. Inclusion of alternative techniques that are beyond the modification of size, coating, and charge has the ability to enhance the level of drug accumulation within pathological tissue. This review discusses and examines the methods that are being developed to improve the targeting of pathological tissues and cells by macromolecular therapeutics. We examine the innovative techniques being developed by researchers in this field and discuss the potential problems that must be overcome to ensure the successful transition of nanomedicine into the clinic.
Challenges to the EPR effect
High interstitial fluid pressure
Interstitial fluid pressure (IFP) in tumor tissue is a significant barrier that prevents the penetration of therapeutic agents.10 The elevation of IFP that is commonly seen in tumor tissue has been found to occur as a result of numerous factors. Firstly, the fenestrated vasculature observed in cancerous and inflamed tissues allows plasma fluid and proteins to leak from the capillaries. Secondly, the high protein content of the interstitial space increases the colloidal pressure and impedes the movement of extravasated nanoconstructs. Thirdly, compression of lymphatic vessels as a result of the pressure of the growing tumor results in a reduction of interstitial fluid drainage and thereby a net gain in IFP.11 Finally, IFP is also increased as a response to contraction of the interstitial space, an effect mediated by stromal fibroblasts.12 All of these factors play a key role in elevating the IFP of tumor tissue and consequently reducing the penetration of therapeutic macromolecules. Therefore, there is the potential to target these pathological factors and in turn reduce the IFP, thereby increasing the quantity of cancerous cells exposed to the nanomedicine.
A well investigated mechanism for decreasing IFP involves normalization of the vasculature in order to reduce extravasation of plasma fluid and proteins. This has previously been demonstrated by the use of DC101, a vascular endothelial growth factor (VEGF) receptor-2 antibody, to normalize the vasculature of subcutaneous MCaIV tumors. Treatment with DC101 40 mg/kg increased the penetration of TRITC-BSA (molecular weight 71,000 Da)13 from 7.26 μm to 11.23 μm from the blood vessel one hour post administration. This enhanced infiltration was suggested to be the consequence of treatment with DC101 lowering IFP by approximately 45%.14 Repeated administration of a VEGF monoclonal antibody (six doses of 100 μg per mouse) was reported to decrease IFP in tumors by up to 74% in a mouse model of colorectal adenocarcinoma.15 However, given that no investigation into the correlation between IFP and the integrity of the vasculature has been conducted, other potential mechanisms, such as tumor cell death or a reduction in microvessel density, may account for the decreased IFP. Promisingly, a 73% reduction in IFP was observed in four patients with rectal carcinoma treated with bevacizumab, a humanized monoclonal antibody targeting VEGF-A,16 indicating that an efficient reduction in IFP is achievable in the clinic. However, normalization of blood vessels and thereby reduced IFP is also expected to significantly reduce extravasation, because vascular normalization also reduces permeability of the vasculature to macromolecules such as albumin by up to 51%.14 Furthermore, the temporal window for efficacy of these drugs is narrow, as little as 6 days, and may show a great deal of heterogeneity depending on the patient and tumor being treated.17
Although normalization of lymphatic vessels may appear to be a suitable method for decreasing IFP, pursuit of this technique is likely to produce unfavorable results and cause more damage than what would be observed in tissues with compressed lymphatics. This is due to the observation that it is through functional lymphatics that tumor cells metastasize, so normalization of these vessels may increase the risk of metastasis.18
IFP may also be lowered via pharmacological modulation of stromal fibroblasts, because these cells are responsible for increased pressure via contraction of the interstitial space. Simulation of the platelet-derived growth factor receptor (PDGFR) has been shown to increase IFP via contraction of stromal cells, the integrins of which interact with the extracellular matrix.19–21 Administration of the PDGFR-β inhibitor, STI-571, was shown to decrease IFP in colon carcinoma by 32%20 and in thyroid carcinoma by 58%,22 while imatinib, a PDGFR inhibitor, reduced IFP in a mouse model of non-small-cell lung cancer by 28%.21 Inhibition of PDGFR activity, while appearing to be less effective than vascular normalization in terms of lowering IFP, does not result in reduced vascular fenestrations, thereby preserving extravasation of macromolecules.22
Despite the presence of elevated IFP, our view is that this pathological feature does not negate the extravasation of nanoconstructs in tumor tissue. Elevated IFP primarily acts on the venous end of tumor vessels because this site maintains a lower hydrostatic pressure than that at the arterial end. Without continuous blood circulation involving venous return in the tumor tissue, growth and survival of tumor cells would not be possible. Furthermore, variations in IFP measured in human tumors range from −1 mmHg to 94 mmHg, making it unreasonable to generalize the role of elevated IFP to all types of human cancer.23
Tissue penetration
Tissue penetration is a significant barrier to the efficacy of a nanomedicine. Although tumor and immune cells are capable of endocytosing these constructs, the macromolecule must first penetrate a number of layers to reach these cells. Due to the presence of the extracellular matrix and dense “rings” of cells around blood vessels, where the supply of nutrients and oxygen necessary for cellular growth is most abundant, the ability of nanomedicines to penetrate to cells beyond the blood vessel is significantly impaired. Consequently, the anticancer efficacy of macromolecules is often restricted to the cells surrounding the blood vessels.
An example of the importance of tissue penetration in successful delivery of nanoconstructs is delivery of macromolecules to brain tissue. The issue of effective utilization of nanomedicine for the treatment of brain disorders is particularly challenging due to the diverse properties of the central nervous system. The blood–brain barrier provides a formidable challenge to delivering therapeutics to treat pathologies of the central nervous system. This is primarily due to the ability of the blood–brain barrier to block passive diffusion of large or hydrophilic constructs and effectively efflux small molecules as a protective mechanism. In certain brain tumors, such as glioblastoma multiforme, the blood–brain barrier does show a degree of disruption, allowing macromolecules to penetrate into the central nervous system.24,25 However, infiltration of macromolecules into this tissue is impeded by the inability of large molecular weight constructs to diffuse easily through the interstitial space. Furthermore, glioblastoma multiforme cells are highly invasive, and the presence of even a small number of remaining cells makes post-surgical recurrence essentially universal within this patient population.26 The residual cells that remain in the brain are supported by the existing vasculature and due to the lack of neoangiogenic vessels in the spreading tumor lesion, the blood–brain barrier still prevents chemotherapeutics from reaching the invasive cell population, and therefore continues to pose a considerable challenge to the effective treatment of central nervous system tumors.
These factors have traditionally made treatment of central nervous system tumors difficult and resulted in the application of systemic antibodies, macromolecular drug carriers, and cytolytic viruses being negatively impacted.27 For these reasons, convection-enhanced delivery (CED), which involves the utilization of a positive pressure gradient applied during an infusion, has been investigated as a possible mechanism to overcome these challenges. This technique is intended to enhance the distribution of therapeutic molecules and constructs into the brain whilst circumventing potentially harmful systemic exposure (Figure 2A). The use of this procedure is currently being investigated for the treatment of central nervous system pathologies such as glioblastoma multiforme and Parkinson’s disease.27,28
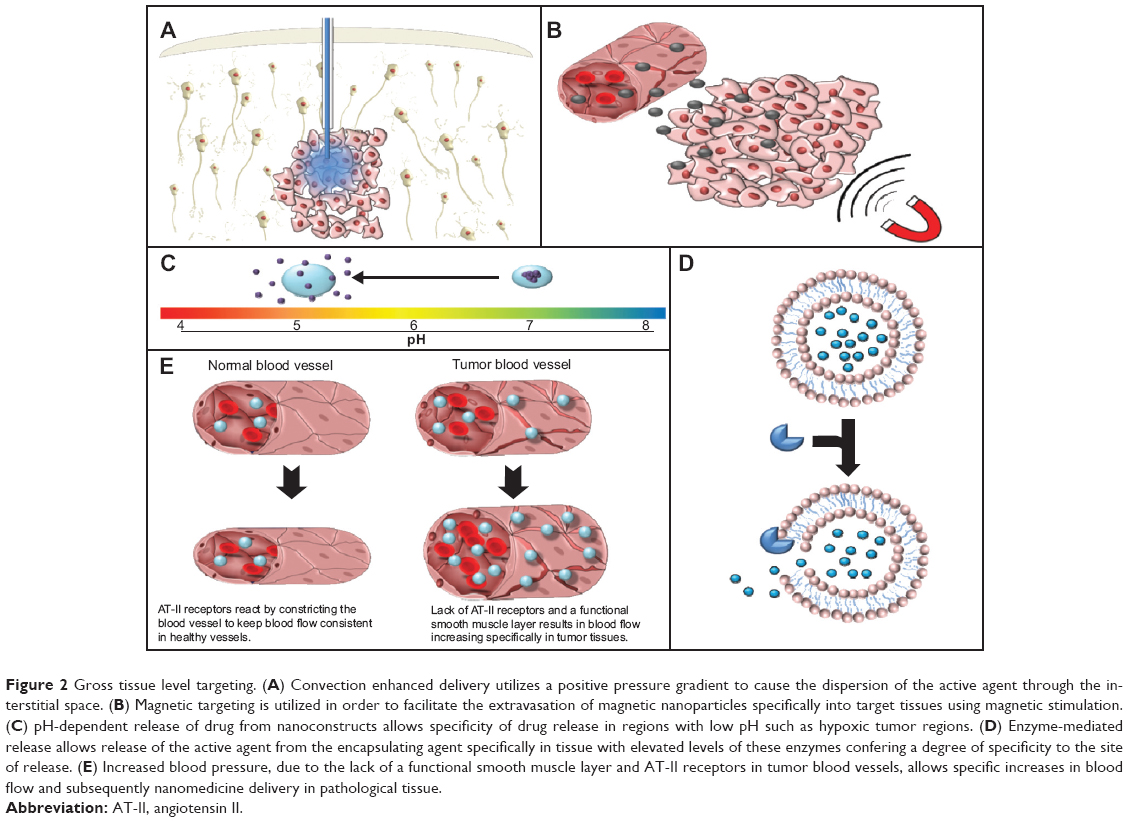 | Figure 2 Gross tissue level targeting. (A) Convection enhanced delivery utilizes a positive pressure gradient to cause the dispersion of the active agent through the interstitial space. (B) Magnetic targeting is utilized in order to facilitate the extravasation of magnetic nanoparticles specifically into target tissues using magnetic stimulation. (C) pH-dependent release of drug from nanoconstructs allows specificity of drug release in regions with low pH such as hypoxic tumor regions. (D) Enzyme-mediated release allows release of the active agent from the encapsulating agent specifically in tissue with elevated levels of these enzymes confering a degree of specificity to the site of release. (E) Increased blood pressure, due to the lack of a functional smooth muscle layer and AT-II receptors in tumor blood vessels, allows specific increases in blood flow and subsequently nanomedicine delivery in pathological tissue. |
The use of CED has the potential to facilitate the efficient distribution of macromolecular constructs through the interstitial space in a manner not possible by simple bolus injection. Clear evidence of CED-mediated improvement has been demonstrated, with one study showing a 115-fold increase in volume of distribution of 50 nm lipid nanocapsules when compared with a classical injection of the same nanoparticles.29
When considering the use of CED, nanomedicine also allows for increased retention and volume of distribution within the central nervous system, whilst also reducing toxicity to cells in the central nervous system. For example, administration of liposomal irinotecan or topotecan into the central nervous system of Sprague-Dawley rats via CED has been seen to cause a 65.6-fold and 15-fold increase, respectively, in half-life relative to that of the free drug.30,31 Similarly, liposomal and micellar doxorubicin administered via CED showed a 4.6-fold and 3.9-fold increase in the volume of distribution when compared with the free drug.32
Although the potential to apply drugs directly to the central nervous system and thereby avoid systemic exposure is an attractive idea, CED is still a technique that requires a good degree of refinement beyond the current methodology. More research into CED needs to be conducted before consistent and efficacious results can be achieved in the clinic. For example, movement of constructs through the central nervous system is defined by the characteristics of the tissue, wherein gray matter shows isotropic transport, and movement through white matter is dependent on the density and direction of the axons. Transport through tumor tissue is also highly complex, with macromolecules showing differential distribution depending on the homogeneity of the tumor tissue itself.33 In addition to this, factors such as preinfusion sealing time, rate of infusion, size of the delivery cannula, and concentration and leakage of the infusate into the subarachnoid space or surgical cavity must also be considered when applying this technique.34,35
Nonvascular tumor tissue
Although tumors must have a blood supply to continue growth because of their demand for oxygen and nutrients, the degree of vascularization varies considerably depending on the tumor being treated. For example, the microvessel density in hepatocellular carcinoma and colorectal carcinoma may be up to 127.2 vessels per field and 145 vessels per field, respectively.7 Conversely, squamous cell carcinoma and stage I–II non-small-cell lung cancer may have a microvessel density of only 14.8 vessels per field and 23.6 vessels per field, respectively.7 In tumor tissues where the microvessel density is low, the capacity to deliver nanomedicine effectively is significantly reduced, given that the distance that the macromolecule will have to diffuse through is significantly increased. Furthermore, the absence of an adequate microvessel structure prevents the supply of a sufficient amount of blood to the tumor tissue, resulting in hypoxia, acidosis, and oxidative stress, all of which lead to development of a necrotic region. These factors make the pursuit of systemically applied nanomedicine for nonvascular tumor tissue insufficient.
Liver and spleen accumulation
The liver and spleen have large endothelial fenestrations (~100 nm and 5 μm, respectively),36,37 so macromolecules will extravasate into these tissues. However, the extent to which these macromolecules accumulate within the liver and spleen is determined by several parameters. Greater accumulation of macromolecules within the liver is sometimes observed in preclinical studies despite the larger fenestration size in the spleen,7 a distribution pattern likely to be a consequence of the high blood flow through the liver relative to the spleen (1.8 mL versus 0.09 mL per minute in mice, 13.8 mL versus 0.63 mL per minute in rats, and 1,450 mL versus 77 mL per minute in humans).38 Previous research has shown that the degree of accumulation of macromolecules within these organs may be further influenced by the nature of the construct. Modulation of construct parameters such as size, shape, and charge7,39 may therefore alter the accumulation of these macromolecules in various organs and prevent the development of unwanted effects. For example, cylindrical silicone particles have been shown to accumulate in the liver with a quantity two-fold that of spherical and quasihemispherical particles and five-fold that of discoidal particles.39 Nevertheless, it has been demonstrated that nanomedicines can accumulate and be retained in hepatic tumors at a concentration higher than that of the surrounding healthy liver,3 an effect which can be attributed to differential lymphatic drainage in healthy and cancerous tissues.
Improvement of the EPR effect at the tissue level
Magnetic nanoparticles
Although targeting mechanisms utilizing tumor-specific receptor ligands confer a degree of specificity to tissue accumulation, receptors that may be overexpressed in pathological conditions are often also expressed in healthy tissues. Therefore, the use of these ligands leaves open the possibility of increasing the delivery of nanoconstructs to not only pathological target tissue but also to healthy tissues. Magnetic nanoparticles allow targeting of a wide variety of tissues via application of a magnetic field to specifically increase nanoparticle accumulation within a specific and restricted area. Due to the atypical nature of the magnetic stimulus, the application of a magnetic field to a specific section of tissue has the potential to offer a greater degree of specificity as regards tissue accumulation (Figure 2B).
The design of magnetic nanoparticles is complex, requiring consideration of a number of factors. Magnetic nanoparticles are typically metal-based, which limits the number of materials suitable for application. Iron nanoparticles, such as Fe3O4 and gamma-Fe2O3, are the most commonly utilized, and are well tolerated due to the body’s ability to store iron effectively. In comparison, metals such as nickel and neodymium are not well tolerated by the body, so are avoided in the design of magnetic nanoparticles. The size of the construct must be carefully considered beyond the ability to escape renal filtration and selectively extravasate from the tumor or inflamed blood vessels. For example, the construct must be small enough to pass safely through the capillaries and not precipitate in response to gravitational force, but must also be large enough to overcome the hemodynamic force of the blood flow and to have a suitable magnetic susceptibility.
The premise of magnetic targeting has been previously demonstrated in a rodent model of glioblastoma multiforme, where accumulation of iron oxide nanoparticles administered to rats bearing orthotopic tumors was measured in the presence or absence of an external magnetic field. This study showed an 11.5-fold greater accumulation of iron oxide nanoparticles in tumor tissue when the animals were concomitantly subjected to a localized magnetic field (0.4 T) relative to control animals.40 Furthermore, in animals exposed to magnetic targeting, accumulation of iron oxide nanoparticles in tumor tissues was 9.5-fold higher than accumulation within the contralateral brain,40 indicating significant tissue specificity.
Magnetic targeting typically involves the use of external magnets, which have a variety of drawbacks. Primarily, external magnets limit the ability of magnetized nanoparticles to be targeted to deep tissues. This drawback may be overcome by the use of surgically implanted magnets, alone or in combination with external magnets, to attract the magnetized nanoparticles to deeper tissues. Small implants placed directly into the target tissues may be magnetized via application of an external magnetic field, thereby creating an internal magnetic field that may be significantly more effective than an external field, particularly in the case of deep tissues. However, whilst appealing, this option does require that the patient be suitable for and consent to this type of invasive surgery.
Furthermore, due to the size of the human body in comparison with animal models, the strength of the magnetic field will need to be scaled up in order to be successful in a human due to the increased working distance. This may result in the applied magnetic strength exceeding the 8 T considered safe by the US Food and Drug Administration.41
Ultrasound
Ultrasound has been utilized for a number of years in a wide variety of pathological conditions to increase the uptake of therapeutics, both small and large molecular weight. The mechanism by which ultrasound increases the cellular uptake of therapeutics, although not indisputable, is thought to be due to membrane disruption. This disturbance results in increased permeability to macromolecular agents and the drugs they release in response to ultrasound.42 Ultrasound serves as a noninvasive, highly manipulable method to disrupt cell membranes and enhance capillary permeability. Most importantly, the effects of ultrasound are able to be tuned in order to affect deep tissues that may not be possible, practical, or safe to reach with surgical methods.
The effect of ultrasound on drug uptake by cells has been previously investigated in a mouse model of intraperitoneal ovarian cancer. In these studies, the uptake of micelles loaded with doxorubicin was significantly improved following application of ultrasound (30 seconds with 1 mHz)42,43 and was sustained for at least 8 hours.42 However, it must be noted that there has been an indication that areas not subject to direct sonication also show evidence of an increased concentration of doxorubicin, indicating nonspecific effects of ultrasound that could be potentially detrimental.42 In addition to these findings, the extended use of ultrasound has the potential to adversely affect the integrity of the nanomedicine formulation. This ultrasound-induced breakdown has been utilized by some researchers to induce site-specific drug release, as opposed to increased internalization or extravasation of the whole nanoconstruct. For example, administration of liposomal cisplatin to mice bearing peritoneal J6456 lymphoma lesions showed that 120 seconds of low frequency ultrasound application induced release of 70% of the encapsulated cisplatin, representing a 23.3-fold increase compared with the release in the absence of ultrasound.44
Ultrasound can also be utilized to increase the permeability of tissue barriers. As discussed previously, the blood–brain barrier is a formidable obstacle to drug delivery into the central nervous system. A potential method to overcome this barrier is to disrupt the blood–brain barrier temporarily and therefore allow for the selective entry of macromolecules into the sonicated area of the brain. Creation of this temporary disruption to the blood–brain barrier is achieved by a process known as cavitation. This technique uses microbubbles that collapse under ultrasonic pressure to generate mechanical stress that can then act to transiently increase the permeability of the blood–brain barrier.45,46 This method is already being examined in order to treat pathologies such as glioblastoma multiforme and Alzheimer’s disease.47–49 Utilization of ultrasound in animals larger than mice may, however, be problematic due to the effect of cranial bone attenuation and scattering. In a porcine model of ultrasound-induced blood–brain barrier disruption, ie, 1 mHz for 30 minutes, the increase in Evans blue dye extravasation in ultrasound-treated portions of the brain increased by ~30% relative to nontreated portions.50 Unfortunately, the groups were highly heterogeneous and the effect was abolished 120 minutes following sonication.50
Exploitation of systemic physiology
Blood pressure
A particular characteristic of tumor vasculature is the poor regulation of blood pressure. Examination of this feature has shown the involvement of several factors, including a lack of a functional smooth muscle layer around the blood vessels,51,52 a significantly reduced density of angiotensin II (AT-II) receptors53–55 and bradykinin receptors,56 and an increased concentration of nitric oxide,57 presumably as a consequence of increased levels of inducible nitric oxide synthase58,59 within tumors.
These factors contribute to the efficacy of AT-II-induced hypertension, because AT-II may be used to increase blood pressure and therefore the quantity of blood flowing specifically through the tumor vasculature, while minimally affecting healthy vasculature (Figure 2E). This tissue-specific effect occurs because healthy vasculature, in response to intravenously administered AT-II, will contract the smooth muscle layer surrounding the blood vessels. This reduction in the size of the blood vessel lumen results in a systemic increase in blood pressure. Conversely, tumor vasculature has fewer AT-II receptors, a defective smooth muscle layer, and a higher concentration of nitric oxide.60 These parameters mean that, while healthy tissues may respond appropriately to AT-II and maintain the same blood flow, the blood flow through tumor vasculature will increase as the vessel lumen enlarges in response to increased blood pressure.
The use of AT-II has been demonstrated to be highly efficacious at enhancing the selective delivery of nanomedicine to tumor tissue while increasing the tumor blood flow by 2–6-fold relative to normal tissues.61 The increase in blood flow will cause a greater amount of drug to pass through the tumor vasculature, which in turn will also augment the EPR effect. Enhancement of the EPR effect has been demonstrated by administration of the 51Cr-labeled nanomedicine known as styrene-co-maleic-acid polymer conjugated to neocarzinostatin (SMANCS) following AT-II-induced hypertension. This treatment resulted in a 1.3–3-fold increase in tumor accumulation of SMANCS, depending on blood pressure.62 Remarkably, use of AT-II also showed the potential to decrease the quantity of SMANCS in healthy tissues because constriction of healthy blood vessels decreases the size of the endothelial fenestrations.62 The degree of selectivity for tumor tissue achieved using this process is remarkable and constitutes the first example of this type of manipulation, thereby providing a platform for advancement of this technique and the development of others.
Enzyme-mediated targeting and release
Enzymes serve essential functions, and as a consequence, are typically tightly regulated by the cell. In tumors and inflamed tissues, the expression and/or activity of enzymes can be significantly different due to alterations in cellular metabolism, proliferation, and invasion. Enzyme targeting is an intriguing mechanism that could be used in two distinct ways to increase the delivery of nanoconstructs to tumor tissue.
Firstly, nanoconstructs could be conjugated to specific substrates that are targeted by enzymes overexpressed in pathological tissue. This may be utilized to allow for selective accumulation of nanomedicine at the therapeutic site. An example of successful use of an enzyme-targeting system is chlorotoxin. Chlorotoxin is a 36 amino acid peptide derived from the venom of the deathstalker scorpion. Despite the name, garnered from early experiments that appeared to suggest chlorotoxin affected cellular chloride channels, this peptide works by selectively binding to surface-bound matrix metalloproteinase 2, resulting in internalization of matrix metalloproteinase 2. Chlorotoxin has been found to react with membranous matrix metalloproteinase 2, which is specifically expressed in a wide variety of tumor tissues, including glioblastoma multiforme, pilocytic astrocytoma, small cell lung carcinoma, and Ewing’s sarcoma.63 Chlorotoxin targeting produces a significant increase in tumor localization of nanoparticles in mouse models of glioblastoma multiforme64,65 and specific localization of radioactive iodine conjugates in humans with glioblastoma multiforme lesions in Phase I/II trials.66,67 This method of targeting shows immense promise; however, no extensive Phase III clinical trials involving chlorotoxin-conjugated nanomedicine have been carried out in humans to date.
Secondly, enzymes may also be used to confer tissue specificity to the action of a nanomedicine by exploiting enzymes to promote the specific release of small molecular weight drugs from the carrier in the target tissue (Figure 2D). These small molecular weight drugs may then diffuse through solid tissue and into the cell. Although this mechanism does not strictly improve accumulation of the nanomedicine at the tumor site, it is a mechanism by which the enhanced accumulation made possible through utilization of the EPR effect can be coupled with site-specific drug release following either extravasation or endocytosis. A particular example of nanoconstructs showing an enzyme-specific response is the synthesis of liposomes that are sensitive to degradation by secreted phospholipase A2. Secreted phospholipase A2 hydrolyses phospholipids to form arachidonic acid and lysophospholipids, ie, the process which is physiologically responsible for inflammation and thrombosis. Secreted phospholipase A2 shows increased expression in a range of tumor types (including esophageal adenocarcinoma, prostate cancer, and hepatocellular carcinoma)68–70 and inflammatory conditions such as psoriasis and rheumatoid arthritis.71,72 Liposomes sensitive to degradation by secreted phospholipase A2 have been synthesized with the purpose of producing a long circulating nanomedicine that can be selectively degraded at pathological sites.73,74 Curiously, the degree of liposome degradation by secreted phospholipase A2 increases with the quantity of poly(ethylene glycol) (PEG)-lipid conjugates incorporated into the liposome, in a charge-dependent manner.73,74 Unfortunately, to the authors’ knowledge, studies to validate the efficacy of this system in vivo have not been published.
Currently, the research into enzyme-mediated delivery and/or release has predominantly focused on the treatment of cancer, although the potential for utilization in the treatment of inflammatory conditions is present. Cleavage of a formulation to release a payload must be carefully undertaken, given that both the uncleaved construct and the cleaved carrier must be nontoxic and the cleaving enzyme must be specific so as to assure that the toxic payload will not be delivered to nontargeted tissues.
pH targeting
Nanoparticle modification via pH targeting is one of the major methods utilized for targeted drug delivery using the external stimuli of pH change. This technique makes use of the variable pH gradient seen across different tissues or subcellular compartments to allow for site-specific drug release.
Cancer cells
The pH of the tumor microenvironment is known to be acidic (as low as pH 5.7). Rapidly growing tumor cells accumulate lactic acid as a result of their high rate of glucose metabolism, insufficient blood supply, poor lymphatic drainage, and reduced rates of oxidative phosphorylation.75 Thus, the acidic pH of the tumor microenvironment can be harnessed for pH-dependent drug delivery (Figure 2C). The role of the acidic environment in increased uptake of nanoparticles was first shown by Maeda et al who observed that SMANCS, under acidic conditions, showed a ten-fold increase in tumor cell binding.76 Paclitaxel-loaded acrylate polymers with a pH-sensitive 2,4,6-trimethoxybenzaladehyde linker have also been developed and shown to be stable at neutral pH but cleaved at acidic pH, leaving the hydroxyl groups of the polymer exposed. This transformation causes swelling of the nanoparticles and subsequent release of almost all paclitaxel within 24 hours, compared with the nonexpansile nanoparticles which showed rapid release independent of pH. The expansile pH-sensitive nanoparticles loaded with paclitaxel were shown to inhibit rapid growth of Lewis lung carcinoma tumors compared with non-pH-sensitive nanoparticles, which were associated with a nonsignificant reduction in tumor size in a 14-day study performed in C57B1/6 mice.77
Polymers with other acid-labile bonds have also been developed so as to specifically release drug from the polymer in the acidic conditions of the tumor environment. Hydrazone linkages are one such example, and have been used widely for preparation of hyaluronic hydrogels for pH-responsive drug release. These hydrazone-linked hyaluronic acid hydrogels release 60% of their linked immunoglobulin G in 8 hours at pH 5, while 60% release is achieved at 400 hours at pH 7.4.78 A major challenge to utilizing low pH for targeting tumor tissues is the lack of an efficient blood supply for delivery of the nanoconstructs to the hypovascular tumor center. This is further complicated by the impedance of particulate convection in the extracellular space as described in the section on tissue penetration. A combinational approach using this strategy is thus a plausible solution.
Intracellular delivery
Rapid endosomal acidification occurs during the endocytosis process due to ATPase-mediated proton influx. This activity results in a drop in pH levels to <6 in early endosomes, sorting endosomes, and multivesicular bodies.79 Subsequently, pH-sensitive liposomes have been developed to release their content in response to acidic pH. The first liposomes developed containing the pH-sensitive lipid palmitoyl homocysteine showed release of a fluorescent marker in a manner that was inversely proportional to the pH of the serum.80 Furthermore, pH-sensitive liposomes, generated by surface modification of egg yolk phosphatidylcholine liposomes with MGlu-Dex, have been shown to be taken up efficiently by dendritic cells to deliver entrapped ovalbumin molecules into the cytosol. Subcutaneous administration of MGlu-Dex-modified liposomes loaded with ovalbumin to mice induced antigen-specific humoral and cellular immunity more effectively than the unmodified liposomes loaded with ovalbumin.81
Nanoparticles are often coated with stealth PEG layers, which are stable in the circulation but released in endosomes. PEG layer shedding helps in nanoparticle endosomal escape by reducing the steric and electrostatic hindrance from the PEG layer.82 These PEG layers are grafted onto the nanoparticles with pH-sensitive linkers favoring drug release.83 This endosomal acidification is used as a trigger for endosomal escape by a mechanism hypothesized to occur via the “proton sponge” effect. This procedure involves nanoparticles absorbing protons at endosomal pH to maintain the pH, causing an increase in osmotic pressure in the endosomal compartment, followed by disruption of the plasma membrane and subsequent release of nanoparticles into the cytoplasm.84 For example, polyhistidine is known to have endosomal membrane disruption activity induced by the “proton sponge” mechanism. This activity causes disruption of the compartment membrane and release of doxorubicin into the cytosol, thus producing much higher in vitro and in vivo anticancer activity.85 Lysosomal escape is also an important step towards achieving nuclear targeting. For example, layered double hydroxide nanorods have shown endosomal escape through deacidification of layered double hydroxide nanoparticles followed by translocation to the nucleus. Since nuclear targeting of layered double hydroxide nanorods is rapid and requires an active process, it has been suggested that the active transport occurs via a microtubule-mediated trafficking process.86
Polymers containing amine group monomers are often used for intracellular delivery due to pH buffering in endosomes. Reducible polymers of poly(amido ethylenimine) (PEI) were developed for intracellular delivery of small interfering RNA (siRNA) for VEGF. These PEI polymers contain multiple disulfide linkages, which can be cleaved into amidoamine monomers in the reductive endosomal environment and therefore allow release of siRNA into the cytoplasm. Subsequently, PEI polymeric nanoparticles showed two-fold higher suppression of VEGF than linear PEI formulations.87 However, the cationic nature of polyamine nanoparticles leads to increased affinity towards the negatively charged cell membrane, so these nanoparticles tend to have toxicity issues, such as activation of apoptotic pathways.88
Gastrointestinal tract
An excellent example of a pH gradient that may be exploited to trigger release from nanoconstructs is the gastrointestinal tract. The parietal cells of the stomach lining secrete acids and digestive enzymes such as pepsin that are essential for denaturation of ingested proteins, so the stomach has a pH as low as 1.2.89 The small intestine contains bile salts and other basic enzymes such as pancreatin, resulting in a pH of around 6.5 in this section of the gastrointestinal tract.89 It is also important to note that pH varies throughout the gastrointestinal tract in the fasted and fed state.90
Release of drug from polymers in response to a change in pH can occur in various ways. Release of drugs by pH-dependent swelling of a polymer has previously been demonstrated using insulin-loaded poly(methacrylic acid) (PMAA)-PEG diblock copolymers to achieve a swelling ratio of 40–90-fold depending on copolymer composition and PEG graft length. At pH 7.4, the copolymer is in the swollen state and releases 90% of its loaded insulin while the remaining 10% is released at pH 1.2 in the complexed state.91 Acrylic-based polymers such as PMAA retain their complexed state in the stomach due to protonation of carboxyl groups, while in the intestine these nanoparticles swell due to the increased pH, causing carboxyl ionization and breakage of hydrogen bonds.92
Release of drugs via pH-dependent dissolution of polymers has extensive applications for targeting specific areas of the gastrointestinal tract and for this reason has already been commercialized. PMAA copolymer modifications, such as polyethylacrylate (PMAA-PEA, commercially available as Eudragit L100-55) dissolve at pH >5.5 and are therefore suitable for release in the duodenum, while polymethacrylate (PMAA-PMA, commercially available as Eudragit S100), dissolves at pH >7 and is therefore more suitable for ileal drug release or to increase the bioavailability of therapeutics such as cyclosporin.93
Another principle of drug release in response to pH change is the surface charge reversal of nanoparticles. Mesoporous silica is one such nanoparticle that is surface-functionalized with different densities of positively charged functional trimethylammonium groups. These trimethylammonium groups facilitate loading of anionic drugs such as sulfasalazine (an anti-inflammatory drug for inflammatory bowel disease) at pH <3. However, in conditions of higher pH, such as 7.4, a partial negative surface charge on the nanoparticles is generated by deprotonation of silanol groups, causing electrostatic repulsion and release of the drug.94 Use of chitosan nanoparticles loaded with heparin for the treatment of Helicobacter pylori infection has also been demonstrated to have significant promise. These constructs are stable at the lower pH of the stomach along the gastric epithelium but, when in contact with H. pylori infection along the gastric epithelium (pH 7.4), deprotonation of chitosan occurs, which weakens the electrostatic interactions and leads to collapse of the nanoparticles and release of heparin.95
Exploiting inflammatory mediators
In inflammatory conditions, the EPR effect is also seen to develop, with vascular permeability induced via contraction of the inflammatory cells lining the capillaries (Figure 3). This pathological response is induced by expression of histamine, bradykinin, leukotrienes, and serotonin in the inflamed tissue and results in increased intraendothelial gaps (Figure 3).96–99 These fenestrations allow extravasation of nanoparticles specifically into the inflamed tissue, as has been previously demonstrated in the case of polystyrene nanoparticles in the rodent trinitrobenzenesulfonic acid model of colitis.100 Furthermore, in inflammatory conditions induced by infection, the pathogen itself may secrete factors that increase the permeability of blood vessels.101,102 The inflammation also causes expression of inflammatory biomarkers, such as reactive oxygen species, that allow the active and regulated release of drug encapsulated in nanomedicines that are sensitive to these biomarkers (Table 1).
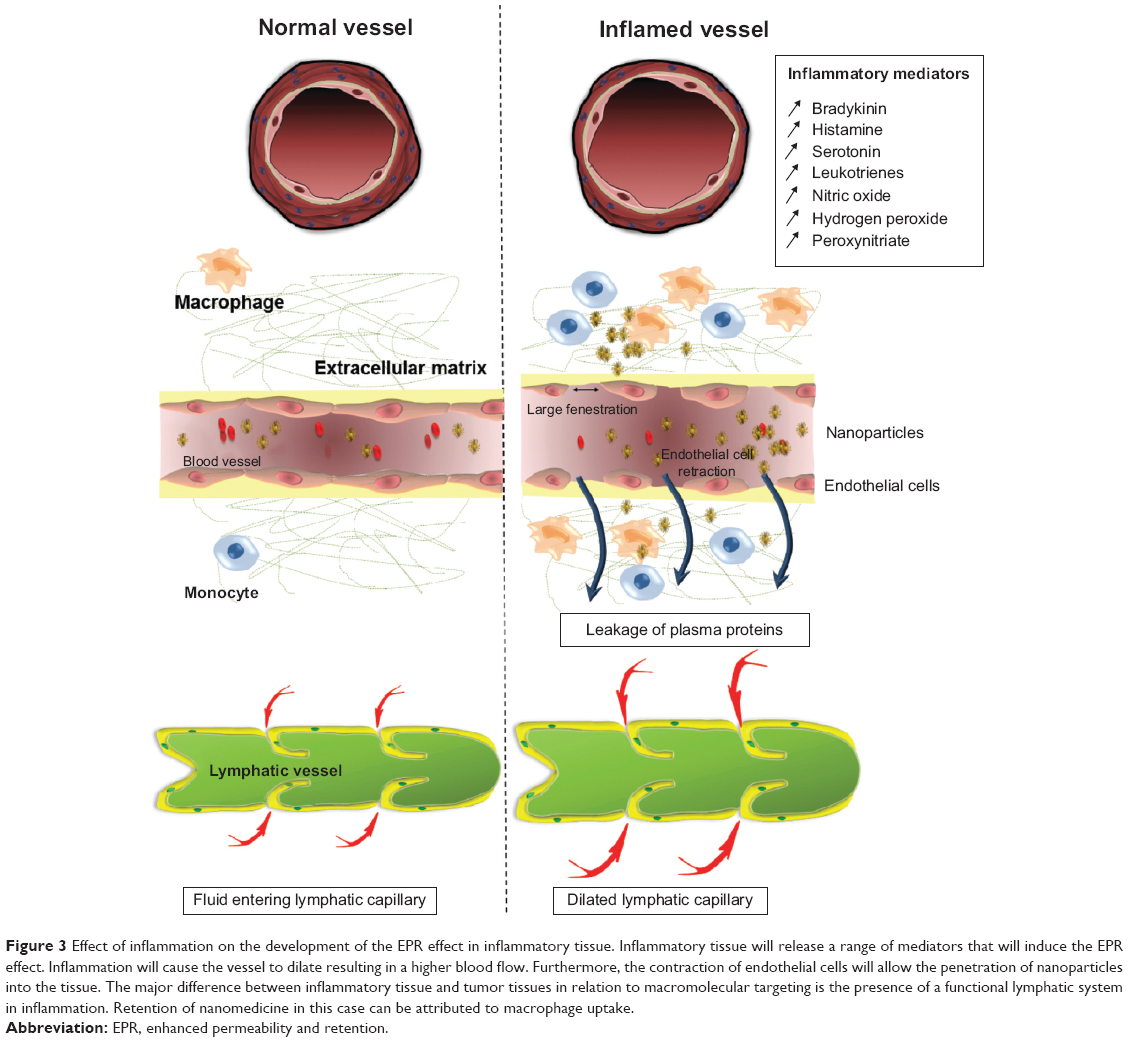 | Figure 3 Effect of inflammation on the development of the EPR effect in inflammatory tissue. Inflammatory tissue will release a range of mediators that will induce the EPR effect. Inflammation will cause the vessel to dilate resulting in a higher blood flow. Furthermore, the contraction of endothelial cells will allow the penetration of nanoparticles into the tissue. The major difference between inflammatory tissue and tumor tissues in relation to macromolecular targeting is the presence of a functional lymphatic system in inflammation. Retention of nanomedicine in this case can be attributed to macrophage uptake. |
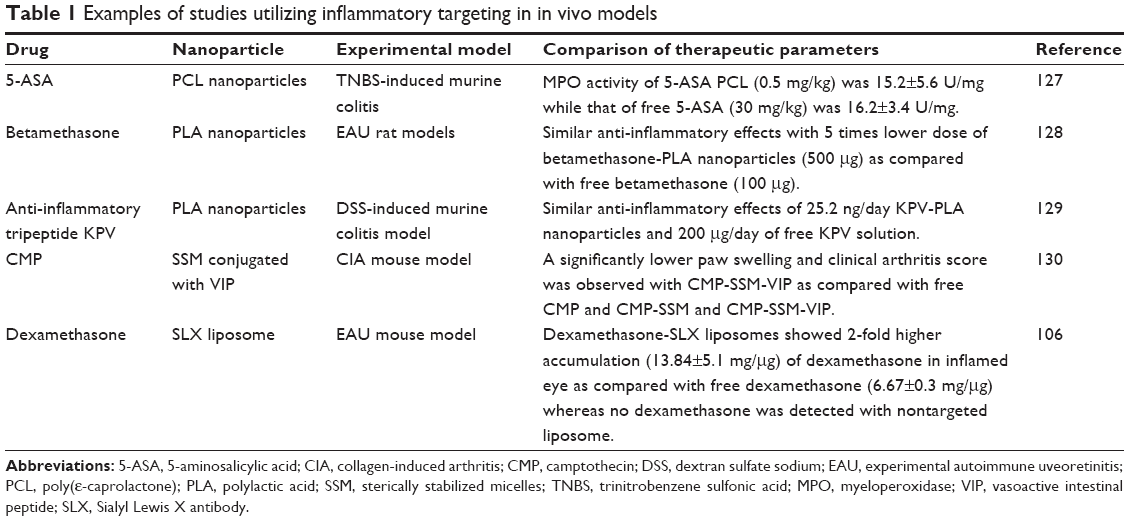 | Table 1 Examples of studies utilizing inflammatory targeting in in vivo models |
The use of siRNA against proinflammatory cytokines is a highly effective option for the treatment of intestinal inflammation. However, administration of siRNA is associated with an increased risk of infection, lymphoma, and cardiac dysfunction due to systemic depletion of proinflammatory cytokines. For this reason, application of siRNA cannot be systemic, making use of targeted nanomedicine a particularly attractive alternative. The use of thioketal nanoparticles, prepared from poly-1,4-phenyleneacetone dimethylene thioketal and possessing reactive oxygen species-sensitive thioketal linkages, has been pursued for targeted delivery of tumor necrosis factor alpha (TNF-α) siRNA to inflamed intestinal tissue. Since the inflamed intestinal tissue contains high concentrations of reactive oxygen species generated by activated phagocytes, use of these thioketal nanoparticles would allow TNF-α siRNA to be specifically targeted to the site of inflammation (Figure 4B). Oral administration of thioketal nanoparticles loaded with TNF-α siRNA showed diminished levels of TNF-α messenger RNA in the colon in a murine model of ulcerative colitis.103
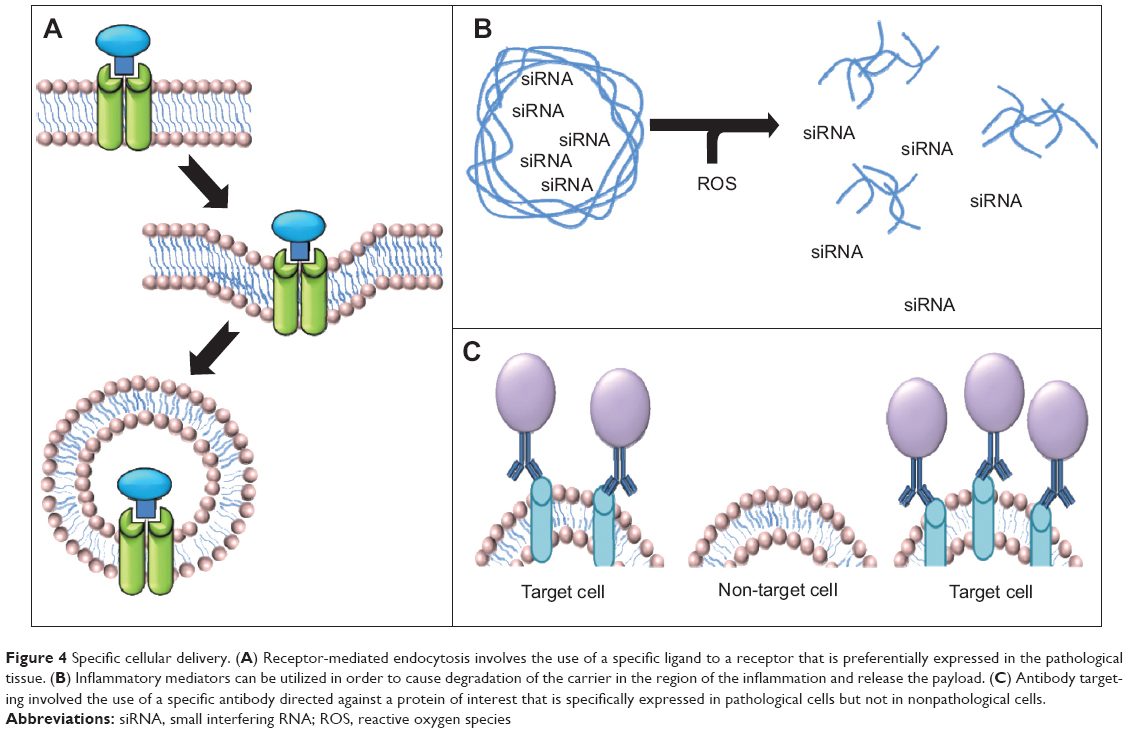 | Figure 4 Specific cellular delivery. (A) Receptor-mediated endocytosis involves the use of a specific ligand to a receptor that is preferentially expressed in the pathological tissue. (B) Inflammatory mediators can be utilized in order to cause degradation of the carrier in the region of the inflammation and release the payload. (C) Antibody targeting involved the use of a specific antibody directed against a protein of interest that is specifically expressed in pathological cells but not in nonpathological cells. |
Inflamed tissues recruit leukocytes via expression of cell adhesion molecules on the cell surface in an effort to remove pathogens from the tissue. These cell adhesion molecules bind to the sialyl Lewis X (SLX) tetrasaccharide which is present on the plasma membrane of polymorphonuclear leukocytes104 and is therefore considered to be a promising target for drug delivery at sites of inflammation.105 This observation has led to the development of intravenously administered SLX-conjugated liposomes encapsulating dexamethasone, which resulted in a two-fold higher accumulation of dexamethasone in the inflamed eye of a murine experimental autoimmune uveoretinitis model when compared with intravenous injection of free dexamethasone.106 Nontargeted liposomes loaded with dexamethasone resulted in no detectable level of dexamethasone in the inflamed eye using this model.106 SLX conjugated to liposomes encapsulating the dyes CY5 or CY3 showed accumulation in inflamed tissue on in vivo fluorescent imaging in a collagen-antibody induced arthritis model.107 Histopathological sections showed fluorescence in the hyperplastic synovium of periarticular soft tissue and pannus invasion with inflammatory cells in the collagen-antibody induced arthritis mouse model. On administration of targeted SLX-liposome with Cy3, accumulation of the fluorescence signal was observed in the synovial tissue after 24 hours, while no accumulation was seen in control mice administered untargeted liposomes.107
Exploiting unique cell characteristics at the tumor level
Folic acid receptor
Tumor cells often show altered expression of receptors due to transformed metabolism or an altered cell replication rate. Targeting upregulated receptors that endocytose their ligands following binding may allow increased uptake of nanomedicines (Figure 4A). There is a wide range of receptor targeting mechanisms being utilized today, including transferrin receptor-mediated, low density lipoprotein receptor-mediated, and folic acid receptor (FR)-mediated endocytosis.108–110
Folate has a high affinity for the FR, which is often overexpressed on human tumor cells and taken up by receptor-mediated endocytosis following binding to the receptor.111,112 In this way, the interaction between folate and the FR can be used for tumor-specific drug delivery.113 There are two different glycosyl phosphatidylinositol-anchored isoforms of the FR, ie, FR-α, which is found on about 40% of human epithelial cancers, and FR-β, which is expressed on activated macrophages in association with chronic inflammatory diseases.114,115 Overexpression of FRs has been observed in a variety of tumor types, including ovarian and endometrial cancers, where FR overexpression occurs in 90% of cases.111
Encapsulation of drugs in folate-targeted nanomedicine has been pursued in a variety of nanoconstructs micelles and liposomes.110,116 In vivo, folate targeting of liposomes has shown the ability to increase the quantity of nanomedicine associated with J6456 lymphoma cells by 17-fold relative to nontargeted liposomes.117 It must be noted that activated macrophages also express the FR, so it is possible that the therapeutic activity of the liposomes is partially due to targeted elimination of FR-expressing tumor-associated macrophages.118
High expression of FRs suggests that the folic acid requirements of the cell are high and that appropriate chemotherapeutics may be utilized in order to fully exploit this characteristic. For example, methotrexate is utilized because of its ability to inhibit dihydrofolate reductase, an enzyme that converts dihydrofolate to tetrahydrofolate, before it is converted to the folate cofactor methylene tetrahydrofolate.119 Therefore, folic acid targeting (such as in PAMAM dendritic polymers conjugated with methotrexate) has been used to increase the concentration of free methotrexate in sensitive tumor cells. This approach allows reduction of methotrexate toxicity and improvement of methotrexate efficacy relative to the nontargeted construct.120
Antibodies
The use of antibodies for specific cell targeting of pathological cells is perhaps the technique most reminiscent of Ehrlich’s “magic bullet” idea due to the sheer diversity of molecules which may be targeted by the variable domains of the antibodies. Antibodies may be engineered to have higher sensitivity and specificity for their targets than receptor ligands. Counterintuitively, the high affinity of antibodies can impede effective tissue delivery through a phenomenon known as the “binding site barrier”. This term is used to describe the high affinity binding of antibodies to target cells close to the extravasation site which prevents the antibodies, and their associated nanomedicines, from penetrating deep into tumor tissue. As a consequence, antibodies must be engineered to have a high enough affinity so that the antibody is still specific and able to effectively target the nanomedicine while not having an affinity high enough to prevent diffusion.121
Antibody targeting has two potential mechanisms for increased efficacy. Firstly, the use of antibodies may increase the quantity of the nanomedicine that reaches the pathological tissue; for example, an antibody to CD22 increases the quantity of doxorubicin in non-Hodgkin lymphoma-bearing mice by 80%.122 The second method by which the efficacy of antibody-targeted nanomedicines may be enhanced does not involve increased delivery of the construct to gross tissue but rather increased delivery to specific cell types within the tissue (Figure 4C). For example, no difference in gross localization of doxorubicin liposomes in tumor tissue was observed relative to whether or not the liposome was targeted with an antibody to the epidermal growth factor (EGF) receptor. However, the efficacy of EGF receptor-targeted liposomes was significantly greater in mice bearing MDA-MB-231 and U87 tumors. These data indicate that internalization of the liposomes specifically by cells targeted by the EGF receptor antibody was the determining factor for the increased efficacy of the construct, as opposed to increased accumulation at the tumor site.123 This same phenomena was also described by Kirpotin et al who reported that nontargeted and HER-2 targeted immunoliposomes showed comparable accumulation in tumor xenografts, but that only HER-2 targeted immunoliposomes localized primarily in tumor cells, while nontargeted immunoliposomes showed localization within the stroma and tumor-associated macrophages.124
Regardless of the sensing and targeting advantages of antibodies for delivering a specific anticancer agent, the presence of antibodies on the cell surface can lead to antibody-dependent cell-mediated cytotoxicity. This biological mechanism makes antibody targeting even more appealing as a therapeutic strategy.
There are, however, a number of drawbacks to studies that examine the use of antibody-targeted nanomedicines. A common experimental flaw observed when utilizing antibody targeting is that nontargeted antibodies serving as a control to assess the effect of antibody conjugation on biodistribution and pharmacokinetics are often not included. Moreover, because of financial restraints, many researchers fail to humanize their antibodies before preclinical in vivo testing. A further problem with this method of targeting is related to the heterogeneous nature of the tumor tissue itself. In tumors, even those which express a high level of the antigen towards which the antibody is directed, there will be a fraction of cells that do not express the antigen and so these cells will evade antibody targeting. Consequently, these cells survive and repopulate the tumor, with recurrence of tumors that are refractory to initial treatment. Tumors may also shed their antigen into the stroma or plasma, resulting in the antibodies being bound to a target that was not present on the cell surface and thereby defeat the purpose of using antibody targeting.
Combinational approaches
The treatment of cancer using a combination of chemotherapeutics is often the preferred method to improve the efficacy of treatment. In the same way, a combination of techniques to improve delivery and/or cell specificity may further increase the efficacy of the system in the target tissue. The combination of a number of the above techniques has been used by researchers keen to improve the efficacy of their systems beyond what is possible using a single technique. This approach has merit but is in the early stages of development.
An example of a combinatorial approach currently being investigated is the combination of CED and receptor targeting. CED combined with specific receptor targeting is an attractive idea due to the potential of CED for gross tissue delivery while further utilizing receptor targeting for specific cellular delivery. This approach has been utilized for the treatment of glioblastoma multiforme, wherein antibodies against an EGF receptor mutant (EGFRvIIIAb) were conjugated with iron oxide nanoparticles (EGFRvIIIAb-IONPs). Mice with orthotopic glioblastoma multiforme lesions treated using CED with EGFRvIIIAb-IONPs had a 15% and 10.5% longer survival time than mice treated with IONs or EGFRvIIIAb, respectively.125 Furthermore, boronated dendrimers conjugated to EGF were developed to target EGFR-expressing glioblastoma multiforme. In mice with EGFR-expressing glioblastoma multiforme lesions, CED resulted in 47.4% of ID/g being present in the lesion relative to 33.2% of ID/g following simple injection at 24 hours post administration. Unfortunately, there was no control performed in this study to compare boronated dendrimers without EGF.126
Conclusion
The majority of the nanoconstructs designed for drug targeting to date have relied intensively on the EPR effect, eschewing consideration of the diverse factors related to human tumors, such as doubling time, vascular density, and preferential uptake of nanomedicine by off-target organs. Furthermore, consideration of the EPR effect as a universal phenomenon within a given patient population or even within the pathological tissue of a single patient has resulted in a disparity between successful preclinical studies and limited clinical transition. The emerging targeting strategies being developed to enable increased specificity of delivery of macromolecular agents on subcellular, cellular, and gross tissue levels are increasingly showing their potential and may serve as an essential component in all future macromolecular drug design. Furthermore, close understanding of the biological nature of the tumor being targeted is essential for the design of nanoconstructs and the appropriate targeting mechanism. Although the EPR effect alone shows the potential to significantly improve the efficacy of medicine directed towards certain pathologies, the current research highlights the means by which this may be further enhanced.
Acknowledgments
The authors gratefully acknowledge the support of Professor Hiroshi Maeda. The EPR effect was first described and extensively studied by Professor Maeda’s group. The authors would also like to thank Dominic McCann for editing the manuscript. This work was supported by departmental fund no.; (PL. 108403.01.S. LM) to KG and an HRC Emerging Researcher first grant (PL. 108360.01.P. LM), University of Otago research grant (PL. 10942.01.R. LM), and AG305 for ST from the Department of Pharmacology and Toxicology, Otago University.
Disclosure
The authors report no conflicts of interest in this work.
References
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1): 6387–6392. | ||
Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000; 156(4):1363–1380. | ||
Iwai K, Maeda H, Konno T. Use of oily contrast medium for selective drug targeting to tumor: enhanced therapeutic effect and X-ray image. Cancer Res. 1984;44(5):2115–2121. | ||
Konno T, Maeda H, Iwai K, et al. Selective targeting of anti-cancer drug and simultaneous image enhancement in solid tumors by arterially administered lipid contrast medium. Cancer. 1984;54(11):2367–2374. | ||
Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60(16):4324–4327. | ||
Jeon BH, Jang C, Han J, et al. Profound but dysfunctional lymphangiogenesis via vascular endothelial growth factor ligands from CD11b+ macrophages in advanced ovarian cancer. Cancer Res. 2008;68(4): 1100–1109. | ||
Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; where is the missing link? J Control Release. 2012;164(3):265–275. | ||
Safra T, Muggia F, Jeffers S, et al. Pegylated liposomal doxorubicin (Doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11(8):1029–1033. | ||
O’Brien ME, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of PEGylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15(3):440–449. | ||
Netti PA, Hamberg LM, Babich JW, et al. Enhancement of fluid filtration across tumor vessels: implication for delivery of macromolecules. Proc Natl Acad Sci U S A. 1999;96(6):3137–3142. | ||
Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004; 427(6976):695. | ||
Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. | ||
Bingaman S, Huxley VH, Rumbaut RE. Fluorescent dyes modify properties of proteins used in microvascular research. Microcirculation. 2003;10(2):221–231. | ||
Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736. | ||
Lee CG, Heijn M, di Tomaso E, et al. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000;60(19):5565–5570. | ||
Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10(2):145–147. | ||
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | ||
Padera TP, Kadambi A, di Tomaso E, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296(5574): 1883–1886. | ||
Uutela M, Wirzenius M, Paavonen K, et al. PDGF-D induces macrophage recruitment, increased interstitial pressure, and blood vessel maturation during angiogenesis. Blood. 2004;104(10):3198–3204. | ||
Pietras K, Ostman A, Sjoquist M, et al. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001;61(7):2929–2934. | ||
Vlahovic G, Rabbani ZN, Herndon JE, 2nd, Dewhirst MW, Vujaskovic Z. Treatment with Imatinib in NSCLC is associated with decrease of phosphorylated PDGFR-beta and VEGF expression, decrease in interstitial fluid pressure and improvement of oxygenation. Br J Cancer. 2006;95(8):1013–1019. | ||
Pietras K, Rubin K, Sjoblom T, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62(19):5476–5484. | ||
Jain RK. Delivery of molecular medicine to solid tumors: lessons from in vivo imaging of gene expression and function. J Control Release. 2001;74(1–3):7–25. | ||
Reichman HR, Farrell CL, Del Maestro RF. Effects of steroids and nonsteroid anti-inflammatory agents on vascular permeability in a rat glioma model. J Neurosurg. 1986;65(2):233–237. | ||
Ambruosi A, Khalansky AS, Yamamoto H, Gelperina SE, Begley DJ, Kreuter J. Biodistribution of polysorbate 80-coated doxorubicin-loaded [14C]-poly(butyl cyanoacrylate) nanoparticles after intravenous administration to glioblastoma-bearing rats. J Drug Target. 2006;14(2): 97–105. | ||
Konishi Y, Muragaki Y, Iseki H, Mitsuhashi N, Okada Y. Patterns of intracranial glioblastoma recurrence after aggressive surgical resection and adjuvant management: retrospective analysis of 43 cases. Neurol Med Chir (Tokyo). 2012;52(8):577–586. | ||
Allard E, Passirani C, Benoit JP. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials. 2009;30(12): 2302–2318. | ||
Bankiewicz KS, Eberling JL, Kohutnicka M, et al. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol. 2000;164(1):2–14. | ||
Vinchon-Petit S, Jarnet D, Paillard A, Benoit JP, Garcion E, Menei P. In vivo evaluation of intracellular drug-nanocarriers infused into intracranial tumours by convection-enhanced delivery: distribution and radiosensitisation efficacy. J Neurooncol. 2010;97(2):195–205. | ||
Noble CO, Krauze MT, Drummond DC, et al. Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer Res. 2006;66(5):2801–2806. | ||
Saito R, Krauze MT, Noble CO, et al. Convection-enhanced delivery of Ls-TPT enables an effective, continuous, low-dose chemotherapy against malignant glioma xenograft model. Neuro Oncol. 2006;8(3):205–214. | ||
Inoue T, Yamashita Y, Nishihara M, et al. Therapeutic efficacy of a polymeric micellar doxorubicin infused by convection-enhanced delivery against intracranial 9L brain tumor models. Neuro Oncol. 2009;11(2):151–157. | ||
Saito R, Bringas JR, McKnight TR, et al. Distribution of liposomes into brain and rat brain tumor models by convection-enhanced delivery monitored with magnetic resonance imaging. Cancer Res. 2004;64(7):2572–2579. | ||
Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100(3):472–479. | ||
Chen MY, Lonser RR, Morrison PF, Governale LS, Oldfield EH. Variables affecting convection-enhanced delivery to the striatum: a systematic examination of rate of infusion, cannula size, infusate concentration, and tissue-cannula sealing time. J Neurosurg. 1999;90(2):315–320. | ||
Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14. | ||
Horn T, Henriksen JH, Christoffersen P. The sinusoidal lining cells in “normal” human liver. A scanning electron microscopic investigation. Liver. 1986;6(2):98–110. | ||
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. | ||
Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141(3):320–327. | ||
Chertok B, Moffat BA, David AE, et al. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials. 2008;29(4):487–496. | ||
Chakeres DW, de Vocht F. Static magnetic field effects on human subjects related to magnetic resonance imaging systems. Prog Biophys Mol Biol. 2005;87(2–3):255–265. | ||
Gao ZG, Fain HD, Rapoport N. Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. J Control Release. 2005;102(1):203–222. | ||
Rapoport NY, Christensen DA, Fain HD, Barrows L, Gao Z. Ultrasound-triggered drug targeting of tumors in vitro and in vivo. Ultrasonics. 2004;42(1–9):943–950. | ||
Schroeder A, Honen R, Turjeman K, Gabizon A, Kost J, Barenholz Y. Ultrasound triggered release of cisplatin from liposomes in murine tumors. J Control Release. 2009;137(1):63–68. | ||
Kodama T, Tomita Y, Koshiyama K, Blomley MJ. Transfection effect of microbubbles on cells in superposed ultrasound waves and behavior of cavitation bubble. Ultrasound Med Biol. 2006;32(6):905–914. | ||
Huang SL. Liposomes in ultrasonic drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1167–1176. | ||
Choi JJ, Wang S, Brown TR, Small SA, Duff KE, Konofagou EE. Noninvasive and transient blood–brain barrier opening in the hippocampus of Alzheimer’s double transgenic mice using focused ultrasound. Ultrason Imaging. 2008;30(3):189–200. | ||
Raymond SB, Treat LH, Dewey JD, McDannold NJ, Hynynen K, Bacskai BJ. Ultrasound enhanced delivery of molecular imaging and therapeutic agents in Alzheimer’s disease mouse models. PLoS One. 2008;3(5):e2175. | ||
Ting CY, Fan CH, Liu HL, et al. Concurrent blood–brain barrier opening and local drug delivery using drug-carrying microbubbles and focused ultrasound for brain glioma treatment. Biomaterials. 2012;33(2):704–712. | ||
Xie F, Boska MD, Lof J, Uberti MG, Tsutsui JM, Porter TR. Effects of transcranial ultrasound and intravenous microbubbles on blood– brain barrier permeability in a large animal model. Ultrasound Med Biol. 2008;34(12):2028–2034. | ||
Konerding MA, Steinberg F, Streffer C. The vasculature of xenotransplanted human melanomas and sarcomas on nude mice. I. Vascular corrosion casting studies. Acta Anat (Basel). 1989;136(1):21–26. | ||
Konerding MA, Steinberg F, Streffer C. The vasculature of xenotransplanted human melanomas and sarcomas on nude mice. II. Scanning and transmission electron microscopic studies. Acta Anat (Basel). 1989;136(1):27–33. | ||
Andrade SP, Beraldo WT. Pharmacological reactivity of neoplastic and non-neoplastic associated neovasculature to vasoconstrictors. Int J Exp Pathol. 1998;79(6):425–432. | ||
Kohzuki M, Tanda S, Hori K, et al. Endothelin receptors and angiotensin II receptors in tumor tissue. J Cardiovasc Pharmacol. 1998;31 Suppl 1: S531–S533. | ||
Tozer GM, Shaffi KM, Prise VE, Bell KM. Spatial heterogeneity of tumour blood flow modification induced by angiotensin II: relationship to receptor distribution. Int J Cancer. 1996;65(5):658–663. | ||
Wu J, Akaike T, Hayashida K, et al. Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int J Cancer. 2002;98(1):29–35. | ||
Kennovin GD, Flitney FW, Hirst DG. “Upstream” modification of vasoconstrictor responses in rat epigastric artery supplying an implanted tumour. Adv Exp Med Biol. 1994;345:411–416. | ||
Moochhala S, Chhatwal VJ, Chan ST, Ngoi SS, Chia YW, Rauff A. Nitric oxide synthase activity and expression in human colorectal cancer. Carcinogenesis. 1996;17(5):1171–1174. | ||
Klotz T, Bloch W, Volberg C, Engelmann U, Addicks K. Selective expression of inducible nitric oxide synthase in human prostate carcinoma. Cancer. 1998;82(10):1897–1903. | ||
Dworkin MJ, Carnochan P, Allen-Mersh TG. Effect of continuous regional vasoactive agent infusion on liver metastasis blood flow. Br J Cancer. 1997;76(9):1205–1210. | ||
Suzuki M, Hori K, Abe I, Saito S, Sato H. A new approach to cancer chemotherapy: selective enhancement of tumor blood flow with angiotensin II. J Natl Cancer Inst. 1981;67(3):663–669. | ||
Li CJ, Miyamoto Y, Kojima Y, Maeda H. Augmentation of tumour delivery of macromolecular drugs with reduced bone marrow delivery by elevating blood pressure. Br J Cancer. 1993;67(5):975–980. | ||
Lyons SA, O’Neal J, Sontheimer H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia. 2002;39(2):162–173. | ||
Huang R, Ke W, Han L, Li J, Liu S, Jiang C. Targeted delivery of chlorotoxin-modified DNA-loaded nanoparticles to glioma via intravenous administration. Biomaterials. 2011;32(9):2399–2406. | ||
Sun C, Veiseh O, Gunn J, et al. In vivo MRI detection of gliomas by chlorotoxin-conjugated superparamagnetic nanoprobes. Small. 2008;4(3):372–379. | ||
Mamelak AN, Rosenfeld S, Bucholz R, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24(22):3644–3650. | ||
Hockaday DC, Shen S, Fiveash J, et al. Imaging glioma extent with 131I-TM-601. J Nucl Med. 2005;46(4):580–586. | ||
Ying Z, Tojo H, Komatsubara T, et al. Enhanced expression of group II phospholipase A2 in human hepatocellular carcinoma. Biochim Biophys Acta. 1994;1226(2):201–205. | ||
Jiang J, Neubauer BL, Graff JR, et al. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am J Pathol. 2002;160(2):667–671. | ||
Lagorce-Pages C, Paraf F, Wendum D, Martin A, Flejou JF. Expression of inflammatory secretory phospholipase A2 and cytosolic phospholipase A2 in premalignant and malignant Barrett’s oesophagus. Virchows Arch. 2004;444(5):426–435. | ||
Lin MK, Farewell V, Vadas P, Bookman AA, Keystone EC, Pruzanski W. Secretory phospholipase A2 as an index of disease activity in rheumatoid arthritis. Prospective double blind study of 212 patients. J Rheumatol. 1996;23(7):1162–1166. | ||
Andersen S, Sjursen W, Laegreid A, Volden G, Johansen B. Elevated expression of human nonpancreatic phospholipase A2 in psoriatic tissue. Inflammation. 1994;18(1):1–12. | ||
Andresen TL, Davidsen J, Begtrup M, Mouritsen OG, Jorgensen K. Enzymatic release of antitumor ether lipids by specific phospholipase A2 activation of liposome-forming prodrugs. J Med Chem. 2004;47(7): 1694–1703. | ||
Jørgensen K, Vermehren C, Mouritsen OG. Enhancement of phospholipase A2 catalyzed degradation of polymer grafted PEG-liposomes: effects of lipopolymer-concentration and chain-length. Pharm Res. 1999;16(9):1491–1493. | ||
Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–8930. | ||
Oda T, Sato F, Maeda H. Facilitated internalization of neocarzinostatin and its lipophilic polymer conjugate, SMANCS, into cytosol in acidic pH. J Natl Cancer Inst. 1987;79(6):1205–1211. | ||
Griset AP, Walpole J, Liu R, Gaffey A, Colson YL, Grinstaff MW. Expansile nanoparticles: synthesis, characterization, and in vivo efficacy of an acid-responsive polymeric drug delivery system. J Am Chem Soc. 2009;131(7):2469–2471. | ||
Tian WM, Zhang CL, Hou SP, et al. Hyaluronic acid hydrogel as Nogo-66 receptor antibody delivery system for the repairing of injured rat brain: in vitro. J Control Release. 2005;102(1):13–22. | ||
Yamashiro DJ, Fluss SR, Maxfield FR. Acidification of endocytic vesicles by an ATP-dependent proton pump. J Cell Biol. 1983;97(3): 929–934. | ||
Yatvin MB, Kreutz W, Horwitz BA, Shinitzky M. pH-sensitive liposomes: possible clinical implications. Science. 1980;210(4475): 1253–1255. | ||
Yuba E, Tajima N, Yoshizaki Y, Harada A, Hayashi H, Kono K. Dextran derivative-based pH-sensitive liposomes for cancer immunotherapy. Biomaterials. 2014;35(9):3091–3101. | ||
Romberg B, Hennink WE, Storm G. Sheddable coatings for long-circulating nanoparticles. Pharm Res. 2008;25(1):55–71. | ||
Cerritelli S, Velluto D, Hubbell JA. PEG-SS-PPS: reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules. 2007;8(6):1966–1972. | ||
Behr JP. The proton sponge: a trick to enter cells the viruses did not exploit. CHIMIA International Journal for Chemistry. 1997; 51(1–2):1–2. | ||
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–418. | ||
Xu ZP, Niebert M, Porazik K, et al. Subcellular compartment targeting of layered double hydroxide nanoparticles. J Control Release. 2008; 130(1):86–94. | ||
Hoon Jeong J, Christensen LV, Yockman JW, et al. Reducible poly(amido ethylenimine) directed to enhance RNA interference. Biomaterials. 2007;28(10):1912–1917. | ||
Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11(6):990–995. | ||
Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos. 1995;16(5):351–380. | ||
Dressman J, Berardi R, Dermentzoglou L, et al. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res. 1990;7(7): 756–761. | ||
Peppas NA. Devices based on intelligent biopolymers for oral protein delivery. Int J Pharm. 2004;277(1–2):11–17. | ||
Colombo P, Sonvico F, Colombo G, Bettini R. Novel platforms for oral drug delivery. Pharm Res. 2009;26(3):601–611. | ||
Dai J, Nagai T, Wang X, Zhang T, Meng M, Zhang Q. pH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine A. Int J Pharm. 2004;280(1):229–240. | ||
Lee CH, Lo LW, Mou CY, Yang CS. Synthesis and characterization of positive-charge functionalized mesoporous silica nanoparticles for oral drug delivery of an anti-inflammatory drug. Adv Funct Mater. 2008; 18(20):3283–3292. | ||
Lin YH, Chang CH, Wu YS, Hsu YM, Chiou SF, Chen YJ. Development of pH-responsive chitosan/heparin nanoparticles for stomach-specific anti-Helicobacter pylori therapy. Biomaterials. 2009;30(19): 3332–3342. | ||
Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. | ||
Majno G, Shea SM, Leventhal M. Endothelial contraction induced by histamine-type mediators: an electron microscopic study. J Cell Biol. 1969;42(3):647–672. | ||
Hulstrom D, Svensjo E. Intravital and electron microscopic study of bradykinin-induced vascular permeability changes using FITC-dextran as a tracer. J Pathol. 1979;129(3):125–133. | ||
Lampugnani MG, Dejana E. Interendothelial junctions: structure, signalling and functional roles. Curr Opin Cell Biol. 1997;9(5): 674–682. | ||
Lamprecht A, Schäfer U, Lehr CM. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18(6):788–793. | ||
Kamata R, Yamamoto T, Matsumoto K, Maeda H. A serratial protease causes vascular permeability reaction by activation of the Hageman factor-dependent pathway in guinea pigs. Infect Immun. 1985;48(3): 747–753. | ||
Matsumoto K, Yamamoto T, Kamata R, Maeda H. Pathogenesis of serratial infection: activation of the Hageman factor-prekallikrein cascade by serratial protease. J Biochem. 1984;96(3):739–749. | ||
Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater. 2010;9(11):923–928. | ||
Foxall C, Watson SR, Dowbenko D, et al. The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J Cell Biol. 1992;117(4):895–902. | ||
Ulbrich H, Eriksson EE, Lindbom L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol Sci. 2003;24(12):640–647. | ||
Hashida N, Ohguro N, Yamazaki N, et al. High-efficacy site-directed drug delivery system using sialyl-Lewis X conjugated liposome. Exp Eye Res. 2008;86(1):138–149. | ||
Minaguchi J, Oohashi T, Inagawa K, Ohtsuka A, Ninomiya Y. Transvascular accumulation of Sialyl Lewis X conjugated liposome in inflamed joints of collagen antibody-induced arthritic (CAIA) mice. Arch Histol Cytol. 2008;71(3):195–203. | ||
Song L, Li H, Sunar U, et al. Naphthalocyanine-reconstituted LDL nanoparticles for in vivo cancer imaging and treatment. Int J Nanomedicine. 2007;2(4):767–774. | ||
Xu L, Pirollo KF, Tang WH, Rait A, Chang EH. Transferrin-liposome-mediated systemic p53 gene therapy in combination with radiation results in regression of human head and neck cancer xenografts. Hum Gene Ther. 1999;10(18):2941–2952. | ||
Hong G, Yuan R, Liang B, Shen J, Yang X, Shuai X. Folate-functionalized polymeric micelle as hepatic carcinoma-targeted, MRI-ultrasensitive delivery system of antitumor drugs. Biomed Microdevices. 2008;10(5):693–700. | ||
Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41(2):147–162. | ||
Zhang B, Li Y, Fang CY, et al. Receptor-mediated cellular uptake of folate-conjugated fluorescent nanodiamonds: a combined ensemble and single-particle study. Small. 2009;5(23):2716–2721. | ||
Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2002;54(5):675–693. | ||
Zhao X, Li H, Lee RJ. Targeted drug delivery via folate receptors. Expert Opin Drug Deliv. 2008;5(3):309–319. | ||
Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13(3):256–262. | ||
Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56(8):1177–1192. | ||
Shmeeda H, Mak L, Tzemach D, Astrahan P, Tarshish M, Gabizon A. Intracellular uptake and intracavitary targeting of folate-conjugated liposomes in a mouse lymphoma model with up-regulated folate receptors. Mol Cancer Ther. 2006;5(4):818–824. | ||
Turk MJ, Waters DJ, Low PS. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 2004;213(2):165–172. | ||
Osborn MJ, Freeman M, Huennekens FM. Inhibition of dihydrofolic reductase by aminopterin and amethopterin. Proc Soc Exp Biol Med. 1958;97(2):429–431. | ||
Kukowska-Latallo JF, Candido KA, Cao Z, et al. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005;65(12):5317–5324. | ||
Adams GP, Schier R, McCall AM, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750–4755. | ||
Tuscano JM, Martin SM, Ma Y, Zamboni W, O’Donnell RT. Efficacy, biodistribution, and pharmacokinetics of CD22-targeted pegylated liposomal doxorubicin in a B-cell non-Hodgkin’s lymphoma xenograft mouse model. Clin Cancer Res. 2010;16(10):2760–2768. | ||
Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65(24): 11631–11638. | ||
Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. | ||
Hadjipanayis CG, Machaidze R, Kaluzova M, et al. EGFRvIII antibody-conjugated iron oxide nanoparticles for magnetic resonance imaging-guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res. 2010;70(15):6303–6312. | ||
Yang W, Barth RF, Adams DM, et al. Convection-enhanced delivery of boronated epidermal growth factor for molecular targeting of EGF receptor-positive gliomas. Cancer Res. 2002;62(22):6552–6558. | ||
Pertuit D, Moulari B, Betz T, et al. 5-amino salicylic acid bound nanoparticles for the therapy of inflammatory bowel disease. J Control Release. 2007;123(3):211–218. | ||
Sakai T, Kohno H, Ishihara T, et al. Treatment of experimental autoimmune uveoretinitis with poly (lactic acid) nanoparticles encapsulating betamethasone phosphate. Exp Eye Res. 2006;82(4):657–663. | ||
Laroui H, Dalmasso G, Nguyen HTT, Yan Y, Sitaraman SV, Merlin D. Drug-loaded nanoparticles targeted to the colon with polysaccharide hydrogel reduce colitis in a mouse model. Gastroenterology. 2010; 138(3):843–853.e842. | ||
Koo OMY, Rubinstein I, Önyüksel H. Actively targeted low-dose camptothecin as a safe, long-acting, disease-modifying nanomedicine for rheumatoid arthritis. Pharm Res. 2011;28(4):776–787. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.