Back to Journals » Neuropsychiatric Disease and Treatment » Volume 18
Myasthenia Gravis and Lambert-Eaton Myasthenic Syndrome: New Developments in Diagnosis and Treatment
Authors Pascuzzi RM ![]() , Bodkin CL
, Bodkin CL
Received 19 September 2022
Accepted for publication 24 November 2022
Published 22 December 2022 Volume 2022:18 Pages 3001—3022
DOI https://doi.org/10.2147/NDT.S296714
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Robert M Pascuzzi, Cynthia L Bodkin
Indiana University School of Medicine, Indiana University Health, Indianapolis, IN, USA
Correspondence: Robert M Pascuzzi, Indiana University School of Medicine, Indiana University Health, Indianapolis, IN, 46202, USA, Email [email protected]
Abstract: “Myasthenia Gravis is, like it or not, the neurologist’s disease!” (Thomas Richards Johns II, MD Seminars in Neurology 1982). The most common disorders in clinical practice involving defective neuromuscular transmission are myasthenia gravis (MG) and Lambert-Eaton myasthenic syndrome (LEMS). The hallmark of weakness related to malfunction of the neuromuscular junction (NMJ) is variability in severity of symptoms from minute to minute and hour to hour. Fatigable weakness and fluctuation in symptoms are common in patients whether the etiology is autoimmune, paraneoplastic, genetic, or toxic. Autoimmune MG is the most common disorder of neuromuscular transmission affecting adults with an estimated prevalence of 1 in 10,000. While LEMS is comparatively rare, the unique clinical presentation, the association with cancer, and evolving treatment strategies require the neurologist to be familiar with its presentation, diagnosis, and management. In this paper we provide a summary of the meaningful recent clinical developments in the diagnosis and treatment of both MG and LEMS.
Keywords: Myasthenia Gravis, Lambert-Eaton Myasthenic Syndrome, disorders of neuromuscular transmission
Myasthenia Gravis
The Clinical Presentation and the Natural Course of MG
Fatigable or variable weakness is a hallmark of myasthenia gravis (MG). Women are more commonly affected than men by about 2:1. The initial symptoms of MG can occur in childhood as well as in the elderly. While no age group is spared, the initial onset in females tends to peak in the 3rd decade compared with males in which the onset tends to be most common in the 5th and 6th decades.1
Initial symptoms are most often referable to the eyes. About half of all patients experience either fluctuating ptosis or binocular diplopia as the first symptoms. Within one month of onset of symptoms, 80% of all MG patients will have some degree of ocular involvement.1 Presenting symptoms of generalized weakness, leg weakness or bulbar symptoms each account for about 10% of the patients.1 Patients lack sensory symptoms, and do not have prominent myalgia nor do they have neuropathic pain. On examination patients may demonstrate variable abnormalities of extraocular movement (with normal pupillary reflexes), ptosis, nasal speech, flaccid dysarthria and/or variable weakness with manual muscle strength testing. However, at times the patient’s examination may be completely normal at the time of their clinic visit. Patients presenting with minute to minute or hour to hour variation in neuromuscular function should be considered for the diagnosis of MG. Muscle stretch reflexes, sensory exam, and cognitive testing are normal. While patients describe fatigable or fluctuating symptoms of weakness they are not sleepy or drowsy and such symptoms should direct the clinician to alternative diagnoses.
About 10% of MG patients have their symptoms restricted to the eyes on a long-term basis (referred to as “ocular myasthenia”) and never develop more widespread involvement. These patients are less likely to be seropositive than those with “generalized MG”.1 About 10–20% of patients experience a long-term spontaneous remission usually within the first year of symptoms.1,2 Grob studied 300 patients with generalized MG who were followed between 1940 and 1958 (prior to the widespread use of therapeutic measures other than cholinesterase inhibitors), thus providing a window into the “natural course” of MG. In this series, 30% died, 26% were improved, 21% were unchanged, 13% experienced a spontaneous long-term remission, and the remaining 10% experienced progressive worsening over time.1 From the initial onset of symptoms patients typically experienced progression or worsening of symptoms with fluctuations over the early years of the disease. In this series and others, for patients who present with pure ocular symptoms and remain restricted to ptosis and diplopia for 1 year, there is a high probability that they will likely remain pure ocular on a long-term basis. The initial 5 years of MG tend to be the most volatile from a clinical perspective with exacerbations and progression as well as risk for myasthenic crisis. With time, the natural course is to gradually stabilize and in many patients there is spontaneous improvement over 5–10 years.1,2
Grob further observed a number of clinically important features of MG. While ocular symptoms were the most common initial manifestations, about 10% presented with dysarthria or dysphagia (bulbar), 10% had initial symptoms in the arms and legs, 10% were noted to have generalized weakness at onset, and although uncommon, 1% had their initial presentation involving diaphragm and breathing. At one month into the course of illness about 40% of patient continued to demonstrate pure ocular symptoms, while 40% had progressed to have more generalized weakness. At one month 10% had symptoms restricted to the limbs and 10% to the bulbar muscles. While the majority of patients presenting with ocular symptoms go on to develop more extensive non-ocular involvement, the initial year represents the most likely time frame (about 90% who develop generalized MG do so in the first year of the disease). In general, the maximal severity of symptoms (70% of patients) typically occurs within the initial 3 years. The possibility of a long-term spontaneous remission (13% in Grob’s series and 10–22% in other reports) typically occurs early (in the initial year or two) if it is going to occur. These data are useful in discussing prognosis and expectations with patients. The variable natural course of the disease is also important to consider when interpreting temporally-related changes (improvement) in patient symptoms in the setting of specific therapies.1
Oosterhuis studied the natural course of MG in 73 patients from Amsterdam over 40 years (1926–1965). In this series the disease progressed to maximal severity within the initial 7 years in 87% of patients. Eighteen patients (29%) died. Eight had associated thymoma. Patients commonly experience improvement including clinical remission. Overall, 16 patients (22%) experienced long-term clinical remission, and an additional 18% reached a clinical status of marked improvement (prednisone was also used in three of these patients). Moderate improvement was noted in 16%, and an additional 16% were unchanged, with two patients becoming worse. Since the majority of these patients were treated with cholinesterase inhibitors and no other immune therapy or thymectomy, these observations provide additional illumination of the variability and “natural course” of myasthenia.2
Validating the Diagnosis
Acetylcholine Receptor Antibodies (AChR-Ab)
In autoimmune MG the vast majority of patients display auto-antibodies that impair neuromuscular transmission. The diagnostic value of these serologies serve as an anchor for validating the clinical suspicion of MG. Approximately 80–85% of patients who present with MG have antibodies that bind to the nicotinic acetylcholine receptor (AChR).3 Serological testing for AChR-antibodies has both a high sensitivity (80–85%) and also a high specificity (false positives are rare). The AChR binding antibody is detectable in the vast majority of patients with MG (elevated in about 80% with rare false positives). The muscle specific kinase (MuSK) antibody is present in about 4% of all patients and the low density lipoprotein receptor-related protein (LRP4) antibody is present in roughly 1%. In general, patients have one of these antibodies and not a combination. There is a limited correlation between the magnitude of the elevation of these antibodies and clinical severity. The “level” of antibody does not necessarily predict the severity of symptoms and patients in remission generally remain seropositive. As such, the serological studies are largely for diagnostic use and are not used for monitoring of disease severity or response to treatment.3
MuSK Myasthenia
About 4% of MG patients have elevated levels of antibodies to muscle specific kinase (MuSK). Muscle specific kinase is a transmembrane protein located at the postsynaptic component of the neuromuscular junction that facilitates the clustering of AChRs. The clustering of AChRs is necessary in neuromuscular transmission, the achievement of postsynaptic action potentials, optimal sodium conductance, and the eventuality of muscle contraction.4 Regarding the overall sensitivity of this antibody in MG most series indicate that about one-fourth of patients who do not have AChR-ab will have MuSK-ab. The presence of MuSK-ab carries important clinical value. Patients with MuSK tend to be more difficult to diagnose. They are most often female and become symptomatic in their teens or 20s. These patients commonly have severe bulbar weakness, and often display striking involvement of neck extensor muscles and respiratory involvement. Such patients may develop static or “fixed” deficits that do not fluctuate. From a diagnostic testing standpoint, they have less likelihood of responding to cholinesterase inhibitors (edrophonium) and are less likely to demonstrate a decremental response to repetitive stimulation on neurophysiological testing. They do not have associated thymoma. Regarding therapy, they often respond poorly to cholinesterase inhibitors, are less likely to benefit from prednisone and most other immunosuppressive drugs, and they do not benefit from thymectomy (thus should not be treated with thymectomy). On the other hand, the MuSK MG patients respond very favorably to rituximab and also to plasma exchange.
LRP4
Myasthenia patients who are seronegative for AChR and MuSK antibodies are referred to as having “double negative” disease. In these patients a minority have antibodies to the low-density lipoprotein receptor-related protein 4 (LRP4).5 About 1–2% of all patients with autoimmune MG have detectable LRP4 antibody. These patients do not have thymoma or thymic hyperplasia. The LRP4 patients are more commonly female and younger in age of onset compared with non-LRP4 “double negative” patients. In general, these patients have mild symptoms, clinical involvement is frequently purely ocular, and the patients tend to improve with cholinesterase inhibitors and if needed, corticosteroids. Clinical experience is ongoing regarding the question of specificity such that caution is needed in applying seropositivity as evidence for establishing a clinical diagnosis of MG.3
Anti-Agrin
Agrin is a basal lamina protein with two distinct isoforms. At the neuromuscular junction agrin binds to LRP4, which in turn is an activator of MuSK, then resulting in clustering of AChR as a necessity for optimal neuromuscular transmission.
Among MG patients who are found to be negative for AChR, MuSK, and LRP4 antibodies (referred to as “triple negative”) a small percentage have anti-agrin antibodies.6 The majority of patients having positive agrin serology are also positive for AChR, MuSK, or LRP4 antibodies.7
Electrophysiological Testing
Electrophysiological testing is an anchor in validation of the diagnosis of MG and is essential in the 10–15% of patients who are seronegative. In addition to securing evidence for MG the nerve conduction studies and EMG needle exam can provide evidence for a collage of alternative neuromuscular causes of weakness. Lambert-Eaton myasthenic syndrome, motor neuron disease, neuropathies, and myopathies can be detected and characterized with electrophysiological testing. The standard test for a defect of neuromuscular transmission involves repetitive nerve stimulation of a motor nerve at a low frequency of 2–3 per second. The presence of a decrement of the compound muscle action potential of at least 10% establishes a defect of neuromuscular transmission. The overall sensitivity in MG is approximately 50% (somewhat greater if testing muscles that are clinically weak, and lesser sensitivity in patients with pure ocular disease). Specificity of repetitive stimulation is more limited as patients with motor neuron disease and those with peripheral neuropathy can display a decrement. Thus, the findings must be considered within the clinical context of the patient presentation. The most sensitive neurophysiological test is single fiber EMG (SFEMG). While more difficult to perform and requiring more extensive examiner training the properly performed SFEMG has an overall sensitivity exceeding 90%. In contrast to serological testing, repetitive stimulation and SFEMG correlate with severity of weakness.8 Neurophysiological testing in MuSK patients is more likely to show small “myopathic” voluntary motor units on needle exam. Also, MuSK patients are more likely to demonstrate myotonic discharges on needle exam compared with AChR-ab positive patients.9
Edrophonium Test
While performed far less commonly in the current era of serological testing for MG the most immediate and readily accessible confirmatory test for MG remains the edrophonium (Tensilon) test. In performing this test, it is best to identify one or two objectively weak muscles to monitor. The most reliable and practical endpoints include ptosis, ophthalmoparesis, and a variety of other cranial abnormalities. There are risks of giving cholinesterase inhibitors intravenously including bradycardia, syncope, muscarinic cholinergic symptoms of abdominal cramps and diarrhea. As such, it is best to administer edrophonium in a controlled clinical setting capable of managing syncope, abdominal cramps, hypotension, or respiratory failure. In patients who present with severe dyspnea, defer the test until their airway has been secured. The following is a suggested approach to conducting an optimal edrophonium test in an adult patient:
- Establish intravenous (IV) access.
- Prepare IV atropine (0.4 mg) to be readily available in the event of symptomatic bradycardia or gastrointestinal (GI) side effects of abdominal cramps and diarrhea.
- Draw up in a 1 mL syringe edrophonium 10 mg (1 mL), administer 1 mg (0.1 mL) as a “test dose” and monitor heart rate for bradycardia. Assuming no significant side effects after 1 minute, administer an additional 3 mg. The majority of patients who are going to improve with edrophonium will do so within 30–60 seconds. If improvement occurs then the test is complete and additional infusion is not indicated.
- In the absence of significant clinical improvement after 1 minute, administer additional 3 mg. Wait 1 additional minute and if no improvement occurs then give the final 3 mg. At any point in the test, should the patient experience muscarinic cholinergic symptoms such as gastrointestinal cramps, increased salivation, or perspiration, or in the event the patient displays fasciculations then one can assume that the patient has received sufficient edrophonium to result in improved strength if it is going to occur, and at that juncture the infusion should be stopped and the test ended.
- When there is concern for examiner bias or for a “placebo effect”, the test is best performed placebo-controlled and double-blinded. To do this a 1 mL control syringe containing either saline, atropine (0.4 mg), or nicotinic acid (10 mg) is administered. Improved strength from edrophonium lasts for just a few minutes. When clinical improvement is clear-cut, the test can be considered “positive”. On the other hand, when the clinical improvement is slight or borderline, one should consider the test “negative”. The edrophonium test may need to be repeated if the results are equivocal. Estimates of the overall sensitivity of the edrophonium test approximate 80–90%. Specificity is limited, as case reports of a positive test result are documented in a variety of other neuromuscular diseases including botulism, motor neuron disease, pituitary tumor, Lambert-Eaton myasthenic syndrome, Guillain–Barré syndrome, various brainstem disorders, and in lesions of the cavernous sinus.
- Selected patients may be candidates for a “neostigmine test” owing to a longer duration of effect. This alternative allows for a longer observation period to establish presence or absence of clinical improvement (and is particularly helpful in testing children). In conducting a “neostigmine test”, 0.04 mg per kg is administered intramuscularly or 0.02 mg per kg intravenously (one time only).8,10
Oral Cholinesterase Inhibitor Challenge
In patients with possible MG for whom an intravenous edrophonium test would be useful but impractical or not available there exists the option of performing an oral cholinesterase inhibitor challenge as a diagnostic test. Similar to the edrophonium test, one or more identifiable weak muscle groups (ptosis, ophthalmoplegia, dysarthria, dysphonia, etc.) are tested and documented. The patient is given a single 60 mg pyridostigmine oral tablet. And then re-examined in 1 hour. Most patients with MG improve after 30–45 minutes, and the improvement persists for about 3–4 hours. This pharmacological challenge, although not as immediate in its indication of a pharmacological restoration of neuromuscular transmission, can provide similar pharmacological evidence in support of the diagnosis.
Ice Pack Test
Heat makes neuromuscular transmission worse while cooling makes transmission better. As such, patients with ptosis or diplopia may show clinical improvement in their exam after application of an ice pack. This test is most useful in patients with significant ptosis. The test involves the application of covered ice to the eyes for 2–5 minutes (the ice needs to be covered in order to protect against ice burns). Sensitivity and specificity have been variable.8 Giannoccaro et al evaluated the icepack test in 155 patients, 102 with ocular myasthenia and 53 with other (non-MG) diagnoses. The IPT had a sensitivity of 86% and a specificity of 79%. This was similar to the sensitivity and specificity of single fiber EMG (sensitivity of 94% and specificity of 79%).11
Disease Classification And Outcome Measures
Classification: Given the wide range of clinical manifestations including distribution of involved muscle groups and severity of weakness it has long been recognized to classify patients with qualitative and descriptive systems. Such classification systems allow for better understanding of the disease as well as serving to guide treatment protocols depending on the subtypes in question. For example, a patient with pure ocular myasthenia may have different treatment strategies than a patient with severe generalized MG who is on a mechanical ventilator. Classification based on antibody status has proven important given the differences in response to immune therapies as well as relationship to thymic pathology and indications for thymectomy. Among the many classification systems devised over the past 50 years the Myasthenia Gravis Foundation of America (MGFA) Classification has evolved as the most widely utilized in clinical practice and in research. Published in 2000, with a focus on conducting clinical trials the Myasthenia Gravis Recommendations for Clinical Research Standards Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America defined the MGFA Classification to reflect localization and severity of disease. Class I refers to pure ocular MG, Class II mild generalized, Class III moderate generalized, Class IV severe generalized, and Class V intubation (myasthenic crisis). Additional sub-classification is based on the predominant distribution of weakness (with axial-extremity predominance being subclass “a” and bulbar predominance being subclass “b”). This classification system is not linear and while it does describe patients it does not serve as a useful measure of outcome in clinical trials.67
Outcome Measures: The Quantitative Myasthenia Gravis (QMG) scale provides a quantitative and objective tool for severity of disease. Established in 1998, the QMG quantifies the presence and severity of extraocular muscle weakness, ptosis, facial muscle strength, dysphagia, dysarthria, proximal limb strength, hand involvement, neck strength, and respiratory status. Each item is scored from 0–3 with total score of 0–39 (the higher the score the more severe the weakness). In general, a change of 2–3 points on this scale is considered to be meaningful.68,74
The MG Activity of Daily Living (MG-ADL) scale was developed in 1999, and is a patient survey of clinical symptoms. Eight items are scored from 0–3 (total score 0–24) addressing ocular, speech, chewing, swallowing, respiratory, and proximal upper and lower extremities strength. In general, a 2-point change in score is considered to be meaningful.69,74
Another patient-reported outcome tool, the MG Quality of Life 15-items (QOL15) was developed in 2008 with 15 questions related to ocular, swallowing, speech, proximal limb strength, mobility, personal grooming, work, social life, activities, fluctuation, and psychological status. Questions are scored by the patient from 0–2 (total score 0–30). This tool has applications for clinical management and follow-up of individual patients as well as for outcome measures in clinical trials.70,71,74
The MG Composite (MGC) scale (developed in 2008) combines high-yield items from the QMG, the MG-ADL, and also Manual Muscle Testing. The clinical exam includes evaluation of ocular, neck, and proximal limb strength. There are 4 MG-ADL items reported by patients (speech, chewing, swallowing, and breathing). The range of total score is 0–50 (higher being more severe). In general, a 3-point change in score is considered to be meaningful.72–74
Post Intervention Status: To better define clinical status of patients with MG following initiation of treatment the MGFA Task Force further established standardized patient outcome definitions (“Post Intervention Status”).67 These definitions allow for a framework for capturing, and communicating patient status during individual management as well as in the setting of clinical trials. Complete stable remission, pharmacologic remission, minimal manifestations (four categories), as well as the assessment of relative change in clinical status (improved, unchanged, worse, exacerbation, and death from MG) are each defined.67
Treatment of Autoimmune MG
Patient education is an essential first step in the clinical management. Since the disease is so highly variable in distribution of muscle groups affected, and can be limited (ocular) or generalized, and can often progress over the initial years, and given the spectrum of very mild to life-threatening weakness, it behooves the physician to review these considerations with the patient and family. The Myasthenia Gravis Foundation of America and Muscular Dystrophy Association provide thorough and accurate informational resources for patients and families. A critical component of patient management is to recognize the potential for life-threatening weakness (respiratory or bulbar) prompting the need for immediate hospitalization and intensive care monitoring and management. For patients who are overtly short of breath, choking or struggling to swallow, or those with rapidly progressing symptoms an urgent evaluation with hospitalization is indicated. Pulmonary function should be monitored assiduously with assistive devices for neuromuscular respiratory failure (noninvasive mechanical ventilation) and a low threshold for intubation. Often, for a patient presenting with new or increasing symptoms including crisis it is possible to identify a precipitating cause such as recent surgery, new medication exposure, hyperthyroidism (10% of patients with MG develop thyroid disease), active infection, and any other major physical or psychological stress.
Cholinesterase Inhibitors
Pharmacological options are numerous and selection should be individualized based on the patient’s symptoms and co-morbidities. In virtually all patients the initial treatment is a reversible cholinesterase inhibitor (CEI), typically pyridostigmine (Table 1). While generally very safe and without any significant long-term side effects or organ injury, it behooves the clinician to recognize that excessive CEI can on occasion result in significant reversible generalized weakness referred to as cholinergic weakness. Cholinergic weakness is very uncommon in patients taking oral cholinesterase inhibitors, but more likely in the setting of parenteral administration of CEI and therefore should be considered in patients with severe generalized weakness including those suspected of having a myasthenic crisis (Table 2).
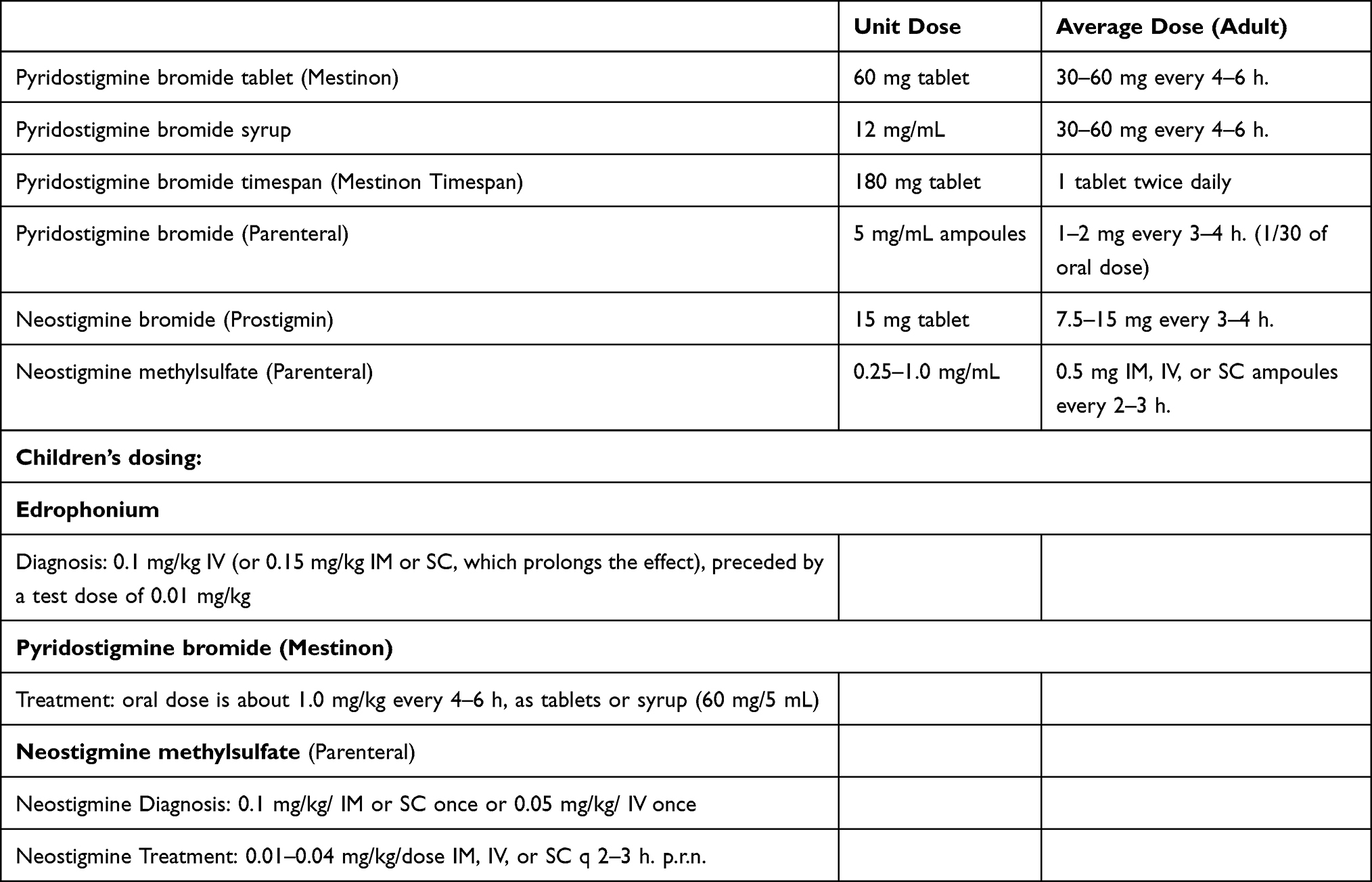 |
Table 1 Myasthenia Gravis Treatment with Cholinesterase Inhibitors – Options and Dosing |
 |
Table 2 Myasthenic Crisis and Cholinergic Crisis – Clinical Differentiation |
Immunotherapy
Corticosteroids
Since the 1970s corticosteroids have been extensively used and broadly viewed as an excellent treatment for patients having moderate or severe disease. Prospective controlled double blind trial support for use of corticosteroids is remarkably limited. Fifty years of clinical experience, including patient compliance, even in the setting of the major toxicity risks with long-term corticosteroid exposure provides support for concluding that corticosteroids represent an anchor for management of patients with moderate and severe symptomatology. While there is no consensus on precise dosing of corticosteroids, there are several generalizations that are widely accepted regarding use in treatment of MG, as follows:
- For severe MG begin with 60–80 mg/day of prednisone.
- Overall, 80% of patients achieve marked improvement (or eventual remission), while 5% have no improvement. Absence of improvement with high-dose corticosteroids over several months should lead to a reconsideration of the diagnosis.
- The time course for improvement varies but in general most patients begin to improve in the first 1–2 weeks of treatment. Improvement is gradual and tends to continue for the subsequent 3–9 months and then plateau (having reached their point of maximal improvement).
- Fifty percent of patients have an “early exacerbation” from administration of high-dose corticosteroids. This is a temporary increase in symptoms starting 1–2 days following initiation of corticosteroids and typically lasts for 3–4 days. While this transient early exacerbation is usually mild, about 10% of patients with early exacerbation can develop bulbar or respiratory weakness requiring ICU hospitalization. For this reason, it is prudent to consider hospitalization of patients with moderate to severe MG for beginning high-dose steroid treatment under observation. Should there be a life-threatening worsening of symptoms then immediate steps for management can be taken.
- Some clinicians favor a lower dose of initiation with gradual increase which appears to reduce (though not eliminate) the risk for early exacerbation. A typical initiation dose of alternate day 25 mg and increasing by 12.5 mg every 3rd dose to a maximum dose of 100 mg on alternate days (or until the patient experiences satisfactory improvement). With this approach the clinical improvement is slower and may not be apparent for several weeks to a month or two. A low-dose alternate day schedule (beginning with 10 mg alternate day prednisone) has been associated with benefit superior to that of placebo in one prospective controlled trial in patients with ocular myasthenia.
- The pace of steroid reduction in patients with MG. In general, optimal corticosteroid use is long-term and if a course of corticosteroids is limited to several weeks or several months or when the rate of reduction in dose is too rapid there is a predictable loss of control of MG and patients are at risk for a major flareup of their disease. For this reason corticosteroids should be slowly tapered at about 10 mg every 1–2 months when above 20 mg/day and slower taper below 20 mg/day. If symptoms should rebound as prednisone is reduced then a corticosteroid sparing option is added (with the goal of facilitating the further reduction on steroid dose, reducing overall toxicity from steroid exposure).12,13
- While long-term corticosteroid therapy is highly effective in controlling MG the complications of chronic corticosteroid use are considerable and it is essential in such patients to have meticulous monitoring of weight, blood pressure, blood glucose, bone health, infection risk, cataracts, glaucoma, and effects on facial and body appearance, as well as impact of mood, sleep, and behavior.
Nonsteroidal Immunosuppressive Medication Options
For patients in whom corticosteroids are contraindicated, or those in whom corticosteroids are ineffective, or leading to concerns of toxicity, a steroid sparing agent is typically initiated. Among the many such drugs azathioprine, tacrolimus, mycophenolate mofetil, methotrexate, and cyclosporine are appropriate options. Cyclosporine administration was associated with clinical improvement in a double-blind placebo controlled trial.14 Following very favorable anecdotal reports of benefit, a controlled double blind trial of mycophenolate mofetil revealed no clear benefit after 3 months compared with those receiving placebo. In this study new patients with seropositive MG were randomized to mycophenolate and prednisone 20 mg/day or to placebo with prednisone 20 mg daily. Both groups improved to a similar degree over 3 months.15 It has been speculated that the time course for clinical benefit from mycophenolate therapy may in many patients require more than 3 months exposure as one explanation for this negative result. The impact of 20 mg daily prednisone in stabilizing and improving MG over 3 months serves as a logical variable in patients in both groups improving.15 Another prospective controlled trial studied patients with MG who were believed to be corticosteroid dependent and were randomized to mycophenolate and placebo with a subsequent attempt to force-reduce corticosteroid dose over 9 months (presuming that mycophenolate provides a steroid-sparing effect). In this study both groups were equally able to reduce corticosteroid dose to an equivalent degree.16 In some centers tacrolimus is often utilized for patients with refractory MG, even though trials have not demonstrated major benefit.17,18 Methotrexate is another immunosuppressive option, however a prospective trial demonstrated no clear-cut corticosteroid sparing effect over a 12-month period.19
Complement Inhibitors
A variety of mechanisms exist by which acetylcholine receptor antibodies lead to impairment of neuromuscular transmission. The simplest mechanism is the capacity for these antibodies to directly “block” the receptor from acetylcholine (blocking antibodies). A more common mechanism involves the cross-linking of two neighboring receptors on the muscle membrane thereby changing their confirmation at the endplate, leading to more rapid turnover or degradation of the receptors and an overall reduction in the number of functional receptors (referred to as “modulating antibodies”). A third mechanism involves the binding of antibodies to the endplate and attracting complement to the post-synaptic membrane. Complement then directly leads to lysis and destruction and thus impaired neuromuscular transmission. The presence of complement-mediated lysis of the post-synaptic membrane has provided an interventional opportunity to treat AChR-ab positive MG patients using complement-inhibiting agents. Eculizumab is a monoclonal antibody that blocks C5 complement. This drug was originally FDA approved for another complement-mediated disorder, paroxysmal nocturnal hemoglobinuria, and has subsequently undergone extensive investigation as a treatment modality for patients with relatively severe MG. Eculizumab binds to the human terminal complement protein C5, which results in inhibition of the enzymatic cleavage of C5 to C5a and C5b, and in doing so prevents the C5a-generated attraction of proinflammatory cells to the NMJ. Eculizumab was tested in a 6-month randomized, double-blind, placebo-controlled trial in 125 AChR-ab positive refractory generalized MG patients. While the primary outcome endpoints did not meet statistical significance, a number of secondary outcomes provided evidence for meaningful clinical benefit. As such, eculizumab received FDA approval for MG in 2017. Observed benefits included the following. Serious clinical exacerbations of MG occurred in only six (10%) of patients in the eculizumab-treated group while in the placebo arm 15 (24%) occurred. Similarly, rescue treatment was required in six (10%) of those receiving eculizumab compared with 12 (19%) in those on placebo. Additional observations included a favorable safety profile, the drug being generally well-tolerated, and related improvement in measures of strength/function, quality of life, and activities of daily living.20
Open-label extension documented long-term efficacy and safety (1200 mg every 2 weeks for a median duration of 22.7 months in 117 patients). It should be noted that there were no cases of meningococcal meningitis. Clinical exacerbations were 75% less than patients had experienced during the 12 months prior to initiation of eculizumab. And the improvements in functional, strength, quality of life, and activities of daily living were sustained. Overall, in this open-label phase 56% of patients reached a clinical status of “minimal manifestations” or a “pharmacological remission”. Patients who had initially been in the placebo arm of the study improved following initiation of open-label.21
Additional long-term follow-up of these study patients established the percent of patients achieving a clinical state of “minimal symptom expression”. The initial study at week 26, reported more eculizumab-treated patients achieving “minimal symptom expression” than placebo (MG-ADL: 21.4% vs 1.7%; and MG-QOL15: 16.1% vs 1.7%) both highly significant differences. In the open-label extension phase, the percent of patients in the placebo/eculizumab group who reached a state of “minimal symptom expression” improved after adding eculizumab treatment and this improvement persisted through 130 weeks of open-label eculizumab (MG-ADL: 1.7 to 27.8%; MG-QOL15: 1.7 to 19.4%). And at the extension study week 130, the proportions of patients in the eculizumab/eculizumab and placebo/eculizumab groups reporting “minimal symptom expression” were similar (MG-ADL: 22.9% and 27.8%, respectively; MG-QOL15: 14.3% and 19.4%, respectively). In general, the drug was well-tolerated and consistent with previous reports.22
Given the mechanism of action of eculizumab, patients should be given meningococcal vaccination in advance of initiation in order to reduce the risk of meningococcal meningitis. Since complement is not known to play a significant role in the pathogenesis of MuSK MG, there is no role for use of these agents in MuSK MG patients.
Ravulizumab is a subsequent modification of eculizumab. Ravulizumab has been engineered to bind to the FcRn thus promoting the recycling of the monoclonal antibody leading to a longer half-life. Recent studies of this modified C5 complement inhibitor have demonstrated a favorable and relatively safe performance in MG patients in multicenter clinical trials.25 As such, the FDA approved this drug for the treatment of MG in April 2022. One advantage in the usage of this agent is the longer duration of clinical benefit. Thus, instead of a maintenance infusion every other week it appears that this newer agent is associated with a sustained clinical benefit well beyond the 2 week duration associated with eculizumab. Ravulizumab, as with eculizumab requires meningococcal vaccination prior to initiation to reduce the risk of infection. Patients are dosed with a 1 hour intravenous infusion, followed by a second infusion in 2 weeks. After that, the infusions are scheduled every 8 weeks thus providing the alternative for patients to require less frequent infusions compared with eculizumab.25 An FcRn antagonist (see below) should not be used with ravulizumab, as this would shorten the half-life.
Another promising approach to complement inhibition is zilucoplan. While not FDA approved at the time of this writing, the drug has completed phase 223 and phase 3 testing in patients with generalized MG with results reported at the International Conference on Myasthenia and Related Disorders (which took place on May 10–12, 2022). Zilucoplan is a subcutaneous self-administered macrocytic peptide that binds to complement protein 5 and prevents cleavage. In a recently completed placebo-controlled double-blind trial testing daily injections (174 patients randomized) for 12 weeks patients showed meaningful benefit compared with placebo (these patients had AChR-ab positive generalized MG). The drug demonstrated a favorable safety profile, thus raising the possibility for patients with AChR-ab positive MG to be treated with a self-administered daily subcutaneous injection.24
Rituximab
Rituximab binds to CD20 antigen on B cells and has been widely used in a broad variety of autoimmune disorders and various malignancies. Its role in the treatment of myasthenia gravis has been the subject of extensive clinical application, with anecdotal studies in patients with AChR-ab positive MG and MuSK MG. In the majority of MuSK-ab positive patients rituximab is generally considered to be markedly effective and is viewed as one of the “drugs of choice” for treating MuSK patients with moderate to severe disease. Hehir conducted a prospective controlled double-blind trial in MuSK-ab positive MG patients using the “Myasthenia Gravis Status and Treatment Intensity” (MGSTI) for primary outcome (MGSTI measures MGFA post-intervention status and also patients’ needs for supplemental immune therapy). At the median follow-up of 3.5 years, the majority (58%, 14/24) of those receiving rituximab reached the primary efficacy goal, while only 16% (5/31) of patients in the control group met this target. Regarding additional immune drug needs, only 29% of the rituximab patients required prednisone (with a mean daily dose of 4.5 mg). In the control group prednisone was required in 74% (with an average daily dose of 13 mg). This study provides Class IV evidence for benefit of rituximab in MuSK-ab positive MG patients.26
Rituximab use in patients with AChR-ab positive myasthenia is generally less predictable with much of the evidence of clinical benefit being uncontrolled and anecdotal. There is consensus that rituximab works in selected refractory generalized AChR-ab positive patients but with a lower rate of success compared with the MuSK patients.27–29 In a retrospective study from Austria of 56 patients treated with rituximab 25% had MuSK-ab positive MG, 70% were AChR-ab positive, and 5% were seronegative.28 After 3 months, 14 of 53 (26.4%) achieved remission. Median follow-up was 20 months, at which point 42.9% of patients were in remission (with an additional 25% reaching a status of minimal manifestations). The remission rate was 71% in the MuSK-ab positive patients while of those patients with AChR-ab positivity 36% were in remission. In this study rituximab was well-tolerated without major complications.28
In a prospective randomized placebo-controlled trial of the steroid-sparing impact of rituximab the BeatMG trial studied the effect of rituximab on 52 mild to moderately severe AChR-ab positive MG patients with generalized MG who were taking corticosteroids (≥15 mg/day). The two primary outcomes were the proportion of subjects demonstrating ≥75% reduction in daily prednisone dose at 12 months (without clinical worsening) and also safety. Patients were randomized equally to rituximab or placebo. Primary steroid-sparing outcome (≥75% reduction in daily prednisone dose at 12 months) was observed in 60% of the rituximab patients and 56% of those on placebo. Rituximab was well-tolerated and there were no safety issues observed in this study.66
A comprehensive 2021 review from the Canadian Agency for Drugs and Technologies in Health of published experience with rituximab in generalized MG (one systematic review with meta-analysis and nine non-randomized studies) reached the following conclusions:30
while rituximab appeared to be associated with improvement in patients with myasthenia gravis, definitive conclusions were not possible and that the findings needed to be interpreted with caution.30
Neonatal Fc Receptor-Targeted Therapy for MG
Of the many new strategies being developed as novel treatment for MG there is major focus on the neonatal Fc receptor (FcRn). The FcRn plays a major role in the maintenance of serum IgG levels. The FcRn is present in endothelial and myeloid cells and is expressed throughout the full lifespan. Essentially the FcRn serves to rescue, sequester, and recycle IgG and thus prevents early degradation. The FcRn facilitates a longer “half-life” and greater overall blood levels of IgG. With small molecule or monoclonal antibody directed against and thus inhibiting the FcRn there is a resulting overall reduction in the levels of IgG including pathogenic IgG (such as AChR-ab). A recently completed prospective controlled clinical trial in MG using efgartigimod (a small molecule) indicates a clear-cut favorable clinical response in the majority of those patients with AChR-ab positive generalized MG. This phase 3 trial was conducted at 56 neuromuscular centers in 15 countries in North America, Europe, and Japan. Patients were at least 18 years old, had generalized MG with Myasthenia Gravis Activities of Daily Living (MG-ADL) score of at least 5. Patients received efgartigimod (10 mg/kg) or placebo, given as four weekly infusions per cycle and then repeated as needed (depending on clinical response and no earlier than 8 weeks after initiation of the previous cycle). In this study 167 patients (84 in the efgartigimod group and 83 in the placebo group) were enrolled, randomly assigned, and treated. Of those, 129 (77%) were AChR-ab positive, of which more of those in the efgartigimod group were MG-ADL responders (44 [68%] of 65) in cycle 1 than in the placebo group (19 [30%] of 64; p<0.0001). Treatment-emergent adverse events were similar in the treatment and the placebo groups. There were no deaths. This study provides strong evidence that efgartigimod is well tolerated and clinically beneficial in patients with generalized AChR-ab positive MG. As such, this drug became FDA approved in December 2021 and is clearly a significant new advance for managing patients with autoimmune MG.31 While the drug is FDA approved for patients with AChR-ab the clinical trial included a smaller number of patients with MuSK-ab (as well as seronegative) with favorable results.31
A monoclonal antibody, rozanolixizumab has completed phase 232 and phase 333 clinical trials with favorable results. This drug is not FDA approved at the time of this writing but preliminary reports of benefit and safety add further support to the approach to managing IgG-positive MG patients by using FcRn receptor inhibition.32,33
Plasmapheresis
Plasma exchange (plasmapheresis or PLEX) has been utilized to treat severe, acute, or refractory MG since 1980. This logical approach to antibody-mediated disease (including those present in MG) has been a mainstay of treatment for patients hospitalized with myasthenic exacerbation or crisis. A standard course of treatment involves placement of a pheresis catheter, one total plasma volume exchange every other day for 5 exchanges. Rationale for the alternate day schedule stems from the presence of about 50% of one’s antibodies being in the intravascular compartment and the other half being extravascular. Thus, if a total plasma exchange is conducted then half of the offending antibodies are ostensibly removed. It takes about 48 hours for antibodies to re-equilibrate between extravascular and intravascular compartments. So, with each additional exchange a smaller and smaller number (percent) of pathogenic antibodies are removed. The alternate day schedule allows for full re-equilibration and optimal efficient extraction from the patient. Improvement is typically seen within a few days to 2 weeks, but only lasting 1–2 months. In general, after the final exchange it takes about 30 days for a patient to re-accumulate their pre-treatment levels of antibodies. Because of the rapid improvement with PLEX, it is commonly used in MG crises. In addition to usage in myasthenic crisis and impending crisis the treatment is indicated for selected patients refractory to or poor candidates for other conventional maintenance therapies and in such patients a permanent pheresis catheter can be placed and maintenance exchanges performed every 1–4 weeks depending on the patient’s response. Additionally, plasma exchange is particularly effective in patients with MuSK-ab positive MG and should be considered for maintenance therapy when other options are ineffective. Complications included line/catheter placement complications, line infection, line occlusion, line displacement proximally, direct exchange-related effects such as blood pressure regulation, bradycardia, electrolyte imbalance, hemolysis and coagulopathy.34,35
Intravenous and Subcutaneous Immunoglobulin
In patients with myasthenic crisis, near crisis, hospitalized for neuro-worsening, and in those refractory to or poor candidates for alternative therapies, and similar to use of plasma exchange, high-dose intravenous immunoglobulin (IVIg) and also subcutaneous immunoglobulin (SCIg) are widely utilized in MG.36–38 Often clinical improvement is seen within days or weeks of initiation. Duration of benefit after a 2 g/kg course of treatment is often a month or two. The dose of 2 g/kg is spread out over 5 days or can be given with longer infusion time over 2–3 days if tolerated. Indications and applications are for treatment of patients with moderate to severe MG. In patients who are poor candidates for other immune therapies or for those who are refractory to other standard treatment modalities Ig should be considered for long-term maintenance therapy.37 Experienced neuromuscular specialists commonly use maintenance IVIg in selected patients with favorable anecdotal reports. There is a paucity of published prospective controlled double-blind evidence for IVIg use for maintenance therapy which leads to challenges with 3rd party coverage, acknowledging the striking financial cost associated with use of Ig as maintenance therapy.
In one report of 52 patients given IVIg for maintenance treatment (who had responded poorly to pyridostigmine, corticosteroids, azathioprine, or a combination) improvement was observed and sustained in 37 patients with treatment maintained for 6 years on average. The investigators characterized the degree of improvement as mild to moderate in general. Patients who seemed to respond most favorably were those with AChR-ab positive serology (including those with higher titers), older patients, and bulbar-onset patients. Maintenance IVIg was associated with lesser requirement for alternative treatments including pyridostigmine, corticosteroids, and azathioprine.39
In general Ig is relatively safe. It is the only powerful immune therapy that does not “suppress” the immune system and predispose to infections often seen in patients with immune suppression. Infusion-related side effects are common and can be limited with pre-medication and slowing of the rate of infusion. Transient flu-like symptoms, myalgias, headache, fever, and chills are not uncommon with IVIg and can be mitigated by decreasing the rate of the infusion and pretreatment with diphenhydramine. Very rare but serious complications ostensibly related to the high protein concentration and resultant increase in serum viscosity include ischemic brain disease (stroke), myocardial infarction, and ischemic bowel. Renal failure is another important consideration and IVIg should be avoided if possible in patients with significant renal insufficiency. In many medical centers patients are initially screened for a selective IgA deficiency to avoid anaphylaxis from IVIg. Aseptic meningitis is a known rare complication of IVIg. Such pre-treatment screening is not uniform and the occurrence of anaphylaxis is exceedingly rare. In comparison with plasma exchange for treatment of severe generalized myasthenia, IVIg appears to be equally effective.37 One report suggest that IVIg may be superior to plasma exchange for pre-treatment of patients MG prior to undergoing thymectomy.40
Increasing evidence and clinical experience suggests that SCIg is equally effective and well-tolerated compared with IVIg. One advantage to using SCIg is the absence of reports of side effects as outlined above related to IVIg infusion (aseptic meningitis, ischemia, headaches, renal failure, etc.).
Exercise
The role of physical exertion, fitness routines, and exercise has been debated given the tendency for MG patients to experience fatigable weakness and logically try to minimize activities that create fatigue. In one recent study of progressive resistance training vs aerobic training, 15 patients with generalized MG were randomized to one or the other with 20 work-out sessions conducted over 8 weeks. Just one patient discontinued participation. Increasing MG symptoms were reported in some patients in both groups (2 reporting increase in bulbar weakness, 3 reporting increase in “fatigue”). No change in the QMG score was found in either group of patients. Patients assigned to the progressive resistance-training sessions had increase in maximal strength and also their functional capacity. These data support the concept that exercise and fitness therapy is not only tolerated by most patients but has the potential for enhancing strength and function.41
Thymectomy
Thymoma is present in about 10% of patients with AChR-ab positive MG. The majority of MG-related thymoma are slowly and locally growing tumors but an important minority have malignant thymoma which can metastasize. Of the 90% of MG patients without thymoma the majority have thymic hyperplasia as seen on microscopic analysis. Only patients with AChR-ab have associated thymic hyperplasia (not those with MuSK-ab or LRP4-ab). For these reasons all patients with suspected autoimmune MG should be screened for thymoma with an imaging study of the chest. It is estimated that 30% of patients who initially present with thymoma are found to have MG.
The association between the thymus gland and myasthenia dates back to 1900 and thymectomy was initially performed by Blalock in the 1930s (results in thymoma patients were favorable leading to this surgery being used in MG patients without thymoma). By the 1940s and 50s thymectomy was considered standard, mostly reserved for younger patients and those with more severe disease. As the reported data were all anecdotal and uncontrolled, disagreement persisted for many decades regarding the value of thymectomy in MG.42 This uncertainty has been effectively resolved following results of a large randomized international multicenter single-blinded controlled trial (MGTX) which provided compelling evidence for clinical benefit from thymectomy in patients with AChR-ab positive generalized MG (thymoma patients were excluded from this study).43,44
In the MGTX 126 patients were randomized to thymectomy plus prednisone or prednisone alone. Duration of symptoms of less than 5 years as well as presence of AChR-ab and Myasthenia Gravis Foundation of America (MGFA) Class II–IV clinical involvement/severity were requirements for enrollment. The initial phase of the trial followed all patients for 3 years. Both groups of patients were given oral prednisone (titrated up to 100 mg on alternate days or until reaching a clinical status of “minimal manifestations”). All thymectomies used the extended trans-sternal procedure. The main outcome measures included patient clinical status and also the total prednisone required to remain well-controlled. Secondary outcomes were measures of serious adverse events, hospitalizations (over the 3 years of the study), and quality of life. Patients who were randomized to thymectomy demonstrated comparative benefit in multiple domains. Their MG symptoms were better (average Quantitative Myasthenia Gravis (QMG) scale: 6.15 vs 8.99) and an overall lesser dose of prednisone was required to maintain clinical improvement (44 mg vs 60 mg on alternate days). Complications were similar in both arms. Furthermore, the average score on the Myasthenia Gravis Activities of Daily Living scale (2.24 vs 3.41), the need for use of azathioprine (17% vs 48%) and those having minimal-manifestation status at month 36 (67% vs 47%) and hospitalizations (9% vs 37%) all favored the thymectomy group.43
A follow-up 2-year extension study (rater-blinded) provided additional consistent data in favor of thymectomy. This extension study included measurements of QMG score, and alternate-day prednisone dose from month 0 to month 60. Of the initial 111 patients who completed the 3-year MGTX, 68 (61%) participated in the 2-year extension (with 50 patients completing the full 60 months). At 60 months, the patients randomized to thymectomy plus prednisone group demonstrated better QMG scores and also the mean alternate-day prednisone doses (24 mg vs 48 mg) compared with subjects in the prednisone-only group. These data provide proof that thymectomy is effective (in patients with AChR-ab positive MG) and establishes expectations for degree and time frame for improvement.44
The American Academy of Neurology Practice Advisory: Thymectomy for Myasthenia Gravis (Practice Parameter Update) states this level B recommendation:45
Clinicians should discuss thymectomy with patients who have AChR ab+ generalized MG and are 18−65 years of age. The discussion should clearly indicate the anticipated benefits and risks of the procedures and uncertainties surrounding the magnitude of these benefits and risks.45
Extended trans-sternal thymectomy is a sizeable operation requiring multiple days of hospitalization. As such, there is an increasing practice to consider a less invasive technique for removing the thymus. Recommendations from the International Consensus Guidance for Management of Myasthenia Gravis regarding the less invasive endoscopic and robotic approaches to thymectomy,37
are increasingly performed and have a good track record for safety in experienced centers. Data from randomized, controlled comparison studies are not available. Based on comparisons across studies, less invasive thymectomy approaches appear to yield similar results to more aggressive approaches.37
For patients with poor control of MG symptoms there is a common practice to treat them with plasma exchange or IVIg just prior to undergoing thymectomy, in an effort to reduce the risk for myasthenic decompensation post-operatively. To address this issue a randomized trial of 24 MG patients compared IVIg (1 g/kg/day for two consecutive days) with plasma exchange (5 liters on alternate days), from 10–30 days prior to undergoing thymectomy. The duration of intubation and duration of surgery favored IVIg as a pre-operative intervention.40
Historically, patients with pure ocular MG have not been commonly treated with thymectomy (unless their imaging suggests the presence of thymoma). However, a meta-analysis of studies evaluating the effect of thymectomy in patients with ocular myasthenia (no thymoma) suggested a potentially favorable response.46
Additional consensus opinions and considerations regarding thymectomy in MG are addressed in the position statement from the International Consensus Guidance for Management of Myasthenia Gravis as follows:
Thymectomy may be considered in patients with generalized MG without detectable AChR antibodies if they fail to respond adequately to IS therapy, or to avoid/minimize intolerable adverse effects from IS therapy”. [And,] Current evidence does not support an indication for thymectomy in patients with MuSK, LRP4, or agrin antibodies.37
A Practical Approach to the Management of Patients with MG
Informed by the broad variations in clinical presentation, immunological characteristics, the nuances of the “natural course of MG” and the recent major advances in therapeutic options, the clinician can create an approach to patient management as outlined below. These general principles are offered as a set of considerations for the practicing physician.
- Be certain of the diagnosis! While MG is a clinical diagnosis there are multiple tools for validation. Be mindful that nonspecific “fatigue” and generalized subjective “weakness” are far different from symptoms in patients with MG, who present with symptoms of loss of function involving specific muscle groups. Patients with nonspecific fatigue/weakness are commonly diagnosed with MG but do not actually have it. Whilst oral cholinesterase inhibitors are remarkably safe medications, all of the other treatment options for MG carry significant clinical risks and/or challenging financial investment.
- Individualize treatment based on patient symptoms, life circumstances, and comorbidities. While mild ptosis and diplopia may not be disabling for most patients, in others it may be critical for their career (such as a pilot or neurosurgeon).
- Pure Ocular MG: if CEI fails then prednisone is the most likely treatment to yield the best results.
- MuSK Myasthenia: these patients respond less favorably to CEI, corticosteroids, immunosuppressive therapy, and IVIG. However, they benefit predictably and disproportionately to rituximab as well as plasma exchange.
- Patients with mild weakness or trivial symptoms (localized or generalized), should all initially be managed with cholinesterase inhibitors (pyridostigmine) (Table 1).
- For moderate to severe weakness (localized or generalized), initial management is with CEI. In patients under age 60 and AChR-positive serology consideration for thymectomy is appropriate. In older patients, thymectomy is usually not performed unless thymoma is suspected. Thymectomy should be performed at a facility with experience in performing the procedure and in managing patients with MG.
- If thymoma is suspected (by chest imaging) thymectomy should be performed, even if their myasthenic symptoms are mild. In patients with pure ocular symptoms thymectomy is typically not performed (unless thymoma is suspected).
- When MG symptoms are poorly controlled on CEI, use immunosuppression. Corticosteroid therapy represents the most predictable and effective choice. In those with severe symptoms, rapidly progression, or life-threatening weakness, start high-dose corticosteroids. In patients who are disabled but stable in which waiting 6 months for clinical response is an option, then a nonsteroidal immunosuppressive drug such as azathioprine is considered.
- For patients with moderate to severe generalized myasthenia who have IgG seropositivity, who are poor candidates for conventional immunosuppressive therapies or who have failed these therapies, treatment with FcRn blocking agents (efgartigimod being the current FDA approved drug) is a logical approach (excellent safety profile, 80% favorable response rate, early onset of improvement in the initial 1–2 weeks, and sustained results for months).
- An alternative to FcRn blocking agents who have AChR binding antibodies is a complement inhibitor (eculizumab or ravulizumab currently FDA approved). Appropriate patients should have moderate to severe generalized MG and be seropositive for AChR binding antibodies.
- Plasma exchange or IVIg are indicated in the following patients:
Adverse Effects of Medications on MG (Medications to Avoid or Use with Caution)
The list of medications reported to adversely impair neuromuscular transmission, or to be associated with observed temporally related worsening of MG symptoms in patients with MG is extensive.47 It behooves the clinician to recognize that almost any medication may possibly result in some degree of neuro-worsening in patients with myasthenia. There are a number of drugs that are better established as having a predictable adverse effect and these are best avoided if at all possible. As such, use of botulinum toxin, chloroquine, quinidine, quinine, and procainamide are contraindicated. Antibiotics are particularly noted to be problematic and in general aminoglycoside antibiotics should be avoided unless needed for a life-threatening infection. The class of fluoroquinolones (ciprofloxacin, levofloxacin, norfloxacin, and moxifloxacin) should also be avoided unless the patient’s infection is life-threatening. Macrolide antibiotics such as azithromycin and telithromycin have significant neuromuscular blocking effects and some MG patients experience severe worsening of MG symptoms. In anesthesiology and critical care medicine the use of nondepolarizing neuromuscular blocking agents such as vecuronium, rocuronium, atracurium, as well as the depolarizing drugs (succinylcholine) can produce prolonged and severe weakness in MG patients.
Commonly prescribed “staples” in general medicine such as statins, beta blockers, and calcium channel blockers are well reported to increase symptoms in occasional patients.
Administration of iodinated contrast dye was reported to cause exacerbation of MG but with changes in the characteristics of iodinated contrast in the 1980s it appears that the overall risk is relatively low.48
While most adverse drug effects reflect an exacerbation of pre-existing MG there are several drugs that can actually induce autoimmune MG de novo. With the use of d-penicillamine (for treatment of Wilson’s disease and rheumatoid arthritis) about 5% of patients develop MG symptoms (and the presence of AChR-ab). In general, these patients experience relatively mild symptoms and upon discontinuation of the drug the majority of patients experience resolution of their MG (and their serological studies revert to normal as well). Another iatrogenic cause for autoimmune MG is α-interferon.47
A more recent association with the development of severe MG is the exposure to Immune Checkpoint Inhibitors (ICI) which can either create or exacerbate MG. The ICI-related MG tends to be relatively severe and challenging to manage. The ICI are increasingly and widely used and needed as standard of care in management of various malignancies. They include ipilimumab, pembrolizumab, nivolumab, durvalumab, atezolizumab, and avelumab. The ICI have a propensity for inducing a variety of immune-mediated disorders which can be severe and require discontinuation of the ICI and use of aggressive immune therapy. Checkpoint inhibitor use in patients with established autoimmune disease can not only result in exacerbation of the pre-existing disorder (in half of such patients) but also can induce a new autoimmune disease in 30%.49 Permanent discontinuation of checkpoint inhibitor treatment may be necessary (and in one study was required in 12% of patients who had underlying autoimmune disease.49
Of all the potential neurological risks from ICI usage, the risk of MG is sufficiently common and potentially severe to justify special attention by the practicing neurologist providing consultation on cancer patients. One retrospective review of 65 MG patients with checkpoint inhibitor exposure illustrated the tendency for a severe and rapidly progressive clinical course of MG. This study also provided evidence for benefit from aggressive management with prompt use of plasma exchange and IVIg.50
Another well-established cause of iatrogenic autoimmune MG is seen in patients who undergo bone marrow transplantation. These patients will occasionally develop MG as part of the chronic graft vs host syndrome and this is important to consider in the evaluation of bone marrow transplant patients who present with symptoms of weakness.47
Transient neonatal myasthenia
Transient neonatal myasthenia is reported to occur in 10–15% of babies born to mothers having autoimmune MG. These neonates are noted to be floppy at birth, or to have a weak cry or suck, and in some cases have respiratory distress. Symptoms are usually recognized immediately upon delivery, or within the first day or two. Respiratory difficulty may require brief mechanical ventilation. Transient neonatal MG is due to the mother’s antibodies crossing the placenta and entering into the baby’s’ circulation towards the end of term. The disease-inducing maternal antibodies are gradually eliminated from the baby’s circulation over a week or two (and replaced by the baby’s natural antibodies). Weakness gradually resolves completely over a few days to 2 weeks, after which the baby is normal. Neonates with severe weakness are treated with oral pyridostigmine 1–2 mg per kg every 4 hours. Kochhar reviewed three case series, totaling 110 cases of babies born to mothers with autoimmune MG in which the type and timing of onset of myasthenic signs were reported and added their local experience with 37 cases. In the total of 147 infants born to women with MG, 15 (10%) developed signs of transient neonatal MG with onset being 1.5 ± 2.6 days (mean ± 3 SD) following birth. Common presenting symptoms/signs were feeding difficulties and reduced muscle tone. Only one of the 147 babies required intubation for respiratory failure. Based on these data the authors suggest a period of only 4 days of routine postnatal observation for infants born to women with MG.51
Congenital myasthenia
Congenital myasthenia defines a diverse collage of rare hereditary non-immunologic disorders of the neuromuscular junction.52 While there are significant differences in the various subtypes, in general, affected patients have life-long and relatively stable fatigable weakness of cranial and limb muscles. Many patients may have been misdiagnosed as limb girdle muscular dystrophy, facioscapulohumeral muscular dystrophy, or as having one of the congenital myopathies. The improved access to affordable genetic testing has dramatically improved the identification of patients with congenital myasthenia gravis. While these disorders are not immune-mediated and do not respond to immune therapy (steroids, thymectomy, and plasma exchange), the majority will respond favorably to cholinesterase inhibitors. Several of the distinctive subtypes are particularly important for therapeutic implications. And knowing the correct mutation in many cases provides the clinician with meaningful options for management. In the “fast channel congenital myasthenic syndrome” the patients display a static or slowly progressive clinical course and tend to respond favorably to a combination of pyridostigmine and amifampridine. With the “slow channel congenital myasthenic syndrome” the patients tend to have progressive weakness over years and develop an endplate myopathy. Unlike most other myasthenic disorders the use of cholinesterase inhibitors tends to be ineffective and actually worsens their symptoms. However, the use of quinidine (contraindicated in all other myasthenic disorders) and fluoxetine, which reduce the duration of acetylcholine receptor channel openings, are both predictably effective treatments for the slow channel syndrome. The “congenital myasthenic syndrome associated with acetylcholine receptor deficiency” tends to be static without progression of weakness over time (and many patient improve with aging). These patients often respond favorably to pyridostigmine, amifampridine, and ephedrine (alone or in combination). Patients with the “endplate acetylcholinesterase deficiency” present as infants or early in childhood with symptoms of generalized fatigable weakness, poor muscle development, and often delayed slow pupillary light responses. These patients do not improve with cholinesterase inhibitors (and may become weaker), but they may improve with albuterol. One form of congenital myasthenia which involves limb and axial (truncal) weakness but spares the cranial muscles is associated with a homozygous or biallelic mutation of Dok-7 gene. Normal formation of neuromuscular synapses requires the presence of normally functioning MuSK and Dok-7 is essential for the activation of MuSK. Albuterol and also amifampridine are reported to be effective in the treatment of patients with “Dok-7 congenital myasthenia”.52
Lambert-Eaton Myasthenic Syndrome
The Clinical Presentation and the Natural Course of LEMS
In 1950 Lee Eaton (clinical neurologist) and Edward Lambert (neurophysiology) from the Mayo Clinic first described patients with fluctuating myasthenic weakness in the setting of lung cancer. Initially referred to as “Eaton-Lambert syndrome”, the name was formally changed to “Lambert-Eaton syndrome” in the early 1980s in recognition of a professional lifetime of work and seminal discovery by Dr. Lambert. Lambert-Eaton myasthenic syndrome (LEMS) is a rare immune-mediated chronic disorder of neuromuscular transmission in which patients typically present with insidious symptoms of symmetrical proximal muscle weakness. The condition is among the classic autoimmune disorders with two distinctive underlying causes. Well known is the form associated with lung cancer. About 50% of LEMS patients have the paraneoplastic form. The malignancy is always small cell cancer and the vast majority arise in the lung. Small cell cancer surface antigens induce a compensatory immune attack with the production of voltage-gated calcium channel antibodies. These voltage-gated calcium channel antibodies cross react with normal channels at nicotinic and also muscarinic pre-synaptic nerve terminals which leads to limitation of calcium influx at motor nerve terminals in response to a nerve action potential triggered depolarization. When these calcium channels are impaired the motor nerve terminal has a limited capacity to generate a sufficient calcium concentration to support normal neuromuscular transmission. Calcium in a sufficient concentration is essential for pre-synaptic vesicular release of ACh and thus the resulting clinical weakness. With sustained stimulation of the motor nerve terminal (as with a high frequency of repetitive nerve action potentials) voltage-gated calcium channels can remain open long enough to achieve better influx of calcium and improvement in neuromuscular transmission, clinical strength, and function. The second distinctive cause for LEMS is that of a primary autoimmune disease (without cancer). These patients tend to be younger, females being more often affected, and they usually have other autoimmune disorders (thyroid, B12, diabetes, etc.). Patients with the paraneoplastic form are usually older, men more than women, and more likely to have a significant history of cigarette smoking.
Proximal lower extremity symptoms are most common in LEMS with patients noting difficulty rising from a chair or a low position, going up and down stairs, and walking. Proximal upper extremity weakness is also common and patients usually have some involvement of cranial muscles (although usually far less severe than in myasthenia gravis). Thus, some patients describe diplopia, ptosis, facial, oropharyngeal, and respiratory involvement. As with MG, patients with LEMS experience fluctuating degrees of weakness. Unlike MG, patients with LEMS may experience improvement in strength with physical exertion (facilitation). The presence of mainly symmetrical proximal weakness often leads to a consideration of myopathy but LEMS patients have normal muscle enzymes. Because this is a disorder of pre-synaptic cholinergic synaptic transmission there are additional symptoms and signals that provide strong clues to the diagnosis. LEMS patients have antibodies directed against voltage-gated calcium channels. Anti-muscarinic presynaptic effects include severe dry mouth (possessed by nearly all LEMS patients). Some patients also describe a metallic taste, some have paresthesia, and others experience myalgia. Bladder and bowel symptoms and other features of impaired autonomic function are seen on occasion.
The examination reveals weakness that on manual muscle strength testing often seems disproportionally mild compared with the patient’s severity of symptomatology. In addition to proximal weakness and mild cranial findings the LEMS patients are typically areflexic. Absent or markedly hypoactive muscle stretch reflexes are normal for patients with LEMS but never seen in patients with myasthenia gravis (in which the reflexes are normal). Absent muscle stretch reflexes in a patient with proximal weakness often leads to a diagnostic question of motor neuropathy such as chronic inflammatory neuropathy which is another reason that many patients can initially be misdiagnosed with other more common neuromuscular conditions. Patients may experience or display on examination an improvement in strength and function with continuous muscle stimulation or exercise. The main presenting symptoms in patients with LEMS are typically proximal lower extremity weakness, difficulty rising from achair and limitations in walking. In addition, about 50% of patients have evidence for bulbar involvement53 and half describe ocular symptoms of ptosis or diplopia.54 Physical examination of sustained grip strength demonstrates a gradual improvement in strength during the first 2–3 seconds of squeezing the hand (referred to as “Lambert’s sign”) (Table 3).
 |
Table 3 Lambert-Eaton Myasthenic Syndrome – Clinical Symptoms and Signs |
These patients require thorough evaluation for associated malignancy with particular attention to the lungs given the high likelihood of small cell carcinoma. While the cancer is usually located in the lungs there are well-described reports of small cell cancer arising in other organs and tissues.55 If the initial evaluation is negative for malignancy, a surveillance re-evaluation should be repeated at 6 month intervals for the next 4 years.56 Symptoms of LEMS may pre-date the diagnosis of cancer by up to 4 years thus justifying assiduous monitoring for malignancy over this time period. One serological marker for the presence of small cell lung cancer is the SOX1 antibody, which is directed against an immunogenic tumor antigen in SCLC. In one study 64% of patients with paraneoplastic LEMS had SOX1 antibodies while none was found in 50 patients without neoplasm. Thus, the presence of these antibodies in a patient’s serum should trigger an aggressive search for SCLC.57
Validating the Diagnosis
The P/Q-type voltage-gated calcium channel IgG antibody is present in well over 90% of LEMS patients and serves as excellent serological evidence to support the clinical diagnosis. Clinical neurophysiology (nerve conduction studies, repetitive stimulation, and electromyography) can confirm the presence of a presynaptic disorder of neuromuscular transmission. On nerve conduction studies the compound muscle action potentials (CMAP) are uniformly low in amplitude and the CMAP demonstrates a decrement to slow rates of repetitive stimulation. Immediately following 10 seconds of brief muscle contraction, there is marked facilitation of the CMAP amplitude (greater than 100% increase) in over 90% of patients. At high rates of repetitive stimulation (20 Hz, 30 Hz, tetanic stimulation), there is usually an incremental response. As high rates of repetitive stimulation are uncomfortable for the patient this part of the assessment is often deferred. Single fiber EMG is markedly abnormal in virtually all patients with LEMS with marked increase in jitter and blocking, although in contrast to myasthenia gravis continued activation of the LEMS muscle may demonstrate reduction in the severity of jitter and blocking.
Treatment of LEMS
Options for treatment of LEMS include treating the malignancy (if paraneoplastic). Pharmacological options include pyridostigmine, and amifampridine, a voltage-gated potassium channel blocker. For patients who are refractory to these options immune suppression is recommended (Table 4).58
 |
Table 4 Lambert-Eaton Myasthenic Syndrome – Approach to Treatment |
Cancer Treatment
In patients with the paraneoplastic form of LEMS successful treatment of the cancer can in some cases be curative of their LEMS. An evaluation of the impact of concurrent LEMS on the survival of patients with SCLC suggests that the presence of LEMS with SCLC conferred a significant survival advantage independently of the other prognostic variables.59
Amifampridine
First-line medical treatment is amifampridine. This is a quaternary ammonium drug that blocks presynaptic voltage-gated potassium channels at the motor nerve terminal resulting in prolonged depolarization which allows for the defective voltage-gated calcium channels to remain open, which in turn allows for increased calcium concentration at the motor nerve terminal (facilitating presynaptic release of ACh). In 1989 a form of this drug (3,4-diaminopyridine) was shown in a prospective controlled trial to be an effective treatment for LEMS.60 As such, the drug has been considered the single most effective drug option for treatment of LEMS. Prior to FDA approval in 2018 patients had been required to receive such medication through approved research centers. In 2018 the FDA approved amifampridine phosphate (Firdapse) for treatment of adults age 17 and older with LEMS.61 A second form of amifampridine (historically referred to as 3,4 diaminopyridine) has also demonstrated benefit in patients with LEMS62 and in 2019 this base form of amifampridine (called Ruzurgi) was approved by the FDA for treatment of juvenile LEMS (age 6 years to less than 17 years). Subsequently, the manufacturer of Firdapse filed a suit against the FDA regarding the approval of Ruzurgi, alleging that the FDA violated the company’s statutory rights to Orphan Drug Exclusivity for the Firdapse brand of amifampridine for the treatment of LEMS (the FDA’s Orphan Drug Exclusivity prevents approval of the same product for the same indication for a 7 year period). In early 2022, the US legal system judged in favor of the manufacturer of Firdapse and as such, the FDA approval of Ruzurgi was rescinded. Thus, there is currently only one FDA-approved form of amifampridine (Firdapse) for the treatment of LEMS. Side effects include transient paresthesia (10%) and very rarely seizures, especially in high doses. The starting daily dose of amifampridine (in adults) is 15–30mg PO daily and spread out to be administered 3–4 times per day. In general, the dose can be increased by 5 mg daily every 3–4 days until optimal response is achieved. Maximal dosing based on FDA approval is 20 mg per single dose and 80 mg daily total.
Pyridostigmine can be an adjunctive and valuable drug for patients with LEMS. The benefits are relatively limited but the quick onset of benefit, favorable safety profile, broad availability, and limited cost combine to make this an important option for selected patients.
LEMS: Immune Therapy
The multitude of immune therapies similar to those employed in the management of myasthenia gravis have all been utilized with some success in the management of LEMS especially in severe or refractory cases. Oral agents including prednisone and azathioprine, as well as immune modulators such as IVIg and rituximab and in some cases antibody removal with plasma exchange are all utilized in patients having limited benefit from amifampridine.58,63,64
Acknowledgment
This presentation is dedicated to the excellence, commitment, and influence of Dr. T.R. Johns, a 20th century pioneer in the care of patients with myasthenia gravis and Lambert-Eaton myasthenic syndrome. He made them better, led his field, and touched those fortunate enough to work by his side. Myasthenia gravis and Lambert-Eaton myasthenic syndrome are, like it or not, the neurologist’s diseases.65
Disclosure
Dr Cynthia Bodkin reports serving as Site PI for MG clinical rials sponsored by RA Pharmaceuticals, and Alexion Pharmaceuticals, outside the submitted work. The authors report no conflicts of interest in this work.
References
1. Grob D, Brunner NG, Namba T. The natural course of myasthenia gravis and effect of therapeutic measures. NYAS. 1981;377:652–669.
2. Oosterhuis HJ. The natural course of myasthenia gravis: a long term follow up study. J Neurol Neurosurg Psychiatry. 1989;52(10):1121–1127. PMID: 2795037; PMCID: PMC1031695. doi:10.1136/jnnp.52.10.1121
3. Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front Immunol. 2020;11:212. doi:10.3389/fimmu.2020.00212
4. Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle Nerve. 2006;33(4):445–461. PMID: 16228970. doi:10.1002/mus.20440
5. Bacchi S, Kramer P, Chalk C. Autoantibodies to low-density lipoprotein receptor-related protein 4 in double seronegative myasthenia gravis: a systematic review. Can J Neurol Sci. 2018;45:62–67. doi:10.1017/cjn.2017.253
6. Zhang B, Shen C, Bealmear B, et al. Autoantibodies to agrin in myasthenia gravis patients. PLoS One. 2014;9:e91816. doi:10.1371/journal.pone.0091816
7. Gasperi C, Melms A, Schoser B, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology. 2014;82(22):1976–1983. doi:10.1212/WNL.0000000000000478
8. Yoganathan K, Stevenson A, Tahir A, Sadler R, Radunovic A, Malek N. Bedside and laboratory diagnostic testing in myasthenia. J Neurol. 2022;269(6):3372–3384. PMID: 35142871; PMCID: PMC9119875. doi:10.1007/s00415-022-10986-3
9. Skolka M, Lamb CJ, Rubin DI, Klein CJ, Laughlin RS. Electrodiagnostic characteristics suggestive of muscle-specific kinase myasthenia gravis. Neurol Clin Pract. 2022;12(3):211–217. doi:10.1212/CPJ.0000000000001166
10. Pascuzzi RM. The edrophonium test. Semin Neurol. 2003;23(1):83–88. PMID: 12870109. doi:10.1055/s-2003-40755
11. Giannoccaro MP, Paolucci M, Zenesini C, et al. Comparison of ice pack test and single-fiber EMG diagnostic accuracy in patients referred for myasthenic ptosis. Neurology. 2020;95(13):e1800–e1806. PMID: 32788239. doi:10.1212/WNL.0000000000010619
12. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15(3):291–298. PMID: 6721451. doi:10.1002/ana.410150316
13. Menon D, Bril V. Pharmacotherapy of generalized myasthenia gravis with special emphasis on newer biologicals. Drugs. 2022;82:865–887. doi:10.1007/s40265-022-01726-y
14. Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. 1993;681:539–551. PMID: 8357194. doi:10.1111/j.1749-6632.1993.tb22937.x
15. Sanders DB, Hart IK, Mantegazza R, et al. An international, Phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71(6):400–406. doi:10.1212/01.wnl.0000312374.95186.cc
16. Cahoon WD
17. Wang L, Zhang S, Xi J, et al. Efficacy and safety of tacrolimus for myasthenia gravis: a systematic review and meta-analysis. J Neurol. 2017;264(11):2191–2200. doi:10.1007/s00415-017-8616-7
18. Yoshikawa H, Kiuchi T, Saida T, Takamori M. Randomised, double-blind, placebo-controlled study of tacrolimus in myasthenia gravis. J Neurol Neurosurg Psychiatry. 2011;82(9):970–977. doi:10.1136/jnnp-2011-300148
19. Pasnoor M, He J, Herbelin L, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology. 2016;87(1):57–64. doi:10.1212/WNL.0000000000002795
20. Howard JF
21. Muppidi S, Utsugisawa K, Benatar M, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. 2019;60:14–24. doi:10.1002/mus.26447
22. Vissing J, Jacob S, Fujita KP, O’Brien F, Howard JF; REGAIN study group. ‘Minimal symptom expression’ in patients with acetylcholine receptor antibody-positive refractory generalized myasthenia gravis treated with eculizumab. J Neurol. 2020;267(7):1991–2001. PMID: 32189108; PMCID: PMC7320935. doi:10.1007/s00415-020-09770-y
23. Howard JF, Nowak RJ, Wolfe GI, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 2020;77(5):582–592. doi:10.1001/jamaneurol.2019.5125
24. Howard JF, Genge A, Hussain Y, et al. Outcomes from raise: a randomized, placebo-controlled, double-blind, phase 3 trial of zilucoplan in generalized myasthenia gravis. mus mgfa Abstracts; 2022.
25. Vu T, Meisel A, Mantegazza R, et al. Efficacy and safety of ravulizumab, a long-acting terminal complement inhibitor, in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: results from the phase 3 CHAMPION MG study. Neurology. 2022;98(18 Supplement):791.
26. Hehir MK, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology. 2017;89:1069–1077. doi:10.1212/WNL.0000000000004341
27. Tandan R, Hehir MK 2nd, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve. 2017;56:185–196. doi:10.1002/mus.25597
28. Topakian R, Zimprich F, Iglseder S, et al. High efficacy of rituximab for myasthenia gravis: a comprehensive nationwide study in Austria. J Neurol. 2019;266:699–706. doi:10.1007/s00415-019-09191-6
29. Di Stefano V, Lupica A, Rispoli MG, Di Muzio A, Brighina F, Rodolico C. Rituximab in AChR subtype of myasthenia gravis: systematic review. J Neurol Neurosurg Psychiatry. 2020;91:392–395. doi:10.1136/jnnp-2019-322606
30. Young C, McGill SC. Rituximab for the Treatment of Myasthenia Gravis: A 2021 Update [Internet]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2021. Available from https://www.ncbi.nlm.nih.gov/books/NBK571915/.
31. Howard JF Jr, Bril V, Vu T, et al; ADAPT Investigator Study Group. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526–536. Erratum in: Lancet Neurol. 2021 Aug;20(8):e5.PMID: 34146511. doi:10.1016/S1474-4422(21)00159-9
32. Bril V, Benatar M, Andersen H, et al; MG0002 Investigators. Efficacy and safety of rozanolixizumab in moderate to severe generalized myasthenia gravis: a phase 2 randomized control trial. Neurology. 2021;96(6):e853–e865. PMID: 33219142; PMCID: PMC8105899. doi:10.1212/WNL.0000000000011108
33. Bril V, Druzdz A, Grosskreutz J, et al. Efficacy and safety of rozanolixizumab in patients with generalized myasthenia gravis: a randomized, multicenter, double-blind, placebo-controlled phase 3 study MGFA abstracts. Neurology. 2022;65:S35. doi:10.1002/mus.27540
34. Usmani A, Kwan L, Wahib-Khalil D, Trivedi J, Nations S, Sarode R. Excellent response to therapeutic plasma exchange in myasthenia gravis patients irrespective of antibody status. J Clin Apher. 2019;34:416–422. doi:10.1002/jca.21694
35. Yamada C, Teener JW, Davenport RD, Cooling L. Maintenance plasma exchange treatment for muscle specific kinase antibody positive myasthenia gravis patients. J Clin Apher. 2015;30:314–319. doi:10.1002/jca.21377
36. Ortiz-Salas P, Velez-Van-Meerbeke A, Galvis-Gomez CA, Rodriguez QJ. Human immunoglobulin versus plasmapheresis in guillain-barre syndrome and myasthenia gravis: a meta-analysis. J Clin Neuromuscul Dis. 2016;18:1–11. doi:10.1097/CND.0000000000000119
37. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87:419–425. doi:10.1212/WNL.0000000000002790
38. Adiao KJB, Espiritu AI, Roque VLA, Reyes J. Efficacy and tolerability of subcutaneously administered immunoglobulin in myasthenia gravis: a systematic review. J Clin Neurosci. 2020;72:316–321.
39. Hellmann MA, Mosberg-Galili R, Lotan I, Steiner I. Maintenance IVIg therapy in myasthenia gravis does not affect disease activity. J Neurol Sci. 2014;338:39–42. doi:10.1016/j.jns.2013.10.043
40. Alipour-Faz A, Shojaei M, Peyvandi H, et al. A comparison between IVIG and plasma exchange as preparations before thymectomy in myasthenia gravis patients. Acta Neurol Belg. 2017;117:245–249. doi:10.1007/s13760-016-0689-z
41. Rahbek MA, Mikkelsen EE, Overgaard K, Vinge L, Andersen H, Dalgas U. Exercise in myasthenia gravis: a feasibility study of aerobic and resistance training. Muscle Nerve. 2017;56:700–709. doi:10.1002/mus.25552
42. McQuillen MP, Leone MG. A treatment carol: thymectomy revisited. Neurology. 1977;27:1103–1106. doi:10.1212/WNL.27.12.1103
43. Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375:511–522. doi:10.1056/NEJMoa1602489
44. Wolfe GI, Kaminski HJ, Aban IB, et al. Long-term effect of thymectomy plus prednisone versus prednisone alone in patients with non-thymomatous myasthenia gravis: 2-year extension of the MGTX randomised trial. Lancet Neurol. 2019;18:259–268. doi:10.1016/S1474-4422(18)30392-2
45. Gronseth GS, Barohn R, Narayanaswami P. Practice advisory: thymectomy for myasthenia gravis (practice parameter update): report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology. Neurology. 2020;94:705–709. doi:10.1212/WNL.0000000000009294
46. Zhu K, Li J, Huang X, et al. Thymectomy is a beneficial therapy for patients with non-thymomatous ocular myasthenia gravis: a systematic review and meta-analysis. Neurol Sci. 2017;38:1753–1760. doi:10.1007/s10072-017-3058-7
47. Mehrizi M, Fontem RF, Gearhart TR, Pascuzzi RM. Medications and myasthenia gravis (a reference for health care professionals) prepared for the myasthenia gravis foundation of America; 2012. Available from: www.myasthenia.org.
48. Mehrizi M, Pascuzzi RM. Complications of radiologic contrast in patients with myasthenia gravis. Muscle Nerve. 2014;50:443–444. doi:10.1002/mus.24254
49. Abdel-Wahab N, Shah M, Lopez-Olivo MA, Suarez-Almazor ME. Use of immune checkpoint inhibitors in the treatment of patients with cancer and preexisting autoimmune disease: a systematic review. Ann Intern Med. 2018;168:121–130. doi:10.7326/M17-2073
50. Safa H, Johnson DH, Trinh VA, et al. Immune checkpoint inhibitor related myasthenia gravis: single center experience and systematic review of the literature. J Immunother Cancer. 2019;7:319. doi:10.1186/s40425-019-0774-y
51. Kochhar PK, Schumacher RE, Sarkar S. Transient neonatal myasthenia gravis: refining risk estimate for infants born to women with myasthenia gravis. J Perinatol. 2021;41:2279–2283. doi:10.1038/s41372-021-00970
52. Vanhaesebrouck AE, Beeson D. The congenital myasthenic syndromes: expanding genetic and phenotypic spectrums and refining treatment strategies. Curr Opin Neurol. 2019;32:696–703. doi:10.1097/WCO.0000000000000736
53. Burns TM, Russell JA, LaChance DH, Jones HR. Oculobulbar involvement is typical with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53:270–273. doi:10.1002/ana.10477
54. Young JD, Leavitt JA. Lambert-Eaton myasthenic syndrome: ocular signs and symptoms. J Neuroophthalmol. 2016;36:20–22. doi:10.1097/WNO.0000000000000258
55. Kennelly KD, Dodick DW, Pascuzzi RM, et al. Neuronal autoantibodies and paraneoplastic neurological syndromes associated with extrapulmonary small cell carsinoma. Neurology. 1997;48:A31–A32.
56. Hong BY, An HJ, Lim SH. Long-Standing Lambert–Eaton myasthenic syndrome caused by undetectable small-cell lung cancer: why we should follow-up LEMS. Diagnostics. 2022;12:1542. doi:10.3390/diagnostics12071542
57. Sabater L, Titulaer M, Saiz A, Verschuuren J, Gure AO, Graus F. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2008;70:924–928. doi:10.1212/01.wnl.0000281663.81079.24
58. Kesner VG, Oh SJ, Dimachkie MM, Barohn RJ. Lambert-Eaton myasthenic syndrome. Neurol Clin. 2018;36:379–394. doi:10.1016/j.ncl.2018.01.008
59. Maddison P, Gozzard P, Grainge MJ, Lang B. Long-term survival in paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2017;88:1334–1339. doi:10.1212/WNL.0000000000003794
60. McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321:1567–1571. doi:10.1056/NEJM198912073212303
61. Oh SJ, Shcherbakova N, Kostera-Pruszczyk A, et al. Amifampridine phosphate (Firdapse((R))) is effective and safe in a phase 3 clinical trial in LEMS. Muscle Nerve. 2016;53:717–725. doi:10.1002/mus.25070
62. Sanders DB, Juel VC, Harati Y, et al. 3,4-diaminopyridine base effectively treats the weakness of Lambert-Eaton myasthenia. Muscle Nerve. 2018;57:561–568. doi:10.1002/mus.26052
63. Anwar A, Saleem S, Ahmed MF, Ashraf S, Ashraf S. Recent advances and therapeutic options in Lambert-Eaton myasthenic syndrome. Cureus. 2019;11(8):e5450. PMID: 31637147; PMCID: PMC6799875. doi:10.7759/cureus.5450
64. Meisel A, Sieb JP, Le Masson G, et al. The European Lambert–Eaton myasthenic syndrome registry: long-term outcomes following symptomatic treatment. Neurol Ther. 2022;11(3):1071–1083. doi:10.1007/s40120-022-00354-8
65. Johns TR, editor. Myasthenia gravis. In: Seminars in Neurology. Vol. 2. StatPearls [Internet]; 1982:3. doi:10.1055/s-002-11126
66. Nowak RJ, Coffey CS, Goldstein JM, et al. (29a) rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: the BeatMG study. Neurology. 2022;98(4):e376–e389. doi:10.1212/WNL.0000000000013121
67. Jaretzki AI, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis recommendations for clinical research standards task force of the medical scientific advisory board of the myasthenia gravis foundation of America. Neurology. 2000;55(1):16–23. doi:10.1212/WNL.55.1.16
68. Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769–772. doi:10.1111/j.1749-6632.1998.tb11015.x
69. Wolfe GI, Herbelin L, Nations SP, Foster B, Bryan WW, Barohn RJ. Myasthenia gravis activities of daily living profile. Neurology. 1999;52:1487–1489. doi:10.1212/WNL.52.7.1487
70. Burns TM, Conaway MR, Cutter GR, Sanders DB. Less is more, or almost as much: a 15-item quality-of-life instrument for myasthenia gravis. Muscle Nerve. 2008;38(2):957–963. doi:10.1002/mus.21053
71. Burns TM, Sadjadi R, Utsugisawa K, et al. International clinimetric evaluation of the MG-QOL15, resulting in slight revision and subsequent validation of the MG-QOL15r. Muscle Nerve. 2016;54(6):1015–1022. doi:10.1002/mus.25198
72. Burns TM, Conaway MR, Cutter GR, Sanders DB. Construction of an efficient evaluative instrument for myasthenia gravis: the MG composite. Muscle Nerve. 2008;38(6):1553–1562. doi:10.1002/mus.21185
73. Burns TM, Conaway M, Sanders DB; MG Composite and MG-QOL15 Study Group. The MG composite: a valid and reliable outcome measure for myasthenia gravis. Neurology. 2010;74(18):1434–1440. doi:10.1212/WNL.0b013e3181dc1b1e
74. Thomsen JLS, Andersen H. Outcome measures in clinical trials of patients with myasthenia gravis. Front Neurol. 2020;11:596382. PMID: 33424747; PMCID: PMC7793650. doi:10.3389/fneur.2020.596382
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.