Back to Journals » International Journal of Nanomedicine » Volume 11
Nanodiamonds coupled with 5,7-dimethoxycoumarin, a plant bioactive metabolite, interfere with the mitotic process in B16F10 cells altering the actin organization
Authors Gismondi A, Nanni V, Reina G, Orlanducci S, Terranova ML ![]() , Canini A
, Canini A
Received 18 September 2015
Accepted for publication 29 October 2015
Published 3 February 2016 Volume 2016:11 Pages 557—574
DOI https://doi.org/10.2147/IJN.S96614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Angelo Gismondi,1 Valentina Nanni,1 Giacomo Reina,2 Silvia Orlanducci,2 Maria Letizia Terranova,2 Antonella Canini1
1Department of Biology, 2Department of Chemical Science and Technology, University of Rome “Tor Vergata”, Rome, Italy
Abstract: For the first time, we coupled reduced detonation nanodiamonds (NDs) with a plant secondary metabolite, citropten (5,7-dimethoxycoumarin), and demonstrated how this complex was able to reduce B16F10 tumor cell growth more effectively than treatment with the pure molecule. These results encouraged us to find out the specific mechanism underlying this phenomenon. Internalization kinetics and quantification of citropten in cells after treatment with its pure or ND-conjugated form were measured, and it was revealed that the coupling between NDs and citropten was essential for the biological properties of the complex. We showed that the adduct was not able to induce apoptosis, senescence, or differentiation, but it determined cell cycle arrest, morphological changes, and alteration of mRNA levels of the cytoskeletal-related genes. The identification of metaphasic nuclei and irregular disposition of β-actin in the cell cytoplasm supported the hypothesis that citropten conjugated with NDs showed antimitotic properties in B16F10 cells. This work can be considered a pioneering piece of research that could promote and support the biomedical use of plant drug-functionalized NDs in cancer therapy.
Keywords: citropten, cytoskeletal structure, plant secondary metabolite, melanoma, internalization kinetics
Introduction
Detonation nanodiamonds (DNDs) are nanoparticles produced by detonation of explosive carbon materials. These compounds are characterized by a stable inert core, an octahedral symmetry, a rigid structure, the ability to form hydrogels, and a reactive surface area.1,2 Since DNDs have been demonstrated to be highly biocompatible, non-cytotoxic, and easily penetrable to mammalian cells, they represent excellent tools for biotechnological and medical applications.3–5 Indeed, nanodiamonds (NDs), conveniently functionalized or not, have been employed as diagnostic probes, antimicrobial agents, tissue scaffolds, protein purification matrixes, fluorescent biolabels, delivery systems for biomolecules, ions, and drugs, biosensors, contrast elements for biological imaging, ingredients of health care products, and anticancer agents.5–7 In particular, the successful use of DNDs in cancer treatment has greatly captured the attention of the scientific community.8–11 Although numerous plant compounds have been widely studied for their antitumor activities,12–15 to date only three papers describe the coupling of natural secondary metabolites with DNDs aimed at enhancing their bioavailability and bioactivity. The first of them illustrated the antimicrobial activity of menthol ((1R,2S,5R)-2-isopropyl-5-methylcyclohexanol)-modified ND particles.16 The other ones, published by our research group,17,18 reported the functionalization of three different types of DNDs with citropten (5,7-dimethoxycoumarin) or quercetin (3,3′,4′,5,6-pentahydroxyflavone), the chemical characterization of these adducts, and their antineoplastic effects on mammalian cancer cells. On the other hand, the association of DNDs with stilbenes (ie, resveratrol), a class of plant polyphenols, was recently proposed, but not yet realized, for the treatment of some pathologies, considering the great anti-radical and cardioprotective properties of these molecules.19 On the basis of our previous research,17 in order to promote and support the possible biomedical use of plant drugs conjugated with NDs in cancer therapy, we investigated the exact molecular mechanism of action of DNDs coupled with citropten. Moreover, for the first time, we performed a study on citropten internalization kinetics and the evaluation of its concentration in mammalian cells after exposure to the pure or ND-functionalized drug.
Materials and methods
ND synthesis and drug loading
NDs (4–5 nm crystallite primary size) were purchased from the Federal Research and Production Center “ALTAI” (Russia) and processed following the strategy widely described by Gismondi et al17 and in Reina et al.18 In particular, for the present experiments, plasma-reduced NDs, obtained as briefly described next, were used. The reduction of the purified DNDs by atomic H was accomplished in a purpose-designed plasma-assisted chemical vapor deposition reactor, where the gas phase was excited by a dual-mode microwave (MW)/radio frequency (RF) plasma.20 The two working frequencies were set at 2.45 GHz (MW) and 13.56 MHz (RF). The power of the RF and MW generators were fixed at 100 W. The chemical vapor deposition chamber was initially evacuated (to a pressure of 10−6 mbar) and then high-purity hydrogen was fluxed up to a final process pressure of 2.5 mbar. Under such experimental conditions, the substrate resulted negatively biased at Vb =250 V. Runs lasting 30 minutes were performed at T=150°C±5°C. The adducts with citropten (C; Sigma-Aldrich Co., St Louis, MO, USA) were prepared by an easy one-step synthetic protocol. Three milliliters of a 1.4 mM 5,7-dimethoxycoumarin solution in phosphate-buffered saline (PBS) was added to a ND dispersion (7 mg/1 mL PBS). The mixture was treated with pulsed ultrasound for 40 minutes. After removal of the supernatant, the solid products were purified from free molecules by cycles of PBS washing and centrifugation until a colorless supernatant was obtained. The obtained light gray-colored powder was finally collected in acetone and dried at room temperature for 24 hours. The total amount of molecules directly adsorbed on the surfaces of ND samples was measured ex post by UV–vis spectroscopy, following the procedure described by Gismondi et al.17 As reported by Reina et al,18 citropten essentially bound to NDs via low polar interactions (ie, p–p stacking), and the amount of the loaded drug was 3.2 μmol of C/mg of ND. In this work, naked and 5,7-dimethoxycoumarin-conjugated reduced DNDs are simply named ND and ND + C, respectively. All ND powders were resuspended in PBS solution for cell experiments.
Cell culture and treatments
Murine melanoma (B16F10) cells were cultivated as reported by Gismondi et al.21 Cells were treated for 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours with ND or functionalized ND + C solutions at two different concentrations (125 μg/mL and 200 μg/mL of culture medium), chosen according to the most interesting and promising data obtained in our previous work.17 Citropten resuspended in dimethyl sulfoxide (DMSO; Sigma-Aldrich Co.), in order to avoid the solvent concentration exceeding 0.1% during the treatment, was used at the final concentration of 400 μM and 640 μM for 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours. Control cells were treated with DMSO (control for citropten) or PBS (control for ND samples) at the same volume of the respective treatment. Doxorubicin (DOX; Sigma-Aldrich Co.), a well-known senescence inducer, was used at 0.2 μM for 72 hours. Phalloidin (PHL; Sigma-Aldrich Co.), a well-known inhibitor of actin depolymerization, was used at 12 μM for 72 hours.
Citropten internalization kinetics and quantification
Cells were plated in 24-well plates and treated with 125 μg/mL of pure ND or ND + C for 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours. The culture medium (1 mL) was collected and centrifuged at 13,000 rpm for 10 minutes to precipitate the NDs. The supernatant was spectrophotometrically (UVICON, Cary 50–Bio Varian) measured at 328 nm, the wavelength of maximum absorbance of the pure citropten, for the quantization of free C in the medium, while the pellet was resuspended in 1 mL of dimethylformamide (DMF, the appropriate solvent for citropten; Sigma-Aldrich Co.) and left under agitation for 24 hours in order to force the separation between the plant drug and ND. This last sample was then centrifuged at 13,000 rpm for 10 minutes to separate the ND residual from DMF containing ND-released citropten, which was analyzed at 328 nm. This purification was repeated twice, and all the absorbance outputs were added up to calculate the total quantity of C coupled with ND in the medium. On the other hand, cells were lysed in high-salt lysis buffer (100 μL), according to Gismondi et al,22 resuspended in a final volume of 1 mL of PBS, and centrifuged at 13,000 rpm for 10 minutes. The supernatant containing free intracellular C was measured at 328 nm. The pellet containing intracellular NDs (functionalized or not with C) was resuspended in DMF for 24 hours under agitation to facilitate the dissociation of C and NDs. This procedure was repeated three times, and all the supernatants were collected after centrifugation at 13,000 rpm for 10 minutes and analyzed at 328 nm. Then, other cells were treated with 400 μM of pure C for 0 hour (actually 1 minute), 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours and subjected to the same procedure as detailed earlier. Control cells treated with DMSO or PBS and ND (according to the case) were analyzed and used as blank samples for the respective measurements. In order to calculate the concentration of free and ND-released citropten in all media and cell samples, the absorbance values at 328 nm were directly compared with a calibration curve properly obtained using an adequate amount of pure C as standard. For the quantification of C concentration per single cell, the amount of plant molecules detected in the whole cell compartment was divided for the number of live cells present in the well. The count of cells was performed by a Neubauer modified chamber in a parallel well subjected to the same treatment of the sample under consideration.
Optical, confocal, and fluorescence microscopy
For morphological analysis, cell samples were directly observed in their plates with a Nikon Eclipse E100 optical microscope. To perform the confocal study, cells were prepared as reported by Gismondi et al.17 Nuclear labeling was carried out by treating the cells for 5 minutes with propidium iodide (PI; Thermo Fisher Scientific, Waltham, MA, USA) 10 μg/mL. The samples were observed by a confocal laser scanning microscope (FV1000 Olympus IX81 Microscopy; Olympus Corporation, Tokyo, Japan, Shinjuku, Tokyo, Japan). PI was revealed in blue by laser excitation at 543 nm; NDs were detected after exiting them by a laser at 635 nm and collecting their reflection in the red channel; while citropten, showed in green, was observed performing a lambda scan with lasers at 405 nm, 543 nm, and 635 nm. During the analysis, different scans of consequent optical sections (0.5 μm) were carried out at 60× magnification combined with 3× optical zoom. Two-dimensional reconstructions were produced by processing cell images using the IMARIS software (Bitplane, Zürich, Switzerland). For fluorescence observations, cells were processed as for confocal analysis. In particular, after fixation and permeabilization, cells were also exposed to a blocking solution (fetal bovine serum 10% in BSA; Sigma-Aldrich Co.) for 1 hour, treated with mouse monoclonal anti-β-actin (anti-ACT) primary antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for 1 hour, subjected to rhodamine TRITC-conjugated goat anti-mouse secondary antibody (Sigma-Aldrich Co.) for 1 hour, and finally stained with a 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich Co.) solution (60 μg/mL) for 5 minutes. Samples were observed by a Leica DMR microscope (Leica Microsystems, Wetzlar, Germany) provided with a Leica DFC 350 FX digital camera, an EBQ 100 isolated fluorescent lamp (Leistungselektronik Jena GmbH, Jena, Germany), UV/FITC/TRITC filters, and 40/63× objectives. The elaboration of the images and their merging were performed by the Leica Qwin Pro image analysis software.
Cell growth study, trypan blue exclusion test, and senescence measurement
Cell growth was measured by a 3-(4,5-dimethyl-thiazol- 2-yl)-2,5-diphenyltetrazolium bromide kit (Sigma-Aldrich Co.) according to the manufacturer’s instruction. Exclusion test to measure the cytotoxicity of the treatments was carried out as reported by Gismondi et al.17 Senescence was analyzed using a specific staining kit (Sigma-Aldrich Co.): the senescent cells, colored in blue, were detected by microscopic observation.
Cytofluorimetric and real-time quantitative polymerase chain reaction analysis
Cell cycle analysis by flow cytometry was carried out as described by Gismondi et al.23 For quantitative polymerase chain reaction (qPCR) investigation, after treatment the cells were harvested and the total RNA was extracted by the Pure Link RNA Mini Kit (Ambion; Thermo Fisher Scientific, Waltham, MA, USA). Then, cDNA was synthesized, PCR amplifications were performed, and the amount of mRNA for each gene was determined, as reported by Iadevaia et al.24 In particular, the primer pairs used in the present work are extensively described in Figure S1.
Western blotting
Proteins were extracted according to the protocol described by Serpinskaya et al,25 with appropriate modifications. Briefly, in order to separate filamentous actin (F) from the globular one (G), cells were lysed in the presence of a specific buffer (50 mM imidazole pH 6.8, 50 mM KCl, 0.5 mM MgCl2, 0.1 mM EDTA, 1 mM EGTA, 4% polyethylene glycol 4000, 1% triton X-100, 1× protease inhibitor cocktail; Sigma-Aldrich Co.) able to maintain intact the cytoskeletal structure, and maintained in ice for 15 minutes. The sample was then centrifuged for 15 minutes at 2,000 rpm. The supernatant containing G-actin was collected in a new Eppendorf tube. The pellet presenting the F-actin form was resuspended in the Laemmli-SDS loading buffer and boiled for 5 minutes at 95°C to destroy the membranes and the cytoskeleton. Protein extracts were quantified, separated on polyacrylamide gel, and analyzed as reported by Gismondi et al.23 Blots were first incubated with mouse monoclonal anti-ACT (Cell Signaling, Danvers, MA, USA) and mouse monoclonal anti-GAPDH (Merck Millipore, Billerica, MA, USA) primary antibodies, and then with horseradish peroxidase-conjugated anti-mouse antibody (Santa Cruz Biotechnology Inc.) in order to reveal protein signals.
Statistical analysis
All the experiments were repeated at least three times, and the results are reported as mean ± standard deviation (SD) of the different independent measurements. Significance value was calculated by one-way analysis of variance test, using the PAST software (P-values <0.05 were considered significant).
Results
NDs coupled with citropten inhibit B16F10 tumor cell growth without inducing cell death
In order to investigate whether the association of the citropten to NDs could modify the bioactivity of the plant compound, the cellular effects induced by pure C were compared with those resulting from cell exposure to an equal concentration of its ND-conjugated form (ND + C). Measurements of drug loading on NDs were carried out to quantify the maximum concentration of plant molecules that could be released under the best conditions during ND + C treatment. In particular, cell exposure to 125 μg and 200 μg of ND + C/mL of culture medium corresponded to a cell treatment with 400 μM and 640 μM solutions of pure C, respectively, according to the drug loading efficiency reported in “Materials and methods” section and widely described by Reina et al.18 By 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, the regulative effects of C, pure (ND), and functionalized (ND + C) NDs on B16F10 tumor cell growth were evaluated. According to the data obtained by Gismondi et al,17 all the treatments were performed for 72 hours at two different concentrations (125 μg/mL and 200 μg/mL for ND samples and 400 μM and 640 μM for pure citropten, as described earlier). Appropriate controls using PBS for pure NDs, NDs for ND-conjugated forms, and DMSO for pure C were always carried out (Figure 1A–C). The results are reported as percentage variation of the cell number of the sample compared to its specific control (considered as unit, 100%). As shown in Figure 1A, ND (200 μg/mL) did not significantly modify the cell growth rate with respect to the control (CNT PBS). Compared to CNT DMSO, pure C at concentrations of 400 μM and 640 μM caused the reduction of cell amount by 87.7% and 91.6%, respectively (Figure 1B).Finally (Figure 1C), ND + C, used at 125 μg/mL and 200 μg/mL, respectively, reduced the cell proliferation by 47.4% and 57.2%, with respect to pure ND treatment at the maximum concentration employed in the experiment (200 μg/mL). The cytotoxicity of each sample was also estimated by counting the dead cells in the exclusion test. With respect to the control (CNT PBS), which showed a minimum toxicity (3.3%), the treatments with CNT DMSO, ND (200 μg/mL), ND + C (125 μg/mL), ND + C (200 μg/mL), C 400 μM, and C 640 μM, respectively, showed toxicity values of 7.3%, 5.2%, 8.8%, 18.1%, 53.3%, and 77% (Figure 1D).
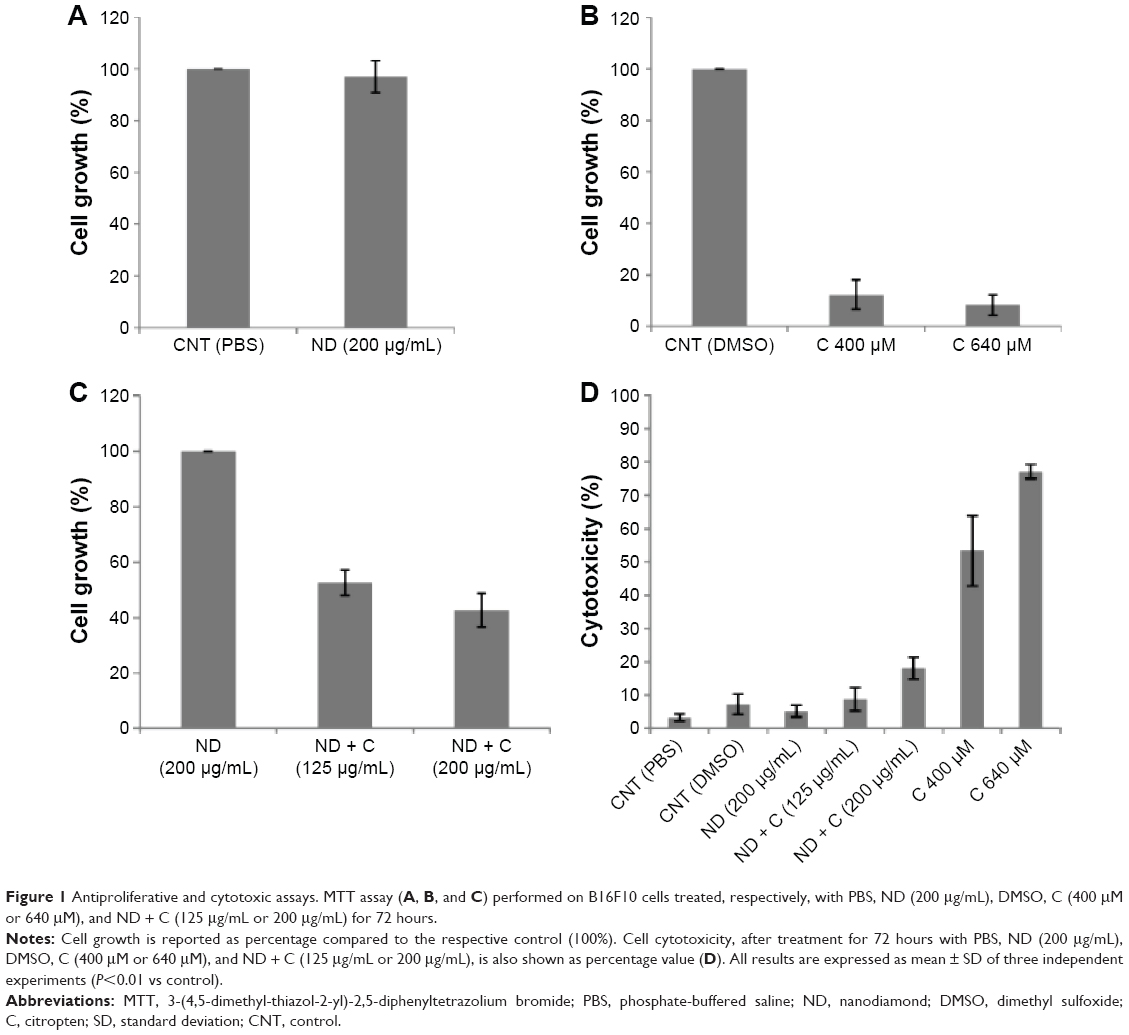 | Figure 1 Antiproliferative and cytotoxic assays. MTT assay (A, B, and C) performed on B16F10 cells treated, respectively, with PBS, ND (200 μg/mL), DMSO, C (400 μM or 640 μM), and ND + C (125 μg/mL or 200 μg/mL) for 72 hours. |
Internalization kinetics and quantification of pure and ND-conjugated citropten in B16F10 cells
B16F10 cells were treated for 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours with 400 μM of pure citropten or with the corresponding amounts of its ND-functionalized form (125 μg/mL). In the treatment with pure C (400 μM), the level of secondary metabolite detected in the culture medium after 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours was 391 μM, 334 μM, 312 μM,322 μM, 333 μM, and 363 μM, respectively. On the other hand, for the same times of treatment, the citropten concentrations found inside the cell compartment was equal to 0 μM, 60 μM, 82 μM, 75 μM, 62 μM, and 29 μM (Figure 2A). After exposing the cells for 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours to the pure plant drug, the citropten amount per cell was 0 nM, 0.22 nM, 0.35 nM, 0.62 nM, 1.15 nM, and 1.02 nM (Figure 2B). The concentrations of C measured in the cell medium after treatment for 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours with ND + C (125 μg/mL corresponding to 400 μM of pure citropten) were, respectively, 397 μM, 384 μM, 378 μM, 389 μM, 390 μM, and 389 μM. Otherwise, intracellular level of the drug was 0 μM, 11.67 μM, 11.74 μM, 6.25 μM, 3.62 μM, and 2.76 μM after treatments for 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours, respectively (Figure 2C). In this experiment, free citropten was also distinguished from the ND-conjugated one both in the culture medium and in the cell compartment. In particular, in the medium, the free C detectable was 15 μM, 335 μM, 320 μM, 343 μM, 333 μM, and 343 μM, while the concentrations of ND-coupled C were 382 μM, 48 μM, 58 μM, 45 μM, 56 μM, and 45 μM, respectively, at 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours of treatment (Figure 2D). On the other hand, after exposure to the plant metabolite for 0 hour, 6 hours, 8 hours, 24 hours, 48 hours, and 72 hours, respectively, the C concentration per cell was equal to 0 nM, 0.04 nM, 0.04 nM, 0.05 nM, 0.06 nM, and 0.09 nM (Figure 2E).In particular, inside each cell, corresponding to the same treatment times, free C levels were 0 nM, 0.035 nM, 0.038 nM, 0.027 nM, 0.032 nM, and 0.027 nM whereas the amounts of absorbed ND-linked C were 0.006 nM, 0.008 nM, 0.024 nM, 0.033 nM and 0.064 nM (Figure 2F). To verify whether the chemical conjunction between ND and C was necessary for the biological activity of the adduct, the previously described B16F10 cell growth was further analyzed after double treatment with separated ND (125 μg/mL or 200 μg/mL) and C (400 μM or 640 μM) (Figure 2G). Moreover, we carried out the same experiment after pretreating the cells with pure ND (125 μg/mL or 200 μg/mL) for 6 hours, which is the minimal time required for ND internalization in cellsbefore the exposure to pure C (400 μM or 640 μM) (Figure 2H). The results showed how, compared to that with CNT (PBS + DMSO), the treatments ND and C (125 μg/mL + 400 μM), ND and C (200 μg/mL + 640 μM), ND (6 hours) and C (125 μg/mL + 400 μM), and ND (6 hours) and C (200 μg/mL + 640 μM) reduced the proliferation, respectively, by 84.3%, 84.1%, 82.8%, and 84.9%. Trypan blue test was also performed on these samples and the results indicated that all the current treatments were cytotoxic for B16F10 cells as well as pure C treatment (data not shown). Confocal analysis, shown in Figure 3, confirmed the presence of the citropten molecules inside the cell cytoplasm after 72 hours of treatment with pure C (640 μM) or ND + C (200 μg/mL). In these images, the quantization of the green signal, which is directly proportional to the intracellular C amount, was notably higher in C-treated cells compared to those exposed to ND + C.
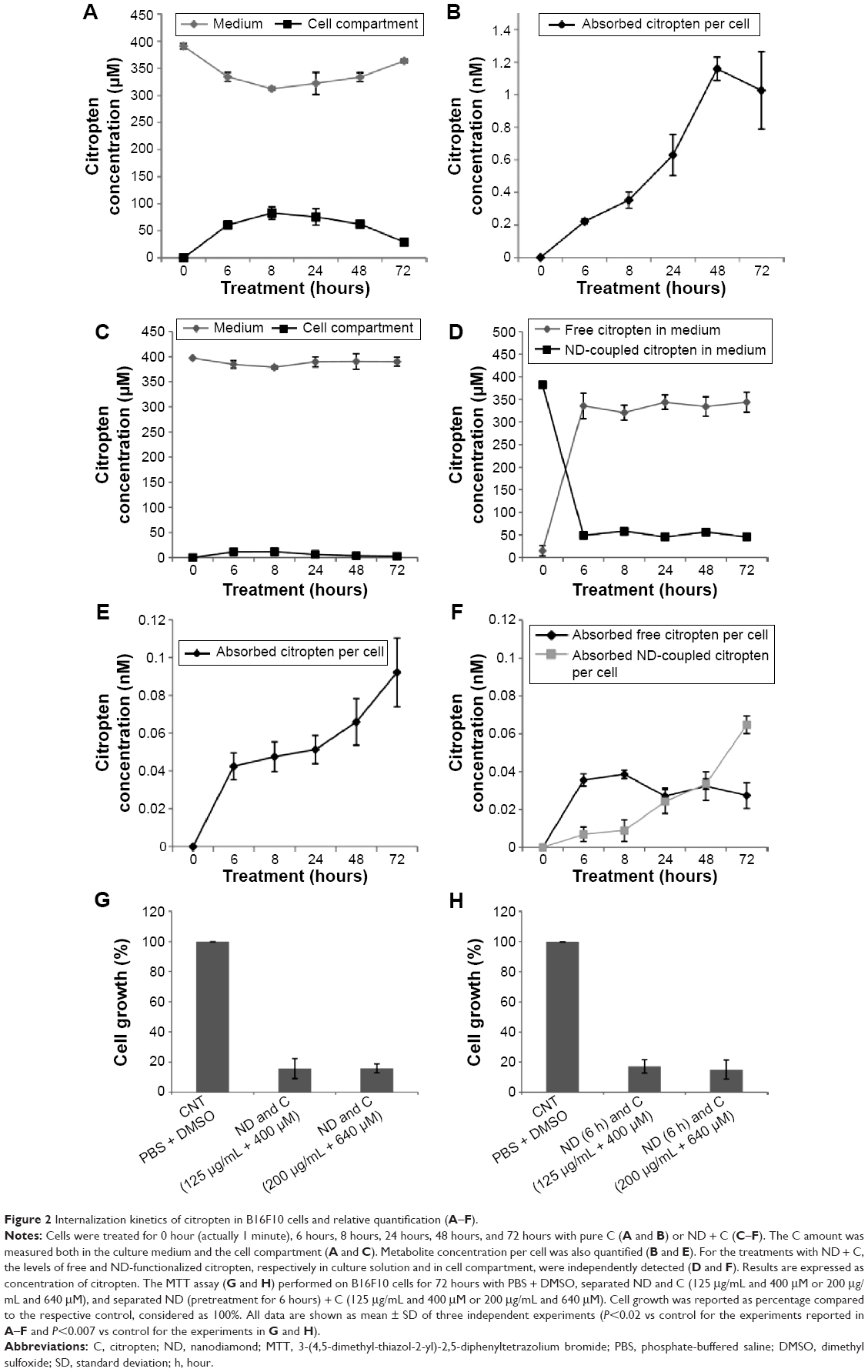 | Figure 2 Internalization kinetics of citropten in B16F10 cells and relative quantification (A–F). |
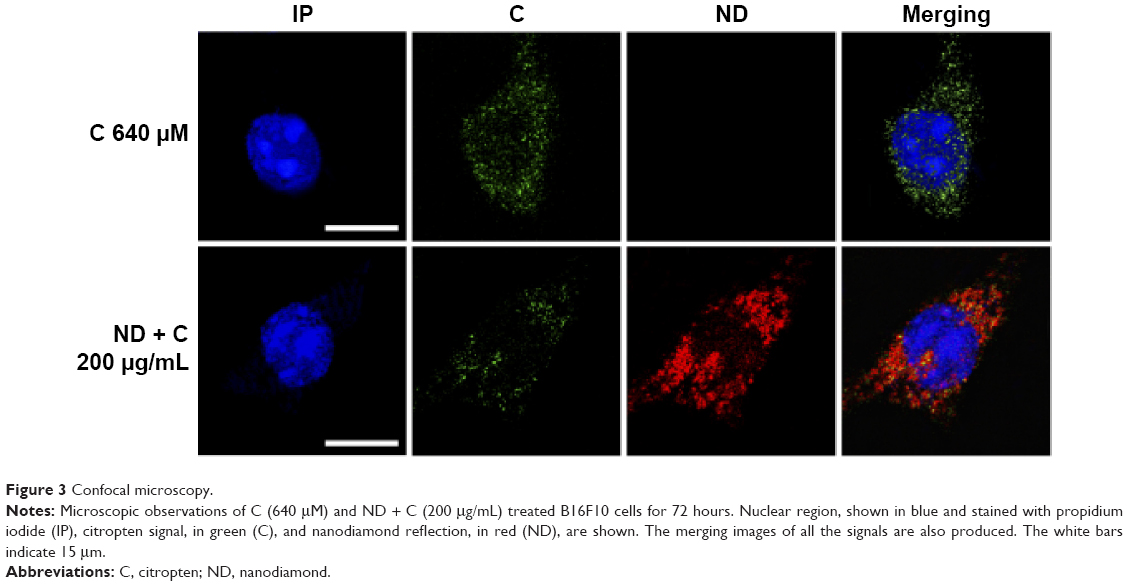 | Figure 3 Confocal microscopy. |
Investigation of (ND + C)’s possible effects on cell senescence, morphology, cycle, and differentiation
By using a specific kit, the senescent cells were stained in blue to be detected by microscopy (Figure S2). After 72 hours of treatment, in all the samples of CNT (PBS), CNT (DMSO), ND (200 μg/mL), ND + C (200 μg/mL), and C (640 μM) (respectively, Figure S2A–E), the number of senescent cells was extremely low, ranging between 0 and 2. On the other hand, cell exposure with DOX, a well-known senescence drug used as positive control, caused the induction of this phenomenon in ~95% of the total cells (Figure S2F). Our attention was then greatly captured by the peculiar phenotypes that B16F10, exposed for 72 hours to pure C (640 μM) (Figure 4C) or ND + C (200 μg/mL) (Figure 4D), showed at the microscopic analysis with respect to control cells treated with PBS solution (Figure 4A) or ND (200 μg/mL) (Figure 4B). In particular, C and ND + C treatments resulted in both a magnification of the cell dimension and the development of very long dendritic protrusions. Moreover, while in the C sample the nuclei appeared clearly condensed and the cytoplasm was strongly tapered, the ND + C specimen presented cells with an essentially round body with several thin filopodia. By cytofluorimetric analysis (fluorescence-activated cell sorting [FACS]) performed after 72 hours of exposure to each treatment, B16F10 distribution in the different phases of the cell cycle was studied (Figure 5). The amount of cells in G0–G1, S, and G2–M phases was, respectively, 60.9%, 10.5%, and 28.6% for the CNT with PBS and 62.8%, 17%, and 20.2% for the ND (200 μg/mL) sample (Figure 5A). The same measurements were also carried out on ND + C (125 μg/mL) and ND + C (200 μg/mL) treated cells. The results, shown in Figure 5B indicate that they presented, respectively, 59.1% and 57.4% of cells in G0–G1, 14.6% and 11.1% in S, and 26.3% and 31.5% in G2–M phases. Moreover, the percentage of cells in the cell cycle phases was estimated to be 57.9% (G0–G1), 12.4% (S), and 29.7% (G2–M) for the sample of CNT with DMSO; 67%, 11.4%, and 21.6% for the treatment with C (400 μM); and, finally, 71.7%, 10.8% and 17.5% for the C (640 μM) specimen (Figure 5C). Real-time PCR was carried out in order to monitor the mRNA levels of the principal genes involved in B16F10 differentiation or in cell cytoskeleton remodeling (Figure S1). Compared to the control cells, pure ND (200 μg/mL) treatment did not produce particular changes in the transcriptional levels of the studied genes (data not shown). The quantity of mRNA for each gene was first normalized for the GAPDH housekeeping gene and then reported as percentage with respect to the ND (200 μg/mL) sample, which was used as control (100%) (Figure 5D). ND + C (125 μg/mL) treatment for 72 hours, compared to ND sample, induced an increase of 8.9%, 8.3%, 51.3%, and 23.8%, respectively, for microphthalmia-associated transcription factor (MITF), tyrosinase (TYR), cofilin-1 (COF), and ACT mRNAs, while it caused a reduction of 2.3%, 24.1%, and 30.1%, correspondingly, for growth-differentiation factor 3 (GDF3), vimentin (VIM), and ανβ3-integrin (INT) genes. No variation was detected for tyrosinase-related protein 1 (TRP), and β-tubulin (TUB) mRNA levels. Otherwise, with respect to ND treatment, cell exposure for 72 hours with ND + C (200 μg/mL) showed an enhancement of MITF, TYR, TRP, GDF3, COF, TUB, and ACT mRNA levels, respectively, of 1.7%, 11.8%, 7.6%, 2.2%, 54.1%, 1.1%, and 12.6%. At the same time, this treatment also resulted in a reduction of 33.8% and 36.1%, respectively, of VIM and INT gene transcription.
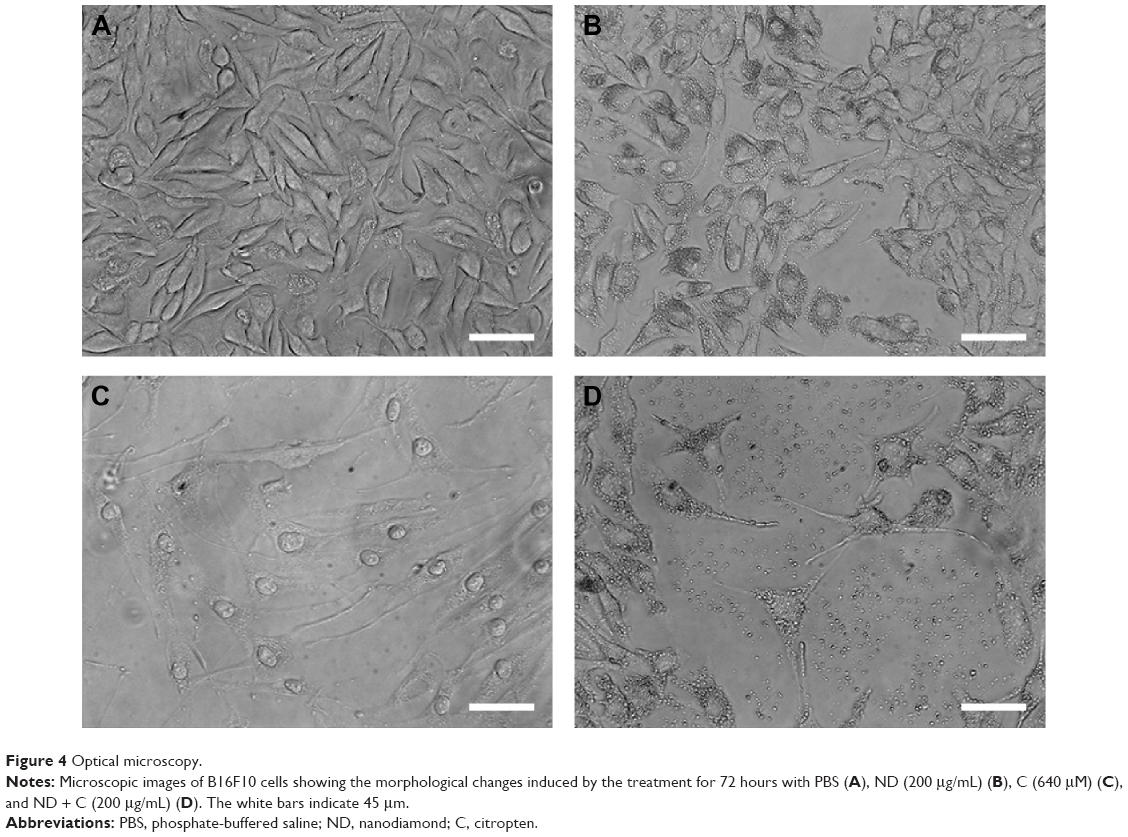 | Figure 4 Optical microscopy. |
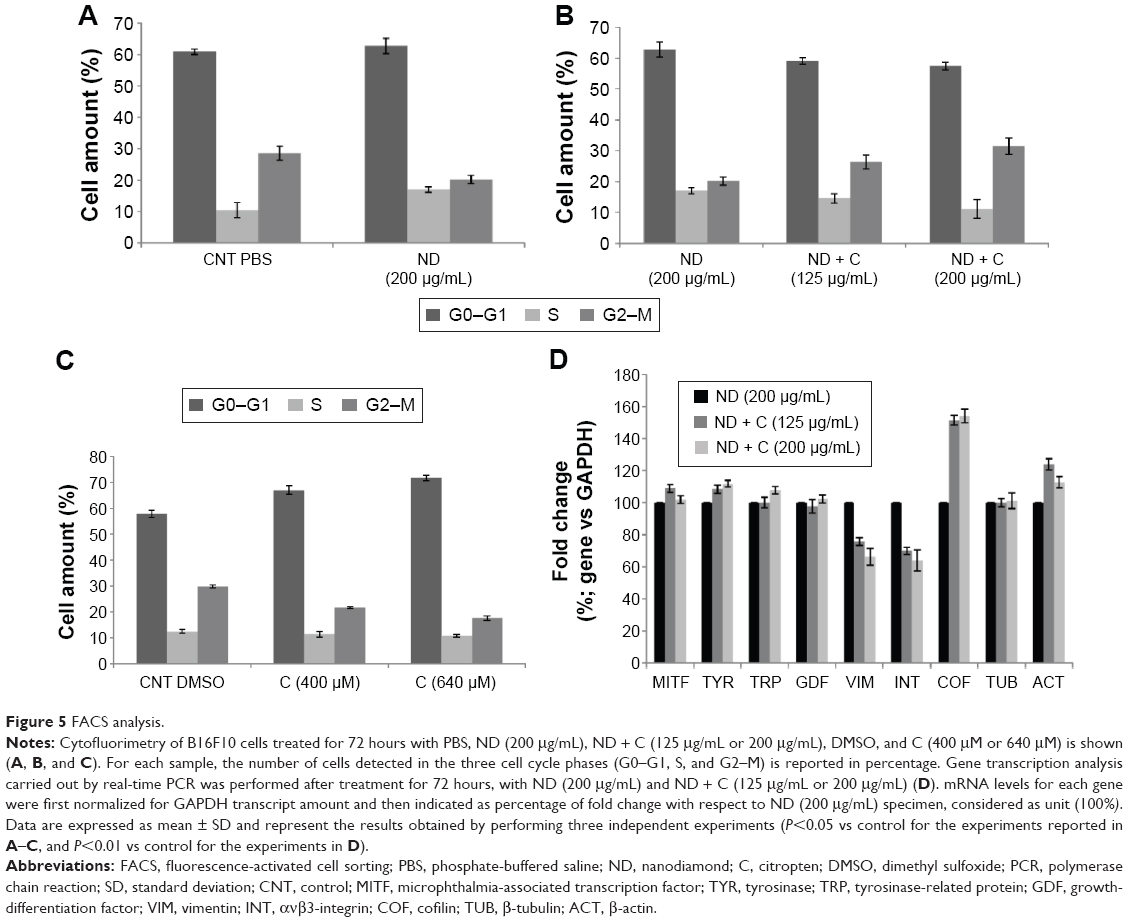 | Figure 5 FACS analysis. |
Citropten-functionalized NDs interfere with cell mitosis by altering actin organization
ACT and the DNA of B16F10 cells were labeled, respectively, in red (by a specific anti-ACT antibody) and in blue (by DAPI) to examine whether the various treatments could induce some modifications on cell actin organization. The immunofluorescence, reported in Figure 6, clearly shows a normal distribution pattern for the ACT in CNT (Figure 6A), ND (200 μg/mL) (Figure 6D), and C (640 μM) (Figure 6O) samples. Moreover, in these specimens, of particular interest was the easily detectable intensification of the red signal on the nuclear region. Indeed, on the contrary, ND + C (200 μg/mL) treated cells (Figure 6G, low magnification, and 6L, high magnification) did not present a similar high concentration of ACT in the proximity of the nuclei although it was widely distributed in the cytoplasm. Finally, the treatment with PHL (Figure 6R), a well-known inhibitor of cell actin depolymerization, resulted in a strong rounding of the cell structure and an accumulation of ACT, probably in filamentous form, on the nuclear area. In this context, the most important result was obtained by DAPI staining. Indeed, while in cells treated with CNT (Figure 6B), ND (200 μg/mL) (Figure 6E), and C (640 μM) (Figure 6P) the nuclear regions showed normal round shapes, in ND + C (200 μg/mL) samples (Figure 6H, low magnification, and M, high magnification) ~30% of the total nuclei appeared blocked in mitosis. In particular, as is visible in Figure 6M, they seemed to be arrested during cell division with pro-metaphase chromosomes. A very similar nuclear phenotype was also individuated in B16F10 cells after treatment with PHL (Figure 6S). The merging of ACT and DAPI signals was also seen in all the samples (Figure 6C, F, I, N, Q, and T). A specific protein extraction was performed to separate the cell filamentous (F) ACT from the monomeric one (G). Western blot analysis of ACT levels, normalized for the GAPDH amount, was carried out both on filamentous (Figure 7A) and monomeric (Figure 7B) fractions of each sample. With respect to the control (CNT PBS, considered 100%), F-actin level increased after 72 hours of treatment with ND (200 μg/mL), C (640 μM), CNT DMSO, and PHL, respectively, by 25.2%, 0.04%, 24.6%, and 142.6%, while it decreased in the presence of ND + C (200 μg/mL) by 58.1% (Figure 7A and C). On the other hand, the exposure of cells to ND (200 μg/mL), C (640 μM), CNT DMSO, and PHL for 72 hours resulted in, in that order, the reduction of G-actin concentration by 0.8%, 10.4%, 10.1%, and 75.4%, compared to control cells (CNT PBS, 100%); the treatment with ND + C (200 μg/mL) only induced an accumulation of G-actin of 52.4% (Figure 7B and D).
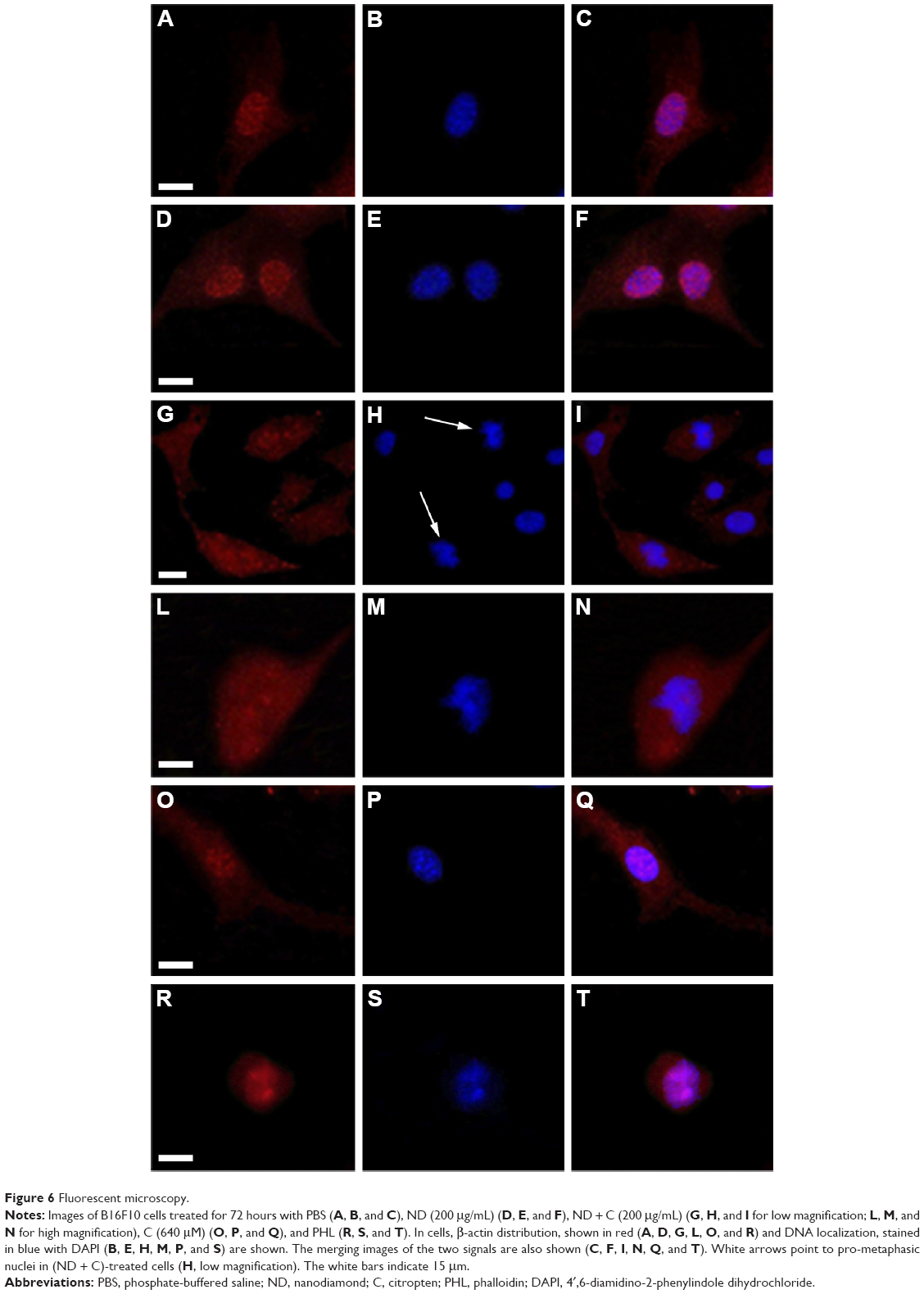 | Figure 6 Fluorescent microscopy. |
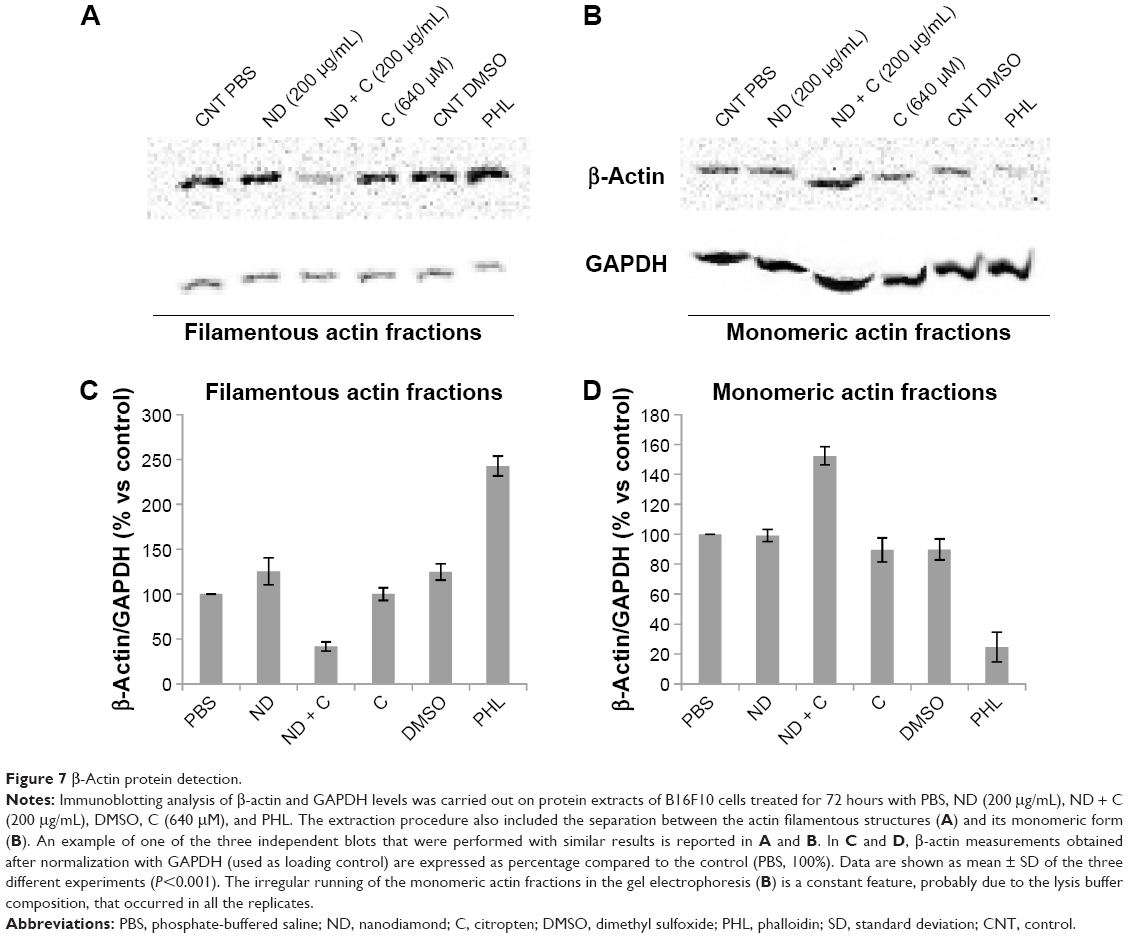 | Figure 7 β-Actin protein detection. |
Discussion
In this work, we decided to focus our attention on the analysis of the biological properties of plasma-reduced NDs conjugated with citropten (ND + C) on B16F10 murine melanoma cells. We demonstrated that pure ND treatment did not even minimally influence the tumor cell growth (Figure 1A and C), confirming literature data,3–5 and that the reduced proliferation rate of B16F10 due to pure citropten exposure was highly associated with cell death induction, while its ND-conjugated form did not result in any significant cytotoxicity although it inhibited cell proliferation (Figure 1B and D). All these data clearly suggested that C coupled with ND was able to act on tumor cells by a different cell mechanism with respect to its pure form. In order to elucidate how this phenomenon could happen and whether it was dependent on the amount of citropten present in the cells, the internalization kinetics and the intracellular concentration of the plant molecule in B16F10 cells were measured after treatment with the various samples. Spectrophotometric analysis performed on cells exposed to pure C for different times (0–72 hours) revealed that citropten introduced in the culture solution was initially absorbed in the cell compartment and then released again into the medium (Figure 2A), probably after the drug’s induction of cell death and consequent destruction of the cellular structure. Indeed, the pro-apoptotic effects of citropten are well documented in the literature.26–32 In the cited works, the antiproliferative effects of the plant molecule on tumor cells due to the alteration of specific signal transduction pathways, such as those of mitogen-activated protein kinase (MAPK) and PI3K/Akt (important in cancer survival and proliferation), suggested the use of citropten as a potential chemotherapy drug. On the other hand, the quantification of C levels per cell (Figure 2B), which rapidly increased up to reach a sort of plateau at 48–72 hours of treatment, corroborated the finding that the plant molecules were accumulated in the cell cytoplasm but not metabolized during the time. Indeed, the citropten re-excreted into the medium did not show a different absorbance spectrum with respect to its initial form. Finally, the nanomolar level (0.2–1.2 nM) of C measured in single cells indicated how this molecule presented a low cell bioavailability compared to the initial treatment concentration (400 μM). This result, in association with previous cytotoxicity data, suggested that, in spite of its low bioavailability, citropten was able to induce apoptosis even at very low doses. On the contrary, after exposure to the ND + C solution, the amount of citropten detected in the whole cell compartment (Figure 2C) and in each distinct cell (Figure 2E) was less (0.02–0.1 nM) than that detected after the previous treatment with pure C. Indeed, the major fraction of citropten was always identifiable in the culture solution (Figure 2C), probably because the release mechanism between ND and functionalized molecule was highly influenced by the pH value and protein concentration of the cell culture medium.33,34 In particular, we observed that the most citropten was rapidly released from the NDs in the medium (Figure 2D). On the other hand, we observed how the level of free C in each cell cytoplasm was maintained constant during the exposure time course while its ND-coupled form increased in concentration (Figure 2F), supporting the idea that ND, through a not-yet-known mechanism, was able to inhibit/modulate the continuous uptake of free citropten into the cells. In addition, the reduced amount of free citropten detected in (ND + C)-treated cells is responsible for its inability to activate apoptosis with respect to B16F10 exposed to pure C. Confocal microscopic investigations (Figure 3) confirmed the presence of citropten inside the cell compartment and its different concentrations in C and (ND + C)-treated cells, which were in conformity with our previous observations. Moreover, in the ND + C sample, merging the image between ND and C cell disposition visibly showed the presence of the plant metabolite in areas where the ND was absent, demonstrating the existence of free C in the cytoplasm. The occurrence of free C in the cytoplasm of (ND + C)-treated cells prompted us to examine whether the functionalization between the plant molecule and ND was absolutely indispensable for the activation of its antiproliferative properties on B16F10. Cell growth and cytotoxicity assays performed after double treatment with separate ND and C (Figure 2G) or pretreating the cells with pure ND for 6 hours before the exposure to pure C (Figure 2H) established the necessity of the chemical conjugation of the citropten to ND to carry out its peculiar bioactivity, namely inhibition of cell growth without toxicity. Since we demonstrated that citropten functionalized with ND was not able to induce cell oxidative stress, toxicity, and apoptosis as the pure C treatment,17 in the present work we investigated the specific molecular mechanism underlying the inhibition of B16F10 cell growth after exposure to the ND + C sample. We excluded the well-known phenomenon of senescence,35 since the number of senescent cells detectable after ND + C treatment was nil (Figure S2). During this analysis, the microscopic observations showed how the phenotypes of ND + C and C cell samples were very peculiar in profile and dimension with respect to the various controls (Figure 4). In particular, while in C-treated cells the nuclear regions appeared highly condensed, which is a typical feature of a trigged apoptotic process,36 the ND + C specimen was characterized by a fundamentally round shape from which extremely thin dendrite-like structures grew, similar but not totally identical to differentiated B16F10 cells.37 We continued then with the cell cycle analysis (Figure 5A–C). Although pure ND treatment, compared to the control, resulted in a small accumulation of the cells in the S phase, it could not be considered significant since the same sample did not even minimally result in any alteration of the cell proliferation rate or toxicity. On the contrary, ND + C induced, in a dose-dependent manner, considerable cell cycle arrest in the G2–M phase, justifying the correlated inhibition of cell growth. The most interesting results were obtained with cells exposed to pure C; the plant compound strongly caused a G0–G1 phase arrest, as documented well in the literature,26 indicating that citropten conjugation with ND changed its effect on the cell cycle. All these results suggested the possibility that ND + C could induce differentiation in B16F10. In order to clarify this hypothesis, the mRNA levels of the principal genes involved in murine melanocyte differentiation (MITF, GDF3, TYR, and TRP) were monitored (Figure 5D), but no significant change was detected, excluding the probability that NDs coupled with citropten had properties of differentiation. However, the abnormal phenotypes produced by ND + C treatment suggested that a specific mechanism of cell shape remodeling had been activated. For these reasons, the amounts of the transcripts of cytoskeleton-associated genes (VIM, INT, COF, ACT, and TUB) were measured by qPCR (Figure 5D). We found that VIM and INT gene expressions were reduced in B16F10 cells after treatment with ND + C. It could be explained because these genes, which regulate cell motility, proliferation, differentiation, and apoptosis, are highly interconnected.38 This result appeared very interesting since VIM, a type III cytoskeletal intermediate filament essential for the creation of the scaffold that allows actin to carry out its work during the mitosis, could be strongly connected to the cytoskeleton organization.39,40 Moreover, we also observed a great upregulation of COF mRNA synthesis (Figure 5D) whose product is a key protein that promotes F-actin depolymerization, providing actin monomers (G-actin) to the cytoplasmic pool.41 Since qPCR analysis clearly indicated that the bioactivity of citropten-conjugated NDs on B16F10 cells was strongly linked to the cytoskeletal organization and to actin remodeling, immunofluorescence analyses were performed to monitor actin and DNA intracellular disposition (Figure 6). ND-associated citropten was able to alter the correct actin distribution around the nucleus, which is a cytoskeletal feature which in normal cells is indispensable for the occurrence of mitosis.42,43 Moreover, ND + C cells only showed a great percentage of pro-metaphasic nuclei, suggesting that the mitotic process was slowed down or inhibited at that stage. B16F10 cells treated with PHL, a fungal bicyclic heptapeptide able to bind F-actin and prevent its depolymerization,44 showed, as with the ND + C sample, altered cell morphology, irregular actin distribution, condensed chromosomes, and mitotic arrest. The similarity of these phenotypes suggested the possibility that ND + C treatment could influence cell actin organization. On the other hand, in the ND + C sample the existence of some nuclei with a normal phenotype could be due to the fact that these cells probably had not yet entered the mitotic process or that the citropten–ND adducts had not yet reached their target in the cytoplasm. Finally, cell F- and G-actin were separated and detected (Figure 7). We observed that PHA treatment, as expected, resulted in an accumulation of F-actin and a consequent reduction of G-actin concentration. On the other hand, to our great surprise, the (ND + C)-treated cells showed the contrary effect: F-actin levels were reduced in favor of those of G-actin. This result showed that NDs coupled with citropten would have acted on B16F10 cells as inhibitors of actin polymerization, promoting the accumulation of its monomeric form. This event was also in concert with our previous cytofluorimetric evidence; indeed, in the literature it was reported how actin depolymerization arrested yeast cells in the G2–M phase by preventing their nuclear division.45 Velasco-Velázquez et al46 demonstrated that the coumarin derivative 4-hydroxycoumarin was able to disorganize cytoskeletal actin in B16F10 melanoma cells without altering its gene expression. Moreover, recently it was shown47 that the fluorophore coumarin-6 coupled with solid lipid nanoparticles was able to penetrate in A30 human alveolar cells and spread out in the cytoplasm depending on the actin cytoskeletal structure, suggesting the possible physicochemical affinity and the consequent direct interaction between coumarin compounds and actin. All these evidences confirmed our results and encouraged us to propose the following action model of ND + C treatment in B16F10 cells (Figure S3): NDs regulate the internalization of citropten in the cells, allowing C to reach low intracellular concentration not sufficient to induce apoptosis. The ND + C chemical adduct interacts with monomeric actin and sequesters it from the cytoplasmic pool. The low intracellular level of available G-actin causes the continuous depolymerization of F-actin.48 Since the newly synthesized monomers are similarly trapped by ND + C, in order to recover the lack of free actin, the transcription of COF mRNA is enhanced. However, this rescue process is insufficient and, consequently, the transcription of the principal interactor of the F-actin, VIM, and its partner INT is reduced. At this point, actin disorganization causes the mitotic arrest in the G2–M phase. In particular, it occurs immediately before the metaphase since at this specific step the VIM presence and a high quantity of F-actin are essential for the separation of duplicated chromosomes at the spindle poles, as widely reported.40 The most important result of the present work was the discovery that the conjugation of NDs with citropten changed the proapoptotic property of the pure secondary metabolite in antimitotic activity. This antitumoral mechanism, induced by the use of ND as a plant drug carrier, could extraordinarily reduce the adverse effects that the actual toxic chemotherapies (ie, cisplatinum) usually inflict on healthy tissues. Indeed, it would limit the drug’s bioactivity only to tumor cells, characterized by uncontrolled proliferation, excluding the differentiated ones that are mitotically inactive. According to all these evidences, the current study can be considered a piece of promotional research that encourages and supports the application of bioactive natural molecules coupled with NDs for antineoplastic therapeutic purposes.
Acknowledgments
The authors thank the Center of Advanced Microscopy “PB Albertano” of the Department of Biology of the University of Rome “Tor Vergata” for the microscopic analysis and Dr Valentina Iannizzotto for her assistance in fluorescence microscopic analysis. This research was supported by the grant “Uncovering Excellence 2014” of the University of Rome ‘Tor Vergata’ for the project entitled “NDDrugCell”.
Disclosure
The authors report no conflicts of interests in this work.
References
Puzyr AP, Baron AV, Purtov KV, et al. Nanodiamonds with novel properties: a biological study. Diam Relat Mater. 2007;16(12):2124–2128. | ||
Mochalin VN, Shenderova O, Ho D, Gogotsi Y. The properties and applications of nanodiamonds. Nat Nanotechnol. 2012;7(1):11–23. | ||
Horie M, Komaba LK, Kato H, Nakamura A, Yamamoto K, Endoh S. Evaluation of cellular influences induced by stable nanodiamond dispersion; the cellular influences of nanodiamond are small. Diam Relat Mater. 2012;24:15–24. | ||
Kaur R, Badea I. Nanodiamonds as novel nanomaterials for biomedical applications: drug delivery and imaging systems. Int J Nanomedicine. 2013;8:203–220. | ||
Schrand AM, Hens SAC, Shenderova O. Nanodiamond particles: properties and perspectives for bioapplications. Crit Rev Solid State. 2009;34(1–2):18–74. | ||
Singh R, Nalwa HS. Medical applications of nanoparticles in biological imaging, cell labeling, antimicrobial agents, and anticancer nanodrugs. J Biomed Nanotechnol. 2011;7(4):489–503. | ||
Malmsten M. Inorganic nanomaterials as delivery systems for proteins, peptides, DNA, and siRNA. Curr Opin Colloid Sci. 2013;18(5): 468–480. | ||
Chen M, Pierstorff ED, Lam R, et al. Nanodiamond-mediated delivery of water-insoluble therapeutics. ACS Nano. 2009;3(7):2016–2022. | ||
Li X, Shao J, Qin Y, et al. TAT-conjugated nanodiamond for the enhanced delivery of doxorubicin. J Mater Chem. 2011;21(22):7966–7973. | ||
Xiao J, Duan X, Yin Q, Zhang Z, Yu H, Li Y. Nanodiamonds-mediated doxorubicin nuclear delivery to inhibit lung metastasis of breast cancer. Biomaterials. 2013;34(37):9648–9656. | ||
Man HB, Kim H, Kim HJ, et al. Synthesis of nanodiamond–daunorubicin conjugates to overcome multidrug chemoresistance in leukemia. Nanomed Nanotechnol. 2014;10(2):359–369. | ||
Romagnolo DF, Selmin OI. Flavonoids and cancer prevention: a review of the evidence. J Nutr Gerontol Geriatr. 2012;31(3):206–238. | ||
Shah U, Shah R, Acharya S, Niyati A. Novel anticancer agents from plant sources. Chin J Nat Med. 2013;11(1):16–23. | ||
Fulda S, Efferth T. Selected secondary plant metabolites for cancer therapy. World J Tradit Chin Med. 2014;1:24–28. | ||
Song YH, Sun H, Zhang AH, Yan GL, Han Y, Wang X-J. Plant-derived natural products as leads to anti-cancer drugs. J Med Plant Herbal Ther Res. 2014;2:6–15. | ||
Turcheniuk V, Raks V, Issa R, et al. Antimicrobial activity of menthol modified nanodiamond particles. Diam Relat Mater. 2015;57:2–8. | ||
Gismondi A, Reina G, Orlanducci S, et al. Nanodiamonds coupled with plant bioactive metabolites: a nanotech approach for cancer therapy. Biomaterials. 2015;38:22–35. | ||
Reina G, Orlanducci S, Gay S, et al. Routes to control the chemical potential and to modulate the reactivity of nanodiamond surfaces. In: Cheng CL, Moran DA, Nemanich RJ, Swain GM, editors. MRS Proceedings; 2015; Vol. 1734; Cambridge: Cambridge University Press: mrsf14-1734-r03-10. | ||
Angjellari M, Peçuli A. Coupling of detonation nanodiamonds (DNDs) with stilbenes: the resveratrol case. Albanian J Pharm Sci. 2014;1(1): 42–54. | ||
Orlanducci S, Guglielmotti V, Cianchetta I, et al. One-step growth and shaping by a dual-plasma reactor of diamond nanocones arrays for the assembling of stable cold cathodes. Nanosci Nanotechnol Lett. 2012;4(3):338–343. | ||
Gismondi A, Serio M, Canuti L, Canini A. Biochemical, antioxidant and antineoplastic properties of Italian saffron (Crocus sativus L.). Am J Plant Sci. 2012;3(11):1573–1580. | ||
Gismondi A, Caldarola S, Lisi G, et al. Ribosomal stress activates eEF2K-eEF2 pathway causing translation elongation inhibition and recruitment of terminal oligopyrimidine (TOP) mRNAs on polysomes. Nucleic Acids Res. 2014;42(20):12668–12680. | ||
Gismondi A, Canuti L, Impei S, et al. Antioxidant extracts of African medicinal plants induce cell cycle arrest and differentiation in B16F10 melanoma cells. Int J Oncol. 2013;43(3):956–964. | ||
Iadevaia V, Caldarola S, Biondini L, et al. PIM1 kinase is destabilized by ribosomal stress causing inhibition of cell cycle progression. Oncogene. 2010;29(40):5490–5499. | ||
Serpinskaya AS, Denisenko ON, Gelfand VI, Bershadsky AD. Stimulation of actin synthesis in phalloidin-treated cells: evidence for autoregulatory control. FEBS Lett. 1990;277(1):11–14. | ||
Alesiani D, Cicconi R, Mattei M, Montesano C, Bei R, Canini A. Cell cycle arrest and differentiation induction by 5,7-dimethoxycoumarin in melanoma cell lines. Int J Oncol. 2008;32(2):425–434. | ||
Chen J, Zhang XD, Proud C. Dissecting the signaling pathways that mediate cancer in PTEN and LKB1 double-knockout mice. Sci Signal. 2015;8(392):pe1. | ||
Chuang JY, Huang YF, Lu HF, et al. Coumarin induces cell cycle arrest and apoptosis in human cervical cancer HeLa cells through a mitochondria- and caspase-3 dependent mechanism and NF-kappaB down-regulation. In Vivo. 2007;21(6):1003–1009. | ||
Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. | ||
Liu H, Wang Y, Sharma A, et al. Derivatives containing both coumarin and benzimidazole potently induce caspase-dependent apoptosis of cancer cells through inhibition of PI3K-AKT-mTOR signaling. Anticancer Drugs. 2015;26(6):667–677. | ||
Riveiro ME, Moglioni A, Vazquez R, et al. Structural insights into hydroxycoumarin-induced apoptosis in U-937 cells. Bioorg Med Chem Lett. 2008;16(5):2665–2675. | ||
Vázquez R, Riveiro ME, Vermeulen M, et al. Toddaculin, a natural coumarin from Toddalia asiatica, induces differentiation and apoptosis in U-937 leukemic cells. Phytomedicine. 2012;19(8):737–746. | ||
Toh TB, Lee DK, Hou W, et al. Nanodiamond–mitoxantrone complexes enhance drug retention in chemoresistant breast cancer cells. Mol Pharm. 2014;11(8):2683–2691. | ||
Wang X, Low XC, Hou W, et al. Epirubicin-adsorbed nanodiamonds kill chemoresistant hepatic cancer stem cells. ACS Nano. 2014;8(12):12151–12166. | ||
Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63(11):2705–2715. | ||
Toné S, Sugimoto K, Tanda K, et al. Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res. 2007;313(16):3635–3644. | ||
Gismondi A, Lentini A, Tabolacci C, Provenzano B, Beninati S. Transglutaminase-dependent antiproliferative and differentiative properties of nimesulide on B16-F10 mouse melanoma cells. Amino Acids. 2010;38(1):257–262. | ||
Kreis S, Schönfeld HJ, Melchior C, Steiner B, Kieffer N. The intermediate filament protein vimentin binds specifically to a recombinant integrin α2/β1 cytoplasmic tail complex and co-localizes with native α2/β1 in endothelial cell focal adhesions. Exp Cell Res. 2005;305(1):110–121. | ||
Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2014;5(10):816–826. | ||
Hubert T, Vandekerckhove J, Gettemans J. Unconventional actin conformations localize on intermediate filaments in mitosis. Biochem Biophys Res Commun. 2011;406(1):101–106. | ||
Hotulainen P, Paunola E, Vartiainen MK, Lappalainen P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16(2):649–664. | ||
Woolner S, O’Brien LL, Wiese C, Bement WM. Myosin-10 and actin filaments are essential for mitotic spindle function. J Cell Biol. 2008;182(1):77–88. | ||
Lu H, Zhao Q, Jiang H, et al. Characterization of ring-like F-actin structure as a mechanical partner for spindle positioning in mitosis. PLoS One. 2014;9:e102547. | ||
Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105(4):1473–1478. | ||
Rupeš I, Webb BA, Mak A, Young PG. G2/M arrest caused by actin disruption is a manifestation of the cell size checkpoint in fission yeast. Mol Biol Cell. 2001;12(12):3892–3903. | ||
Velasco-Velázquez MA, Agramonte-Hevia J, Barrera D, et al. 4-Hydroxycoumarin disorganizes the actin cytoskeleton in B16–F10 melanoma cells but not in B82 fibroblasts, decreasing their adhesion to extracellular matrix proteins and motility. Cancer Lett. 2003;98(2):179–186. | ||
Rivolta I, Panariti A, Collini M, et al. A biophysical model of intracellular distribution and perinuclear accumulation of particulate matter. Biophys Chem. 2011;158(2):134–140. | ||
Freeman WH. The dynamics of actin assembly. In: Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J, editors. Molecular Cell Biology. 4th ed. New York, NY: W. H. Freeman; 2000:Section 18.2. |
Supplementary materials
 | Figure S1 The names of the primers used in qPCR assay, their nucleotide sequences and corresponding references, were reported. |
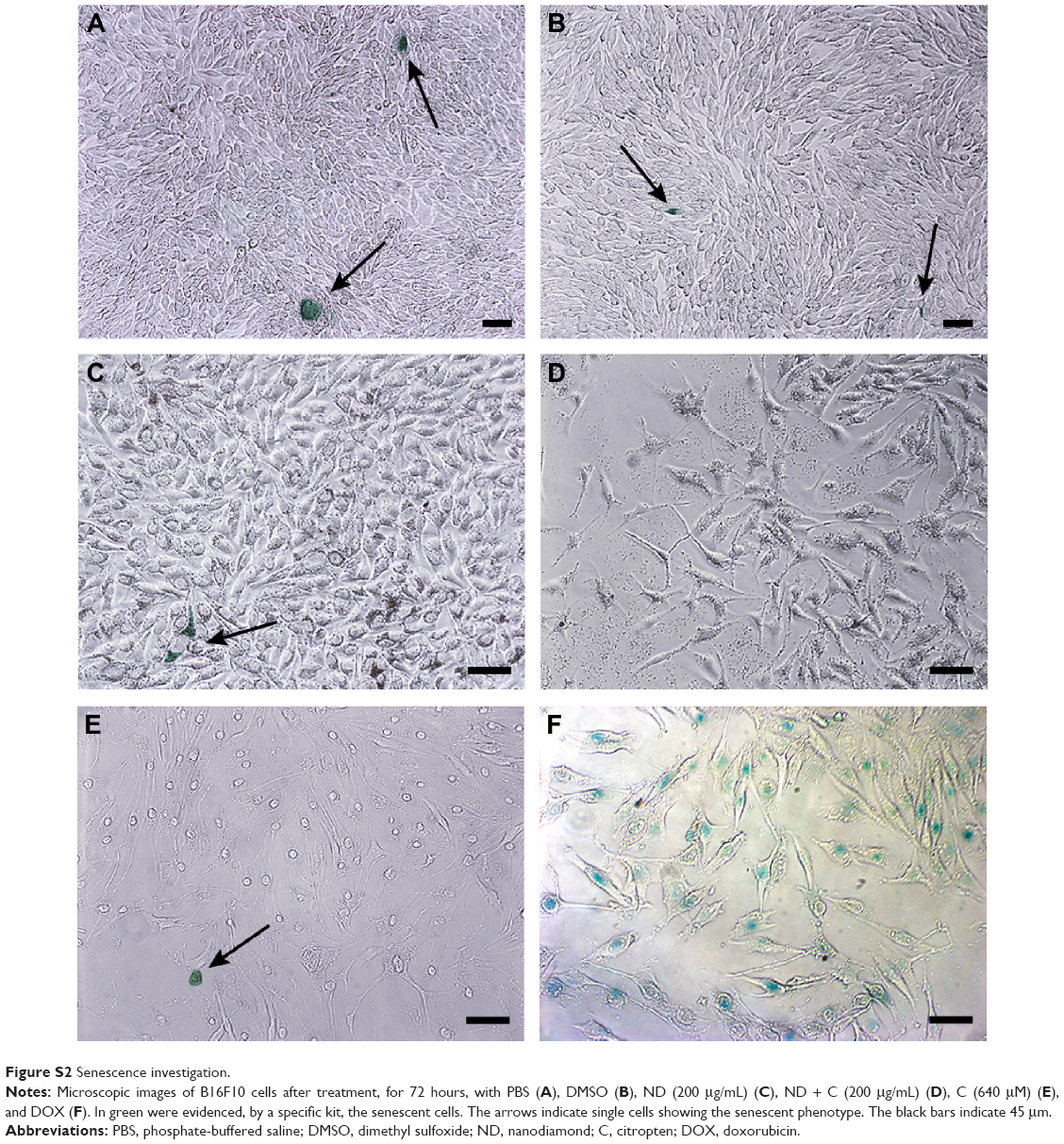 | Figure S2 Senescence investigation. |
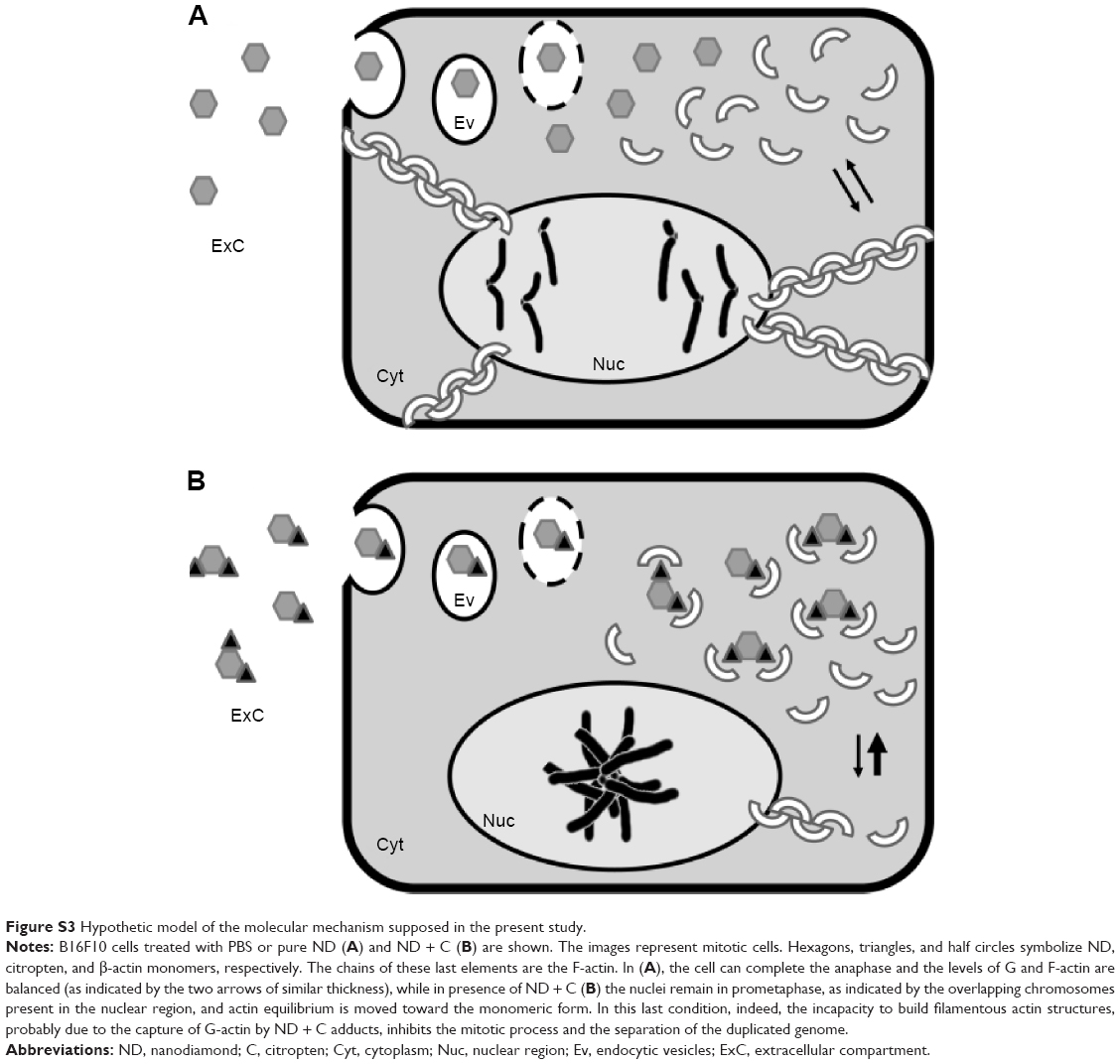 | Figure S3 Hypothetic model of the molecular mechanism supposed in the present study. |
References
Mansky KC, Sulzbacher S, Purdom G, et al. The microphthalmia transcription factor and the related helix-loop-helix zipper factors TFE-3 and TFE-C collaborate to activate the tartrate-resistant acid phosphatase promoter. J Leukoc Biol. 2002;71:304–310. | ||
Vetrini F, Auricchio A, Du J, et al. The microphthalmia transcription factor (Mitf) controls expression of the ocular albinism type I gene: link between melanin synthesis and melanosome biogenesis. Mol Cell Biol. 2004;24:6550–6559. | ||
Ashworth S, Teng B, Kaufeld J, et al. Cofilin-1 inactivation leads to proteinuria–studies in zebraflsh, mice and humans. PLOS One. 2010;5:e12626. | ||
Simard MJ, Chabot B. Control of hnRNP AI alternative splicing: an intron element represses use of the common 3′ splice site. Mol Cell Biol. 2000;20:7353–7362; (GenBank: NM_007393.4). | ||
Blavier L, Lazaryev A, Dorey F, Shackleford GM, DeClerck YA. Matrix metalloproteinases play an active role in Wntl-induced mammary tumorigenesis. Cancer Res. 2006;66:2691–2699. | ||
Ehira N, Oshiumi H, Matsumoto M, Kondo T, Asaka M, Seya T. An embryo-specific expressing TGF-β family protein, growth-differentiation factor 3 (GDF3), augments progression of BI6 melanoma. J Exp Clin Cancer Res. 20I0;29:135. | ||
Pan Y, Zhong LJ, Zhou H, et al. Roles of vimentin and 14-3-3 zeta/delta in the inhibitory effects of heparin on PC-3M cell proliferation and B16-F10-luc-G5 cells metastasis. Acta Pharmacol Sin. 2012;33:798–808. | ||
Kusam S, Vasanwala FH, Dent AL. Transcriptional repressor BCL-6 immortalizes germinal center-like B cells in the absence of p53 function. Oncogene. 2004;23:839–844. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.