")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Nanofabrication of methylglyoxal with chitosan biopolymer: a potential tool for enhancement of its anticancer effect
Authors Pal A, Talukdar D, Roy A, Ray S, Mallick A, Mandal C, Ray M
Received 27 November 2014
Accepted for publication 31 January 2015
Published 12 May 2015 Volume 2015:10(1) Pages 3499—3518
DOI https://doi.org/10.2147/IJN.S78284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Aparajita Pal,1,* Dipa Talukdar,1,* Anirban Roy,1 Subhankar Ray,2 Asish Mallick,3 Chitra Mandal,3 Manju Ray1
1Department of Biophysics, Bose Institute, Kolkata, India; 2Department of Biochemistry, University of Calcutta, Kolkata, India; 3Cancer Biology and Inflammatory Disorder Division, Council of Scientific and Industrial Research (CSIR)-Indian Institute of Chemical Biology, Kolkata, India
*These authors contributed equally to this work
Purpose: The normal metabolite methylglyoxal (MG) specifically kills cancer cells by inhibiting glycolysis and mitochondrial respiration without much adverse effect upon normal cells. Though the anticancer property of MG is well documented, its gradual enzymatic degradation in vivo has prompted interest in developing a nanoparticulate drug delivery system to protect it and also to enhance its efficacy.
Materials and methods: MG-conjugated chitosan nanoparticles (Nano-MG) were prepared by conjugating the carbonyl group of MG with the amino group of chitosan polymer (Schiff’s base formation). Nano-MG were characterized in detail using the dynamic light scattering method, zeta potential measurement, Fourier transform infrared spectroscopy, and transmission electron microscopic analysis. Amount of MG anchored to Nano-MG, stability of Nano-MG, and in vitro release of MG from Nano-MG were estimated spectrophotometrically. Ehrlich ascites carcinoma (EAC) cells, human breast cancer cell line HBL-100, and lung epithelial adenocarcinoma cell line A549 were used as test systems to compare Nano-MG with bare MG in vitro. Cytotoxicity to EAC cells was evaluated by the trypan blue dye exclusion test, and cell viability of HBL-100 and A549 cells were studied using 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT) assay. Apoptosis of HBL-100 cells was assessed by flow cytometry and confocal microscopy. In vivo studies were performed on both EAC cells inoculated and also in sarcoma-180-induced solid tumor-bearing Swiss albino mice to assess the anticancer activity of Nano-MG in comparison to bare MG with varying doses, times, and administrative routes.
Results: Fourier transform infrared spectroscopy revealed the presence of imine groups in Nano-MG due to conjugation of the amino group of chitosan and carbonyl group of MG with diameters of nanoparticles ranging from 50–100 nm. The zeta potential of Nano-MG was +21 mV and they contained approximately 100 µg of MG in 1 mL of solution. In vitro studies with Nano-MG showed higher cytotoxicity and enhanced rate of apoptosis in the HBL-100 cell line in comparison with bare MG, but no detrimental effect on normal mouse myoblast cell line C2C12 at the concerned doses. Studies with EAC cells also showed increased cell death of nearly 1.5 times. Nano-MG had similar cytotoxic effects on A549 cells. In vivo studies further demonstrated the efficacy of Nano-MG over bare MG and found them to be about 400 times more potent in EAC-bearing mice and nearly 80 times more effective in sarcoma-180-bearing mice. Administration of ascorbic acid and creatine during in vivo treatments augmented the anticancer effect of Nano-MG.
Conclusion: The results clearly indicate that Nano-MG may constitute a promising tool in anticancer therapeutics in the near future.
Keywords: nano-methylglyoxal, anticancer agent, C2C12, HBL-100, A549, EAC, sarcoma-180, apoptosis
Introduction
Mortality due to cancer is rising at an alarming rate; cancer is the third leading cause of death worldwide and is projected to claim 13.1 million lives by 2030, which is nearly double the 7.6 million cancer-related deaths in 2008. Although chemotherapy is an important mode of cancer treatment, it had the disadvantage of widespread adverse side effects.1 This has prompted interest in the development of a specific, targeted anticancer drug with reduced toxicity. Advancements in nanotechnology may aid with the development and design of next-generation drugs with more efficient targeting and delivery strategies that are lacking in existing conventional chemotherapy.
The anticancer property of methylglyoxal (MG) has been well known for a long time.2–5 The metabolic pathway of this “enigmatic ketoaldehyde” has been firmly established and is in the biochemical map of intermediary metabolism.6 The beauty of MG, a normal metabolite, lies in the fact that it kills exclusively cancer cells by inhibition of glycolysis and mitochondrial respiration, leaving no adverse effect on normal cells.7,8 A detailed pharmacokinetic and toxicological study of MG confirmed that it is apparently devoid of any toxic effect.9 MG can also activate macrophages10 via superoxide and nitrite production through the MAPK/NF-κB signaling pathway.11 These findings paved the way for anticancer drug development using MG as a key component.12,13 A major problem with using MG as an anticancer drug is that MG, being a normal metabolite, is rapidly degraded by various enzymes present in the body.6,14,15 Hence, shielding MG with convenient nanoparticles may prevent this in vivo degradation, making it more competent as an anticancer drug.
Nanoparticles are taken up by various cells more readily and efficiently than larger micromolecules16 and are often used an an effective means of transport and potent delivery.17 Chitosan, a nontoxic cationic polysaccharide, generally derived from chitin by alkaline deacetylation, is widely used as a versatile, soft tissue-compatible particle for its biocompatibility, biodegradability, and natural origin.18,19 It has also been reported that chitosan nanoparticles inhibit the proliferation of human gastric carcinoma cell line MGC-803.20 Nanoparticles made of MG-loaded chitosan, prepared by Schiff’s base conjugation between chitosan and MG designated as Nano-MG,21 are expected to have sustained release of MG and hence improve its efficacy.
For more than a decade, various drugs such as paclitaxel,22 doxorubicin,23 5-fluorouracil,24 and dexamethasone25 have been successfully formulated using nanomaterials. Studies have shown regression of solid tumor and increased survival rates with doxorubicin-loaded chitosan nanoparticles in tumor-implanted rats,26 and that paclitaxel-loaded chitosan and glyceryl monooleate nanoparticles have excelled in sustained-release drug delivery.27 These findings inspired us to nanofabricate MG with chitosan biopolymer, thereby minimizing the amount of MG used and increasing its efficiency by prolonging its circulation time in blood.28
In the present study, we demonstrate the preparation of MG-loaded chitosan nanoparticles, their characterization, and their anticancer activity both in vitro and in vivo. These Nano-MG were more efficient and prepared in a more convenient way than our previously made Nano-MG.21 This preparation also takes less time, thus making it more acceptable for production in pharmaceutical industries. The in vitro effect of Nano-MG was tested on HBL-100, A549, and Ehrlich ascites carcinoma (EAC) cell lines. Ascorbic acid and creatine were administered in combination with Nano-MG and bare MG in murine models bearing EAC and sarcoma-180 cells to increase efficiency, as evidenced in our earlier observations.29
Materials and methods
MG (40% aqueous solution, w/v), low-molecular-weight chitosan, 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT), Sephadex G-50, trypan blue, cell culture media, and fetal bovine serum were purchased from Sigma Aldrich Co. (St Louis, MO, USA). C2C12, HBL-100, and A549 cell lines were obtained from American Type Culture Collection ([ATCC] Manassas, VA, USA). Filter paper (0.2 μm) was procured from Merck Millipore (Billerica, MA, USA). An annexin V–propidium iodide (PI) kit was purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Hoechst 33342 stain was procured from Calbiochem (La Jolla, CA, USA). All other chemicals and biochemicals were of analytical grade and purchased from Merck Millipore.
Synthesis of nanoparticles
Nano-MG were prepared by the method described previously,21 with some modifications. In brief, initially a 100 mL solution of 0.3% low-molecular-weight chitosan (~40 kD) in dilute acetic acid (34.8 mM) was prepared by sonication for 45 minutes at 37°C. To the above solution, 1 mL of MG was added under stirring condition followed by addition of 1 mL Tween 80 after 1 hour of stirring. Stirring was continued for another hour before adding 0.4 mL of 20% sodium sulfate solution dropwise. To crosslink the nanoparticles, 10 μL of 25% glutaraldehyde solution was added, with stirring for another 30 minutes. Finally, 1 mL of 10% sodium metabisulfite was added and the solution was stirred for another 30 minutes and kept overnight at room temperature. The Nano-MG solution was passed through a Sephadex G-50 column (20×1 cm) to eliminate small molecules using normal saline as eluent. The solution was strained through 0.2 μm-pore-size filter paper and preserved at room temperature for further characterization and therapeutic studies.
The same protocol was repeated with the chitosan molecules to prepare void chitosan nanoparticles (without MG) without sodium sulfate (to avoid precipitation of nanoparticles). This was designated as Nano-Chitosan.
Characterization
Fourier transform infrared (FT-IR) spectra of Nano-Chitosan and Nano-MG were recorded on an FT-IR spectrometer using a high-sensitivity pyroelectric detector (Spectrum 100; PerkinElmer Inc., Waltham, MA, USA). The sample pellets were prepared by grinding the lyophilized nanoparticles with KBr in a mortar at 1:100 ratios (w/w). The tablets were recovered with a clip and were analyzed in the wave number region from 4,000–400 cm−1.
The size distributions of these two nanoparticles in aqueous medium were determined directly using the dynamic light scattering technique using a Zetasizer 3000 (Malvern Instruments, Malvern, UK) in the automatic analysis mode at 25°C. The zeta potential of these two samples was also measured using the same Malvern Zetasizer instrument at the same temperature.
The sizes, ie, diameter and morphological images of Nano-Chitosan and Nano-MG were examined and recorded using a high-resolution transmission electron microscope (JEM 2010; JEOL, Tokyo, Japan) operating at an accelerating voltage of 200 kV. The samples were prepared by drop-casting on a carbon-coated copper grid and dried.
MG content anchored in Nano-MG was estimated spectrophotometrically using an ultraviolet-visible spectrophotometer (Cary Varian 50 scan UV–Vis optical spectrometer equipped with Cary Win UV software). The sample solution was treated with 5 M perchloric acid solution so that MG was released from the nanoparticles. It was then derivatized with 1,2-diaminobenzene to produce 2-methylquinozaline and was estimated spectrophotometrically by measuring the absorbance at 336 nm.9 The concentrations of perchloric acid and 1,2-diaminobenzene in the reaction mixture were 0.5 M and 1.8 mM, respectively. Similarly, five known concentrations of MG and their corresponding absorbances were recorded at 336 nm. A linear standard curve was obtained plotting known MG concentration versus absorbance. This was used to calculate the amount of MG conjugated to chitosan in Nano-MG. The authenticity of the method was confirmed by enzymatic assay of MG.30
For stability study of Nano-MG, MG content was measured at every month up to 6 months. Each time, a dialysis of Nano-MG solution was done against 0.9% NaCl solution for 1 hour. The amount of MG in both the dialyzed solution and dialysate was estimated spectrophotometrically as mentioned previously.
To study in vitro release of MG, 250 mL of Nano-MG was taken in a dialysis bag and was dialyzed against 1,000 mL of water at two different pH levels (pH 5.5 and pH 6.8). Aliquots of 1 mL were withdrawn from the release media (dialysate) at various time intervals and replenished with an equivalent amount of fresh water after each collection. Amounts of MG present in the withdrawn aliquots were estimated by the ultraviolet-visible spectrophotometer as mentioned previously.
Efficacy study
In vitro study
Cell culture
Mouse normal myoblast cell line (C2C12), human breast cancer cell line (HBL-100), and human lung cancer cell line (A549) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 μg/mL), streptomycin (100 μg/mL), and gentamycin (100 μg/mL) and grown in a 37°C humidified incubator containing 5% CO2.
Phase-contrast imaging
In all cases, cells were treated with various concentrations of MG (500, 750, and 1,000 μM) or Nano-MG (25, 37.5, and 50 μM) and kept for another 24 or 48 hours. The images were taken at 20× with an Olympus CKX-41 microscope (Olympus Corporation, Tokyo, Japan).
Cell viability analysis by MTT assay
The principle of MTT assay is based on the ability to cleave the tetrazolium ring of MTT by the mitochondrial dehydrogenase enzyme of viable cells forming dark blue insoluble formazan, which accumulates in living cells. The number of viable cells is directly proportional to the amount of the formazan product created. This formazan is then solubilized in a suitable solvent and absorbance is measured.31
For this assay, cells in their log phase were seeded overnight in colorless DMEM in 96-well tissue culture plates, each well having nearly 10,000 cells. The cells were then incubated with increasing concentrations of Nano-MG (6.25, 12.5, 25, 37.5, 50, 62.5, 75, 87.5, and 100 μM) and bare MG (125, 250, 500, 1,000, 1,250, 1,500, 1,750, and 2,000 μM) for different time intervals at 37°C. The medium was then replaced with 100 μL (0.5 mg/mL) MTT per well and further incubated for 4 hours. The insoluble formazan was dissolved in dimethyl sulfoxide (100 μL/well) and absorbance was measured using a 96-well plate reader (ELx 800; Biotek, Winooski, VT, USA) at 490 nm. The percentage of viability was calculated compared with the untreated control (considered as 100% viable).
Apoptosis assay by flow cytometry
HBL-100 cells were seeded overnight in six-well tissue culture plates at a density of 5×105 cells/well and were treated with various concentrations of bare MG (500, 750, and 1,000 μM) and Nano-MG (25, 37.5, and 50 μM) for 24 and 48 hours. Cells were trypsinized and doubly stained using an annexin V–fluorescein isothiocyanate (FITC) and PI kit (Cell Signaling Technology kit no 6592S) according to the manufacturer’s protocol for flow cytometric analysis of apoptotic populations. Briefly, 2 μL of annexin V–FITC and 8 μL of PI were added to the binding buffer in which the cells were suspended. Apoptosis data were acquired using a BD FACS Verse flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed using BD FACS DIVA software within 1 hour.
Confocal microscopy analysis
HBL-100 cells were harvested on cover slips in DMEM supplemented with 10% fetal bovine serum, penicillin, streptomycin, and gentamycin. After treatment with MG or Nano-MG for 24 hours, cells were gently washed with phosphate-buffered saline (PBS) and treated with 3.7% formaldehyde and kept in a 37°C incubator for 15 minutes. Cells were again washed with PBS, and 0.1% Triton X-100 was added to each cover slip and kept on a rocker for 15 minutes, followed by addition of Hoechst 33342 stain at a final concentration of 1 μg/mL. The samples were incubated at room temperature in the dark for 20 minutes and then were washed thrice very gently with PBS on a rocker. The cover slips were dried and mounted on glass slides with a drop of DPX mounting media and stored at 4°C before observation under a Leica model DM 900 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany). Digital images were captured with a cool (−25°C) charge-coupled device camera controlled with MetaMorph software (Universal Imaging, Bedford Hills, NY, USA).
In vitro study with EAC cells
EAC cells were maintained in the intraperitoneal cavity of mice.32 Briefly, cells were harvested for 8–10 days post-inoculation in female Swiss albino mice before being collected from the peritoneal cavity in 3 mL of normal saline (0.9% NaCl) and centrifuged (2,000× g) for 5 minutes at 4°C. The resulting pellets were washed again in normal saline before they were re-suspended in the requisite amount of PBS at pH 7.4. Cells (3×103) were treated with various combinations of 0.17 mM Nano-MG, 1 M MG, and 1 M ascorbic acid or with Nano-Chitosan in a final reaction volume of 1 mL and incubated at 37°C for 2 hours. The amount of chitosan in the Nano-Chitosan was the same as in 1 mL of 0.17 mM Nano-MG. A control tube with only EAC cells was maintained in a similar condition. Viable and dead cells were evaluated using the trypan blue dye exclusion method.33 To 1 mL of cell suspension, 0.1 mL of 0.4% trypan blue dye was added for staining before loading on a hemocytometer and counting under a microscope. In the case of Nano-Chitosan, it was found that, after 2 hours’ incubation at 37°C, 95% of EAC cells remained viable with normal morphology (data not shown), which is almost comparable to the control tube (97%). Hence, further in vitro cell culture studies with Nano-Chitosan were not performed.
In vivo study
Animal model
Swiss albino female mice aged 4–6 weeks and weighing ~20 g were maintained under standard laboratory conditions. The animal handling and animal experimental protocols were approved by the Institutional Animal Ethics Committee of Bose Institute, Kolkata, India.
Tumor induction by EAC cell line
EAC cells were maintained in the intraperitoneal cavity of mice as previously described.32 Cells were syringed out in normal saline from EAC -inoculated mice, resuspended in normal saline, and washed, then the cells were inoculated again into other healthy normal mice by intraperitoneal injection.
Induction of solid tumor by sarcoma-180 cell line
Sarcoma-180 cells were maintained in vivo by intraperitoneal inoculation into mice.21 For in vivo experiments, the sarcoma-180 cells that were maintained in the intraperitoneal cavity were inoculated intramuscularly in the left hind leg for the development of solid tumor. The tumor volumes of treated and untreated mice were measured with slide calipers.
Animal treatment
The experimental mice were inoculated with either EAC or sarcoma-180 cells. The mice that were inoculated with EAC cells in the peritoneal cavity received either bare MG or Nano-MG via two different routes, either intraperitoneal or intravenous, through the lateral tail vein. Mice bearing the sarcoma-180 cell-induced solid tumor were treated intravenously. The mice were divided into the following groups: 1) control group, which was inoculated with either EAC or sarcoma-180 cells and received only normal saline as placebo but no drug; 2) EAC or sarcoma-180 cell-inoculated group receiving various doses of either Nano-MG or MG (in sterile saline); 3) EAC or sarcoma-180 cell-inoculated group treated with Nano-Chitosan; and 4) normal mice treated with Nano-Chitosan. The EAC or sarcoma-180 cell-inoculated group receiving various doses of either Nano-MG or MG was fed orally with ascorbic acid and creatine as supplements. Each group contained six mice and received a single dose per day. The detailed treatment protocol with MG and Nano-MG is described in Table 1. Mice treated with Nano-Chitosan (~15 mg/kg body weight/day) had neither any anticancer activity nor any toxic effect at this dose (results not presented), which is in accordance with our previous observation.21
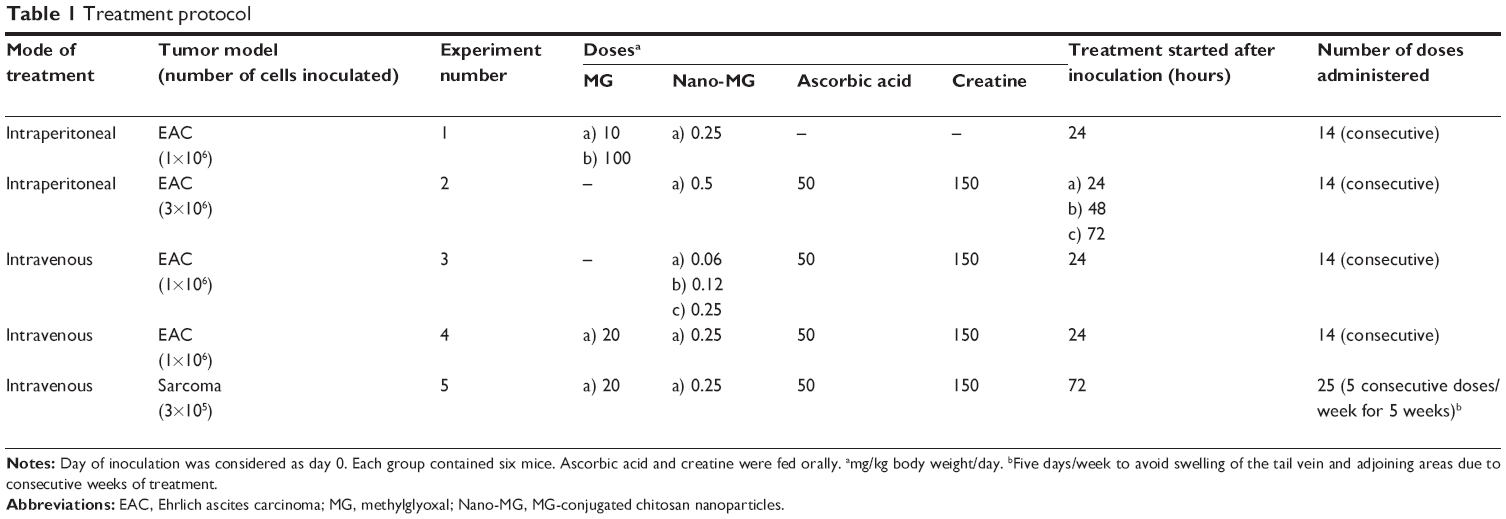 | Table 1 Treatment protocol |
Statistical analysis
Experiments were performed at least three times for in vitro and four times for in vivo studies. Results are expressed as mean ± standard deviation or Student’s t-test for significance (P<0.05 was considered significant).
Results
Physical characterization of the nanoparticles
The sizes of nanoparticles were measured by dynamic light scattering, which showed the polydispersed nature of the particles. Most of the particles had a diameter between 50 and 100 nm. A typical result showed that most of the Nano-MG particles had a diameter of approximately 74 nm with a polydispersity index value of 0.18 (Figure 1A), whereas most of the Nano-Chitosan particles were approximately 68 nm in diameter, having a polydispersity index value of 0.31 (Figure 1B).
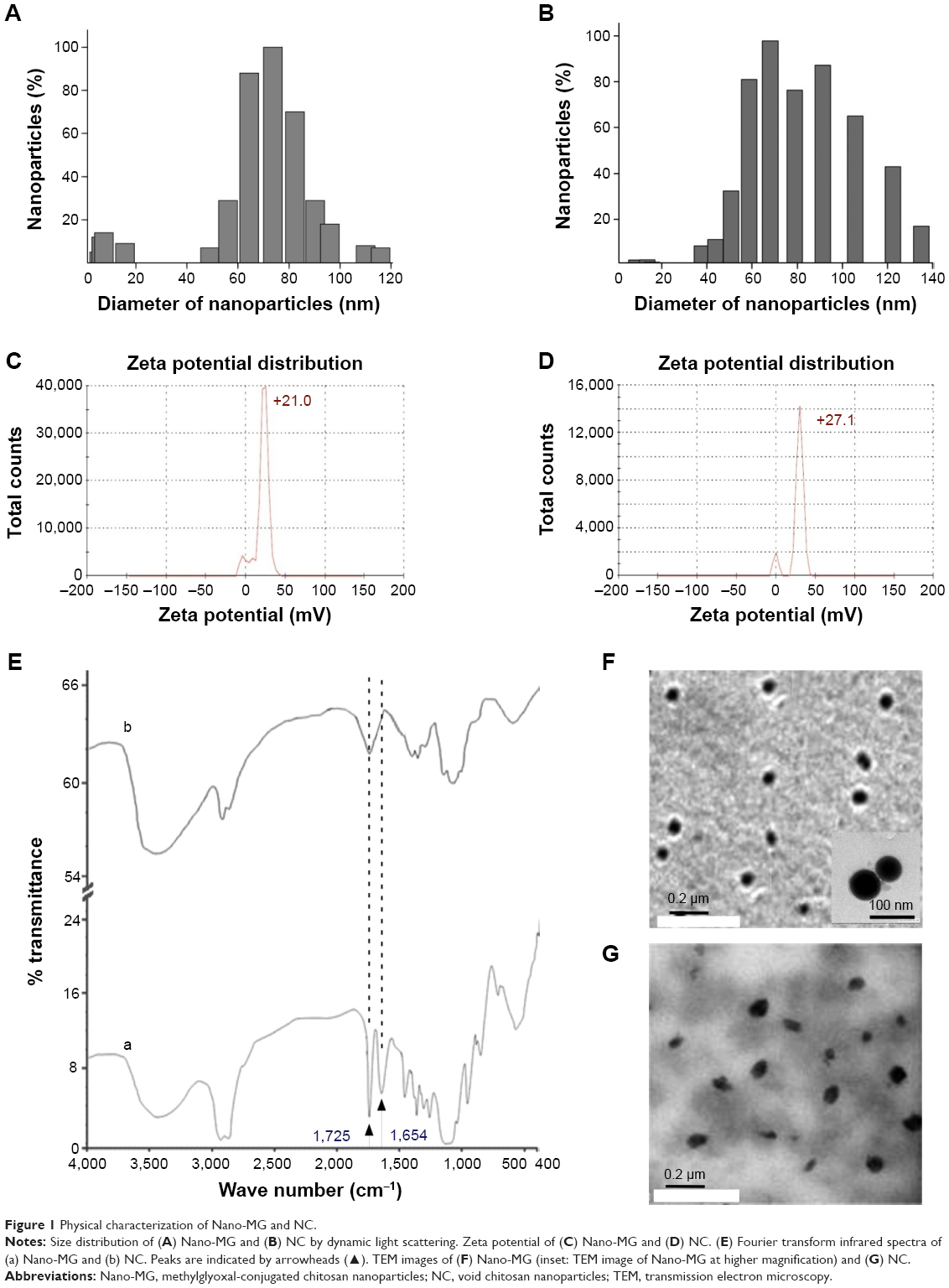 | Figure 1 Physical characterization of Nano-MG and NC. |
The zeta potential of Nano-MG was +21mV (Figure 1C) and that of Nano-Chitosan +27.1 mV (Figure 1D), which indicates the conjugation of MG in chitosan nanoparticles.
FT-IR spectroscopic analysis was done with both Nano-Chitosan and Nano-MG. Comparison between the infrared spectra of Nano-MG (Figure 1Ea) and Nano-Chitosan (Figure 1Eb) reveals a strong peak at 1,725 cm−1 which corresponds to the carbonyl group of MG, clearly indicating the CH3CO stretch. A new peak at 1,654 cm−1 corresponding to the imine group (–N=C<) formed due to Schiff’s base formation between the –NH2 group of chitosan and >C=O group of MG was also detected (Figure 1E).
Transmission electron microscope data of these two nanoparticles clearly show that the nanoparticle structure was almost spherical in shape and that the size distribution was similar to that found in the dynamic light scattering studies (Figure 1F and G). The amount of MG anchored in drug-loaded nanoparticles was calculated from a standard curve and was found to be ≈ 100 μg/mL.
In vitro release assay of MG from Nano-MG
In vitro release assay of free MG from Nano-MG revealed that, at pH 5.5, approximately 60% of MG was released within 10–12 hours, but, at pH 6.8, it remained quite stable (Figure 2). As the intracellular pH of cancer cells is acidic, it may be assumed that there will be an accelerated release of MG from Nano-MG at low pH.
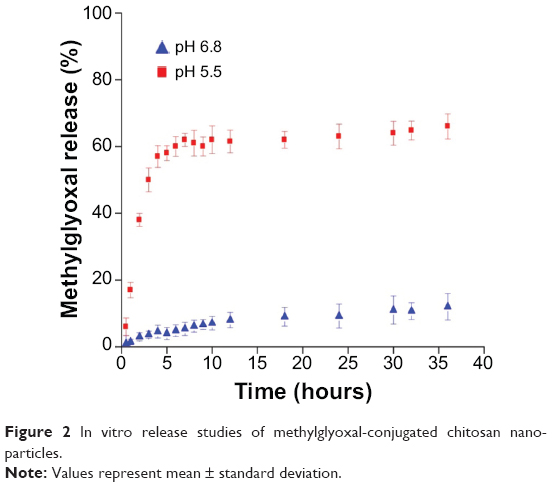 | Figure 2 In vitro release studies of methylglyoxal-conjugated chitosan nanoparticles. |
Stability study
The amount of MG bound to Nano-MG was estimated spectrophotometrically at 1-month intervals up to 6 months after Nano-MG was prepared. Nano-MG remained stable for up to 6 months at room temperature in 0.9% sodium chloride solution (pH ≈ 7.4).
Efficacy study
Effect of MG and Nano-MG on cell viability (in vitro studies)
Phase-contrast imaging and MTT assay
Significant morphological changes appeared in MG- and Nano-MG-treated HBL-100 cells as observed at 20× under a phase-contrast microscope. With increases in the doses of MG and Nano-MG, the changes appeared greater and indicated greater loss of viability compared to untreated control cells (Figure 3A).
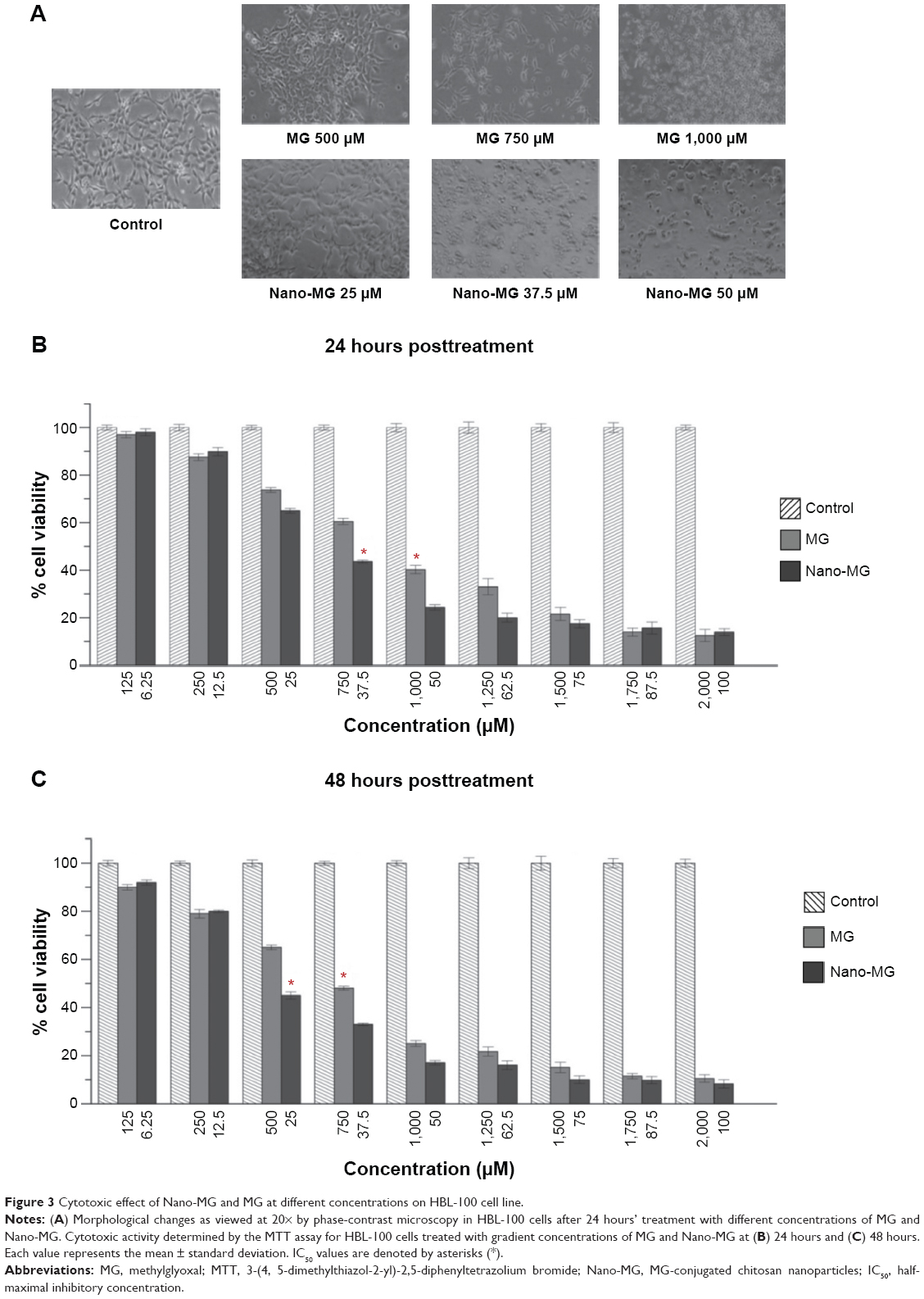 | Figure 3 Cytotoxic effect of Nano-MG and MG at different concentrations on HBL-100 cell line. |
MTT assay was performed to determine cell viability. Results from the MTT assay were in accordance with the phase-contrast images and demonstrated that both MG and Nano-MG were detrimental and significantly cytotoxic towards HBL-100 in a dose-dependent manner as observed 24 hours (Figure 3B) and 48 hours (Figure 3C) after treatment. Interestingly, the potency of Nano-MG was found to be far greater at the higher doses. MG in its nano form (6.25, 12.5, and 25 μM) was 20 times as potent as MG (125, 250, and 500 μM) in its bare form. At higher doses of Nano-MG (37.5 and 50 μM), the effectiveness was found to be as high as 35–40 times more compared to bare MG (750 and 1,000 μM). The potency or effectiveness is considered in respect to the relative amount of MG in its bare or nano form. The half-maximal inhibitory concentration (IC50) of MG and Nano-MG for HBL-100 was 1,000 μM and 37.5 μM, respectively, after 24 hours (Figure 3B). After 48 hours of treatment, the IC50 of MG and Nano-MG was found to be 750 μM and 25 μM, respectively (Figure 3C).
To determine the time of initiation of cell death, a time-dependent study was performed, which demonstrated the advent of cellular decease as early as 2 hours at higher doses of both MG and Nano-MG (Figure S1).
The cytotoxic nature of Nano-MG was not only restricted to the HBL-100 cell line, but A549 also showed a sharp decrease in cell viability with Nano-MG, which again demonstrates the anticancer activity of Nano-MG (Figure S2).
Flow cytometry analysis
We speculated after observing cell death by MTT assay that apoptosis was being induced by MG and Nano-MG treatments. We used annexin V-conjugated FITC and PI for analyzing early and late apoptotic cells. We observed a dose-dependent increase in the number of early and late apoptotic cells (Figure 4A). Treatment with 500, 750, and 1,000 μM bare MG showed 11.9%, 16%, and 18.8% of cells were early apoptotic (annexin V–FITC+/PI−) and 2.1%, 1.4%, and 4.3% of cells were late apoptotic (annexin V–FITC+/PI+), respectively (Figure 4A, middle row). Nano-MG treatment showed 27.9%, 32.9%, and 48.4% of cells were early apoptotic and 1.8%, 1.8%, and 2.3% of cells were late apoptotic at 25, 37.5, and 50 μM, respectively (Figure 4A, bottom row). Supporting our observed cell viability data, we found that MG encapsulated in chitosan is more effective at a far lower dose than in its bare form.
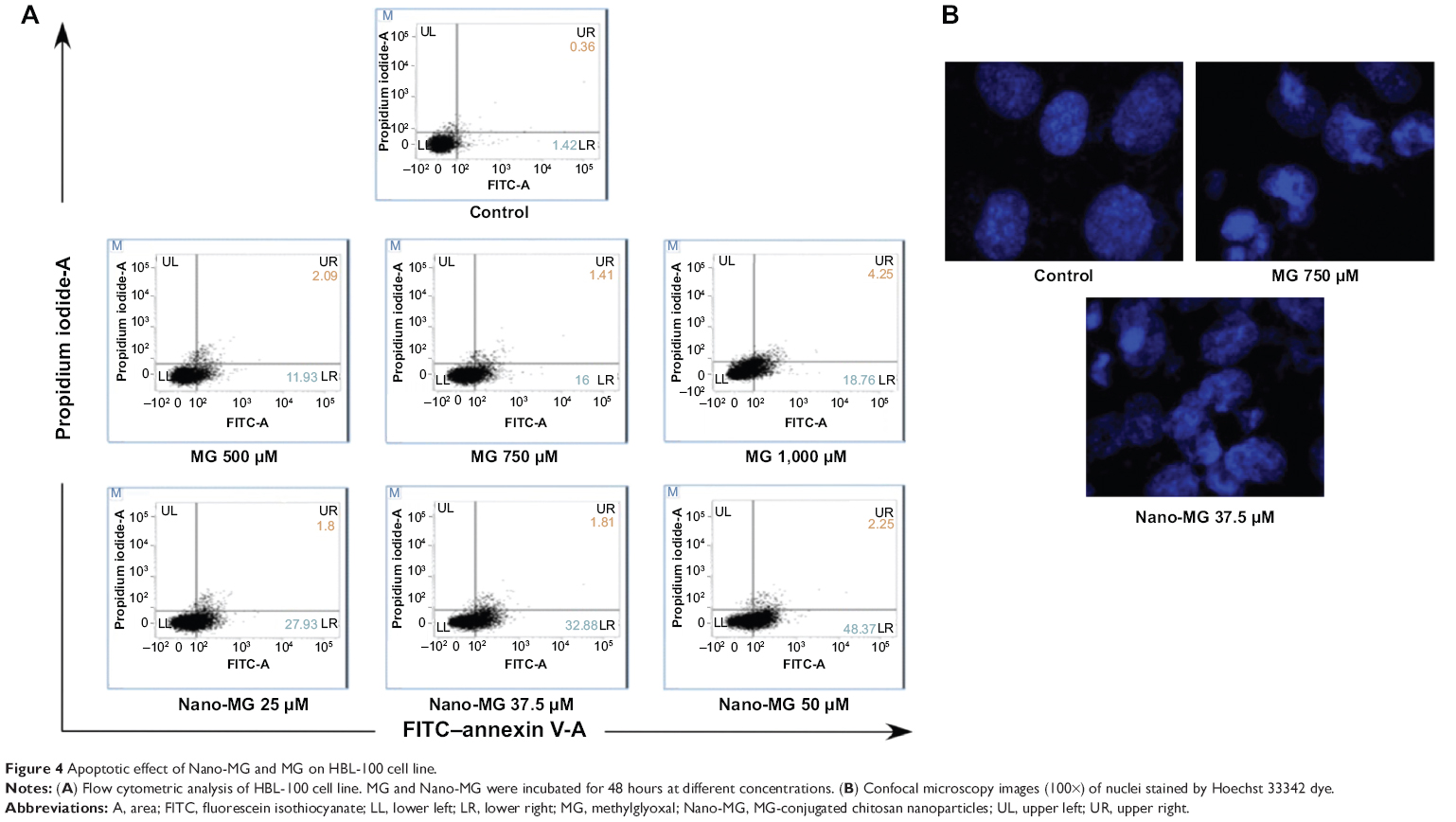 | Figure 4 Apoptotic effect of Nano-MG and MG on HBL-100 cell line. |
Confocal microscopy analysis
Confocal microscopy revealed apoptotic nuclei after MG and Nano-MG treatments in HBL-100 cells. It was clear from the images that nuclear destruction occurred after treatment with 750 μM of MG and 37.5 μM of Nano-MG (Figure 4B), which supports our previous results from flow cytometric analysis (Figure 4A). Confocal microscopy studies indicated an indirect proof of MG and Nano-MG uptake.
Effect of MG and Nano-MG on C2C12 (normal mouse myoblast) cell line
Though cytotoxic to malignant cell lines, these compounds had little toxic effect on normal cell line C2C12 (normal mouse myoblasts) as examined under phase-contrast microscopy (Figure 5A) and supported by MTT analysis. Approximately 90% of cells remained viable even when treated with 1,000 μM MG (Figure 5B) or 50 μM Nano-MG (Figure 5C).
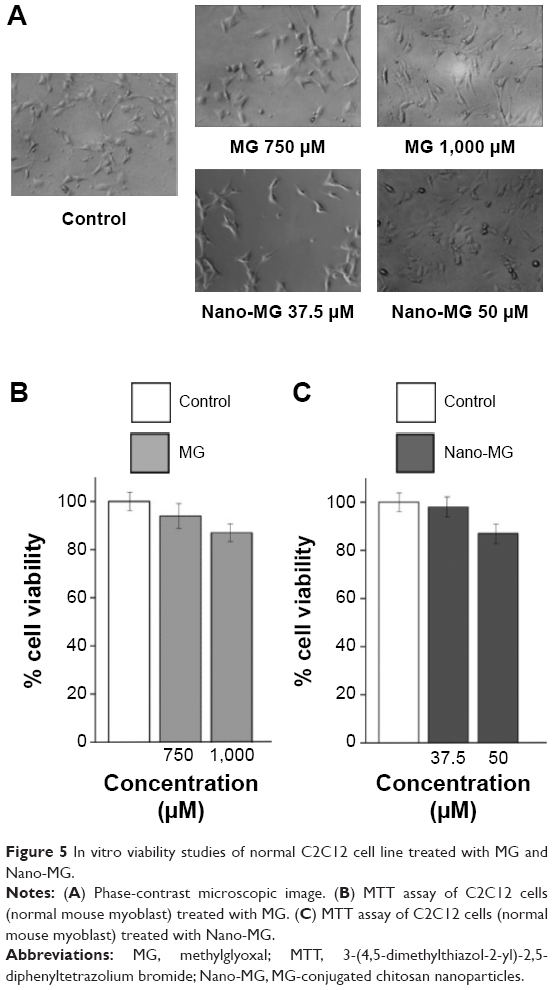 | Figure 5 In vitro viability studies of normal C2C12 cell line treated with MG and Nano-MG. |
Effect of MG and Nano-MG on EAC cells
The maximum inhibitory effect was observed when Nano-MG were combined with ascorbic acid, and it appeared that Nano-MG were by far more potent than bare MG. Nano-MG- and ascorbic acid-treated cells showed nearly 80% cell death and lost their transparency in comparison with MG and ascorbic acid, which showed nearly 50% cell death. In contrast, about 97% of the control or untreated cells were viable (Figure 6).
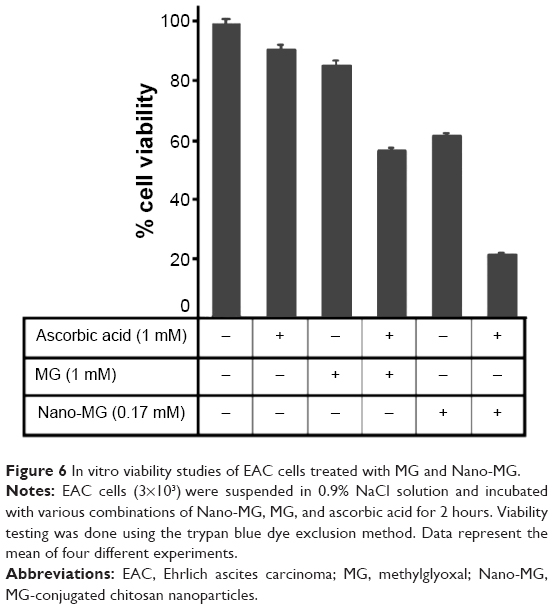 | Figure 6 In vitro viability studies of EAC cells treated with MG and Nano-MG. |
Effect of MG and Nano-MG on tumor inhibition (in vivo studies)
Effect of MG and Nano-MG on EAC-induced in vivo ascites
Nano-MG (0.25 mg/kg) administered at a 40 times lower dose than MG (10 mg/kg) via an intraperitoneal route demonstrated 300 times more potency in destroying malignant cells. To attain comparable efficacy with MG, a dose of 100 mg/kg must be administered by the same route, which is 400 times more than the amount of Nano-MG (Figure 7A and B).
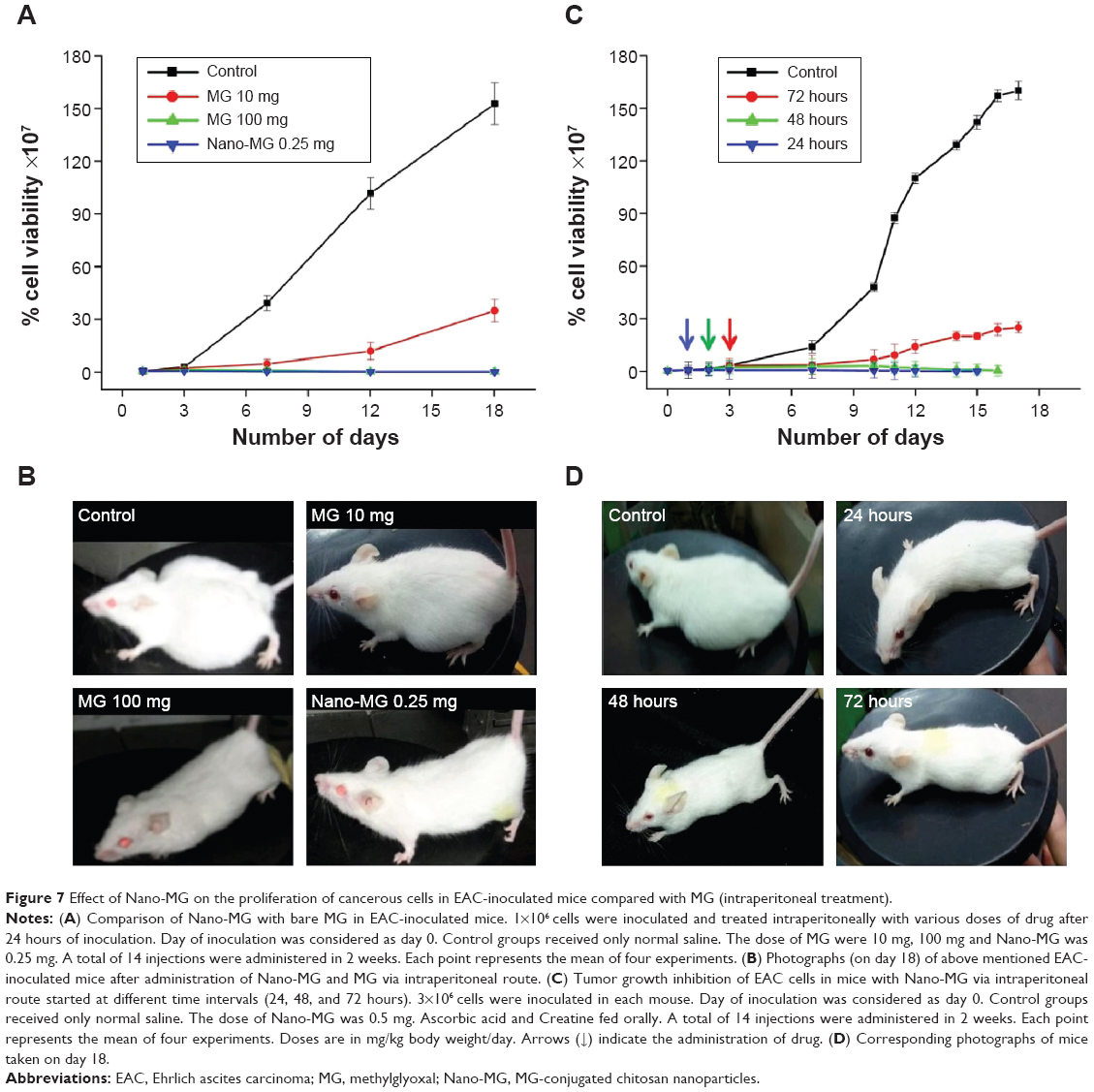 | Figure 7 Effect of Nano-MG on the proliferation of cancerous cells in EAC-inoculated mice compared with MG (intraperitoneal treatment). |
The efficacy of Nano-MG was assessed by varying the starting day of intraperitoneal treatment (24, 48, 72 hours) post-inoculation. The results indicate a comparable prognosis when treatment starts after 24 and 48 hours (Figure 7C and D).
A dose-dependent study of MG or Nano-MG administered via the intravenous route through the lateral tail vein of EAC-inoculated mice confirmed the efficacy of Nano-MG (Figure 8A). EAC-inoculated mice received 14 consecutive doses of Nano-MG (0.25 mg/kg) or 80 times higher doses of bare MG (20 mg/kg) via the intravenous route. Treatment with Nano-MG showed 300–400 times more effectiveness in comparison to bare MG, even at such a low dose (Figure 8B and C). It may be concluded, therefore, that Nano-MG are 300–400 times more effective than bare MG.
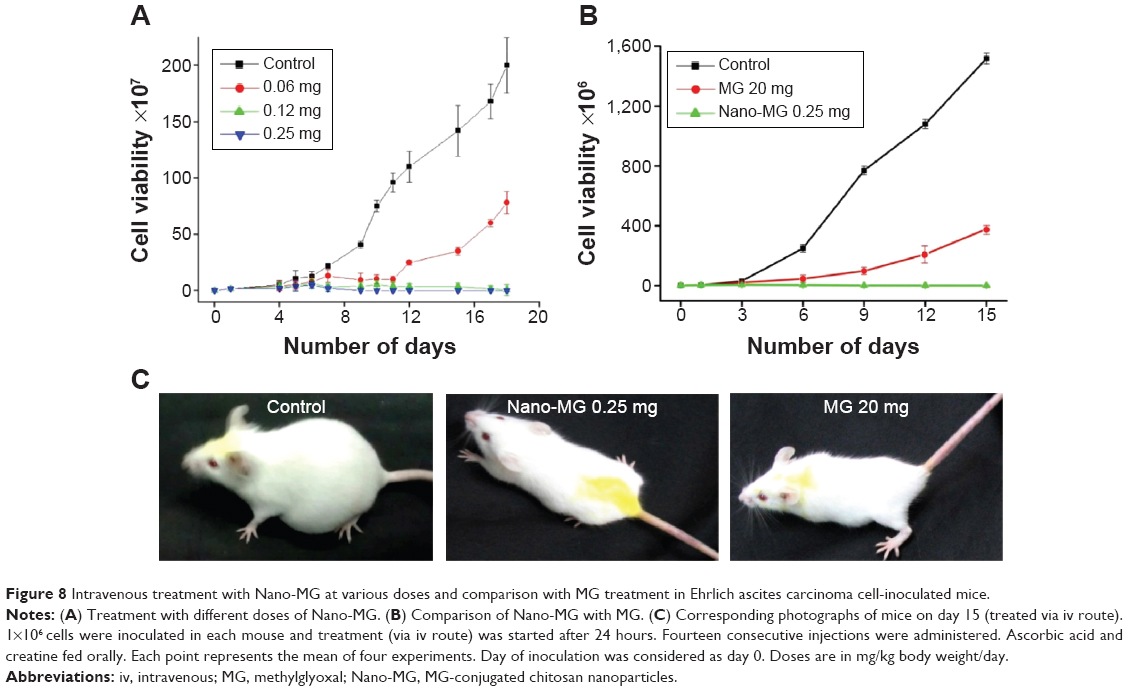 | Figure 8 Intravenous treatment with Nano-MG at various doses and comparison with MG treatment in Ehrlich ascites carcinoma cell-inoculated mice. |
Inclusion of ascorbic acid and creatine in the treatment regimen markedly enhanced the anticancer effect of Nano-MG up to 20 times, which supports our previous observation that creatine and ascorbic acid have a synergistic effect on MG-based anticancer treatment (Table S1).29 The regression of EAC cells in mice by intravenous treatment with Nano-MG was 20 times more compared to bare MG at an 80 times lower dose (Table S1).
Effect of MG and Nano-MG on sarcoma-180 cell line-induced solid tumor
Solid tumor developed by sarcoma-180 cells showed gradual regression when treated with Nano-MG (MG content 80 times lower than bare MG) or MG via the intravenous route compared to untreated mice. The tumor size of Nano-MG-treated mice was approximately 7.5 times less than that of controls (receiving normal saline) and approximately three times less than in bare MG-treated mice after 35 days of treatment (Figure 9A and B). Ascorbic acid and creatine were fed orally to mice treated with MG or Nano-MG.
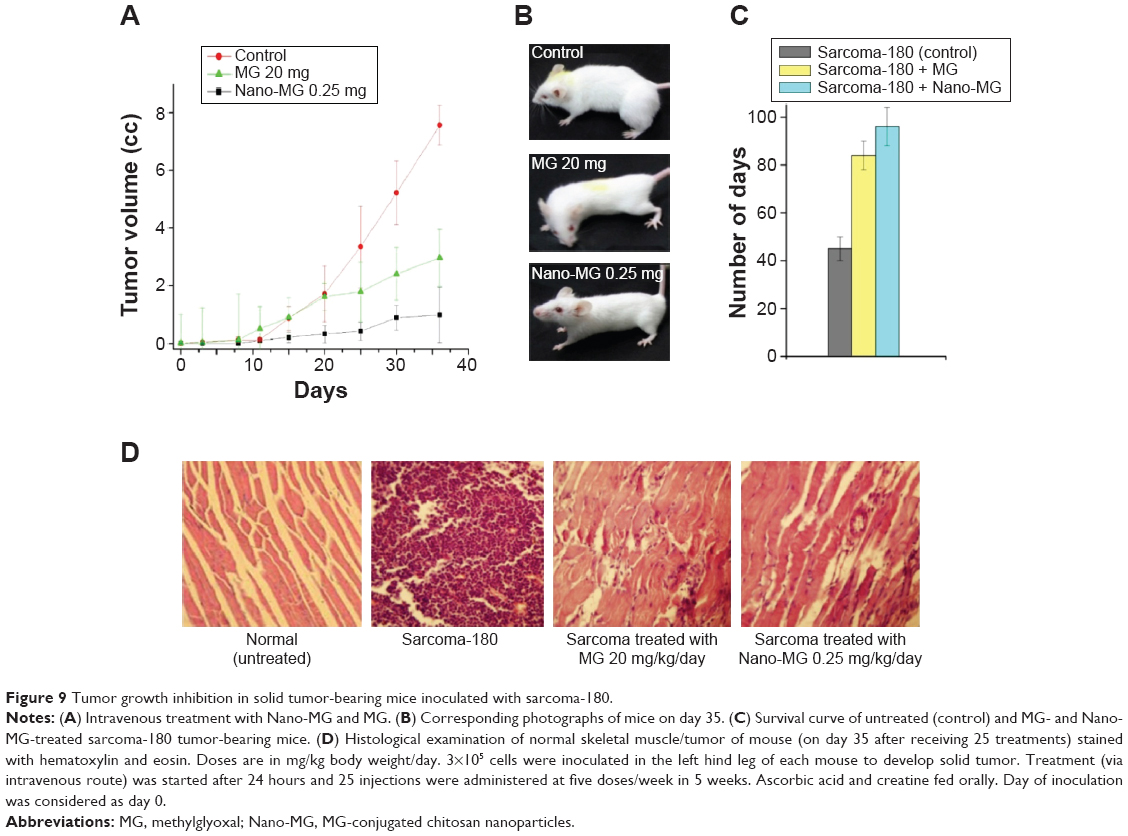 | Figure 9 Tumor growth inhibition in solid tumor-bearing mice inoculated with sarcoma-180. |
An increase in survivality of sarcoma-180-bearing mice was apparent in the Nano-MG-treated group compared to the control or bare MG-treated counterparts (Figure 9C).
Histological analysis of tumor tissue cross-sections showed degenerating fibers, whereas treatment with Nano-MG revived the normal tissue architecture to a large extent (Figure 9D).
Discussion
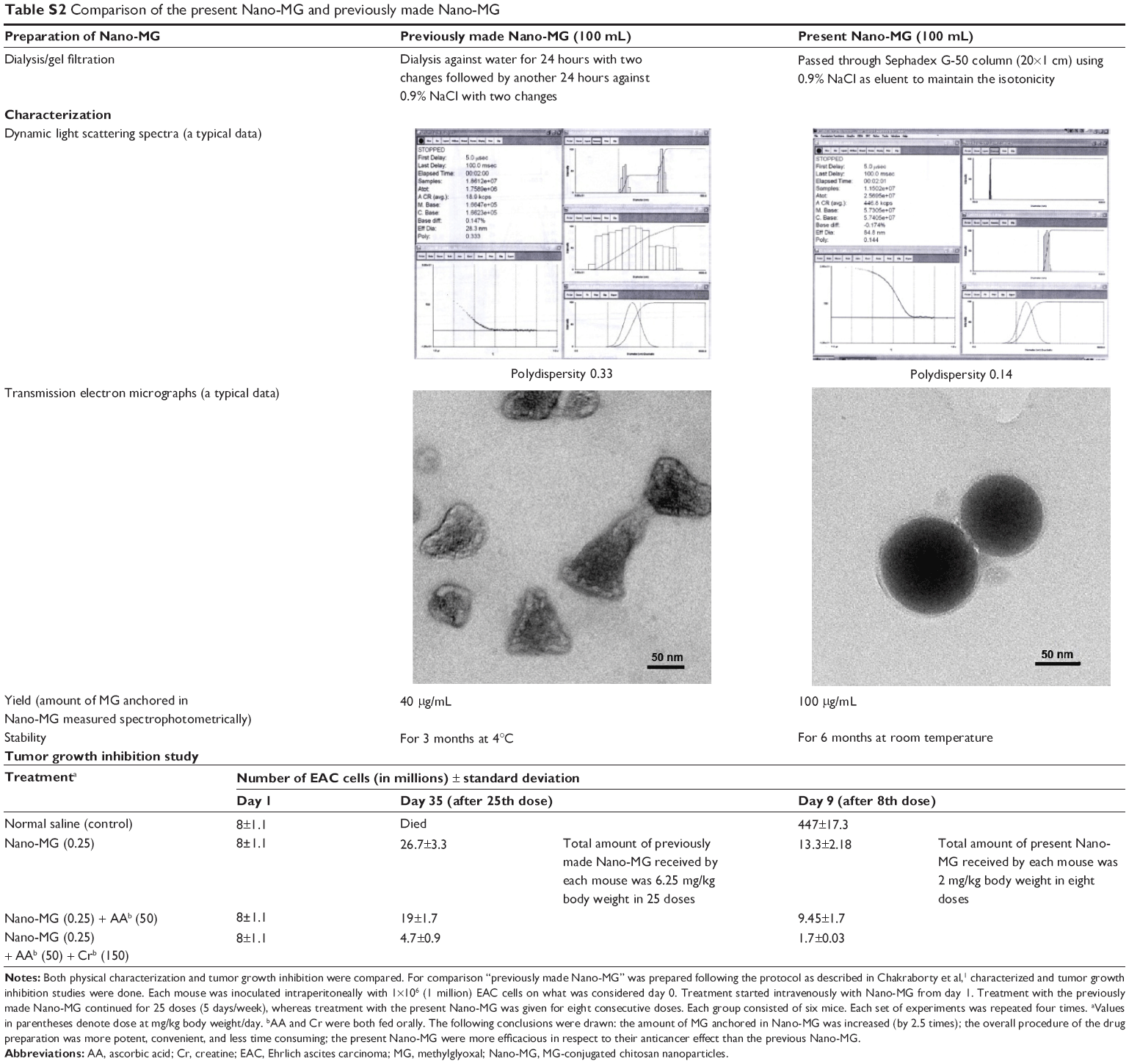
Favorable characteristics of chitosan such as nontoxicity, biodegradability, and biocompatibility and the potent anticancer property of MG justify the synthesis of MG-conjugated chitosan nanoparticles. The Nano-MG in this study were more potent, stable, and easier to prepare than previously made Nano-MG21 (Table S2) and proved to be a more effective cancer therapeutic drug. It is convincingly demonstrated (Table S2) that this Nano-MG killed three times more EAC cells (treating each mouse with eight consecutive doses receiving total 2 mg/kg body weight) compared to the previous Nano-MG (with 25 doses, 5 days/week, receiving in total 6.25 mg/kg body weight).21
Physical characterization showed that stable nanoparticles had a diameter up to 100 nm, which may facilitate extravasation from systemic circulation to tumor tissue. Chitosan acts as a carrier for controlled and sustained intracellular release of MG, thereby may help to retain it within cells and provide protection from in vivo enzymatic degradation making Nano-MG more tumoricidal. Increased release of MG from Nano-MG at lower pH ensures higher activity of Nano-MG in the acidic intracellular environment of cancer cells.
The in vitro efficacy of Nano-MG over MG was also studied. Phase contrast-imaging revealed cytotoxicity, which was further confirmed by cell viability data obtained by the MTT assay, indicating its heightened efficacy on HBL-100 and A549 cell lines with no detrimental effect on C2C12 cells. This also supports our previous observation that MG can kill cancer cells by depleting energy production through inhibition of both glycolysis8,34 and mitochondrial respiration35,36 of malignant cells without affecting normal cells.7,35,36 The apoptotic potential of Nano-MG has also been established in this study. HBL-100 cells were shown to be nearly twice more prone to apoptosis when treated with Nano-MG where the amount of MG was 40 times lower than MG in bare form. The strong apoptotic potential of MG is indicative of an underlying signaling regulation, which should make for interesting study in future. Any cancer therapeutic is incomplete unless it is tested in an animal model. Therefore, a series of experiments was performed in a mouse model to confirm the efficacy of Nano-MG in vivo. In EAC-bearing mice, this formulation based on Nano-MG showed an excellent result in comparison with bare MG. Drug administration was performed via both intraperitoneal and intravenous routes. Nano-MG were more effective (nearly 400 times) than bare MG in the case of intraperitoneal treatment and ≈ 40 times more effective in intravenous treatment at an 80 times lower dose. The intravenous route is preferable from the therapeutic point of view.
It is very much critical for an anticancer drug to be able to combat solid tumor. The Nano-MG-based drug formulation also showed an excellent result in the case of solid tumor (in the sarcoma-180 model). Intravenous treatment with this drug reduced the tumor size nearly three times more than bare MG treated and eight times more than in the untreated mice.
It has also been reported that creatine and its analog cyclocreatine have anticancer properties.37,38 Moreover, it is reported in the literature that ascorbic acid augments the anticancer effects of MG.9 Considering these facts and the beneficial effects of creatine and ascorbic acid, we included them in the treatment regimen along with Nano-MG. In every case of our in vivo treatments, ascorbic acid and creatine acted synergistically with Nano-MG and bare MG in agreement with our previous observation.29
Conclusion
In a nutshell, we can conclude that Nano-MG prepared by nanofabrication of chitosan polymer with MG has excellent anticancer activity that is augmented by creatine and ascorbic acid. This formulation is a potential candidate for the development of better anticancer therapeutics and might open up a new avenue in the treatment of cancer.
Acknowledgments
This research is funded by the Department of Science and Technology (DST) Nanomission, Government of India; Council of Scientific and Industrial Research (CSIR), Government of India; University Grants Commission (UGC), Government of India; and Lifecare Innovations Private Limited, Gurgaon, India.
Disclosure
The authors report no conflicts of interest in this work.
References
Corrie PG. Cytotoxic chemotherapy: clinical aspects. Medicine. 2008;36(1):24–28. | ||
Együd LG, Szent-Györgyi A. Cancerostatic action of methylglyoxal. Science. 1968;160:1140. | ||
Apple MA, Greenberg DM. Arrest of cancer in mice by therapy with normal metabolites. II. Indefinite survirors among mice treated with mixtures of 2-oxopropanal (NSC-79019) and 2,3-dihydroxypropanal (NSC67934). Cancer Chemother Rep. 1968;52(7):687–696. | ||
Conroy PJ. Carcinostatic activity of methylglyoxal and related substances in tumour-bearing mice. Ciba Found Symp. 1978;67:271–300. | ||
Elvin P, Slater TF. Anti-tumour activity of novel adducts of ascorbic acid with aldehydes. Eur J Cancer Clin Oncol. 1981;17:759–765. | ||
Ray M, Ray S. Methylglyoxal: from a putative intermediate of glucose breakdown to its role in understanding that excessive ATP formation in cells may lead to malignancy. Curr Sci. 1998;75:103–113. | ||
Biswas S, Ray M, Misra S, Dutta DP, Ray S. Selective inhibition of mitochondrial respiration and glycolysis in human leukaemic leukocytes by methylglyoxal. Biochem J. 1997;323:343–348. | ||
Halder J, Ray M, Ray S. Inhibition of glycolysis and mitochondrial respiration of Ehrlich ascites carcinoma cells by methylglyoxal. Int J Cancer. 1993;54:443–449. | ||
Ghosh M, Talukdar D, Ghosh S, Bhattacharyya N, Ray M, Ray S. In vivo assessment of toxicity and pharmacokinetics of methylglyoxal. Augmentation of the curative effect of methylglyoxal on cancer-bearing mice by ascorbic acid and creatine. Toxicol Appl Pharmacol. 2006;212:45–58. | ||
Bhattacharyya N, Pal A, Patra S, Haldar AK, Roy S, Ray M. Activation of macrophages and lymphocytes by methylglyoxal against tumor cells in the host. Int Immunopharmacol. 2008;8:1503–1512. | ||
Pal A, Bhattacharya I, Bhattacharya K, Mandal C, Ray M. Methylglyoxal induced activation of murine peritoneal macrophages and surface markers of T lymphocytes in sarcoma-180 bearing mice: involvement of MAP kinase, NF-kappa beta signal transduction pathway. Mol Immunol. 2009;46:2039–2044. | ||
Talukdar D, Ray S, Das S, Jain AK, Kulkarni A, Ray M. Treatment of a number of cancer patients suffering from different types of malignancies by methylglyoxal-based formulation: a promising result. Cancer Ther. 2006;4B:205–222. | ||
Talukdar D, Ray S, Ray M, Das S. A brief critical overview of the biological effects of methylglyoxal and further evaluation of a methylglyoxal-based anticancer formulation in treating cancer patients. Drug Metabol Drug Interact. 2008;23:175–210. | ||
Inoue Y, Kimura A. Methylglyoxal and regulation of its metabolism in microorganisms. Adv Microb Physiol. 1995;37:177–227. | ||
Racker E. The mechanism of action of glyoxalase. J Biol Chem. 1951;190:685–696. | ||
Suri SS, Fenniri H, Singh B. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007;2:16. | ||
Chen SC, Wu YC, Mi FL, Lin YH, Yu LC, Sung HW. A novel pH-sensitive hydrogel composed of N,O-carboxymethyl chitosan and alginate cross-linked by genipin for protein drug delivery. J Control Release. 2004;96:285–300. | ||
Dev A, Binulal NS, Anitha A, et al. Preparation of poly(lactic acid)/chitosan nanoparticles for anti-HIV drug delivery applications. Carbohydr Polym. 2010;80(3):833–838. | ||
Kumar MN, Muzzarelli RA, Muzzarelli C, Sashiwa H, Domb AJ. Chitosan chemistry and pharmaceutical perspectives. Chem Rev. 2004;104:6017–6084. | ||
Qi LF, Xu ZR, Li Y, Jiang X, Han XY. In vitro effects of chitosan nanoparticles on proliferation of human gastric carcinoma cell line MGC803 cells. World J Gastroenterol. 2005;11(33):5136–5141. | ||
Chakrabarti A, Talukdar D, Pal A, Ray M. Immunomodulation of macrophages by methylglyoxal conjugated with chitosan nanoparticles against Sarcoma-180 tumor in mice. Cell Immunol. 2014;287:27–35. | ||
Fonseca C, Simões S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83(2):273–286. | ||
Yoo HS, Lee KH, Oh JE, Park TG. In vitro and in vivo anti-tumor activities of nanoparticles based on doxorubicin-PLGA conjugates. J Control Release. 2000;68(3):419–431. | ||
Bhadra D, Bhadra S, Jain S, Jain NK. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int J Pharm. 2003;257(1–2):111–124. | ||
Panyam J, Labhasetwar V. Sustained cytoplasmic delivery of drugs with intracellular receptors using biodegradable nanoparticles. Mol Pharm. 2004;1(1):77–84. | ||
Bisht S, Maitra A. Dextran-doxorubicin/chitosan nanoparticles for solid tumor therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:415–425. | ||
Trickler WJ, Nagvekar AA, Dash AK. A novel nanoparticle formulation for sustained paclitaxel delivery. AAPS Pharm Sci Tech. 2008;9(2):486–493. | ||
Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5:1909–1917. | ||
Patra S, Ghosh A, Roy SS, et al. A short review on creatine-creatine kinase system in relation to cancer and some experimental results on creatine as adjuvant in cancer therapy. Amino Acids. 2012;42:2319–2330. | ||
Cooper RA. Methylglyoxal synthase. Methods Enzymol. 1975;41: 502–508. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. | ||
Ray M, Halder J, Dutta SK, Ray S. Inhibition of respiration of tumor cells by methylglyoxal and protection of inhibition by lactaldehyde. Int J Cancer. 1991;47:603–609. | ||
Bekesi JG, Molnar Z, Winzler RJ. Inhibitory effect of d-glucosamine and other sugar analogs on the viability and transplantability of ascites tumor cells. Cancer Res. 1969;29:353–359. | ||
Ray M, Basu N, Ray S. Inactivation of glyceraldehyde-3-phosphate dehydrogenase of human malignant cells by methylglyoxal. Mol Cell Biochem. 1997;177:21–26. | ||
Ray S, Biswas S, Ray M. Similar nature of inhibition of mitochondrial respiration of heart tissue and malignant cells by methylglyoxal. A vital clue to understand the biochemical basis of malignancy. Mol Cell Biochem. 1997;171:95–103. | ||
Ray S, Dutta S, Halder J, Ray M. Inhibition of electron flow through complex I of the mitochondrial respiratory chain of Ehrlich ascites carcinoma cells by methylglyoxal. Biochem J. 1994;303:69–72. | ||
Miller EE, Evans AE, Cohn M. Inhibition of rate of tumor growth by creatine and cyclocreatine. Proc Natl Acad Sci U S A. 1993;90:3304–3308. | ||
Kristensen CA, Askenasy N, Jain RK, Koretsky AP. Creatine and cyclocreatine treatment of human colon adenocarcinoma xenografts: 31P and 1H magnetic resonance spectroscopic studies. Br J Cancer. 1999;79(2):278–285. |
Supplementary materials
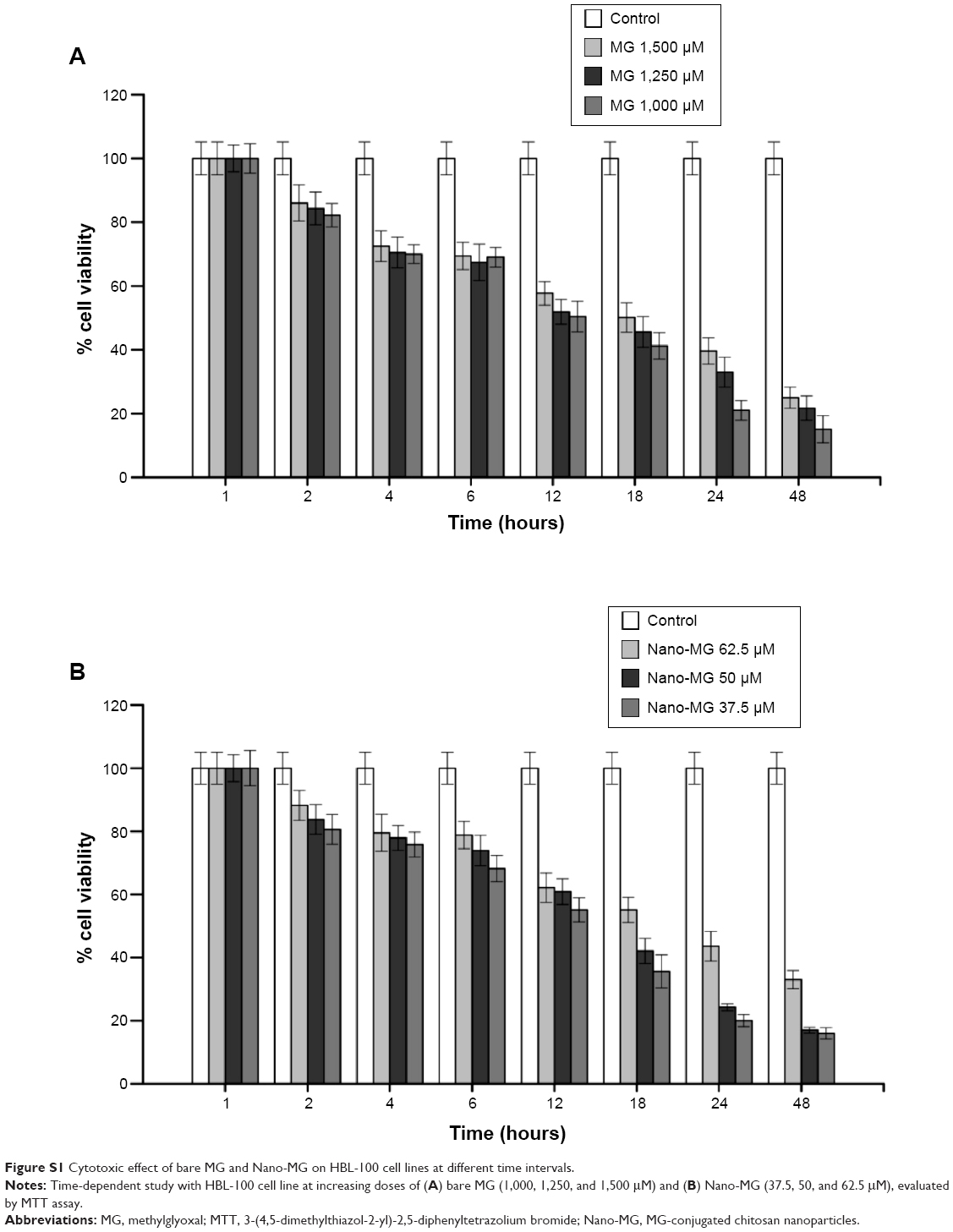 | Figure S1 Cytotoxic effect of bare MG and Nano-MG on HBL-100 cell lines at different time intervals. |
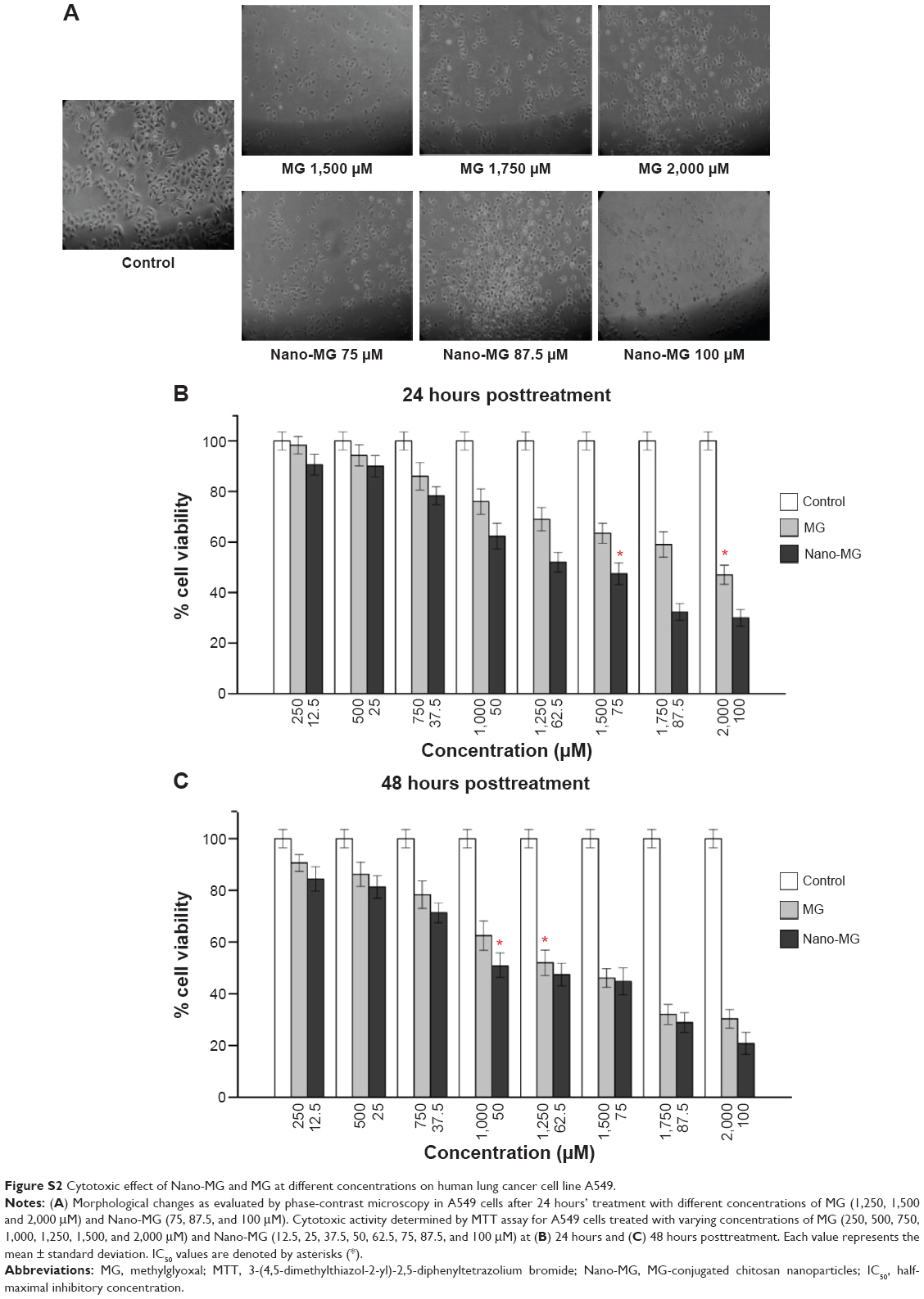 | Figure S2 Cytotoxic effect of Nano-MG and MG at different concentrations on human lung cancer cell line A549. |
 | Table S1 Efficacy study of Nano-MG (at various doses), ascorbic acid, and creatine and comparison with bare MG |
 | Table S2 Comparison of the present Nano-MG and previously made Nano-MG |
Reference
Chakrabarti A, Talukdar D, Pal A, Ray M. Immunomodulation of macrophages by methylglyoxal conjugated with chitosan nanoparticles against Sarcoma-180 tumor in mice. Cell Immunol. 2014;287:27–35. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.