Back to Archived Journals » Oncolytic Virotherapy » Volume 4
Myxoma virus attenuates expression of activating transcription factor 4 (ATF4) which has implications for the treatment of proteasome inhibitor–resistant multiple myeloma
Authors Dunlap K, Bartee M, Bartee E
Received 7 August 2014
Accepted for publication 27 August 2014
Published 13 January 2015 Volume 2015:4 Pages 1—11
DOI https://doi.org/10.2147/OV.S72372
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Katherine M Dunlap, Mee Y Bartee, Eric Bartee
Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, USA
Abstract: The recent development of chemotherapeutic proteasome inhibitors, such as bortezomib, has improved the outcomes of patients suffering from the plasma cell malignancy multiple myeloma. Unfortunately, many patients treated with these drugs still suffer relapsing disease due to treatment-induced upregulation of the antiapoptotic protein Mcl1. We have recently demonstrated that an oncolytic poxvirus, known as myxoma, can rapidly eliminate primary myeloma cells by inducing cellular apoptosis. The efficacy of myxoma treatment on proteasome inhibitor–relapsed or –refractory myeloma, however, remains unknown. We now demonstrate that myxoma-based elimination of myeloma is not affected by cellular resistance to proteasome inhibitors. Additionally, myxoma virus infection specifically prevents expression of Mcl1 following induction of the unfolded protein response, by blocking translation of the unfolded protein response activating transcription factor (ATF)4. These results suggest that myxoma-based oncolytic therapy represents an attractive option for myeloma patients whose disease is refractory to chemotherapeutic proteasome inhibitors due to upregulation of Mcl1.
Keywords: drug resistance, oncolytic
Introduction
Multiple myeloma (MM) is a clonal plasma cell malignancy that is newly diagnosed in over 24,000 patients annually.1,2 Historically, the prognosis for MM patients was grim; however, the recent development of chemotherapeutic proteasome inhibitors (PIs), such as bortezomib (BTZ), has begun to improve these outcomes.3–5 PIs block degradation of misfolded proteins, which increases the protein-folding load in the endoplasmic reticulum (ER). This subsequently induces a lethal unfolded protein response (UPR) in MM cells, due to their inherently high translational burden.6,7 Unfortunately, while PIs have significantly improved the outcomes of many MM patients, disease relapse remains a major clinical challenge,8 and the prognosis for these relapsed patients is grim. Therefore, alternative treatments, particularly ones effective against relapsed or refractory disease, are required.
One proposed alternative is the use of cancer tropic replicating viruses, an approach known as oncolytic virotherapy. This approach is especially attractive for relapsed/refractory patients since oncolytic viruses often eliminate infected malignant cells through mechanisms distinct from those used by traditional chemotherapeutics and can therefore overcome drug resistance in a variety of cancer models.9–11 A variety of oncolytic viruses have been shown to have therapeutic potential for treating MM including: reovirus, vesicular stomatitis virus, vaccinia virus, and measles.12–20 Indeed, systemic injection of attenuated measles virus was recently shown to cause disease remission in an MM patient suffering from relapsed disease,20 demonstrating that oncolytic therapy can offer clinical benefit to patients suffering from relapsed MM.
Our lab is interested in the clinical potential of an oncolytic virus known as myxoma (MYXV). MYXV is a member of the Poxviridae, whose natural tropism is tightly restricted to lagomorphs (rabbits).21 Injection of MYXV into European rabbits causes a highly lethal disease known as myxomatosis. However, the virus is completely nonpathogenic in any other known species and is unable to replicate in or kill normal cells in vivo due to its susceptibility to the host innate inflammatory response.22,23 In contrast to its benign nature in normal tissues, MYXV has demonstrated significant oncolytic potential against a wide variety of solid tumors including: glioma, medulloblastoma, gallbladder cancer, melanoma, pancreatic cancer, and rhabdoid tumors.24–32 Despite these promising preclinical results, MYXV infection is generally nonlytic, and the virus is often outperformed by other oncolytic poxviruses in vitro.33 Therefore, identifying the mechanisms through which MYXV mediates its oncolytic effects remains a critical question.
We have recently reported that treatment of any of four established human MM cell lines, as well as a number of MM patient samples, with MYXV resulted in a complete loss of malignant cell viability within 6 hours. This oncolysis was specific to MM cells as normal hematopoietic cells were not affected by viral treatment. These effects were strong enough that ex vivo MYXV treatment could successfully prevent MM relapse in animal models of contaminated autologous stem cell transplant.34 Due to these promising results, we wanted to determine the mechanisms involved in MYXV-based treatment of human MM as well as explore the possibility of using MYXV as a novel treatment for PI-refractory MM.
Materials/subjects and methods
Cell lines and reagents
BSC40 and U266 cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). BTZ-sensitive (Dox40) and -resistant (Dox40BTZ) human MM cells35 were a kind gift from Dr Bei Liu at the Medical University of South Carolina. BSC40 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) plus 10% fetal bovine serum plus 1× penicillin-streptomycin-L-glutamine (Mediatech, Inc., Manassas, VA, USA). U266, Dox40, and Dox40BTZ cells were cultured at concentrations below 5×105 cells/mL in Roswell Park Memorial Institute (RPMI)-1644 with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) plus 20% fetal bovine serum plus 1× penicillin-streptomycin- l-glutamine. Cell viability was performed by CellTiter® 96 Non-Radioactive Cell Proliferation Assay (MTT) (Promega Corp, Fitchburg, WI, USA) and read on a FLUOstar Optima Plate reader (BMG LABTECH GmbH, Ortenberg, Germany). The following antibodies were used: from Cell Signaling Technology, Inc. (Danvers, MA, USA), Mcl1 (5453) activating transcription factor (ATF)4 (11815), CHOP (CCAAT/-enhancer-binding protein homologous protein) (2895), Actin (8457), and p-eIF2α (3398); and from Santa Cruz Biotechnology (Dallas, TX, USA), protein kinase RNA-like ER kinase (PERK) (13073), eIF2α (11386), and ATF6 (22799).
Virus amplification, purification, and infection
vMYX-GFP (recombinant MYXV strain Lausanne expressing green fluorescent protein [GFP] under a viral synthetic early/late promoter) was a kind gift from Dr Grant McFadden.36 The virus was amplified in BSC40 cells as previously described.37 In short, cells were infected with vMYX-GFP at a multiplicity of infection (MOI) =0.05, and then virus was allowed to replicate for 72 hours. Cells were removed from plates, mechanically lysed and the resulting supernatant clarified through a 36% sucrose pad. The resulting pellet was then further purified through a discontinuous (40%, 36%, 32%, 28%, and 24%) sucrose gradient. Virus banding at the 40%/36% interface was pelleted, resuspended in phosphate-buffered saline (PBS), and stored at −80°C.
Unless otherwise noted, experiments were carried out by infecting cells at a concentration of 1×107 cells/mL for 30 minutes using MOI =10. After the infection period, media was added to dilute cells to a concentration of 1×106 cells/mL. Drug treatments (1 μM Brefeldin [Acros Organics, Pittsburgh, PA, USA], 200 nM thapsigargin [Acros Organics], 50 μg/mL cytosine arabinoside [Sigma-Aldrich Corp, St Louis, MO, USA], 10 μM MG132 [Selleckchem, Houston, TX, USA], 12.5 nM BTZ [LC Laboratories, Woburn, MA, USA], or 10 nM GSK2606414 [Calbiochem®, Merck KGaA, Darmstadt, Germany]) were performed post-vMYX-GFP infection, by adding drug to the media when the cells were diluted. All experiments were controlled with mock infection, which refers to parallel treatment of cells with a PBS vehicle control.
Immunoblot analysis
For immunoblot analysis, cell were lysed in 1× Laemmli buffer and then separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE gels). Proteins were then transferred to Immobilon®-P PVDF membrane (EMD Millipore, Billerica, MA, USA), using wet transfer. Following transfer, membranes were blocked in 5% milk in TBS plus 1% Tween® 20. Membranes were incubated with primary and secondary antibodies in 5% milk for 30 minutes. Following each antibody incubation, membranes were washed three times with tris-buffered saline (TBS) plus 1% Tween 20. Quantitation was done using densitometry on Photoshop CS6.
Quantitative real-time polymerase chain reaction (qPCR)
RNA was purified using an RNeasy kit (Qiagen, Venlo, the Netherlands), and complementary (c)DNA generated as previously described.23 qPCR was performed using SYBR® Green PCR Core Reagents on a StepOne™ Real-Time PCR System (Invitrogen; Life Technologies Corp, Carlsbad, CA, USA). The generated data was normalized to 18sRNA and plotted as percent expression. The primers used for the qPCR were as follows:
- CHOP_Fw 5′GACTGAGGAGGAGCCAGAAC/ CHOP_Rv 5′ACCACTCTGTTTCCGTTTCC
- ATF4_Fw 5′AATACAACTGCCCTGTTCCC/ ATF4_Rv 5′CGTGAGAAGCCTGAATGAGA
- 18s_Fw 5′TAAAGGAATTGACGGAAGGG/ 18s_Rv 5′CTGTCAATCCTGTCCGTGTC.
XBP-1 splicing
RNA from cells was purified with an RNeasy kit (Qiagen) and cDNA synthesized as above.23 Subsequently, a fragment from the XBP-1 gene was amplified by PCR using the following primers: XBP1_Fw 5′AAACAGAGTAGCAGCTCAGACTGC/ XBP1_Rv 5′TCCTTCTGGGTAGACCTCTGGGAG. The resulting PCR product was digested with Pst1 at 37°C overnight (Fermentas, Pittsburgh, PA, USA). Pst1-digested and -undigested samples were electrophoresed on an 8% DNA acrylamide gel and then visualized with SYBR Gold (Invitrogen; Life Technologies Corp).
Polysome profile
A total 2.5×107 U266 cells per condition were infected as described above. At 12 hours postinfection, cells were pelleted, resuspended in TMK100 lysis buffer (10 mM Tris pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% Triton X-100, and 0.5% deoxycholate, in diethyl pyrocarbonate [DEPC]-treated water; 2 mM dithiothreitol [DTT] and 100 μg/mL cyclohexamide added fresh just before use), and homogenized with a 26 gauge needle before placing on ice for 5 minutes. Samples were centrifuged at 10,000× g for 5 minutes at 4°C, the supernatant then layered on top of a (50%, 40%, 30%, 20%, and 10%) discontinuous sucrose gradient, and centrifuged at 35,000 rpm for 3 hours at 4°C. After centrifugation, the gradient was separated into 25 fractions of 400 μL each. RNA was extracted from each fraction using TRIzol® (Invitrogen; Life Technologies Corp), and the localization of various ribosomal subunits in each fraction was determined using electrophoresis. RNA fractions were then combined into transfer (t)RNA, 40s, 60s, and polysome samples based on the electrophoretic profile, and cDNA was synthesized from each fraction as above. Fragments from individual genes were then amplified by PCR from each fraction and products visualized on an 8% DNA acrylamide gel stained with SYBR SAFE (Invitrogen; Life Technologies Corp). The primers used were as follows:
- ATF4_Fw 5′GCATGGTTTCCAGGTCATCT/ ATF4_Rv 5′AGTCCCTCCAACAACAGCAA
- UBC_Fw 5′TTGCCTTGACATTCTCGATG/ UBC_Rv 5′ATCGCTGTGATCGTCACTTG.
Results
MYXV kills PI-resistant MM cells
To assess the feasibility of MYXV virotherapy on PI-resistant MM cells, we asked whether drug resistance affected the loss of MM cell viability induced by MYXV treatment. Dox40 and Dox40BTZ cells35 were infected with MYXV at MOIs, and then cell viability was analyzed 24 hours postinfection, using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. We observed that MYXV treatment caused equal losses of viability from both PI-sensitive and -resistant Dox40 MM cells (Figure 1), suggesting that MYXV treatment can overcome the resistance to PI-based chemotherapy developed by some MM cells.
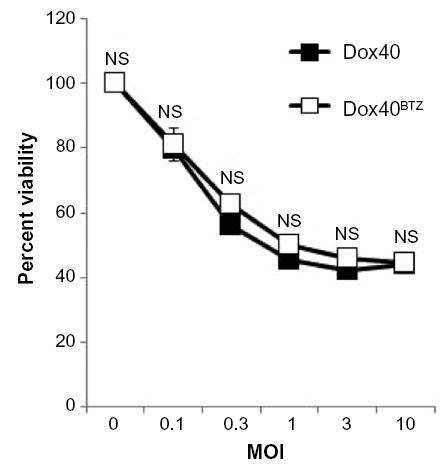 | Figure 1 MYXV eliminates bortezomib-resistant human MM cells. Dox40 or Dox40BTZ cells were infected with MYXV at the indicated MOIs. After 24 hours, cellular viability was measured using the MTT assay. No significant differences in the killing of Dox40 or Dox40BTZ were observed. |
MYXV simultaneously induces and inhibits the UPR in human MM cells
Since PIs eliminate MM cells by inducing a lethal UPR,6,7,38 and resistance to PI treatment is often mediated by changes in this pathway,39 we next asked how MYXV treatment might affect the UPR in human MM cells. We therefore investigated the expression of the UPR death effector molecule, CHOP, following infection with MYXV in either the presence or absence of an existing UPR response. U266 MM cells, which are highly susceptible to MYXV treatment,34 were mock-treated or infected with MYXV in either the presence or absence of the UPR-inducer brefeldin-A (BreA). At 6 hours after infection, cells were harvested and CHOP expression was assayed by immunoblot (Figure 2A). Consistent with an inactive UPR, we observed that mock-treated U266 cells displayed virtually no expression of CHOP. Cells infected with MYXV displayed slightly increased CHOP expression; however, this expression was significantly lower than that observed in cells treated with BreA. Interestingly, cells infected with MYXV failed to induce high levels of CHOP even when treated with BreA. Since CHOP is predominantly regulated transcriptionally, we next asked how MYXV affected expression of the CHOP messenger (m)RNA (Figure 2B). Consistent with our results for the CHOP protein, we observed that expression of the CHOP mRNA was low in mock-treated cells and that infection with MYXV induced this expression only slightly. In contrast, treatment with BreA induced expression of the CHOP mRNA to much more robust levels; however, this induction was prevented by MYXV infection. These data are consistent with MYXV inducing and subsequently inhibiting the UPR.
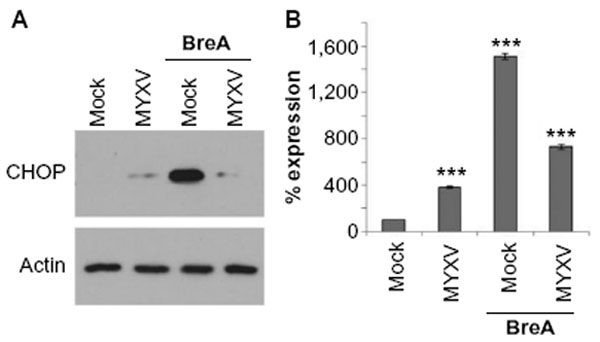 | Figure 2 MYXV prevents expression of CHOP. U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 1 μM BreA. Six hours after infection, cells were harvested and analyzed for either (A) CHOP and actin protein expression via immunoblot or (B) expression of CHOP mRNA using qPCR. ***P<0.005 using Student’s t-test. |
MYXV differentially affects each arm of the UPR
The UPR is comprised of three major pathways, each mediated by a single ER-resident stress-sensing protein.40,41 Activation of any of these pathways can result in CHOP expression. Since our result indicated that MYXV both induced and inhibited the UPR, we next asked whether viral infection might differentially affect each of these pathways. Human U266 MM cells were infected with MYXV and then either mock-treated or treated with BreA. At 6 hours postinfection, the activation of each arm of the UPR was analyzed.
The first arm of the UPR is mediated by the stress sensor inositol-requiring protein-1 (IRE1).42,43 ER stress results in dimerization and phosphorylation of IRE1, which in turn, excises 26nt from the mRNA of the transcription factor XBP1. This excision causes a frame shift in the XBP1 mRNA, allowing for the translation of the active XBP1s isoform, which transcribes genes involved in protein folding, quality control, and ER-associated degradation (ERAD). To assess activation of the IRE1 arm of the UPR, we therefore assayed whether this splicing event had taken place. Total cellular RNA was reverse-transcribed into cDNA, and then a fragment surrounding the spliced region of XBP1 was amplified using PCR. The sensitivity of this PCR fragment to digestion with Pst1, a unique restriction site found within the 26nt region of the mRNA, which is removed upon splicing, was then determined (Figure 3A). We observed that in mock-treated cells, the majority of the XBP1 PCR product was sensitive to digestion with Pst1, indicating that the mRNA was not being spliced by activated IRE1. In contrast, in cells treated with BreA, significant amounts of XBP1 PCR products were resistant to Pst1 digestion, indicating that BreA treatment induced activation of IRE1 and subsequent splicing of the XBP1 mRNA. This Pst1-resistant PCR product was not observed following infection with MYXV, and infection did not significantly prevent the appearance of this resistant product following BreA treatment, suggesting that MYXV neither activates nor inhibits the IRE1 arm of the UPR.
 | Figure 3 MYXV differentially activates each arm of the UPR. MYXV-treated U266 cells were harvested 6 hours postinfection. (A) RNA extracted from U266 cells was subjected to qPCR. The resulting PCR product was then digested with Pst1, loaded onto an 8% DNA acrylamide gel, and stained with SYBR® Gold. PCR products derived from XBP1 mRNA, which had undergone IRE1-mediated splicing, contain a Pst1-resistant band of higher molecular weight (arrow). (B) U266 lysate was subjected to treatment with 50 mM DTT prior to loading onto a Laemmli gel. Samples were then immunoblotted for ATF6. Full-length ATF6 (arrow) is lost following activation. (C) U266 lysate was separated on a Laemmli gel and then immunoblotted with antibodies against PERK. Phosphorylated PERK is observed as a high-molecular-weight band (arrow). |
The second arm of the UPR is mediated by the transcription factor ATF6.44,45 ATF6 normally resides in the ER membrane, but when cells undergo translational stress, it is transported to the Golgi by the COPII complex, where it can be cleaved by the site 1 and site 2 proteases. This proteolysis event releases the cytosolic fragment of ATF6, which then traffics to the nucleus as an active transcription factor where it upregulates expression of proteins associated with ERAD.44 To assess the activation of this arm of the UPR, we therefore asked whether ATF6 underwent proteolytic cleavage during MYXV infection (Figure 3B). Consistent with an inactive UPR, in mock-treated cells, we observed a band corresponding to full-length ATF6. This band, however, was lost following infection with MYXV or treatment with BreA, suggesting that MYXV activates the ATF6 arm of the UPR.
The third arm of the UPR is mediated by the stress sensor PERK.46 Upon ER stress, PERK is phosphorylated and subsequently phosphorylates the translational regulator eIF2α. This results in global translational arrest, allowing for reduction of the existing protein burden. To assess the activation of this arm of the UPR, we assayed the direct phosphorylation of PERK (Figure 3C) as well as the subsequent phosphorylation of the translation factor eIF2α (Figure 4). Consistent with an inactive UPR, in mock-treated cells, we observed that immunoblot for PERK revealed a single band, consistent with nonphosphorylated PERK. Similar to ATF6, however, infection with MYXV, treatment with BreA, or both, rapidly induced the appearance of a second, higher-molecular-weight band corresponding to directly phosphorylated PERK (Figure 3C). Additionally, infection with MYXV or treatment with BreA rapidly induced phosphorylation of eIF2α (Figure 4A), and treatment with the PERK-specific inhibitor GSK2606414 reduced the abundance of phosphorylated eIF2α in MYXV-infected MM cells (Figure 4B and C). These data suggest that MYXV infection activates the PERK arm of the UPR, leading to subsequent PERK-mediated phosphorylation of eIF2α.
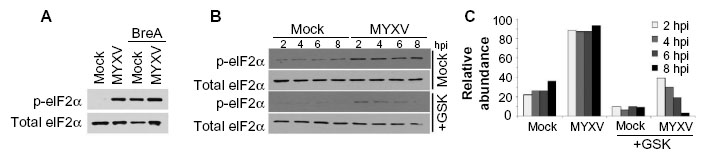 | Figure 4 Phosphorylation of eIF2alpha: (A) U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 1 μM BreA. Six hours after infection, phosphorylation of eIF2α was analyzed using immunoblot. (B) U266 cells were pretreated for 30 minutes with the PERK-specific inhibitor GSK2606414 at a final concentration of 10 nM. Cells were mock-infected or infected with MYXV at MOI =10. At the indicated time points, cells were harvested, and the phosphorylation of eIF2α was analyzed using immunoblot. (C) Densitometry quantitation of data shown in (B). |
MYXV inhibits expression of ATF4
Phosphorylation of eIF2α leads to global translational arrest, allowing for protein refolding. Sustained activation of eIF2α, however, results in preferential translation of ATF4, which is the primary mediator in CHOP-mediated apoptosis during the UPR. Since our data indicated that MYXV infection induced sustained phosphorylation of eIF2α (Figure 4B) but did not cause induction of CHOP (Figure 2), we asked whether viral infection might affect expression of ATF4. Consistent with the UPR being inactive in resting U266 cells, we observed that these cells expressed virtually no ATF4 protein (Figure 5). Interestingly, while infection with MYXV activated the PERK arm of the UPR, no induction of ATF4 protein was observed, and infected cells were unable to induce ATF4 expression following treatment with BreA. These data suggest that, while MYXV activates the PERK arm of the UPR, it subsequently blocks downstream signaling from this pathway by inhibiting expression of ATF4.
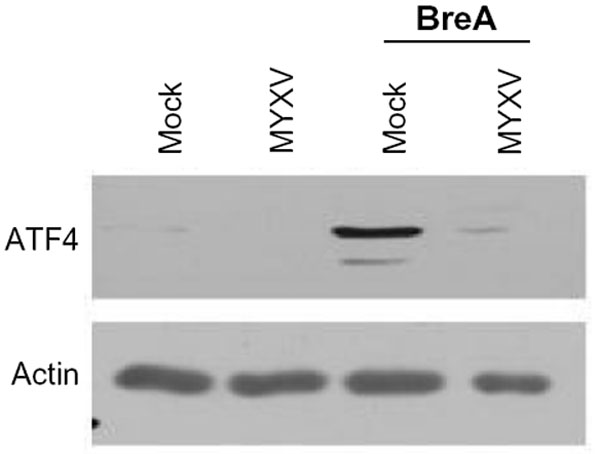 | Figure 5 MYXV inhibits expression of ATF4. U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 1 μM BreA. Six hours after infection, expression of ATF4 was analyzed using immunoblot. |
MYXV blocks ATF4 protein expression through a translational mechanism
ATF4 is a master regulator of the UPR as well as the integrated stress response. As such, it is tightly regulated through a variety of mechanisms, including transcriptional, translational, and posttranslational.47,48 We therefore wished to determine the mechanism through which MYXV infection inhibited expression of the ATF4 protein. To test whether the lack of ATF4 protein was due to rapid degradation, we infected human U266 MM cells with MYXV in the presence or absence of the proteasomal inhibitor MG132 and assayed the induction of ATF4 protein after 6 hours (Figure 6A). We observed that while MG132 alone induced high levels of ATF4 expression, infection with MYXV still prevented this induction, suggesting that the virus did not block ATF4 by inducing rapid protein degradation. We therefore asked whether MYXV infection might prevent ATF4 expression by affecting the expression of the ATF4 mRNA. Human U266 MM cells were infected with MYXV in either the presence or absence of BreA, and the levels of ATF4 mRNA were then measured using qPCR. Consistent with previous reports demonstrating induction of ATF4 mRNA during cellular stress,48 we observed that activation of the UPR by BreA caused a roughly twofold induction of the ATF4 mRNA (Figure 6B). In contrast, MYXV infection induced a much smaller induction of ATF4 mRNA, and this infection was additive with the induction caused by BreA treatment. Additionally, even in mock-treated U266 cells, robust expression of ATF4 mRNA was observed (Figure 6C). Taken together, these data indicate that MYXV infection has only minimal effects on the transcription of ATF4 and that high levels of ATF4 mRNA are found in infected cells, suggesting that MYXV does not prevent ATF4 protein expression through a transcriptional mechanism.
 | Figure 6 MYXV prevents translation of ATF4 mRNA. (A) U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 1 μM BreA or 10 nM MG132. Cells were then either mock-infected or infected with MYXV at MOI =10. Six hours after infection, the expression of ATF4 was analyzed using immunoblot. (B) U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 1 μM BreA. After 6 hours, RNA was harvested, cDNA synthesized, and the expression of ATF4 mRNA was determined using qPCR. (C) Quantitation of qPCR normalized to 18s RNA. (D) U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 1 μM BreA. Twelve hours postinfection, translation of ATF4 or UBCH5 mRNAs was analyzed using polysome profiling. ***P<0.005 and *P<0.05 using Student’s t-test. |
The final known level of regulation of ATF4 expression is at the level of protein translation. To assess whether MYXV affected ATF4 translation, we used polysome analysis to determine the level of translation occurring on the ATF4 mRNA (Figure 6D). We observed that under resting conditions, while the ATF4 mRNA was abundantly expressed, the majority of this mRNA was found in monosome and light polysome fractions, indicating that translation of this mRNA was low. In contrast, treatment of cells with BreA caused a noticeable shift of ATF4 mRNA toward heavier polysome fractions, indicating that this treatment increased gene translation. This shift, however, was blocked in cells infected with MYXV, suggesting that MYXV infection inhibits expression of ATF4 by blocking the translation of its mRNA during activation of the UPR.
MYXV infection depletes Mcl1
One of the primary mechanisms for MM cell resistance to PI-based chemotherapy is the increased expression of an antiapoptotic molecule, Mcl1. Interestingly, Mcl1 is a transcriptional target of ATF4, and depletion of ATF4 during the UPR both prevents expression of Mcl1 and resensitizes resistant MM cells to PI treatment.49,50 Since our data indicated that MYXV treatment effectively killed PI-resistant MM cells and prevented expression of ATF4 during activation of the UPR, we next asked whether viral infection altered expression of Mcl1. Human U266 MM cells were infected with MYXV at MOI =10. At the indicated time points, cells were then harvested and the expression of Mcl1 analyzed by immunoblot. We observed that while resting MM cells expressed high levels of Mcl1, these levels were rapidly reduced following MYXV infection (Figure 7A). To determine whether this depletion also occurred in the context of PI treatment, U266 cells were either mock-treated or infected with MYXV and then treated with 1nM BTZ. At 6 hours postinfection, the expression of both ATF4 and Mcl1 was measured by immunoblot. Consistent with previous literature,49 we observed that treatment of mock-infected MM cells with BTZ resulted in a significant increase in the expression of ATF4 as well as an upregulation of Mcl1. However, in cells infected with MYXV, ATF4 expression was significantly depressed and Mcl1 levels were reduced below baseline (Figure 7B). These data indicate that MYXV infection can alleviate the upregulation of Mcl1 responsible for MM cell resistance to PI therapy.
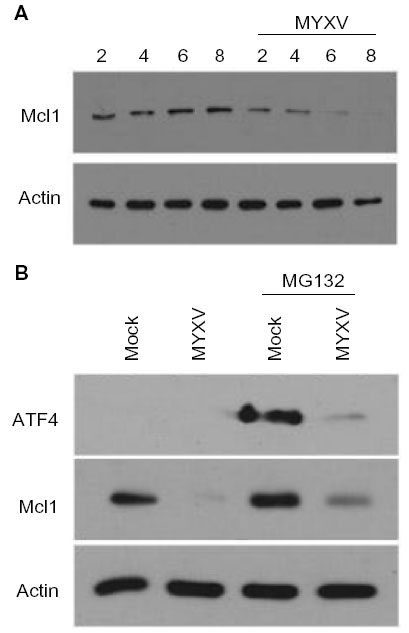 | Figure 7 MYXV prevents expression of Mcl1 during the UPR. (A) U266 cells were either mock-infected or infected with MYXV at MOI =10. At the indicated times postinfection, cells were harvested, and the expression of Mcl1 was analyzed using immunoblot. (B) U266 cells were either mock-infected or infected with MYXV at MOI =10 and subsequently incubated with 10 nM MG132. Six hours postinfection, expression of ATF4 and Mcl1 was analyzed using immunoblot. |
Discussion
Due to their secretion of high levels of immunoglobulin, MM cells are extremely sensitive to drugs or conditions that induce a lethal UPR. This sensitivity is the basis for the novel chemotherapeutics based around the PI BTZ, which has been demonstrating exciting success for the treatment of MM patients. Chemotherapeutic PIs act by inhibiting protein degradation in the proteasome. This causes a backlog of misfolded proteins, which induces a lethal, CHOP-mediated UPR in MM cells. Since MYXV largely represses expression of CHOP (Figures 2), these data suggest that combination therapy with MYXV and BTZ in nonresistant patients might be counterproductive since viral infection would prevent the primary mechanism of action of the chemotherapeutic. We did not observe any synergy between BTZ and MYXV in vitro (data not shown); however, this is likely due to the extremely high sensitivity of MM cells to MYXV-based killing, which renders synergy difficult to measure.
Interestingly, while induction of the UPR is the primary mechanism of killing for chemotherapeutic PIs, this induction is also a primary mediator of cancer cell resistance. This resistance develops when the UPR induced by PIs results in enhanced expression of ATF4. This enhanced ATF4 subsequently results in increased expression of the antiapoptotic protein Mcl1, which in turn, prevents PI-based elimination of MM cells. Interestingly, MYXV infection specifically blocks expression of ATF4, apparently by inhibiting translation of the ATF4 mRNA (Figure 6). In addition to this block, MYXV also depletes the cellular stores of Mcl1, suggesting that MYXV can overcome the development of resistance to PI mediated by Mcl1 overexpression. Importantly, these results were seen in two other MM cell lines (Figure S1), indicating that they are likely applicable to MM in general and not specific to U266 cells. Thus, MYXV might represent an attractive treatment option, either as a monotherapy for PI-resistant patients or as a sensitizing agent for chemotherapeutic PI in relapsed/resistant patients.
In normal cells, the objective of the UPR is to unburden the ER by expanding the mechanisms for protein refolding, removal of misfolded proteins, and by slowing down translation. If these mechanisms are unable to alleviate ER stress in a timely manner, however, the UPR also provides mechanisms, predominantly through induction of the proapoptotic transcription factor CHOP, through which to eliminate the injured cell. MYXV repression of CHOP (Figure 2A) is therefore in line with viral survival strategies that have evolved to combat the prolonged ER stress resulting from the rapid production of viral proteins during infection. Indeed, a variety of other viruses have been shown to modulate the UPR in both positive and negative manners. MYXV appears to predominantly inhibit the UPR by blocking translation of ATF4 (Figure 8). This blockade has the effect of preventing the high levels of CHOP expression needed to drive UPR-mediated apoptosis. Interestingly, ATF4 is a central transcription factor for the integrated stress response as well as the UPR. This suggests that MYXV-infected cells might display altered responsiveness to a variety of stress inducers that activate the integrated stress response. The natural end results of such alterations, however, remain to be determined.
 | Figure 8 Model of MYXV modulation of the UPR. The UPR is primarily comprised of three distinct arms mediated by the ER stress sensors IRE1, ATF6, and PERK. Our results indicate that MYXV infection activates both the ATF6 and PERK arms of the UPR (shown in blue) while having virtually no effect on the IRE1 arm. After activation of PERK, however, MYXV infection subsequently blocks the downstream signaling from this arm by actively inhibiting the translation of ATF4 (shown in red). |
MYXV infection in MM cells leads to activation of PERK and subsequent phosphorylation of eIF2α. This result is consistent with previous reports22 that MYXV induces phosphorylation of eIF2α through a PKR-independent mechanism. However, while treatment with the PERK-inhibitor GSK2606414 clearly dampened the total levels of P-eIF2α in MYXV infected cells, it did not completely eliminate this phosphorylation. Therefore, we are unable to rule out the possibility that one of the other eIF2α kinases also plays a role. For example, GCN2 has been shown to phosphorylate eIF2α during amino acid starvation, which is known to occur during rapid viral infections. Thus, the phosphorylation of eIF2α during MYXV infection and the resulting blockade of ATF4 are likely to be more complex than we currently understand.
In conclusion, our previous results demonstrate that MYXV is a promising new therapeutic option to treat MM. However, the mechanisms through which the virus interacts with MM cells are complex and appear to have significant impact for combination therapy with existing chemotherapeutics.
Acknowledgments
Dr Bartee is supported by a grant from the National Institute of Allergy and Infectious Disease (NIAID; grant number 1K22AI095372-01A1) as well as start-up funding from the Medical University of South Carolina. This work was also supported, in part, by a Hollings Cancer Center Support Grant (grant number P30 CA138313) as well as by the South Carolina Clinical and Translational Research (SCTR) Institute, National Institutes of Health (NIH) (grant number UL1TR000062). We would like to thank Dr Grant McFadden for the gifts of vMYX-GFP, Dr Bei Liu for the Dox40BTZ cells, and Dr Philip Howe for technical assistance with the polysome profiles.
Disclosure
The authors report no conflicts of interest in this work.
References
llscanada.org [homepage on the Internet]. Disease information and support: Myeloma. Leukemia and Lymphoma Society of Canada; 2012 [updated July 10, 2013]. Available from: http://www.llscanada.org/#/diseaseinformation/myeloma/. Accessed October 13, 2014. | |
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011; 364(11):1046–1060. | |
Kurtin SE, Bilotti E. Novel agents for the treatment of multiple myeloma: proteasome inhibitors and immunomodulatory agents. J Adv Pract Oncol. 2013;4(5):307–321. | |
Yaqub S, Ballester G, Ballester O. Frontline therapy for multiple myeloma: a concise review of the evidence based on randomized clinical trials. Cancer Invest. 2013;31(8):529–537. | |
Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–3076. | |
Ling SC, Lau EK, Al-Shabeeb A, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica. 2012;97(1):64–72. | |
Dong H, Chen L, Chen X, et al. Dysregulation of unfolded protein response partially underlies proapoptotic activity of bortezomib in multiple myeloma cells. Leuk Lymphoma. 2009;50(6):974–984. | |
Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. | |
Beljanski V, Hiscott J. The use of oncolytic viruses to overcome lung cancer drug resistance. Curr Opin Virol. 2012;2(5):629–635. | |
Heiber JF, Xu XX, Barber GN. Potential of vesicular stomatitis virus as an oncolytic therapy for recurrent and drug-resistant ovarian cancer. Chin J Cancer. 2011;30(12):805–814. | |
Eberle J, Fecker LF, Hossini AM, Kurbanov BM, Fechner H. Apoptosis pathways and oncolytic adenoviral vectors: promising targets and tools to overcome therapy resistance of malignant melanoma. Exp Dermatol. 2008;17(1):1–11. | |
Thirukkumaran CM, Shi ZQ, Luider J, et al. Reovirus as a successful ex vivo purging modality for multiple myeloma. Bone Marrow Transplant. 2014;49(1):80–86. | |
Thirukkumaran CM, Shi ZQ, Luider J, et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin Cancer Res. 2012;18(18):4962–4972. | |
Naik S, Nace R, Federspiel MJ, Barber GN, Peng KW, Russell SJ. Curative one-shot systemic virotherapy in murine myeloma. Leukemia. 2012;26(8):1870–1878. | |
Naik S, Nace R, Barber GN, Russell SJ. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-β. Cancer Gene Ther. 2012;19(7):443–450. | |
Yarde DN, Nace RA, Russell SJ. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells in vitro but have additive anti-myeloma activity in vivo. Exp Hematol. 2013;41(12):1038–1049. | |
Deng H, Tang N, Stief AE, et al. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia. 2008;22(12):2261–2264. | |
Kawa A, Arakawa S. The effect of attenuated vaccinia virus AS strain on multiple myeloma; a case report. Jpn J Exp Med. 1987;57(1):79–81. | |
Reddi HV, Madde P, McDonough SJ, et al. Preclinical efficacy of the oncolytic measles virus expressing the sodium iodide symporter in iodine non-avid anaplastic thyroid cancer: a novel therapeutic agent allowing noninvasive imaging and radioiodine therapy. Cancer Gene Ther. 2012;19(9):659–665. | |
Russell SJ, Federspiel MJ, Peng KW, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014; 89(7):926–933. | |
Fenner F, Ratcliffe FN. Myxomatosis. Cambridge: Cambridge University Press; 1965. | |
Wang F, Ma Y, Barrett JW, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5(12):1266–1274. | |
Bartee E, Mohamed MR, Lopez MC, Baker HV, McFadden G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J Virol. 2009;83(2):498–511. | |
Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–9990. | |
Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. | |
Weng M, Gong W, Ma M, et al. Targeting gallbladder cancer: oncolytic virotherapy with myxoma virus is enhanced by rapamycin in vitro and further improved by hyaluronan in vivo. Mol Cancer. 2014;13:82. | |
Weng M, Zhang M, Qin Y, et al. Targeting gallbladder carcinoma: bone marrow-derived stem cells as therapeutic delivery vehicles of myxoma virus. Chin Med J (Engl). 2014;127(12):2350–2356. | |
Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. | |
Thomas DL, Doty R, Tosic V, et al. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother. 2011;60(10):1461–1472. | |
Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20(4):759–768. | |
Woo Y, Kelly KJ, Stanford MM, et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329–2335. | |
Wu Y, Lun X, Zhou H, et al. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin Cancer Res. 2008;14(4):1218–1227. | |
Nichols AC, Yoo J, Um S, et al. Vaccinia virus outperforms a panel of other poxviruses as a potent oncolytic agent for the control of head and neck squamous cell carcinoma cell lines. Intervirology. 2014;57(1):17–22. | |
Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Transplant. 2012;18(10):1540–1551. | |
Komina O, Nosske E, Maurer M, Wesierska-Gadek J. Roscovitine, a small molecule CDK inhibitor induces apoptosis in multidrug-resistant human multiple myeloma cells. J Exp Ther Oncol. 2011;9(1):27–35. | |
Johnston JB, Barrett JW, Chang W, et al. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J Virol. 2003;77(10):5877–5888. | |
Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010;Chapter 14:Unit 14A.1. | |
Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100(17):9946–9951. | |
Stessman HA, Baughn LB, Sarver A, et al. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Mol Cancer Ther. 2013;12(6):1140–1150. | |
Cox DJ, Strudwick N, Ali AA, Paton AW, Paton JC, Schröder M. Measuring signaling by the unfolded protein response. Methods Enzymol. 2011;491:261–292. | |
Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. | |
Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318(5852):944–949. | |
Urano F, Bertolotti A, Ron D. IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci. 2000;113 Pt 21:3697–3702. | |
Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33(1):75–89. | |
Wu J, Rutkowski DT, Dubois M, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13(3):351–364. | |
Schröder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med. 2006;6(1):5–36. | |
Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem. 2010;285(43):33165–33174. | |
Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40(1):14–21. | |
Hu J, Dang N, Menu E, et al. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood. 2012;119(3):826–837. | |
Milani M, Rzymski T, Mellor HR, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009;69(10):4415–4423. |
Supplementary material
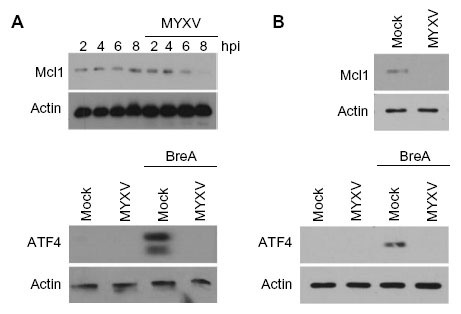 | Figure S1 MYXV blocks ATF4 and Mcl1 expression in RPMI-8226 and MM.1S myeloma cells. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.