Back to Journals » International Journal of Nanomedicine » Volume 14
Nanocarriers and nonviral methods for delivering antiangiogenic factors for glioblastoma therapy: the story so far
Authors Clavreul A, Pourbaghi-Masouleh M, Roger E ![]() , Menei P
, Menei P
Received 16 November 2018
Accepted for publication 8 February 2019
Published 9 April 2019 Volume 2019:14 Pages 2497—2513
DOI https://doi.org/10.2147/IJN.S194858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Anne Clavreul,1,2 Milad Pourbaghi-Masouleh,2,3 Emilie Roger,4 Philippe Menei1,2
1Department of Neurosurgery, CHU, Angers, France; 2CRCINA, INSERM, University of Nantes, University of Angers, Angers, France; 3Division of Drug Delivery and Tissue Engineering, School of Pharmacy, University of Nottingham, Nottingham, UK; 4MINT, INSERM 1066, CNRS 6021, University of Angers, Angers, France
Abstract: Angiogenesis, the formation of new blood vessels, is an essential component of glioblastoma (GB) progression. The development of angiogenesis inhibitor therapy, including treatments targeting vascular endothelial growth factor (VEGF) in particular, raised new hopes for the treatment of GB, but no Phase III clinical trial to date has reported survival benefits relative to standard treatment. There are several possible reasons for this limited efficacy, including VEGF-independent angiogenesis, induction of tumor invasion, and inefficient antiangiogenic factor delivery to the tumor. Efforts have been made to overcome these limitations by identifying new angiogenesis inhibitors that target angiogenesis through different mechanisms of action without inducing tumor invasion, and through the development of viral and nonviral delivery methods to improve antiangiogenic activity. Herein, we describe the nonviral methods, including convection-enhanced delivery devices, implantable polymer devices, nanocarriers, and cellular vehicles, to deliver antiangiogenic factors. We focus on those evaluated in intracranial (orthotopic) animal models of GB, the most relevant models of this disease, as they reproduce the clinical scenario of tumor progression and therapy response encountered in GB patients.
Keywords: antiangiogenic factors, delivery methods, glioblastoma, convection-enhanced delivery devices, implantable polymer devices, nanocarriers, cellular vehicles
Introduction
Glioblastoma (GB) is the commonest and most aggressive primary brain tumor. Despite standard treatment including resection and radiotherapy plus concomitant and adjuvant temozolomide (TMZ), prognosis remains poor, with a median survival of 12–18 months after diagnosis.1,2
GB is highly invasive and is characterized by a high rate of cell proliferation, heterogeneity, necrosis, and an abnormal angiogenic vasculature. This abnormal vasculature contributes to the development of high interstitial fluid pressure within the tumor, preventing the effective delivery of chemotherapy agents to the tumor tissue. This dysfunctional vasculature can also hinder tumor oxygenation, thereby promoting resistance to radiotherapy.3
This key role of the vasculature in treatment resistance has led to interest in GB treatment strategies that interfere with angiogenesis or destroy the existing tumor blood vessel network. The vascular abnormalities observed in GB have been attributed principally to the very high levels of vascular endothelial growth factor (VEGF) produced by tumor cells and tumor-associated stromal and inflammatory cells. VEGF is an angiogenic mitogen that operates by binding to VEGF receptors, triggering endothelial cell proliferation, migration, and the formation of new vessels. The possibility of blocking this key process with angiogenesis inhibitors has raised hopes that it might be possible to inhibit tumor growth, thereby prolonging patient survival. However, Phase III clinical trials involving the systemic administration of the anti-VEGF-A antibody bevacizumab (Avastin) or a pan-VEGF receptor (VEGFR)-2 tyrosine kinase inhibitor (cediranib, Recentin) in patients with recurrent or newly diagnosed GB have yielded disappointing results.4,5 These agents alleviated symptoms and made it possible to reduce steroid dose, but no improvement in overall survival relative to standard treatment was observed. There are several possible reasons for this limited efficacy, including VEGF-independent angiogenesis, induction of tumor invasion, and inefficient antiangiogenic factor delivery to the tumor.
These limitations have led to an intensification of efforts to discover new angiogenesis inhibitors targeting this process via more than one mechanism without inducing tumor invasion, and efforts to develop viral and nonviral delivery methods for local or systemic treatment to improve antiangiogenic activity. Many studies have evaluated these methods in subcutaneous (heterotopic) models of GB. However, these models do not take into account tissue-specific constraints, such as the effects of the blood–brain barrier (BBB) and the brain microenvironment associated with GB therapy. Studies in such models may, therefore, lead to an overinterpretation of the effects of the engineered delivery methods.6 In this review, we present the local or systemic nonviral delivery methods used to increase the activity of antiangiogenic factors, focusing in particular on those evaluated in intracranial (orthotopic) animal models of GB, which are the most relevant, as they closely resemble the human disease in terms of the clinical scenario of tumor progression and treatment response.
Angiogenesis and GB
The tumor requires new blood vessels to provide it with oxygen and nutrients once its volume increases beyond 1–2 mm3.7 Angiogenesis increases the blood supply to the tumor through the development of new vessels from the pre-existing vasculature (Figure 1). This process is regulated by the balance between proangiogenic factors, such as VEGF and fibroblast growth factor-2 (FGF-2), and antiangiogenic factors, such as angiostatin, angiopoietin 2, and endostatin. These factors may be released by the tumor itself or by the surrounding tissues. The blood supply may also be increased by vascular co-option, vascular intussusception, vasculogenic mimicry, and bone marrow-derived vasculogenesis (Figure 1).8–11 Briefly, vascular co-option involves the infiltration of tumor cells into normal tissue and adoption of the pre-existing vasculature. Vessel intussusception is the formation of new vessels by the enlargement and bifurcation of existing vessels. Vasculogenic mimicry is a process in which GB stem-like cells (GSCs) contribute to the formation of tumor blood vessels by differentiating into endothelial cells or pericytes. Bone marrow-derived vasculogenesis involves the recruitment of endothelial progenitor cells (EPCs), mesenchymal stem cells (MSCs), or hematopoietic stem cells to the tumor, their integration into the vessel wall, and their terminal differentiation into endothelial cells (Figure 1).
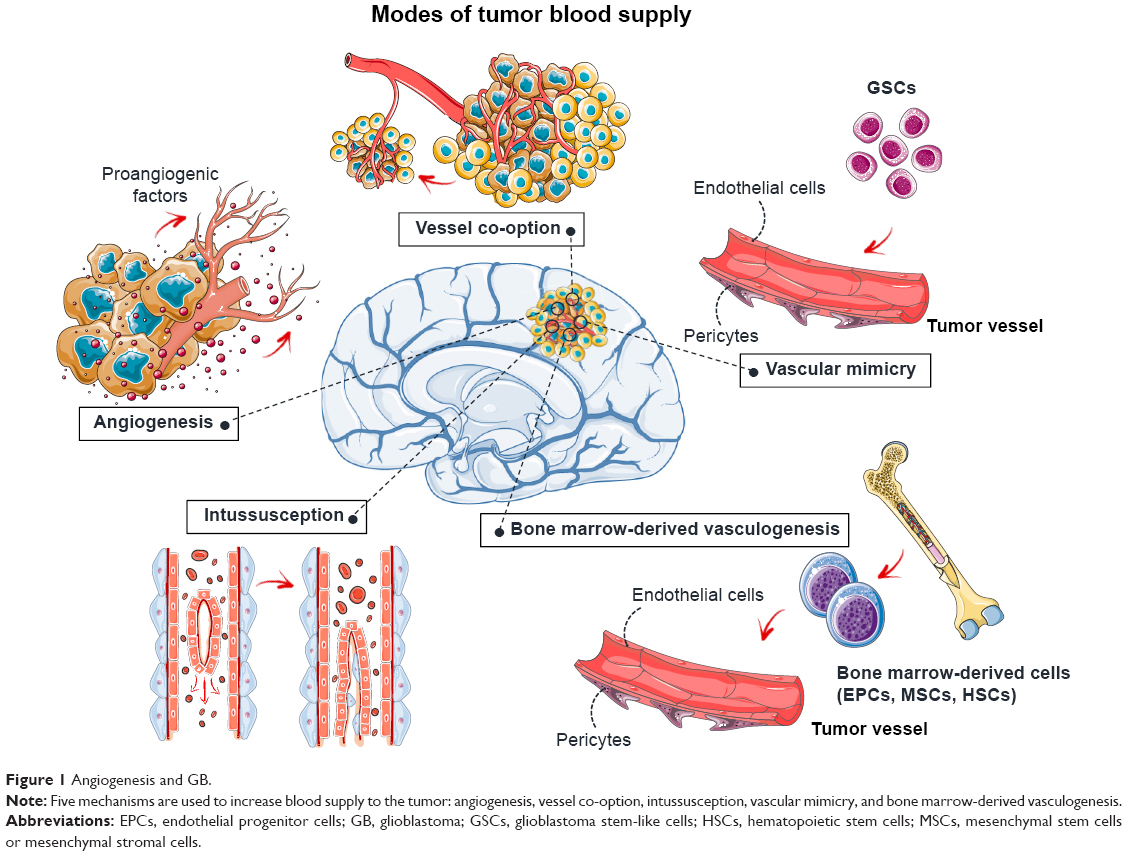 | Figure 1 Angiogenesis and GB. |
The new tumor vasculature is leaky, circuitous, dilated, and saccular, with a haphazard pattern of interconnection.12,13 In particular, the endothelial cells lining the new vessels have an irregular, disorganized morphology and are often poorly connected or overlap. The cells supporting the endothelial cells, the pericytes, are loosely attached or absent, and the basement membrane may be abnormally thick or entirely absent. The physiological consequences of these vascular abnormalities include temporal and spatial heterogeneity in tumor blood flow and oxygenation and an increase in tumor interstitial fluid pressure. These abnormalities and the microenvironment they create fuel tumor progression and decrease the efficacy of chemotherapy, radiotherapy, and immunotherapy.12,13
Antiangiogenic factors and GB
Given the importance of angiogenesis in GB progression and resistance to treatment, efforts have been made to develop novel therapies based on antiangiogenic factors, to achieve tumor regression through starvation. A number of angiogenesis inhibitors have been shown to affect the growth of various tumors, including GB, and this list is continuing to grow. These inhibitors can be classified into four categories (Figure 2).
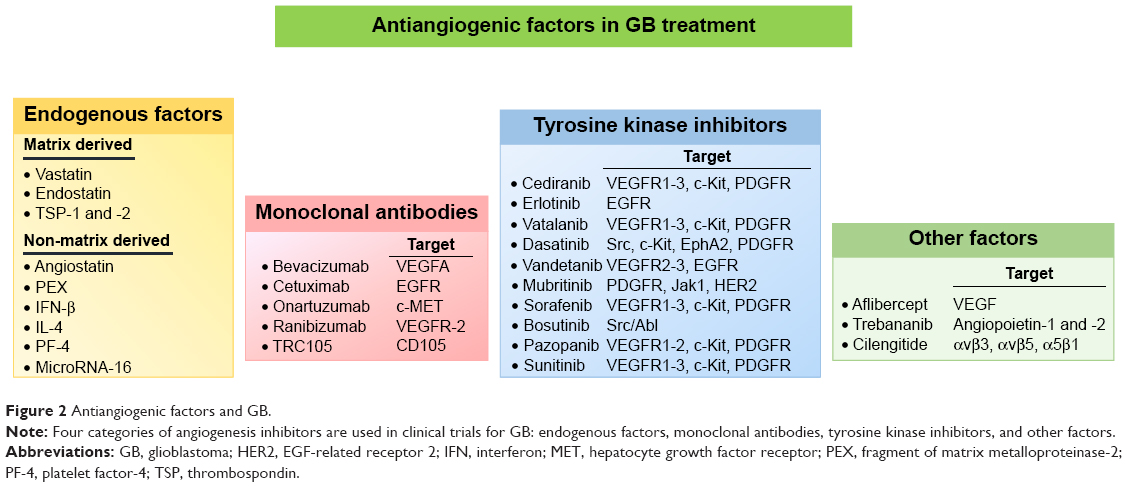 | Figure 2 Antiangiogenic factors and GB. |
Endogenous antiangiogenic factors
Endogenous antiangiogenic factors are molecules with antiangiogenic activity naturally present in body fluids or tissues.14 They are highly attractive candidate drugs because they are relatively non-toxic and well tolerated, with a low risk of drug resistance. They exert their effects through multiple mechanisms, including the induction of endothelial cell apoptosis, inhibition of endothelial cell migration to sites of neovascularization, inhibition of proangiogenic molecules, and changes to the regulation of proangiogenic and antiangiogenic molecules. Some endogenous antiangiogenic factors are secreted by specific cells in different organs. These factors include pigment epithelium-derived factor (PEDF), platelet factor-4 (PF-4), thrombospondin (TSP)-1 and -2, and several members of the interleukin (IL) and interferon (IFN) families, such as IL-4, IL-12, and IFN-β. Others are created by the extracellular proteolytic cleavage of plasma-derived or extracellular matrix proteins, such as angiostatin (fragment of plasminogen), endostatin (fragment of collagen XVIII), PEX (fragment of matrix metalloproteinase-2 [MMP-2]), and vastatin (fragment of collagen VIII). MicroRNAs have also been identified as a new class of endogenous angiogenesis inhibitors. For example, microRNA-16 plays a crucial role in repressing endothelial function and angiogenesis in GB.15 The efficacy of angiostatin, endostatin, PEX, PEDF, and TSP-1 and -2 has been studied in experimental models of malignant gliomas.16,17 However, most of these molecules have not yet been through clinical trials. A Phase I trial was performed to determine the maximum tolerated dose of ABT-510, a TSP-1 mimetic drug, used concomitantly with TMZ and radiotherapy in patients with newly diagnosed GB.18 The addition of ABT-510 to TMZ and radiotherapy was well tolerated by patients with GB, without unexpected adverse or serious adverse events. Progression-free and overall survival results were encouraging in light of historical expectations, but no Phase II trial was conducted.
Antiangiogenic monoclonal antibodies
Antibodies are key immune system components providing protection against infection. Antibodies directed against proangiogenic signaling proteins have been produced and shown to have antitumor activity in preclinical and clinical trials.19 Bevacizumab (Avastin), a recombinant humanized monoclonal antibody that targets VEGF-A, has been extensively studied in GB. However, Phase III trials showed that although the addition of bevacizumab to the standard treatment for newly diagnosed GB prolonged progression-free survival and improved the performance status of patients, it did not increase overall survival.20,21 Onartuzumab, a humanized monovalent monoclonal antibody against c-Met (or hepatocyte growth factor receptor [HGFR]) was recently evaluated in combination with bevacizumab in patients with recurrent GB.22 This antibody inhibits HGF binding and abolishes the ligand-induced phosphorylation of c-Met. However, no evidence of further clinical benefit was observed in comparisons of onartuzumab plus bevacizumab with bevacizumab plus placebo in unselected patients with recurrent GB. Nevertheless, exploratory biomarker analyses have suggested that patients with tumors displaying high levels of HGF or unmethylated O6-methylguanine-DNA methyltransferase expression may benefit from treatment with a combination of these two antibodies. Phase I/II trials with three other monoclonal antibodies (cetuximab [anti-epidermal growth factor receptor (EGFR), Erbitux], tanibirumab [TTAC-0001, anti-VEGR2], and TRC105 [carotuximab, anti-CD105]) either as single agents or in combination with bevacizumab have either recently been completed or are currently underway in patients with recurrent GB (https://ClinicalTrials.gov).
Antiangiogenic tyrosine kinase inhibitors (TKIs)
TKIs have been developed and clinical trials have been performed to assess their ability to block the specific tyrosine phosphorylation (activation) of a panel of angiogenic cell-surface tyrosine kinase receptors, such as VEGFR, bFGFR, platelet-derived growth factor receptor (PDGFR), EGFR, and their downstream intracellular serine/threonine kinases.23 Many of these TKIs are described as “multi-targeted kinase inhibitors” because they target several different kinases. A number of TKIs are being studied in Phase I/II clinical trials in patients with GB24 (Figure 2). To date, limited clinical benefit has been reported following treatment with TKIs in monotherapy or in combination with other approaches for the treatment of GB.24
Other antiangiogenic factors
Other antiangiogenic factors have been developed, including VEGF-Trap (afibercept), a soluble VEGF receptor; AMG 386 (trebananib), which binds angiopoietins Ang1 and Ang2; and cilengitide, a potent αvβ3 and αvβ5 integrin antagonist. However, no improvement was observed in patients with GB treated with these factors in clinical trials.25–27
Effects of antiangiogenic factors on the tumor vasculature
Antiangiogenic agents are thought to have a direct effect on the tumor vasculature. This effect involves endothelial cell apoptosis and ultimately leads to a cytostatic effect on new blood vessel growth and a decrease in tumor perfusion (Figure 3). These effects restrict the supply of oxygen and nutrients to the tumor, causing “tumor starvation”.7 However, it has also been suggested that antiangiogenic agents achieve their antitumor effects by vascular normalization, decreasing the permeability and diameter of blood vessels, thereby increasing tumor perfusion, decreasing interstitial pressure, and promoting tumor oxygenation (Figure 3). Such conditions sensitize the tumor to radiotherapy and increase the exposure of the tumor to cytotoxic drugs during chemotherapy.28 Antiangiogenic agents may also be active against GSCs29–31 and may disrupt proangiogenic signaling from bone marrow-derived cells, thereby abolishing tumor vascularization.32
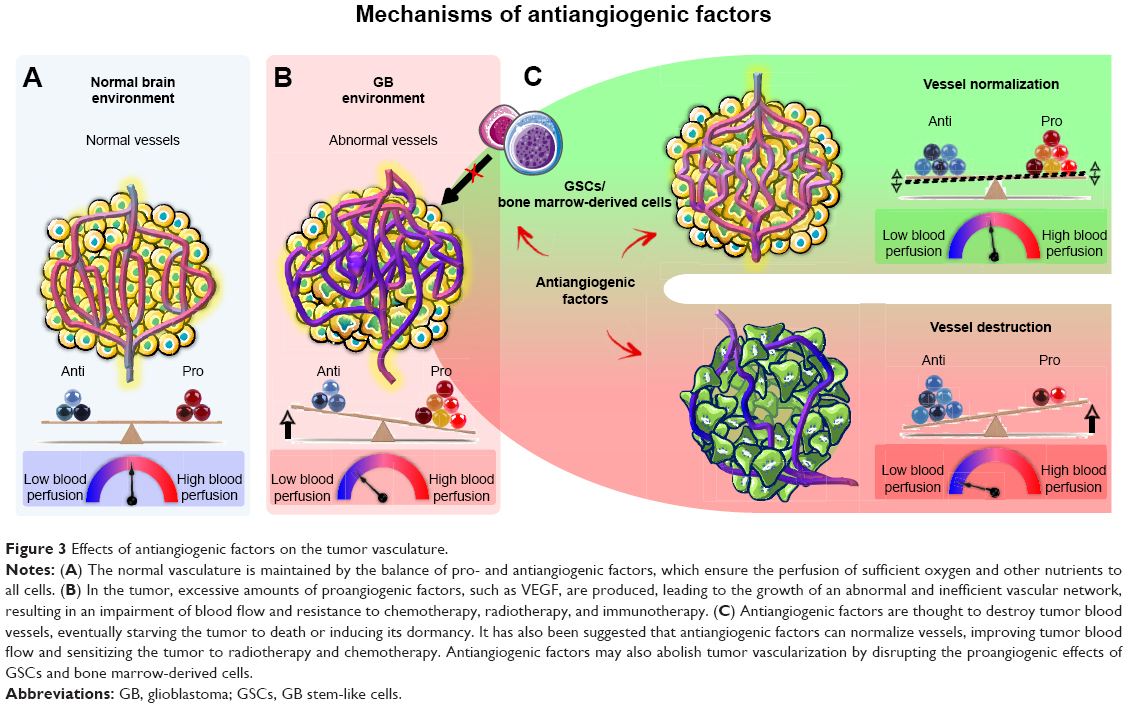 | Figure 3 Effects of antiangiogenic factors on the tumor vasculature. |
Nonviral delivery methods for antiangiogenic factors
Many antiangiogenic factors have been discovered, but, like other anticancer drugs, all face challenges to their use in GB therapy. In particular, most antiangiogenic factors cannot cross the BBB, and do not, therefore, reach therapeutic concentrations in the brain. In GB, the BBB is partially disrupted in the tumor core, but is mostly intact, forming a strong barrier protecting the tumor from the external environment. High systemic levels of antiangiogenic factors are, therefore, required to achieve effective concentrations within the tumor. However, increases in the dose of antiangiogenic agent, or in the frequency and duration of treatment, are often limited by systemic toxicity. For example, the systemic administration of antiangiogenic agents targeting the VEGF pathway has been associated with diverse adverse effects, including cardiovascular and renal complications, hemorrhage, wound complications, gastrointestinal perforation, and reversible posterior leukoencephalopathy syndrome.33 For these reasons, major efforts are currently being made to develop viral and nonviral delivery methods to solve the problem of BBB permeability and to reduce the amounts of antiangiogenic factor required for treatment, thereby decreasing treatment toxicity and adverse effects. We present four main local or systemic nonviral methods for antiangiogenic factor delivery that have been evaluated in orthotopic animal models of GB: convection-enhanced delivery (CED) devices, implantable polymer devices, nanocarriers, and cellular vehicles (Figure 4). The principal results obtained for the use of these devices alone or in combination with chemotherapy or radiotherapy are indicated in Table 1.
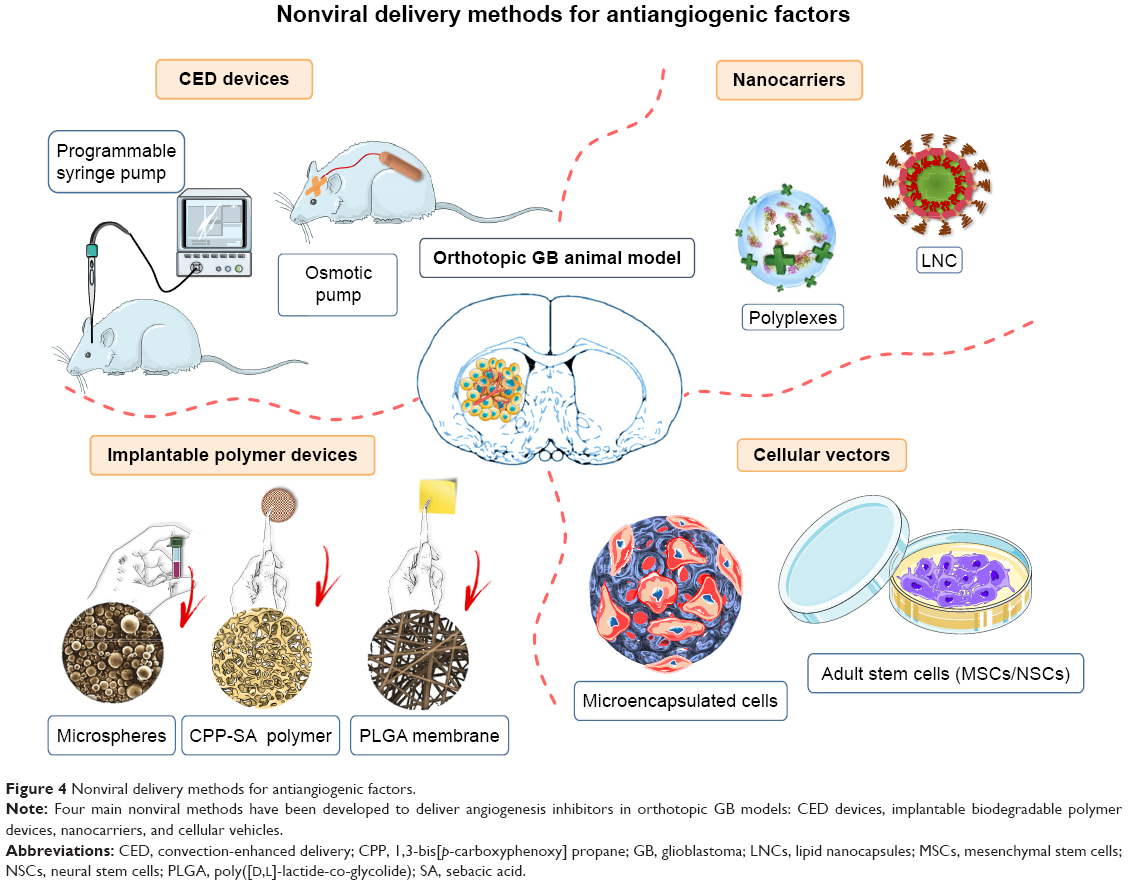 | Figure 4 Nonviral delivery methods for antiangiogenic factors. |
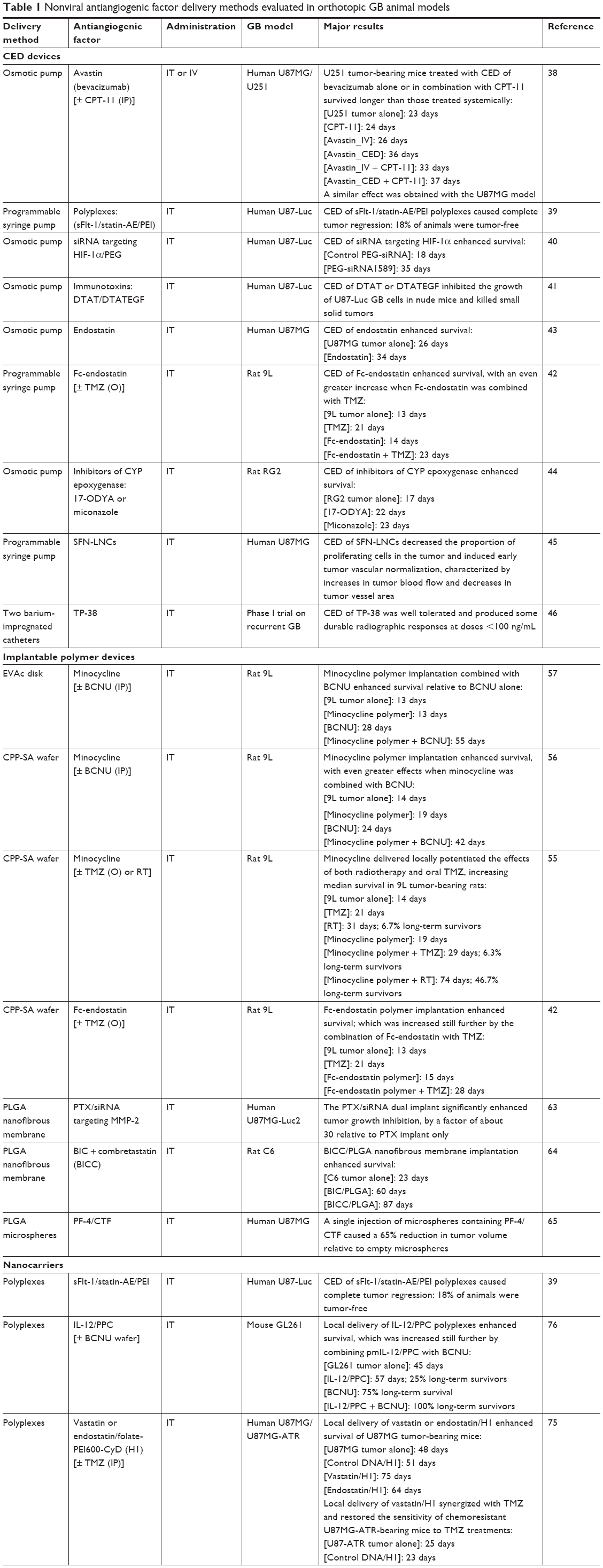 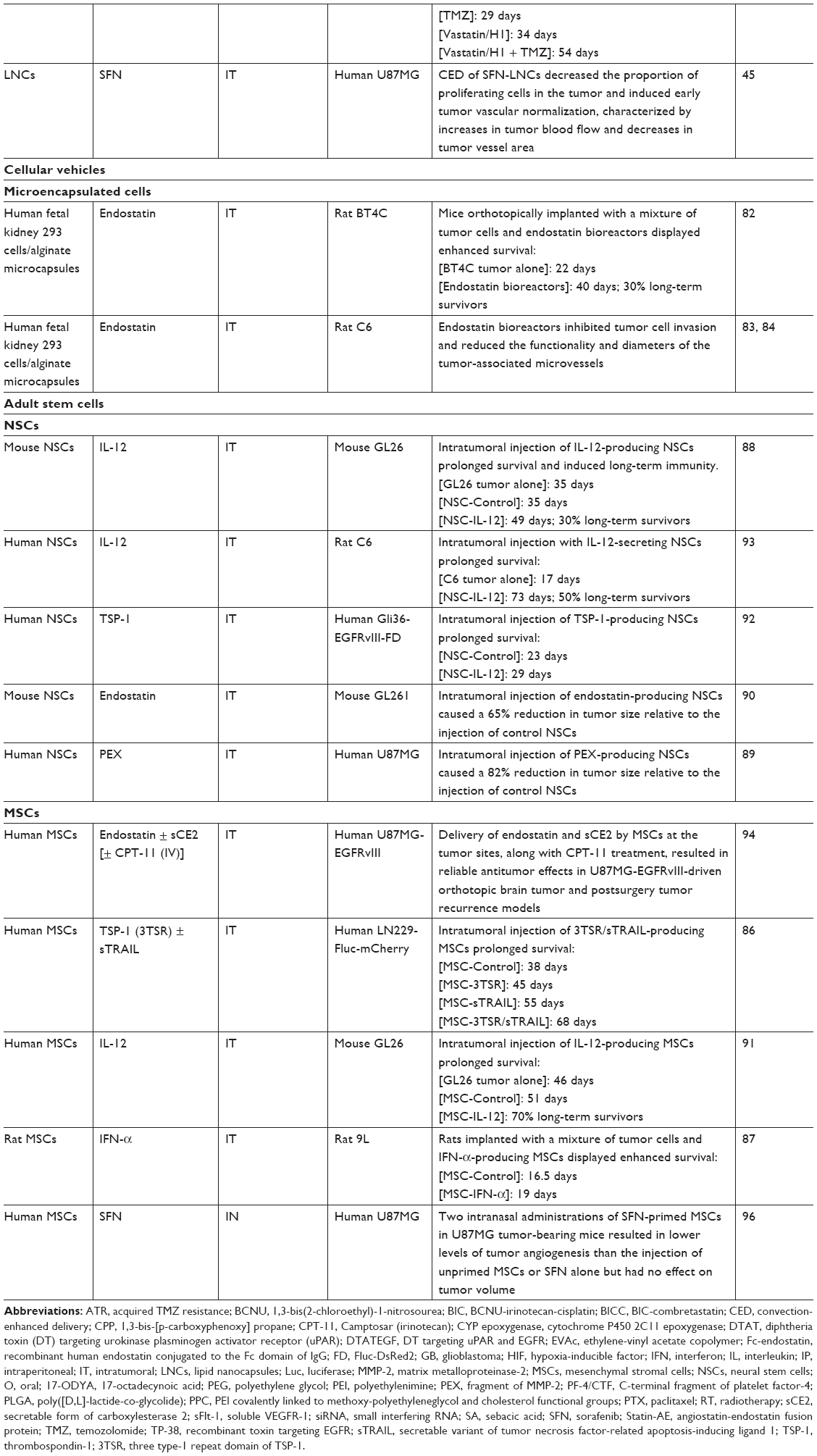 | Table 1 Nonviral antiangiogenic factor delivery methods evaluated in orthotopic GB animal models |
CED devices
In several recent clinical trials on GB, CED has been used for administration of anticancer agents directly into the tumor with a potential to provide concentrations that cannot be achieved with systemic drug delivery.34–37 CED involves the surgical implantation of catheters in the tumor itself or in the surrounding brain parenchyma. The catheters are connected to an infusion pump to create a positive pressure gradient at the catheter tip. This allows for safe, targeted, and reliable homogeneous delivery of small and large molecular weight substances at clinically relevant volumes in a manner that bypasses the BBB. The CED of a mixture of anticancer agent and a surrogate tracer for imaging can now be used to monitor drug distribution in real time by computed tomography or magnetic resonance imaging.36 The preclinical studies performed on animal models in which CED was used to administer angiogenesis inhibitors are listed in Table 1. The CED devices used were based on Alzet osmotic pumps or programmable syringe pumps. Diverse antiangiogenic agents or nanocarriers carrying antiangiogenic factors have been administered in this way in GB-bearing animals: bevacizumab,38 polyplexes (polymer/DNA) encoding a soluble VEGFR-1 (sFlt-1) and an angiostatin-endostatin fusion protein (statin-AE),39 siRNA targeting hypoxia-inducible factor 1α (HIF-1α),40 the immunotoxins, DTAT (diphtheria toxin [DT] targeting urokinase plasminogen activator receptor [uPAR]) and DTATEGF (DT targeting uPAR and EGFR),41 endostatin or endostatin fused to an antibody (IgG) Fc domain (Fc-endostatin),42,43 two mechanistically different inhibitors of the CYP/CYP450 2C11 epoxygenase, 17-octadecynoic acid and miconazole,44 and sorafenib-loaded lipid nanocapsules (SFN-LNCs).45 The administration of these agents by CED has frequently been shown to lead to a significant decrease in capillary formation and tumor size and an increase in animal survival. Wang et al38 demonstrated that bevacizumab administered by CED resulted in longer animal survival than the same drug administered intravenously. Furthermore, combining the CED of angiogenesis inhibitors (bevacizumab or Fc-endostatin) with the systemic administration of chemotherapy agents (CPT-11 or TMZ) was found to be the most effective approach for increasing survival.38,42 One clinical trial of intracerebral infusion by CED was performed with a recombinant toxin, TP-38, targeting the EGFR in patients with recurrent GB.46 The CED of TP-38 was found to be well tolerated at effective doses and encouraging radiographic responses were observed. However, this trial also showed that the potential efficacy of agents delivered by CED in humans might be limited by inconsistent and inefficient infusion in many patients. These technical shortcomings will need to be addressed if CED is to fulfill its therapeutic potential.34,37
Implantable polymer devices
Implantable polymer devices provide a robust technology platform for the local and sustained delivery of drugs directly to the tumor.47,48 Gliadel wafers are currently the only biodegradable polymer implants for intracranial drug delivery approved for GB treatment by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA).49–51 They are made of a polyanhydride copolymer (1,3-bis[p-carboxyphenoxy] propane [CPP] and sebacic acid [SA]), loaded with carmustine (1,3-bis[2-chloroethyl]–1-nitrosourea [BCNU]). Gliadel wafers are implanted in the tumor bed after resection. They provide a controlled local release of BCNU over a period of 2–4 weeks. This delivery system was shown to be superior to the systemic administration of BCNU in the 9L rat glioma model, and its systemic toxicity was limited.52 Clinical studies have reported Gliadel wafers to be effective against both newly diagnosed and recurrent GB.53 The combination of Gliadel wafers with standard radiotherapy plus concurrent and adjuvant TMZ has been shown to increase patient survival by 3–4 months relative to Gliadel wafers or TMZ alone.54 The antiangiogenic agent Fc-endostatin has been loaded onto CPP-SA wafers, and the intracranial implantation of Fc-endostatin wafers was found to prolong survival in 9L tumor-bearing rats.42 A similar effect on survival has been reported for the CED of Fc-endostatin to the tumor.42 Additional survival benefits were observed when the intratumoral delivery of Fc-endostatin via CPP-SA wafers or CED was combined with the oral chemotherapy agent TMZ42 (Table 1). Minocycline, another angiogenesis inhibitor, has also been loaded onto CPP-SA wafers, and the intracranial implantation of minocycline wafers potentiated the effects of systemic chemotherapy agents (BCNU or TMZ) or radiotherapy, increasing the median survival of treated rats55,56 (Table 1). Weingart et al57 incorporated minocycline into ethylene-vinyl acetate copolymer (EVAc), a non-biodegradable polymer. They found that a combination of the local delivery of minocycline from EVAc disks and systemic BCNU increased median survival by 93% relative to BCNU alone in 9L tumor-bearing rats.
Other studies have used poly([d,l]-lactide-co-glycolide) (PLGA) nanofibrous membranes to deliver antiangiogenic factors. PLGA, an FDA- and EMA-approved polymer, is highly biocompatible and biodegradable, and is one of the most attractive polymeric candidates available for the manufacture of devices for drug-delivery and tissue-engineering applications.58,59 PLGA nanofibrous membranes are thinner than Gliadel wafers, can be cut into any size or shape, and can conform completely to the shape of the tissue after tumor removal.60 Various drugs can be incorporated into these membranes easily, and the use of PLGA nanofibrous membranes with different lactic acid/glycolic acid compositions can yield different PLGA degradation profiles, making it possible to control the sequential elution of drugs and their release at therapeutic doses.61,62 PLGA nanofibrous membranes also provide a longer therapeutic period (more than 8 weeks) than Gliadel wafers (2–4 weeks), potentially resulting in a higher therapeutic efficacy and lower resistance. Lei et al63 incorporated paclitaxel (PTX) and polyplexes encoding a siRNA targeting MMP-2 into PLGA nanofibrous membranes. MMP-2 is an essential proteinase regulating brain tumor invasion and angiogenesis. This dual delivery of PTX and MMP-2 siRNA by PLGA nanofibrous membranes was tested in mice with intracranial U87MG-Luc2 xenografts, and was found to have a significantly greater therapeutic efficacy than the delivery of PTX alone, with the two agents acting in synergy. Tseng et al60,64 loaded 50:50 PLGA nanofibers with three chemotherapy agents (BCNU, irinotecan, and cisplatin; BIC/PLGA), and 75:25 PLGA nanofibers with BIC and an antiangiogenic agent (combretastatin phosphate, CA4P; BICC/PLGA). The chemotherapy agents were rapidly released from the 50:50 PLGA nanofibers after implantation, and combretastatin was released from the 75:25 PLGA nanofibers about 2 weeks later. BICC/PLGA nanofibrous membranes implanted in the brains of C6 tumor-bearing rats slowed tumor growth and decreased malignancy more effectively than BIC/PLGA nanofibrous membranes.
One study used PLGA microspheres to deliver an antiangiogenic factor, C-terminal fragment of PF-4 (PF-4/CTF).65 This study showed that the intratumoral injection of PF-4/CTF-loaded microspheres in U87MG tumor-bearing mice decreased tumor volume by about 70%, this effect being accompanied by a significant decrease in angiogenesis and an increase in apoptosis. Microspheres constitute a versatile class of drug carriers for which drug release kinetics can be controlled by modulating microsphere formulation (size, molecular weight, and PLGA composition).66 Due to their size and shape, polymeric microspheres are easy to implant at the tumor site by stereotactic injection, or to inject into the walls of brain tumor resection cavities with a handheld syringe during surgery.67
Encouraging results have been obtained with implantable polymer devices, but further optimization of these devices is required to overcome the specific challenges of localized treatment, including sustainable release over a long period and deeper tissue penetration.68 Polymeric microreservoirs could potentially improve the efficacy of local treatment for brain cancer, as they can deliver large doses of drugs in solid or liquid form. For example, Ong et al69 developed a microdevice consisting of a micromachined polysulfone reservoir and cap. The reservoir was drug-impermeable and consisted of a cylinder of 2 mm in diameter and 2 mm in height. The cap of the device was machined with pores of 300 μm in diameter through which the encapsulated drug could pass. Drug release from this polysulfone microdevice was linear and highly dependent on the number of holes in the cap: the greater the number of holes, the faster the release of the drug. Grayson et al70 produced another form of microreservoir consisting of a polymeric microchip device. These microchips were 1.2 cm in diameter and 480–560 μm thick, and they had 36 reservoirs, each of which could be loaded with a different substance. These devices were made of poly(l-lactic acid) (PLLA) and each reservoir was covered with a PLGA membrane. By altering the molecular mass of the PLGA membrane, drug delivery systems could be developed with the potential to release pulses of different drugs at different times after implantation in a patient.
Nanocarriers
The use of nanocarriers for cancer treatment has been described in a number of studies.71–74 Nanocarriers are particles with at least one dimension in the 1–1,000 nm size range. They can be used for the local or systemic delivery of various agents, such as drugs, radionuclides, imaging agents, and bioactive compounds (siRNA or DNA). These agents can be entrapped in, adsorbed onto, or chemically bonded to the nanocarrier surface. Nanocarriers protect the active agents against enzymatic and hydrolytic degradation, improve their solubility and pharmacokinetic parameters, and control their release. Their composition, size, and other surface characteristics can easily be modified to bypass physiological barriers, including the BBB. Various nanocarriers are available: polymeric, lipid-based, and inorganic.
Two types of nanocarriers have been developed for antiangiogenic therapy: 1) nanocarriers carrying antiangiogenic factors, and 2) vascular-targeted nanocarriers generated by the conjugation of nanocarriers to molecules that bind to angiogenesis-related receptors. These nanocarriers can be used to detect and visualize angiogenesis and/or to direct chemotherapy, phototherapy, or gene therapy to the tumor vasculature. In this review, we present only the nanocarriers used for antiangiogenic factor delivery.
An entire arsenal of nanocarriers carrying antiangiogenic factors has been produced, but few have been tested in orthotopic GB models. We identified three studies reporting the use of polyplexes for the delivery of genes encoding antiangiogenic factors39,75,76 in addition to the work of our laboratory on the use of LNCs for SFN delivery45 (Table 1; Figure 4). Ohlfest et al39 produced polyplexes encoding two antiangiogenic proteins, sFlt-1 and statin-AE. They showed that the CED of these polyplexes in U87MG tumor-bearing mice led to strong antitumor activity characterized by a decrease in tumor vessel density, the inhibition of tumor growth, and tumor elimination in up to 50% of mice (Table 1). In other studies, polyplexes encoding IL-12 and vastatin or endostain were produced.75,76 The intratumoral injection of these antiangiogenic polyplexes in GB-bearing mice led to a significant increase in survival relative to untreated mice, which was increased still further by their combination with chemotherapy agents (BCNU or TMZ) (Table 1). Our laboratory recently produced LNCs for SFN encapsulation, because this multikinase inhibitor is poorly soluble in water, which limits its use for local or systemic administration.45 We showed that the CED of SFN-LNCs in the orthotopic U87MG GB model decreased the proportion of proliferating cells in the tumor and induced early tumor vascular normalization, characterized by an increase in tumor blood flow and a decrease in tumor vessel area (Table 1). This vascular normalization may provide a window of opportunity for enhancing the efficacy of chemotherapy or radiotherapy.
Cellular vehicles
Cellular vehicles, including microencapsulated cells and adult stem cells, have been used to deliver antiangiogenic factors in orthotopic GB models (Table 1; Figure 4).
Microencapsulated cells
Living cells are encapsulated in polymeric microspheres or microcapsules for many biomedical and biotechnological applications, including local and long-term drug delivery and regenerative medicine.77–81 The microencapsulation of cells allows the free exchange of nutrients and waste while excluding agents of the immune system, thereby promoting the survival of the transplanted cells. It also permits the release of therapeutic cell products. A number of different biomaterials, such as alginate, hyaluronic acid, agarose, and other polymers, have been used for encapsulation.
Read et al82 encapsulated human fetal kidney 293 cells transfected with a pCEPPu episomal expression vector encoding human endostatin in sodium alginate microcapsules. They showed that encapsulated endostatin-producing 293 cells remained viable for at least 4 months after intracerebral implantation, and that they continued to produce endostatin after encapsulation and implantation. The spatial distribution of endostatin in the brain/tumor tissue was determined, and this molecule was detected at 1–1.7 mm from the bioreactors, with some more distant deposits (4–6 mm). It was also found in the cerebrospinal fluid. Rats receiving transplants of BT4C glioma cells together with encapsulated endostatin-producing 293 cells survived for 84% longer than controls. The endostatin released from the microcapsules triggered apoptosis, hypoxia, and the formation of large necrotic avascular areas in 77% of the treated tumors, whereas these features were not observed in any of the controls. The apparent biological effects of endostatin were observed for tumors inoculated 1 week before or at the same time as bioreactor implantation. However, no significant effect on animal survival was observed when animals received the treatment 1 week after tumor inoculation. Intravital multifluorescence microscopy showed that the endostatin released from the capsules decreased not only total vascular density, but also the functionality and diameter of blood vessels and tumor cell invasion.83,84
Efforts are currently being made to encapsulate cell lines producing different recombinant proteins capable of interfering with tumor growth and progression. In this way, a “library” of encapsulated cells can be generated, with the aim of tailoring local therapy to specific biological parameters regulating tumor growth. In parallel, optimization is required to achieve sufficient biosafety and biocompatibility for clinical approval.
Adult stem cells
The particular tropism for brain tumors displayed by stem cells, such as neural stem cells (NSCs) and MSCs, has led these cells to being considered potential candidate treatment delivery agents.85 Adult NSCs are found within the subventricular zone and the hippocampal dentate gyri and give rise to neurons and glia. The preparation of sufficient numbers of autologous NSCs for clinical applications remains technically challenging. Fetal brain, adult allogeneic brain, and embryonic stem cells are therefore all currently being considered as possible substitutes for autologous NSCs. MSCs are multipotent cells capable of differentiating into multiple cell types, including chondrocytes, adipocytes, and osteoblasts. They were originally isolated from bone marrow, but can be obtained from many other sources, including peripheral blood, umbilical cord blood, and adipose tissue. MSCs are an attractive alternative to NSCs because they are easy to propagate in vitro and because the implantation of autologous MSCs in patients with GB entails fewer ethical problems. In most studies of antiangiogenic therapy with NSCs or MSCs, these cells are genetically modified to express IL-12, endostatin, IFN-α, TSP-1, or PEX and are delivered directly to the brain tumor.86–94 The treatment of intracranial gliomas with these engineered cells has been shown to inhibit tumor progression, and/or to increase survival in mice (Table 1). Furthermore, the simultaneous delivery of antiangiogenic factors (endostatin or TSP-1) and chemotherapy factors (a secretable form of carboxylesterase 2 [sCE2] along with CPT-11 treatment or a secretable variant of tumor necrosis factor-related apoptosis-inducing ligand [TRAIL]) by MCSs yielded the stronger antitumor responses86,94 (Table 1). We recently showed that MSCs can deliver the chemotherapy drug ferrociphenol and the antiangiogenic TKI SFN to brain tumors without the need for genetic modification.95,96 MSCs can be primed in vitro with SFN, with no effect on viability, and can release 60% of the drug with which they are loaded. The cytostatic activity of the released SFN was entirely conserved, significantly decreasing the survival of human U87MG GB cells and endothelial cells in vitro. We investigated the in vivo effect of SFN-primed MSCs on the orthotopic U87MG GB model, following their intranasal administration. Unlike direct intracranial delivery, intranasal delivery is noninvasive, making the repeated administration of treatment possible. Furthermore, this delivery route appears to be a promising alternative to intravenous injections of MSCs, which generally lead to the entrapment and elimination of MSCs in peripheral organs and a risk of vascular and pulmonary embolization.97,98 The treatment of U87MG tumor-bearing mice with two intranasal administrations of SFN-primed MSCs 4 days apart has been shown to result in lower levels of tumor angiogenesis than the injection of unprimed MSCs or SFN alone, but with no effect on tumor volume. The absence of an effect on tumor volume is probably due to the use of too low a dose of SFN-primed MSCs. The intranasal administration of larger numbers of SFN-primed MSCs may be required for an effect on U87MG growth. No clinical trial has yet been performed to assess the use of MSCs as cellular vehicles for GB therapy. One reason for this may be the widespread use of viral vectors for the production of therapeutic MSCs. The use of viruses is associated with a number of disadvantages, including risks of toxicity, immunogenicity, insertional mutagenesis, and high manufacturing costs.99 Another possible reason is the conflicting data published concerning the use of MSCs in cell-based therapies. The role of MSCs in cancer progression remains a matter of heated debate, but the number of studies reporting a role for these cells in cancer progression is steadily increasing.100–103 Consistent with the findings of these studies, we found that the intranasal administration of unprimed MSCs induced a significant increase in the number of small vessels in the U87MG tumor, which was abolished when MSCs were primed with SFN. Several studies have indicated that MSCs promote angiogenesis by secreting angiogenic factors, such as VEGF, releasing exosomes, recruiting endothelial progenitors, and/or transdifferentiating into endothelial cells.104,105 Our previous studies96,106 showed no effect of unprimed MSCs on tumor volume or the proportion of Ki67+ tumor cells in the human U87MG GB model, but MSCs have been reported to facilitate tumor growth by secreting various anti-inflammatory cytokines and proangiogenic factors.100–103 Furthermore, MSCs can differentiate into cancer-associated fibroblasts (CAFs), which have been detected in the stroma of carcinomas and are known to promote tumor growth.100 In the GB peritumoral environment, we identified MSC-like cells that we named GB-associated stromal cells (GASCs), which had phenotypic and functional properties in common with MSCs and CAFs.107–109 Like unprimed MSCs, their injection into intracranial U87MG tumors had no effect on tumor volume but promoted angiogenesis, increasing the number of small intratumoral vessels.108 In other studies, MSC-like cells were isolated from the GB tumor core and shown to increase angiogenesis, GB cell proliferation, and invasion.110–114 Consistent with these findings, the percentage of GB-associated MSC-like cells has recently been shown to be inversely correlated with overall survival, indicating a role for these cells in promoting the aggressive behavior of GB.115,116 All these data raise questions about the suitability of MSCs as cellular vehicles for the delivery of therapeutic molecules in the context of GB. Even if MSCs are able to deliver the therapeutic agent to the tumor, their protumorigenic and proangiogenic properties may limit the effect of this agent. We need to find ways of guaranteeing the safety of this cellular vector for clinical use. One possibility would be to use a suicide gene or a small molecule to induce senescence in the MSCs after drug delivery. EPCs, which display specific homing to angiogenic sites in vivo, are a potentially attractive alternative to MSCs, but the therapeutic effect of modified EPCs in orthotopic GB models has never been investigated.117–121
Conclusion
Various nonviral delivery methods, including CED devices, implantable polymer devices, nanocarriers, and cellular vehicles, have been developed to improve the efficacy of GB treatment with antiangiogenic factors. All these delivery methods, tested in orthotopic GB models, were found to be effective, but only CED devices have been translated into clinical practice. Studies showing clear-cut advantages of one particular type of antiangiogenic factor delivery method over another in a representative GB model would facilitate the translation of such methods from the bench to the clinic (Figure 5). For such studies, there is an urgent need to identify the angiogenesis inhibitors giving the best response in preclinical studies and to focus on cheap, easily produced biocompatible nonviral delivery methods that could be rapidly transferred into clinical practice. The delivery route is also a major matter of concern that must be addressed. CED devices, implantable polymer devices, and microencapsulated cells are local delivery methods. They have the advantage of providing higher concentrations of antiangiogenic factors in the tumor than systemic delivery methods. However, they require intracranial surgery, which is not compatible with the use of factors for which a regime involving repeated injections is required for an effect on the tumor vasculature. In this case, nanocarriers or adult stem cells, which can be administered systemically, should be favored. Furthermore, it should also be borne in mind that angiogenesis inhibitor therapy alone is not sufficient to counteract the growth of GB, given the aggressiveness of this type of brain cancer and its resistance to various treatments. Further studies will therefore also be required to evaluate and optimize dosing schedules, for the development of effective combinations of the selected antiangiogenic factor delivery method with other treatments, such as chemotherapy, radiotherapy, and phototherapy. All these studies will take time but are necessary to ensure that the selected antiangiogenic factor delivery method is of unequivocal and meaningful clinical benefit to GB patients in the future. Particular attention should be paid to the emerging vascular-targeted nanotheranostic devices consisting of multifunctional nanocarriers for both diagnosis and the delivery of therapeutic agents to the tumor vasculature.122 The results obtained with these systems in preclinical models of GB should be monitored to determine whether they are likely to be more effective than antiangiogenic factor delivery methods (Figure 5).
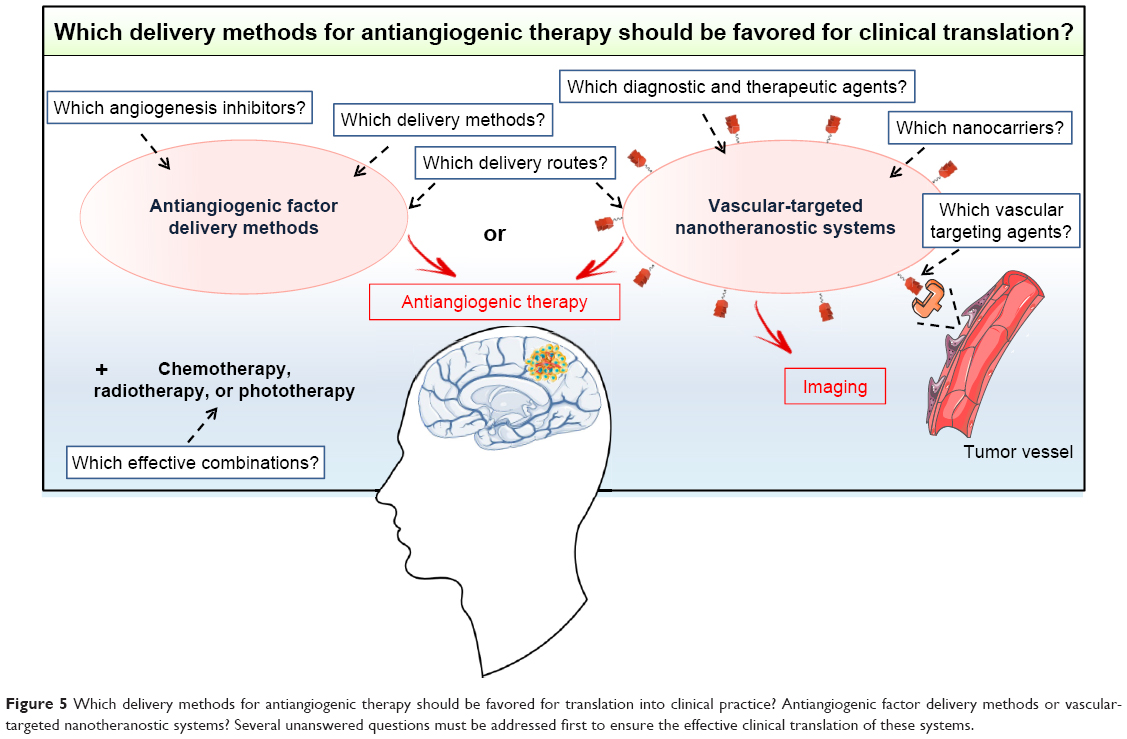 | Figure 5 Which delivery methods for antiangiogenic therapy should be favored for translation into clinical practice? Antiangiogenic factor delivery methods or vascular-targeted nanotheranostic systems? Several unanswered questions must be addressed first to ensure the effective clinical translation of these systems. |
Acknowledgments
This work was supported by grants from the “Fondation de France”, “Ligue Contre le Cancer du Grand-Ouest, Comité Départemental du Maine-et-Loire”, “Association en Avant la Vie”, and the European Commission, Education, Audiovisual and Culture Executive Agency (EACEA), through the NanoFar Erasmus Mundus joint Doctoral program. We thank Professor Franck Boury (Team 17, GLIAD, CRCINA, INSERM 1232, Angers), Doctor Snow Stolnik, and Doctor Cynthia Bosquillon (School of Pharmacy, University of Nottingham, Nottingham, UK) for providing facilities. We also thank Alex Edelman and associates for correcting the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
de Groot J, da R, Batchelor TT. Antiangiogenic therapy for glioblastoma: the challenge of translating response rate into efficacy. Am Soc Clin Oncol Educ Book. 2013. doi:10.1200/EdBook_AM.2013.33.e71. | ||
Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(26):3212–3218. | ||
Khasraw M, Ameratunga M, Grommes C. Bevacizumab for the treatment of high-grade glioma: an update after phase III trials. Expert Opin Biol Ther. 2014;14(5):729–740. | ||
Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W. Glioma: experimental models and reality. Acta Neuropathologica. 2017;133(2):263–282. | ||
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. | ||
Das S, Marsden PA. Angiogenesis in glioblastoma. N Engl J Med. 2013;369(16):1561–1563. | ||
Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181(4):1126–1141. | ||
Plate KH, Scholz A, Dumont DJ. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathologica. 2012;124(6):763–775. | ||
Wang N, Jain RK, Batchelor TT. New directions in anti-angiogenic therapy for glioblastoma. Neurotherapeutics. 2017;14(2):321–332. | ||
Dimberg A. The glioblastoma vasculature as a target for cancer therapy. Biochm Soc Trans. 2014;42(6):1647–1652. | ||
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8(8):610–622. | ||
Rao N, Lee YF, Ge R. Novel endogenous angiogenesis inhibitors and their therapeutic potential. Acta Pharmacol Sin. 2015;36(10):1177–1190. | ||
Chen F, Chen L, He H, et al. Up-regulation of microRNA-16 in glioblastoma inhibits the function of endothelial cells and tumor angiogenesis by targeting Bmi-1. ACAMC. 2016;16(5):609–620. | ||
Gagner J-P, Law M, Fischer I, Newcomb EW, Zagzag D. Angiogenesis in gliomas: imaging and experimental therapeutics. Brain Pathology. 2005;15(4):342–363. | ||
Rege TA, Fears CY, Gladson CL. Endogenous inhibitors of angiogenesis in malignant gliomas: nature’s antiangiogenic therapy. Neuro-Oncol. 2005;7(2):106–121. | ||
Nabors LB, Fiveash JB, Markert JM, et al. A phase 1 trial of ABT-510 concurrent with standard chemoradiation for patients with newly diagnosed glioblastoma. Arch Neurol. 2010;67(3):313–319. | ||
Kong D-H, Kim MR, Jang JH, Na H-J, Lee S. A review of anti-angiogenic targets for monoclonal antibody cancer therapy. Int J Mol Sci. 2017;18(8). pii: E1786. | ||
Chinot OL, Reardon DA. The future of antiangiogenic treatment in glioblastoma. Current Opinion in Neurology. 2014;27(6):675–682. | ||
Gilbert MR, Sulman EP, Mehta MP. Bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(21):2048–2049. | ||
Cloughesy T, Finocchiaro G, Belda-Iniesta C, et al. Randomized, double-blind, placebo-controlled, multicenter phase ii study of onartuzumab plus bevacizumab versus placebo plus bevacizumab in patients with recurrent glioblastoma: efficacy, safety, and hepatocyte growth factor and O6-methylguanine–DNA methyltransferase biomarker analyses. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35(3):343–351. | ||
Gotink KJ, Verheul HMW. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13(1):1–14. | ||
Popescu AM, Purcaru SO, Alexandru O, Dricu A. New perspectives in glioblastoma antiangiogenic therapy. Współczesna Onkologia. 2016;2(2):109–118. | ||
de Groot JF, Lamborn KR, Chang SM, et al. Phase II study of aflibercept in recurrent malignant glioma: a North American brain tumor Consortium study. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29(19):2689–2695. | ||
Reardon DA, Lassman AB, Schiff D, et al. Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer. 2018;124(7):1438–1448. | ||
Weller M, Nabors LB, Gorlia T, et al. Cilengitide in newly diagnosed glioblastoma: biomarker expression and outcome. Oncotarget. 2016;7(12):15018–15032. | ||
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | ||
Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. | ||
Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–152. | ||
Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67(8):3560–3564. | ||
de Groot JF, Piao Y, Tran H, et al. Myeloid biomarkers associated with glioblastoma response to anti-VEGF therapy with aflibercept. Clinical Cancer Research. 2011;17(14):4872–4881. | ||
Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6(8):465–477. | ||
Jahangiri A, Chin AT, Flanigan PM, Chen R, Bankiewicz K, Aghi MK. Convection-enhanced delivery in glioblastoma: a review of preclinical and clinical studies. J Neurosurg. 2017;269(3):191–200. | ||
Mehta AM, Sonabend AM, Bruce JN. Convection-enhanced delivery. Neurotherapeutics. 2017;14(2):358–371. | ||
Ndesendo VM. Convection-enhanced delivery of neurotherapeutics. In: Viness Pillay P, Choonara EPY, editors. Advances in Neurotherapeutic Delivery Technologies. Vol 8. UK: OMICS International; 2015. | ||
Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro-Oncology. 2015;17(suppl 2):ii3–ii8. | ||
Wang W, Sivakumar W, Torres S, et al. Effects of convection-enhanced delivery of bevacizumab on survival of glioma-bearing animals. Neurosurg Focus. 2015;62(3):E8. | ||
Ohlfest JR, Demorest ZL, Motooka Y, et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the sleeping Beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Molecular Therapy. 2005;12(5):778–788. | ||
Gillespie DL, Aguirre MT, Ravichandran S, et al. RNA interference targeting hypoxia-inducible factor 1α via a novel multifunctional surfactant attenuates glioma growth in an intracranial mouse model. J Neurosurg. 2015;122(2):331–341. | ||
Huang J, Yuan D, Liu D, et al. Efficacy of antiangiogenic targeted immunotoxin DTAT and DTATEGF against glioblastoma multiforme. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2014;39(1):1–5. | ||
Grossman R, Tyler B, Hwang L, et al. Improvement in the standard treatment for experimental glioma by fusing antibody Fc domain to endostatin. J Neurosurg. 2011;284(6):1139–1146. | ||
Schmidt NO, Ziu M, Carrabba G, et al. Antiangiogenic therapy by local intracerebral microinfusion improves treatment efficiency and survival in an orthotopic human glioblastoma model. Clin Cancer Res. 2004;10(4):1255–1262. | ||
Zagorac D, Jakovcevic D, Gebremedhin D, Harder DR. Antiangiogenic effect of inhibitors of cytochrome P450 on rats with glioblastoma multiforme. J Cereb Blood Flow Metab. 2008;28(8):1431–1439. | ||
Clavreul A, Roger E, Pourbaghi-Masouleh M, Lemaire L, Tétaud C, Menei P. Development and characterization of sorafenib-loaded lipid nanocapsules for the treatment of glioblastoma. Drug Delivery. 2018;25(1):1756–1765. | ||
Sampson JH, Akabani G, Archer GE, et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro-Oncol. 2008;10(3):320–329. | ||
Chew SA, Danti S. Biomaterial-based implantable devices for cancer therapy. Adv Healthcare Mater. 2017;6(2):1600766. | ||
Wait SD, Prabhu RS, Burri SH, Atkins TG, Asher AL. Polymeric drug delivery for the treatment of glioblastoma. Neuro-Oncology. 2015;17(suppl 2):ii9–ii23. | ||
Basu A, Domb AJ. Recent advances in polyanhydride based biomaterials. Adv Mater Deerfield Beach Fla. 2018;30(41):e1706815. | ||
Bota DA, Desjardins A, Quinn JA, Affronti ML, Friedman HS. Interstitial chemotherapy with biodegradable BCNU (Gliadel) wafers in the treatment of malignant gliomas. Ther Clin Risk Manag. 2007;3(5):707–715. | ||
Brem H, Gabikian P. Biodegradable polymer implants to treat brain tumors. J Control Release. 2001;74(1–3):63–67. | ||
Tamargo RJ, Myseros JS, Epstein JI, Yang MB, Chasin M, Brem H. Interstitial chemotherapy of the 9L gliosarcoma: controlled release polymers for drug delivery in the brain. Cancer Res. 1993;53(2):329–333. | ||
D’Avella D, Dellapuppa A. Safety and efficacy of Gliadel wafers for newly diagnosed and recurrent glioblastomas. Acta Neurochirurgica. 2012;154(8):1379–1381. | ||
Ashby LS, Smith KA, Stea B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: a systematic literature review. World J Surg Onc. 2016;14(1):225. | ||
Bow H, Hwang LS, Schildhaus N, et al. Local delivery of angiogenesis-inhibitor minocycline combined with radiotherapy and oral temozolomide chemotherapy in 9L glioma. J Neurosurg. 2014;11(4):662–669. | ||
Frazier JL, Wang PP, Case D, et al. Local delivery of minocycline and systemic BCNU have synergistic activity in the treatment of intracranial glioma. J Neurooncol. 2003;64(3):203–209. | ||
Weingart JD, Sipos EP, Brem H. The role of minocycline in the treatment of intracranial 9L glioma. J Neurosurg. 1995;48(4):635–640. | ||
Martins C, Sousa F, Araújo F, Sarmento B. Functionalizing PLGA and PLGA derivatives for drug delivery and tissue regeneration applications. Adv Healthcare Mater. 2018;7(1):1701035. | ||
Mir M, Ahmed N, Rehman AU. Recent applications of PLGA based nanostructures in drug delivery. Colloids and Surfaces B: Biointerfaces. 2017;159:217–231. | ||
Tseng Y-Y, Yang T-C, Wang Y-C, et al. Targeted concurrent and sequential delivery of chemotherapeutic and antiangiogenic agents to the brain by using drug-loaded nanofibrous membranes. Int J Nanomedicine. 2017;12:1265–1276. | ||
Hadjiargyrou M, Chiu JB. Enhanced composite electrospun nanofiber scaffolds for use in drug delivery. Expert Opin Drug Deliv. 2008;5(10):1093–1106. | ||
Tseng Y-Y, Wang Y-C, Su C-H, et al. Concurrent delivery of carmustine, irinotecan, and cisplatin to the cerebral cavity using biodegradable nanofibers: in vitro and in vivo studies. Colloids and Surfaces B: Biointerfaces. 2015;134:254–261. | ||
Lei C, Cui Y, Zheng L, Kah-Hoe Chow P, Wang C-H. Development of a gene/drug dual delivery system for brain tumor therapy: potent inhibition via RNA interference and synergistic effects. Biomaterials. 2013;34(30):7483–7494. | ||
Tseng Y-Y, Su C-H, Yang S-T, et al. Advanced interstitial chemotherapy combined with targeted treatment of malignant glioma in rats by using drug-loaded nanofibrous membranes. Oncotarget. 2016;7(37):59902–59916. | ||
Benny O, Kim S-K, Gvili K, et al. In vivo fate and therapeutic efficacy of PF-4/CTF microspheres in an orthotopic human glioblastoma model. Faseb J. 2008;22(2):488–499. | ||
Floyd JA, Galperin A, Ratner BD. Drug encapsulated polymeric microspheres for intracranial tumor therapy: a review of the literature. Adv Drug Deliv Rev. 2015;91:23–37. | ||
Menei P, Capelle L, Guyotat J, et al. Local and sustained delivery of 5-fluorouracil from biodegradable microspheres for the radiosensitization of malignant glioma: a randomized phase II trial. Neurosurgery. 2005;56(2):242–248. | ||
Chakroun RW, Zhang P, Lin R, Schiapparelli P, Quinones-Hinojosa A, Cui H. Nanotherapeutic systems for local treatment of brain tumors. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2018;10(1):e1479. | ||
Ong Q, Hochberg FH, Cima MJ. Depot delivery of dexamethasone and cediranib for the treatment of brain tumor associated edema in an intracranial rat glioma model. J Controlled Release. 2015;217:183–190. | ||
Grayson ACR, Choi IS, Tyler BM, et al. Multi-pulse drug delivery from a resorbable polymeric microchip device. Nat Mater. 2003;2(11):767–772. | ||
Karim R, Palazzo C, Evrard B, Piel G. Nanocarriers for the treatment of glioblastoma multiforme: current state-of-the-art. J Control Release. 2016;227:23–37. | ||
Kumari P, Ghosh B, Biswas S. Nanocarriers for cancer-targeted drug delivery. J Drug Target. 2016;24(3):179–191. | ||
Narayana A. Applications of nanotechnology in cancer: a literature review of imaging and treatment. J Nucl Med Radiat Ther. 2014;05(04):195. | ||
Tan YF, Lao LL, Xiong GM, Venkatraman S. Controlled-release nanotherapeutics: state of translation. J Controlled Release. 2018;284:39–48. | ||
Li Y, Li J, Woo YM, et al. Enhanced expression of Vastatin inhibits angiogenesis and prolongs survival in murine orthotopic glioblastoma model. BMC Cancer. 2017;17(1):126. | ||
Sonabend AM, Velicu S, Ulasov IV, et al. A safety and efficacy study of local delivery of interleukin-12 transgene by PPC polymer in a model of experimental glioma. Anti-Cancer Drugs. 2008;19(2):133–142. | ||
Gasperini L, Mano JF, Reis RL. Natural polymers for the microencapsulation of cells. J R Soc Interface. 2014;11(100):20140817. | ||
Gonzalez-Pujana A, Santos E, Orive G, Pedraz JL, Hernandez RM. Cell microencapsulation technology: current vision of its therapeutic potential through the administration routes. J Drug Delivery Sci Technol. 2017;42:49–62. | ||
Olabisi RM. Cell microencapsulation with synthetic polymers. J Biomed Mater Res. 2015;103(2):846–859. | ||
Opara EC. Applications of cell microencapsulation. Methods Mol Biol Clifton NJ. 2017;1479:23–39. | ||
Shah K. Encapsulated stem cells for cancer therapy. Biomatter. 2013;3(1):e24278. | ||
Read T-A, Sorensen DR, Mahesparan R, et al. Local endostatin treatment of gliomas administered by microencapsulated producer cells. Nat Biotechnol. 2001;19(1):29–34. | ||
Bjerkvig R, Read TA, Vajkoczy P, et al. Cell therapy using encapsulated cells producing endostatin. Acta Neurochir Suppl. 2003;88:137–141. | ||
Read TA, Farhadi M, Bjerkvig R, et al. Intravital microscopy reveals novel antivascular and antitumor effects of endostatin delivered locally by alginate-encapsulated cells. Cancer Res. 2001;61(18):6830–6837. | ||
Roger M, Clavreul A, Venier-Julienne M-C, Passirani C, Montero-Menei C, Menei P. The potential of combinations of drug-loaded nanoparticle systems and adult stem cells for glioma therapy. Biomaterials. 2011;32(8):2106–2116. | ||
Choi SH, Tamura K, Khajuria RK, et al. Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol Ther. 2015;23(2):235–243. | ||
de Boüard S, Guillamo J-S, Christov C, et al. Antiangiogenic therapy against experimental glioblastoma using genetically engineered cells producing interferon-alpha, angiostatin, or endostatin. Hum Gene Ther. 2003;14(9):883–895. | ||
Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, Js Y. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002;62(20):5657–5663. | ||
Kim S-K, Cargioli TG, Machluf M, et al. PEX-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin Cancer Res. 2005;11(16):5965–5970. | ||
Lorico A, Mercapide J, Soloduschko V, et al. Primary neural stem/progenitor cells expressing endostatin or cytochrome P450 for gene therapy of glioblastoma. Cancer Gene Ther. 2008;15(9):605–615. | ||
Ryu CH, Park S-H, Park SA, et al. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum Gene Ther. 2011;22(6):733–743. | ||
van Eekelen M, Sasportas LS, Kasmieh R, et al. Human stem cells expressing novel TSP-1 variant have anti-angiogenic effect on brain tumors. Oncogene. 2010;29(22):3185–3195. | ||
Yang S-Y, Liu H, Zhang J-N. Gene therapy of rat malignant gliomas using neural stem cells expressing IL-12. DNA and Cell Biol. 2004;23(6):381–389. | ||
Yin J, Kim J-K, Moon J-H, et al. hMSC-mediated concurrent delivery of endostatin and carboxylesterase to mouse xenografts suppresses glioma initiation and recurrence. Mol Ther. 2011;19(6):1161–1169. | ||
Clavreul A, Lautram N, Franconi F, Montagu A, Lainé A-L. Targeting and treatment of glioblastomas with human mesenchymal stem cells carrying ferrociphenol lipid nanocapsules. IJN. 2015;10:1259–1271. | ||
Clavreul A, Pourbaghi-Masouleh M, Roger E, Lautram N, Montero-Menei CN, Menei P. Human mesenchymal stromal cells as cellular drug-delivery vectors for glioblastoma therapy: a good deal? J Exp Clin Cancer Res. 2017;36(1):135. | ||
Fischer UM, Harting MT, Jimenez F, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18(5):683–692. | ||
Ramot Y, Steiner M, Morad V, et al. Pulmonary thrombosis in the mouse following intravenous administration of quantum dot-labeled mesenchymal cells. Nanotoxicology. 2010;4(1):98–105. | ||
Glover DJ, Lipps HJ, Jans DA, Safe T. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet. 2005;6(4):299–310. | ||
Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017;16(1):31. | ||
Sherman LS, Shaker M, Mariotti V, Rameshwar P. Mesenchymal stromal/stem cells in drug therapy: new perspective. Cytotherapy. 2017;19(1):19–27. | ||
Shi Y, du L, Lin L, Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16(1):35–52. | ||
Zhang C, Yang S-J, Wen Q, et al. Human-derived normal mesenchymal stem/stromal cells in anticancer therapies. J Cancer. 2017;8(1):85–96. | ||
Bronckaers A, Hilkens P, Martens W, et al. Mesenchymal stem/stromal cells as a pharmacological and therapeutic approach to accelerate angiogenesis. Pharmacol Ther. 2014;143(2):181–196. | ||
Merino-González C, Zuñiga FA, Escudero C, et al. Mesenchymal stem cell-derived extracellular vesicles promote angiogenesis: Potencial clinical application. Front Physiol. 2016;7(99):24. | ||
Roger M, Clavreul A, Sindji L, et al. In vitro and in vivo interactions between glioma and marrow-isolated adult multilineage inducible (Miami) cells. Brain Res. 2012;1473:193–203. | ||
Clavreul A, Etcheverry A, Tétaud C, et al. Identification of two glioblastoma-associated stromal cell subtypes with different carcinogenic properties in histologically normal surgical margins. J Neurooncol. 2015;122(1):1–10. | ||
Clavreul A, Guette C, Faguer R, et al. Glioblastoma-associated stromal cells (GASCs) from histologically normal surgical margins have a myofibroblast phenotype and angiogenic properties. J Pathol. 2014;233(1):74–88. | ||
Clavreul A, Etcheverry A, Chassevent A, et al. Isolation of a new cell population in the glioblastoma microenvironment. J Neurooncol. 2012;106(3):493–504. | ||
Appaix F, Nissou M-F, van der Sanden B, et al. Brain mesenchymal stem cells: The other stem cells of the brain? World J Stem Cells. 2014;6(2):134–143. | ||
Hossain A, Gumin J, Gao F, et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells. 2015;33(8):2400–2415. | ||
Kong BHO, Shin H-DO, Kim SE-H, et al. Increased in vivo angiogenic effect of glioma stromal mesenchymal stem-like cells on glioma cancer stem cells from patients with glioblastoma. Int J Oncol. 2013;42(5):1754–1762. | ||
Lim E-J, Suh Y, Yoo K-C, et al. Tumor-associated mesenchymal stem-like cells provide extracellular signaling cue for invasiveness of glioblastoma cells. Oncotarget. 2017;8(1):1438–1448. | ||
Svensson A, Ramos-Moreno T, Eberstål S, Scheding S, Bengzon J. Identification of two distinct mesenchymal stromal cell populations in human malignant glioma. J Neurooncol. 2017;131(2):245–254. | ||
Shahar T, Rozovski U, Hess KR, et al. Percentage of mesenchymal stem cells in high-grade glioma tumor samples correlates with patient survival. Neuro Oncol. 2017;19(5):660–668. | ||
Yoon S-J, Shim J-K, Chang JH, et al. Tumor mesenchymal stem-like cell as a prognostic marker in primary glioblastoma. Stem Cells International. 2016;2016(1):1–7. | ||
Argyriou AA, Giannopoulou E, Kalofonos HP. Angiogenesis and anti-angiogenic molecularly targeted therapies in malignant gliomas. Oncology. 2009;77(1):1–11. | ||
Ferrari N, Glod J, Lee J, Kobiler D, Fine HA, Marrow-Derived B. Bone marrow-derived, endothelial progenitor-like cells as angiogenesis-selective gene-targeting vectors. Gene Ther. 2003;10(8):647–656. | ||
Nolan DJ, Ciarrocchi A, Mellick AS, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21(12):1546–1558. | ||
Peters BA, Diaz LA, Polyak K, et al. Contribution of bone marrow–derived endothelial cells to human tumor vasculature. Nat Med. 2005;11(3):261–262. | ||
Wong MLH, Prawira A, Kaye AH, Hovens CM. Tumour angiogenesis: its mechanism and therapeutic implications in malignant gliomas. J Clin Neurosci. 2009;16(9):1119–1130. | ||
Sonali, Viswanadh MK, Singh RP, et al. Nanotheranostics: emerging strategies for early diagnosis and therapy of brain cancer. Nanotheranostics. 2018;2(1):70–86. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.