Back to Journals » The Application of Clinical Genetics » Volume 14
Mutations Involved in Premature-Ageing Syndromes
Authors Coppedè F
Received 18 March 2021
Accepted for publication 17 May 2021
Published 2 June 2021 Volume 2021:14 Pages 279—295
DOI https://doi.org/10.2147/TACG.S273525
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Fabio Coppedè
Department of Translational Research and of New Surgical and Medical Technologies, University of Pisa, Pisa, Italy
Correspondence: Fabio Coppedè
Department of Translational Research and of New Surgical and Medical Technologies, University of Pisa, Via Roma 55, Pisa, 56126, Italy
Tel +39 050 2218544
Fax +39 050 2211034
Email [email protected]
Abstract: Premature-ageing syndromes are a heterogeneous group of rare genetic disorders resembling features of accelerated ageing and resulting from mutations in genes coding for proteins required for nuclear lamina architecture, DNA repair and maintenance of genome stability, mitochondrial function and other cellular processes. Hutchinson–Gilford progeria syndrome (HGPS) and Werner syndrome (WS) are two of the best-characterized progeroid syndromes referred to as childhood- and adulthood-progeria, respectively. This article provides an updated overview of the mutations leading to HGPS, WS, and to the spectrum of premature-ageing laminopathies ranging in severity from congenital restrictive dermopathy (RD) to adult-onset atypical WS, including RD-like laminopathies, typical and atypical HGPS, more and less severe forms of mandibuloacral dysplasia (MAD), Néstor-Guillermo progeria syndrome (NGPS), atypical WS, and atypical progeroid syndromes resembling features of HGPS and/or MAD but resulting from impaired DNA repair or mitochondrial functions, including mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) syndrome and mandibuloacral dysplasia associated to MTX2 (MADaM). The overlapping signs and symptoms among different premature-ageing syndromes, resulting from both a large genetic heterogeneity and shared pathological pathways underlying these conditions, require an expert clinical evaluation in specialized centers paralleled by next-generation sequencing of panels of genes involved in these disorders in order to establish as early as possible an accurate clinical and molecular diagnosis for a proper patient management.
Keywords: Hutchinson–Gilford progeria syndrome, Werner syndrome, restrictive dermopathy, mandibuloacral dysplasia, Néstor-Guillermo progeria syndrome, atypical progeroid syndromes
Introduction
Premature-ageing syndromes are a heterogeneous group of rare genetic disorders resembling features of accelerated ageing.1,2 The term “progeria” derives from Greek words meaning “prematurely old”, and most of these syndromes are referred to as segmental progeroid syndromes (SPS) because only some organs and tissues mimic signs of accelerated ageing, including among others graying or loss of hair, lipodystrophy, scleroderma-like skin lesions, atherosclerosis and cardiovascular disease, hearing loss, osteoporosis, type 2 diabetes mellitus and various malignancies at an early age.1–3 Overall, over 100 syndromes showing signs of premature ageing have so far been described, with an estimated worldwide incidence of about 1:50.000.2 They can be categorized into congenital, infantile, and juvenile/adult forms according to the age at onset of the symptoms.2 However, based on the underlying molecular defect, premature-ageing syndromes can be grouped into diseases resulting from altered nuclear lamina architecture, diseases caused by mutations of genes required for DNA repair and maintenance of genome stability, and diseases resulting from diverse pathophysiological processes, such as connective tissue alterations and mitochondrial impairment among others.4 Hutchinson–Gilford progeria syndrome (HGPS) and Werner syndrome (WS), also referred to as childhood- and adulthood-progeria, respectively, are two of the best-characterized SPS as they closely mimic natural ageing, the first resulting from alterations of the nuclear lamina and the latter from impaired DNA repair mechanisms.1 The identification of their genetic basis and the creation of patient’s registers and mutation databases have revealed the existence of a disease spectrum ranging from moderate and mild-severe to very aggressive forms, known as atypical progeroid syndromes.5 Indeed, the recent application of next-generation sequencing (NGS) technologies has allowed us to uncover the causative mutations of several atypical premature-ageing syndromes, albeit in some cases, the genetic cause is still unclear.2–4 Owing to the genetic heterogeneity and the overlapping signs and symptoms among different SPS, NGS of panels of genes involved in these disorders is currently recommended for early molecular diagnosis in specialized centers, paralleled by an interdisciplinary collaboration of medical geneticists, pediatricians and other specialists in order to establish an accurate clinical and molecular diagnosis for a proper patient management.2–4 This review article will focus primarily on the mutations leading to HGPS and to the spectrum of premature-ageing laminopathies ranging in severity from congenital restrictive dermopathy (RD) to adult-onset atypical WS, and including RD-like laminopathies, typical and atypical HGPS, more and less severe forms of mandibuloacral dysplasia (MAD), Néstor-Guillermo progeria syndrome (NGPS) and atypical progeroid syndromes. Other forms of MAD resembling features of atypical progeroid laminopathies, but originating from mutations of genes involved in DNA repair or mitochondrial pathways will also be described.1–5 Furthermore, the article will describe the genetics of WS, the prototypic adult-onset progeria caused by mutations of a gene required for DNA repair and maintenance of genome stability.1 These selected disorders that represent some of the best-characterized SPS are intended to give the reader a complete and updated overview of the genetic heterogeneity underlying these conditions, resulting in a spectrum of more or less aggressive progeroid syndromes with overlapping symptoms and interrelated underlying molecular mechanisms. A summary of the main characteristics of the SPS discussed in this article is shown in Table 1.
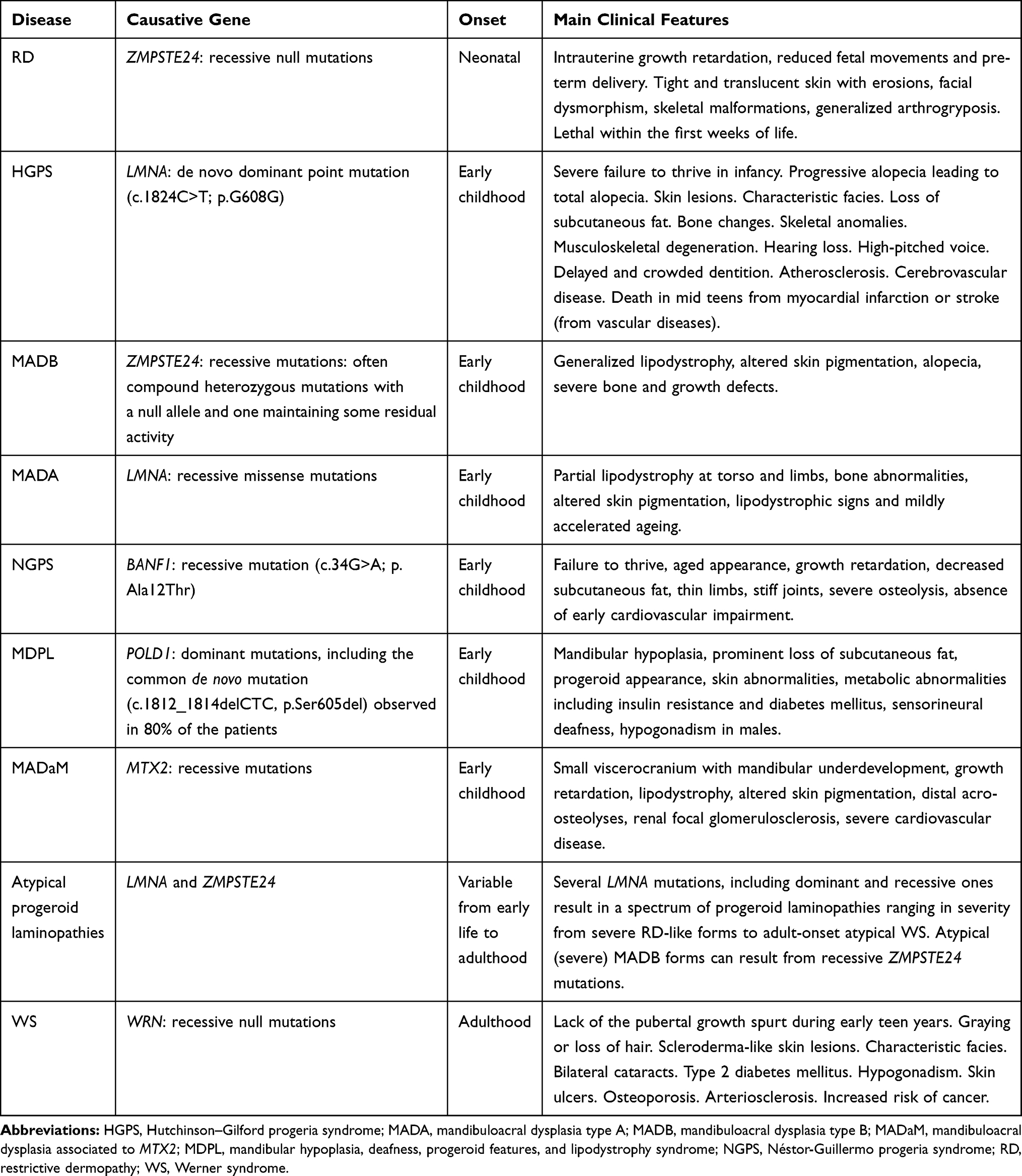 |
Table 1 Main Characteristics of the Premature-Ageing Syndromes Described in the Article |
Progeroid Syndromes Resulting from Alterations of the Nuclear Lamina Architecture
The nuclear lamina is a mesh-like layer of intermediate filaments termed lamins and of lamin-associated proteins underlying the inner nuclear membrane, and represents the structural scaffold of the nuclear envelope, required for maintenance of nuclear shape and structure, and for anchoring chromatin.6 Indeed, the nuclear lamina is essential for the organization of the chromatin structure and for the proper functioning of several nuclear processes including DNA replication and repair, epigenetic mechanisms, regulation of gene expression, cell division and differentiation.6 The LMNA gene codes for A-type lamins (lamins A and C), major components of the nuclear lamina in most differentiated cells, and in 2003 a de novo dominant point mutation in exon 11 of LMNA (c.1824C>T, p.Gly608Gly) was identified as the cause of the majority of HGPS cases.7,8 Lamin A is synthesized as prelamin A and undergoes a four-step process to become a mature protein, that includes farnesylation, methylation and removal of C-terminal residues (Figure 1). The LMNA c.1824C>T mutation results in the activation of a cryptic donor splice site leading to the production of a lamin A isoform, called progerin, containing an internal deletion of 50 amino acids near its C-terminal end.7 This deletion eliminates the site for endoproteolitic cleavage by ZMPSTE24, a zinc metalloproteinase that cleaves the 15 C-terminal amino acids of prelamin A to yield mature lamin A, so that progerin retains its farnesyl moiety at the C-terminus (Figure 1). Retention of the farnesyl group causes progerin to become permanently anchored into the nuclear membrane leading to structural, functional and molecular changes overall leading to a prematurely ageing phenotype.6 Particularly, HGPS cells accumulate structural and mechanical defects of the nuclear lamina with passaging, resulting from the accumulation of progerin and leading to bigger and dysmorphic nuclei with thickening of the nuclear lamina, lobulations, clustering of nuclear pores, and disorganization of chromatin, coupled to changes in DNA methylation and histone tail modifications leading to a widespread transcriptional misregulation.6,9–11 Fibroblasts from HGPS patients also exhibit accumulation of DNA damage, defective DNA repair and chromosomal instability with cell passaging, as well as mitochondrial dysfunction and a faster telomere shortening.12 Several other LMNA mutations causing a progeroid phenotype have so far been described, and are collected in the updated LMNA mutations database (UMD-LMNA: http://www.umd.be/LMNA/).13 These include either progerin-producing mutations or LMNA mutations that do not produce progerin but still cause damages in the nuclear lamina resulting in a disease spectrum of progeroid associated phenotypes ranging from very aggressive to milder forms, including congenital forms resembling RD, mandibuloacral dysplasia type A (MADA), as well as atypical progeroid syndromes or atypical WS.5,6 Damages in the nuclear lamina similar to those caused by LMNA mutations can result from recessive or compound heterozygous mutations in ZMPSTE24, leading to impaired lamin A maturation (Figure 1) and accumulation of uncleaved and farnesylated lamin A precursors.14 Indeed, ZMPSTE24 mutations cause three distinct SPS with increasing disease severity, namely mandibuloacral dysplasia type B (MADB), atypical HGPS, and RD.14–17 Several ZMPSTE24 mutations have been identified in homozygosis or compound heterozygosis in the patients, and disease severity is inversely correlated with the residual protein activity conferred by those mutations.14,17 Although most progeroid laminopathies result from impaired maturation of lamin A caused by LMNA or ZMPSTE24 mutations, a recessive point mutation in the BANF1 gene (p.Ala12Thr) that encodes the barrier to self-integration factor 1 (BAF) causes Néstor-Guillermo progeria syndrome (NGPS), a disorder that partially phenocopies HGPS, although patients have a relatively long lifespan and show no early cardiovascular complications.18,19 BAF is a chromatin-associated protein that also binds to lamins and lamin-associated proteins, thus anchoring chromatin to the nuclear envelope.20,21 Indeed, BAF is required for nuclear envelope dynamics during mitosis and post-mitotic nuclear envelope reassembly, but it is also involved in chromatin epigenetics, chromosome segregation, DNA damage response, and defense against viral infections.20,21 Fibroblasts from NGPS patients show a dramatic reduction in BAF protein levels and abnormalities in the nuclear lamina, including blebs and abnormal distribution of lamin-associated proteins, that can be restored by expression of wild type BAF.18
 |
Figure 1 Schematic representation of prelamin A processing in normal individuals, and in patients with Hutchinson–Gilford progeria syndrome and restrictive dermopathy. Notes: Adapted from Coppedè F. The epidemiology of premature aging and associated comorbidities. Clin Interv Aging. 2013;8:1023–1032.5© 2013 Coppedè. Creative Commons Attribution – Non Commercial (unported, v3.0) License (http://creativecommons.org/licenses/by-nc/3.0/). In cells from healthy individuals, prelamin A undergoes a four-step process to become a mature lamin A, including (1) farnesylation of the cysteine residue on the C-terminal CaaX motif by a farnesyltransferase (FTASE), (2) cleavage of the –aaX terminal residues that can be performed either by RCE1 (Ras converting enzyme 1) or by ZMPSTE24 (Zinc metalloprotease related to Ste24p), (3) carboxymethylation of the C-terminal cysteine residue by ICMT (isoprenylcysteine carboxyl methyltransferase), (4) removal of the C‑terminal 15 residues through cleavage by ZMPSTE24. In Hutchinson–Gilford progeria syndrome (HGPS) the common LMNA (p.G608G) mutation results in the deletion of a 50aa region from prelamin A (in orange) containing the upstream ZMPSTE24 cleavage site, resulting in a shorter farnesylated protein that cannot undergo the last maturation step, termed progerin. In restrictive dermopathy (RD) recessive null mutations of ZMPSTE24 result in lack of ZMPSTE24 activity with subsequent accumulation of uncleaved and farnesylated lamin A precursors. |
Hutchinson–Gilford Progeria Syndrome
HGPS (OMIM #176670) is an extremely rare genetic disorder affecting about 1 in 4–8 million births, but represents one of the best-characterized SPS.5 The onset of disease symptoms usually occurs during the first year of life and the mean life expectancy is 14.6 years, with death primarily occurring from myocardial infarction or stroke resulting from vascular disease.5,6 The disease results from a de novo dominant LMNA mutation and affects both genders equally.5 A visible vein across the nasal bridge and skin lesions are often the first signs observed in HGPS infants, followed by a profound failure to thrive leading to an average final weight of almost 14.5 Kg. Other main symptoms of HGPS include alopecia and loss of subcutaneous fat. Musculoskeletal complications observed in the affected children include loss of muscle mass, stiffness of joints, hip dislocations, skeletal abnormalities and fragile bones. A large cranium in relation to the facial size and a small face with pinched nose, receding mandible, prominent eyes, and scalp veins are the main facial abnormalities. Moreover, common features of HGPS children are a high-pitched voice, a delayed/crowded dentition and protruding ears that lack lobules. Individuals with HGPS do not reproduce, as they are sexually immature. Cognitive and motor functions are preserved, and there is no increased cancer risk.22,23
According to the disease prevalence, there is an estimate of between 350 and 400 HGPS children worldwide at any one time, with 131 identified cases as of January 2021 (http://www.progeriaresearch.org).24 Most of the tested HGPS children (85–90%) carry the heterozygous LMNA c.1824C>T (p.Gly608Gly) mutation and are referred to as classic HGPS.24 Less frequent progerin-producing mutations are found in the remaining cases. They include heterozygous exon 11 c.1822G>A (p.Gly608Ser), c.1821G>A (p.Val607Val), and c.1868C>G (p.Thr623Ser) mutations, as well as heterozygous mutations at the intronic border of exon 11, such as c.1968+1G>A, c.1968+1G>C, c.1968+2T>A, c.1968+2T>C, and c.1968+5G>C, all leading to a less or more aggressive phenotype depending on the resultant final amount of progerin protein that is produced.5,24 For example, both c.1968+1G>A and p.Val607Val mutations result in a strong activation of the aberrant splice site, leading to increased progerin production and a more severe phenotype compared to classic HGPS, whilst the p.Thr623Ser mutation results in the deletion of only 35 amino acids from exon 11 leading to a slowly progressing progeria.5,6
Several atypical progeroid syndromes, also referred to as nonclassic progeria, atypical HGPS or atypical WS, result from LMNA mutations that do not produce progerin.5 These mutations can either be dominant or recessive and alter residues throughout the protein structure with no clear clustering in a single region of lamin A, but still cause damages in the nuclear lamina that overlap with those produced by progerin-producing alleles leading to a spectrum of less or more aggressive progeroid laminopathies all characterized by growth retardation and involving the same body systems (bones, body fat, skin, and hair) as in classic HGPS, but that differ in age at onset, course, and severity of the symptoms with respect to classic HGPS.5,6 More than 20 such mutations have been described, mainly dominant missense ones such as p.Pro4Arg, p.Thr10lle, p.Ala57Pro, p.Leu59Arg, p.Glu111Lys, p.Arg133Leu, p.Asp136His, p.Glu138Lys, p.Leu140Arg, p.Ser143Phe, p.Glu145Lys, p.Glu159Lys, p.Asp300Asn, p.Asp300Gly, p.Asp300His, p.Asn466Lys, p.Glu578Val, p.Cys588Arg, and p.Arg644Cys.5,25 Several of these mutations are far less severe than those causing HGPS and are found in individuals suspected to have WS on clinical grounds, but that lack mutations in the causative gene of classic WS (the WRN gene), denoted as atypical WS.5 More details on atypical WS will be provided in the paragraphs of this review dedicated to WS. Also, recessive LMNA mutations are found in progeroid syndromes, including those causing MADA and detailed in the following paragraphs, as well as others such as the p.Arg435Cys one leading to a more aggressive RD-like progeria.26 A continuously updated list of LMNA mutations causing classic HGPS or atypical progeroid syndromes can be found in the UMD-LMNA mutations database.13 However, not all LMNA mutations cause progeria. Almost 500 different LMNA mutations have so far been described, causing different disorders collectively called primary laminopathies and including cardiomyopathies, muscular dystrophies, lipodystrophies, neuropathies, as well as progeroid syndromes.13
Restrictive Dermopathy
RD (OMIM #275210) is an extremely rare congenital and lethal genodermatosis so far described in less than 100 individuals worldwide.3 The disease is severer than classic HGPS, with signs appearing during gestation and including intrauterine growth retardation with polyhydramnios, reduced fetal movements and pre-term delivery.16 Newborns are characterized by a very tight, thin, and translucent skin with erosions and scaling at flexure sites, associated with typical facial dysmorphism, skeletal malformations and generalized arthrogryposis. Death usually occurs within the first weeks of life resulting from pulmonary hypoplasia leading to respiratory insufficiency.16 In 2004, Navarro and coworkers screened nine individuals with a clinical diagnosis of RD, observing LMNA mutations in two of them, and a common frame-shift mutation in exon 9 of ZMPSTE (c.1085dupT) in all the remaining cases, resulting in a truncated protein lacking the catalytic domain.16 Concerning the two identified LMNA mutations, one patient had the common c.1824C>T mutation in exon 11 but showed an atypical progeroid phenotype ranging in severity between RD and HGPS, and the other patient showed a heterozygous LMNA mutation (c.1968+1G>A) leading to a very aggressive progeria that was diagnosed at birth as RD.16 Subsequent studies have revealed that RD is caused by recessive or compound heterozygous null mutations in ZMPSTE24, such as the common c.1085dupT, resulting in defective processing and nuclear accumulation of prelamin A.27,28 Particularly, almost 30 different ZMPSTE24 mutations have been identified in patients with progeroid syndromes.17 Complete loss-of-function of ZMPSTE24 leads to RD and results from homozygous or compound heterozygous null mutations in ZMPSTE24, while other less severe phenotypes, including severe HGPS or MADB, are associated with a residual partial activity conferred by at least one haploinsufficient allele, resulting in a spectrum of progeroid laminopathies whose severity is inversely correlated with the residual protein activity.14 Null ZMPSTE24 mutations leading to RD include the common c.1085dupT and other small deletions/duplications (c.50delA, c.54dupT, c.209_210delAT, c.295delC, c.584_585delAT, c.591dupT), intron mutations (c.475–2A>G, c.627+1G>C, c.628–2A>G, c.954+2T>A), base substitutions (c.691G>T, c.715G>T, c.826 C>T, c.1020G>A, c.1105C>T, c.1249C>T, c.1385C>T) and a large deletion (r.271_627del; p.Leu91_Leu209del), all observed in homozygosity or compound heterozygosity in the patients and resulting in frame-shift mutations, exon skipping and/or insertion of premature stop codons leading to truncated non-functional proteins.17 Only in a minority of the cases, very severe LMNA mutations, such as the c.1968+1G>A one can lead to a congenital progeria resembling RD.17 Interestingly, a patient with both a homozygous frame-shift ZMPSTE null mutation (c.1204_1225del22) and a heterozygous nonsense LMNA mutation (c.1960C>T) has been described.29 The homozygous mutation in ZMPSTE24, resulting in null activity of the enzyme, should have led to RD, but the presence of the heterozygous nonsense LMNA mutation leading to a truncated prelamin A protein that lacks the C-terminal 11 amino acids and is unable to undergo post-translational modifications, acted as a salvage alteration alleviating the clinical phenotype from RD to HGPS-like.29 Cells from RD patients show several nuclear abnormalities, including increased nuclear size and altered shape, reduced levels of lamins A and C, and mislocalization of lamin-associated proteins.16
Mandibuloacral Dysplasia
Mandibuloacral dysplasia (MAD) is a rare autosomal recessive condition, so far diagnosed in almost 50 children worldwide, and represents a milder form of progeroid laminopathy that can either result from LMNA (MADA) or ZMPSTE24 (MADB) mutations.3,30 The clinical signs of MADA (OMIM #248370) present usually in early childhood (4–5 years) and include mandibular hypoplasia, dental crowding, clavicular resorption, acral osteolysis, skin abnormalities and a mildly accelerated ageing that becomes visible in the second decade of life.30 MADA is also characterized by partial lipodystrophy in the torso and limbs.30 In 2002, Novelli et al observed that MADA was caused by a recessive c.1580G>A mutation (p.Arg527His) in exon 9 of LMNA, opening the way to the search of LMNA mutations in progeroid diseases.31 Indeed, the c.1580G>A substitution is the most common mutation causing MADA.30 However, at least seven less frequent homozygous LMNA mutations have so far been identified in MADA children, including c.1411C>T (p.Arg471Cys), c.1579C>T (p.Arg527Cys), c.1585G>A (p.Ala529Thr), c.1586C>T (p.Ala529Val), c.1620G>A (p.Met540Ile), c.1626G>C (p.Lys542Asn), and c.1940T>G (p.Leu647Arg), all mapping in the C-terminal globular tail which is common to prelamin A and lamin C.30 In addition, compound heterozygous LMNA mutations have been identified, leading to atypical MADA phenotypes.30
MADB (OMIM #608612) results from biallelic mutations in ZMPSTE24,15 and the clinical signs and symptoms can develop by the age of 2 years, defining a more severe phenotype than MADA.30 Indeed, in addition to the signs observed in MADA, generalized lipodystrophy and a more pronounced accelerated ageing are distinctive of MADB.30 In addition to null ZMPSTE24 mutations, various missense ZMPSTE24 mutations resulting in a residual partial activity have so far been described in patients with typical MADB or atypical (severe) MADB phenotypes, including c.281T>C (p.Leu94Pro), c.743C>T (p.Pro248Leu), c.794A>G (p.Asn265Ser), c.1018T>C (p.Trp340Arg), and c.1196A>G (p.Tyr399Cys).17,30 MADB patients frequently carry one of these missense mutations on one allele and a null ZMPSTE24 mutation on the other allele, including the common c.1085dupT mutation, or less frequent frame-shift (c.207_208delCT) or nonsense mutations (c.121C>T, c.1349G>A).17,30 However, a homozygous ZMPSTE24 c.281T>C patient with MADB and congenital myopathy has been described,32 and more recently the screening of a cohort of eight related MADB patients from Suriname revealed that they all resulted homozygous for the c.1196A>G mutation, indicative of a common ancestor.33 ZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin A cleavage activity and/or protein stability, and the resulting disease severity is inversely correlated to the residual protein activity conferred by the mutation.14,34 Indeed, MADB fibroblasts exhibit a significant reduction in lifespan associated with major abnormalities of the nuclear shape and structure.32 Similarly, accumulation of the lamin A precursor protein, a marked alteration of the nuclear architecture, and chromatin disorganization that become more severe in older patients have been observed in cells from MADA patients.35
Néstor-Guillermo Progeria Syndrome
NGPS (OMIM #614008) is a rare autosomal recessive disorder so far described in 3 individuals worldwide.18,19,36 In 2011, the clinical features of the first two identified patients have been described.18,19 The two patients were from unrelated Spanish families and both exhibited normal development until 2 years of age, but from that age onwards they experienced failure to thrive and featured signs of HGPS and/or MAD, including aged appearance, growth retardation, decreased subcutaneous fat, thin limbs, and stiff joints. However, their long survival respect to other childhood-progerias (patients were still alive at 24 and 32 years, respectively), their height of 145 cm or more (respect to an average final height of 110 cm in classic HGPS), the presence of eyebrows and eyelashes, the persistence of scalp hair until the second decade of life, a very severe osteolysis, and the absence of signs of cardiovascular impairment or metabolic complications led to the diagnosis of a novel progeria, that was confirmed at the molecular level by absence of mutations in LMNA or ZMSPTE24 genes, and by the finding of a pathogenic recessive mutation in the BANF1 gene (c.34G>A; p.Ala12Thr).18,19 Recently, a Latin American girl aged 2 years and 8 months, with short stature, poor weight gain, sparse hair, and dysmorphic facial features reminiscent of premature ageing was described, and the genetic screening confirmed the presence of the recessive BANF1 p.Ala12Thr mutation, leading to the third molecularly confirmed diagnosis of NGPS.36 Interestingly, signs of growth retardation in this girl have been noticed earlier than in the two previously described cases, as they started by one year of age.36 BANF1 encodes the BAF protein that anchors chromatin to the nuclear lamina and is essential for the assembly and disassembly of the nuclear envelope during mitosis.20,21 Functional studies on fibroblasts from NGPS patients revealed that the p.Ala12Thr mutation impairs protein stability and results in profound abnormalities in the nuclear lamina.18 However, a subsequent study suggested that rather than altering protein stability, the BANF1 p.Ala12Thr mutation disrupts BAF ability to bind to the DNA resulting in impaired anchoring of chromatin to the nuclear lamina and disruption of the nuclear envelope.37 It was also demonstrated that BAF controls the DNA damage response to oxidative stress via regulation of the DNA repair protein poly [ADP-ribose] polymerase 1 (PARP1), and that cells from NGPS patients have defective PARP1 activity and impaired repair of oxidative DNA lesions.38
Mandibuloacral Dysplasia Resulting from Mutations of POLD1 and MTX2 Genes
In addition to the two major forms of mandibuloacral dysplasia resulting from either LMNA (MADA) or ZMPSTE mutations (MADB), MAD is also a feature of other SPS resembling features of atypical progeroid laminopathies but originating from mutations of genes involved in other pathways.39,40 These include an autosomal dominant form of MAD termed mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) syndrome and caused by mutations in the POLD1 gene encoding the catalytic subunit of DNA polymerase delta,39 and a recently identified severe premature ageing syndrome with clinical features resembling HGPS termed MADaM (mandibuloacral dysplasia associated to MTX2), resulting from homozygous recessive mutations in the MTX2 gene encoding metaxin-2, an outer mitochondrial membrane protein.40
Mandibular Hypoplasia, Deafness, Progeroid Features, and Lipodystrophy Syndrome
MDPL (OMIM #615381) is an autosomal dominant systemic disorder characterized by prominent loss of subcutaneous fat, a characteristic facial appearance and metabolic abnormalities including insulin resistance and diabetes mellitus, sensorineural deafness occurring during the first or second decade of life, and hypogonadism in males.39,41 The affected patients show signs resembling atypical progeroid syndromes, such as mandibular hypoplasia, a beaked nose with bird-like facies, prominent eyes, crowded teeth, a high-pitched voice, growth retardation, joint contractures and skeletal anomalies, as well as muscle and skin atrophy.39,41 These features were reminiscent of MAD, but the absence of LMNA or ZMPSTE24 mutations in the patients coupled to the presence of distinct characteristics, such as sensorineural hearing loss and absence of clavicular hypoplasia and acroosteolysis suggested a novel syndrome whose molecular basis was elucidated in 2013.39,41 Indeed, a recurrent de-novo in-frame mutation in the POLD1 gene (c.1812_1814delCTC, p.Ser605del) was identified as causative of this new progeroid disorder featuring mandibular hypoplasia, deafness and generalized lipodystrophy.39 POLD1 encodes for the catalytic subunit of DNA polymerase delta, a key enzyme responsible for lagging strand DNA synthesis during DNA replication. It possesses both 5ʹ to 3ʹ polymerase and 3ʹ to 5ʹ exonuclease activity, this latter enabling proofreading during DNA replication, and the common p.Ser605del mutation causing MDPL results in the deletion of a single amino acid at the active site leading to impaired polymerase activity.39 Less than 30 patients have so far been described as having POLD1-related MDPL syndrome, and most of them (almost 80%) carry the common p.Ser605del mutation.42–51 Less frequent POLD1 mutations observed in the patients include the p.Arg507Cys one, observed in three patients and affecting the C-terminal part of the exonuclease domain, the p.Ile1070Asn mutation observed in one patient and predicted to impair the zinc finger 2 domain of the protein, and another likely pathogenic mutation in the same domain (p.Glu1067Lys) observed in two related patients with a milder phenotype.42–51 Interestingly, MDPL fibroblasts carrying the recurrent POLD1 p.Ser605del mutation showed nuclear and cellular alterations resembling features observed in other progeroid syndromes, including severe nuclear envelope anomalies, accumulation of prelamin A, altered cell growth, cellular senescence, impaired ability to repair DNA double-strand breaks (DSBs), presence of micronuclei, and an increased rate of telomere shortening, suggesting that the ageing phenotype of MDPL syndrome is associated with an impaired DNA repair capacity.52
Mandibuloacral Dysplasia Associated to MTX2
MADaM (OMIM #619127) has been recently described in seven patients from five families originating from India, Turkey, Algeria, Egypt, and Ecuador, and diagnosed as a severe progeroid form of MAD with clinical features resembling HGPS, including small viscerocranium with mandibular underdevelopment, growth retardation, lipodystrophy, altered skin pigmentation, distal acro-osteolyses, renal focal glomerulosclerosis, and severe cardiovascular disease, but all showing normal cognitive development.40 Patients were first screened for pathogenic mutations in the LMNA, ZMPSTE24, BANF1, and POLD1 genes, but none was found.40 Subsequent NGS approaches in affected members of these families revealed five pathogenic recessive mutations in MTX2, and particularly a c.2T>A missense mutation (p.Met1Lys) in the first family, a c.544–1G>C base substitution within the splice acceptor site in intron 8 in the second family and a c.208+3_208+6del within the splice donor site in intron 4 in the third family, both leading to altered mRNAs resulting in premature termination codons in the protein (respectively, p.Val182Argfs*3 and p.Ala46Valfs*12), a frame-shift mutation in exon 9 in the fourth family (c.603del; p.Tyr202Ilefs*26), and a homozygous 2-bp deletion in exon 6 (c.294_295delTC) resulting in a nonsense mutation (p.Leu99*) in the fifth family. Owing to the clinical and molecular similarities among the affected members, the disease was named MADaM, mandibuloacral dysplasia associated with MTX2.40 MTX encodes metaxin-2, an outer mitochondrial membrane protein involved in protein translocation into mitochondria, in TNF-α-induced apoptosis, as well as in interactions with other mitochondrial proteins responsible for shaping and stabilizing cristae junctions.40,53 Functional studies on primary fibroblasts from MADaM patients revealed that loss of MTX2 resulted in loss of its partner protein metaxin-1 and mitochondrial dysfunction, including mitochondrial network fragmentation, decreased oxidative phosphorylation, resistance to induced apoptosis, increased cell senescence and mitophagy, and reduced proliferation. In addition, secondary nuclear defects, including blebs and herniations resembling those described in HGPS and other progeroid laminopathies, were observed in both MTX2-mutant fibroblasts and mtx-2-depleted C. elegans. However, prelamin A or progerin accumulation was absent in MADaM fibroblasts.40 Therefore, the authors concluded that the striking clinical resemblance among HGPS and MADaM patients is likely related to commonly altered signal transduction pathways, activated upon either nuclear or mitochondrial primary defects and reciprocally impacting each other.40 Indeed, a marked downregulation of mitochondrial oxidative phosphorylation proteins accompanied by mitochondrial dysfunction has been documented in fibroblasts from HGPS patients.54 Similarly, mitochondrial dysfunction and increased production of reactive oxygen species (ROS) have been observed in human fibroblasts subjected to knockdown of LMNA and ZMPSTE24, respectively.55
Werner Syndrome: The Prototypic Adult-Onset Progeria
It is estimated that the genome of every single cell in our body is subjected to a daily average of almost one million lesions that arise from exposure to endogenous and exogenous DNA damaging agents as well as from DNA replication errors.56 These lesions include abasic sites, single-strand breaks (SSBs), double-strand breaks (DSBs), small insertions and deletions, DNA mismatches, DNA adducts, and DNA-protein crosslinks that, if not properly repaired, can drive mutagenesis and/or interfere with DNA transcription and replication.56 Indeed, mammalian cells possess a battery of DNA repair enzymes that work in different pathways to counteract the daily amount of DNA damage.56 The major DNA repair pathways include: base excision repair (BER) that removes DNA bases modified by oxidation, alkylation, and deamination; nucleotide excision repair (NER) that repairs UV photoproducts, DNA crosslinks, and bulky lesions; mismatch repair (MMR) that corrects replication errors; homologous recombination (HR) and non-homologous end-joining (NHEJ) that repair DSBs.56 The association between DNA damage and ageing is documented by extensive evidence in humans showing increased markers of genome instability with advancing age, as well as in age-related diseases, such as cancer, neurodegenerative and cardiovascular diseases, partially due to the fact that the DNA repair capacity may decrease with advancing age.56,57 Indeed, failure to repair DNA damage can lead to mutations transforming normal cells into cancerous ones, can induce a non-proliferative state termed cellular senescence or result in apoptotic cell death.57
Mutations in genes coding for proteins involved in DNA repair and maintenance of genome stability result in different premature-ageing syndromes that are often characterized by increased risk for certain cancers.56,57 WS is one of these disorders and represents the prototypic adult-onset SPS as the affected individuals undergo multiple features of accelerated ageing.58 Indeed, WS is often termed “adult progeria” to distinguish it from HGPS, the “childhood progeria”.5 The disease is an autosomal dominant SPS resulting from null mutations of the WRN gene, coding for a protein (WRN) that plays a critical role in repairing damaged DNA.59 In particular, WRN is a member of the RecQ family of helicases and is regarded as a “caretaker of the genome” allowing the maintenance of genome stability during DNA replication, recombination, repair, and transcription. In addition, WRN is required for telomere maintenance and apoptosis.60 Recessive WRN mutations account for classical WS. However, atypical forms of the disease (atypical WS) are reported, with patients showing no WRN mutations and often an early onset of the symptoms and an accelerated disease progression.61
Classical Werner Syndrome
Classical WS (OMIM #277700) is an autosomal recessive disorder resulting from homozygous or compound heterozygous loss of function mutations in the WRN gene.59,61 Founder mutations leading to genetic clusters have been identified in Japan (disease prevalence ranging from 1 in 20,000 to 1 in 40,000) and in Sardinia (disease prevalence of 1 in 50,000). The disease prevalence in other populations is less clear, but estimated at 1 or a few cases per million individuals.62
WS individuals develop normally during the first decade of life, and the lack of a pubertal growth spurt in teenage years is the first clinical sign, leading to an average final height of 142 cm and a final weight of about 36 Kg. In their 20s WS individuals begin to manifest graying or loss of hair and scleroderma-like skin lesions, followed by bilateral ocular cataracts, type 2 diabetes mellitus, hypogonadism, skin ulcers, and osteoporosis in their 30s. An aged face with beaked nose is common, and fertility is reduced in both genders, albeit they can reproduce.23 In contrast to HGPS individuals, WS individuals show an unusual spectrum of cancers including soft tissue sarcomas, thyroid carcinomas, malignant melanomas, meningiomas, hematologic/lymphoid cancers, osteosarcomas, and other less frequent cancers that often manifest with early age of onset and with the presence of multiple tumors in the same individual.5,63 A few cases of dementia or peripheral neuropathy have been reported in WS subjects, but there is no clear evidence of increased risk for neurodegeneration.5 WS patients usually die at a mean age of 53–54 years, and the primary causes of death are complications resulting from cancer or myocardial infarction resulting from coronary artery atherosclerosis.62
The gene causing WS was identified in 1996 by positional cloning and codes for the WRN protein, a member of the RecQ DNA helicase family with multiple nuclear functions, including DNA dependent ATPase, 3′ → 5′ helicase, 3′ → 5′ exonuclease, and DNA strand annealing activities.59 WRN is a long gene consisting of 35 exons, of which 34 are coding for a protein of 1432 amino acids. More than 80 different pathogenic WRN mutations have been identified in WS patients, mainly resulting in protein truncation with the elimination of the nuclear localization signal at the C-terminus of the protein, and including nonsense mutations, small insertions and deletions leading to frame-shift, and splice site mutations leading to exon skipping.64 Among them, founder mutations are reported in Japanese (c.3139–1G>C, resulting in skipping of exon 26) and Sardinian patients (c.2089–3024A>G, creating a novel exon between exons 18 and 19 and introducing a premature stop codon).65,66 The c.1105C>T mutation creating a stop codon in exon 9 (p.Arg369*) is the most common mutation in non-Japanese populations and the second most common one in Japan (after the c.3139–1G>C one), accounting for almost 18.6% and 15.9% of the cases, respectively, and suggesting that it could represent a mutational hot spot across ethnic groups.64 Other ethnic-specific and potentially founder mutations have been described in Moroccan (c.2179dupT, p.Cys727fs), Dutch (c.3590delA, p.Asn1197fs), Turkish (c.3460–2A>G, resulting in skipping of exon 30), and Asian (c.561A>G, creating a cryptic splice site followed by a frame-shift: p.Lys187fs) populations.67,68 Also, a few missense WRN mutations have been observed in WS patients, mainly regarded as null mutations causing protein instability (p.Lys125Asn and Lys135Glu) or a significant decrease in protein activity (p.Gly574Arg and p.Arg637Trp).67,69,70 Furthermore, deep intronic mutations creating novel exons and three large rearrangements leading to multi-exon deletions and duplications have also been detected in a few patients.64,67 A complete and updated list of WRN mutations can be found at the Werner Syndrome Mutational Database (http://www.pathology.washington.edu/research/werner/database/).71 The WRN protein allows maintenance of genome stability during DNA replication, recombination, repair, and transcription, and is required for telomere maintenance and apoptosis.60 Most of the mutations observed in WS patients result in the absence of a functional WRN protein in nuclei, and cells from WS patients display impaired DNA replication, chromosomal instability, telomere shortening, and premature senescence with cell passing in culture.72
Atypical Werner Syndrome
The International Registry of Werner Syndrome (www.wernersyndrome.org) is located at the Department of Pathology of the University of Washington (Seattle, WA, USA) and has been recruiting patients suspected of having WS since 1988.73 Patients with progeroid signs resembling WS who have been referred to the Registry, but who do not carry WRN mutations and often feature early onset of the symptoms and accelerated progression, have been operationally categorized as “atypical Werner syndrome”.64 Subsequent genetic screening in those patients revealed mutations in LMNA leading to atypical progeroid syndromes in some individuals, as detailed in the previous sections of this review.5,74 Other patients originally diagnosed with WS on clinical grounds were subsequently found to carry POLD1 mutations and referred to as MDLP cases.44 Moreover, other individuals referred to the International Registry of Werner Syndrome and showing a segmental progeroid syndrome characterized by genomic instability and susceptibility toward early onset hepatocellular carcinoma, were found to carry pathogenic biallelic germline mutations in SPRTN, a gene coding for a protein required for DNA replication and replication-related G2/M-checkpoint regulation.75 The study was performed on three patients from two unrelated families presenting with early onset hepatocellular carcinoma, genomic instability and progeroid features. A homozygous 1-bp deletion (c.721delA) predicted to introduce a premature stop codon at amino acid 249 (p.Lys241AsnfsX8) was detected in the first family, whilst the screened member of the second family was compound heterozygous for a c.350A>G missense mutation (p.Tyr117Cys), and a 4-bp deletion (c.717_718+2delAGGT) that predominantly caused intron inclusion, inducing a premature stop codon at amino acid 246 (p.Lys239LysfsX7).75 The analysis of SPRTN in 48 additional patients with suspected WS who were negative for mutations in the WRN or LMNA genes revealed no other SPRTN mutations.75 This study revealed that recessive mutations in SPRTN cause a novel progeroid syndrome termed Ruijs-Aalfs syndrome (OMIM #616200). Cells derived from patients with Ruijs-Aalfs syndrome elicit genomic instability, and it was found that SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks.76 Another patient referred to the International Registry of Werner Syndrome presented with an aged appearance with prematurely gray hair and scleroderma-like skin lesions, spastic paraplegia, and apparent disability.77 Exome sequencing showed a homozygous c.626–1G>C mutation in intron 5 and leading to aberrant splicing of exon 6 of the SAMHD1 gene, a gene which encodes a triphosphohydrolase involved in the regulation of intracellular dNTP pools.77
Other Premature-Ageing Syndromes
In the previous sections, the author provides an updated overview of the genetic heterogeneity underlying some of the best-characterized progeroid syndromes, resulting in a spectrum of more or less severe disorders characterized by both distinctive and overlapping symptoms that range in age at onset from early life to adulthood, and including both classical and atypical forms (Table 1). In addition to these syndromes, many other premature-ageing disorders have been observed in humans, whose main symptoms, genetic heterogeneity and molecular mechanisms cannot be discussed in detail due to space limitations, but that will be briefly described in this section.2–4 Several of these syndromes result from mutations in genes required for DNA repair and maintenance of genomic stability.78–88 Examples include Bloom syndrome and Rothmund-Thomson syndrome resulting from mutations of RecQ helicase family members, and particularly from BLM (RECQL3) and RECQL4 genes, respectively.78–81 Similarly, mutations in genes coding for proteins required for the NER pathway cause distinct genetic photodermatoses characterized by neurological symptoms and premature onset of pathologies that overlap with those associated with old age in humans, including Cockayne’s syndrome, xeroderma pigmentosum, and trichothiodystrophy.82–85 Wiedemann-Rautenstrauch syndrome, is a neonatal progeroid syndrome characterized by severe pre- and postnatal growth retardation, facial dysmorphism, and generalized lipodystrophy with local fatty tissue accumulations, resulting from recessive mutations of the POLR3A gene encoding a subunit of the DNA-dependent RNA polymerase III.86 Ataxia-telangiectasia results from biallelic mutations in the ATM gene, coding a protein involved in DNA damage response and telomere maintenance.87,88 Moreover, dyskeratosis congenita is a genetically heterogeneous disorder showing autosomal recessive, autosomal dominant, and X-linked inheritance, and resulting from mutations of several genes encoding telomerase components leading to a disease characterized by premature telomere shortening, which manifests clinically as the classic triad of reticulate hyperpigmentation, nail dystrophy and leukoplakia, and with increased risk of cancer and other potentially lethal complications, such as bone marrow failure, lung and liver diseases.3,89
Other progeroid syndromes result from impairments in mitochondrial pathways.90,91 For example, Fontaine progeroid syndrome is a fatal neonatal disorder characterized by sparse hair, lipodystrophy, thin skin, osteoporosis, and growth retardation, resulting from de novo missense mutations in SLC25A24, coding for the calcium-binding mitochondrial carrier protein SCaMC-1, and leading to mitochondrial dysfunction in the affected patients.90 Similarly, PYCR-related cutis laxa syndrome is an autosomal recessive disorder characterized by excessive congenital skin wrinkling, a large fontanelle with delayed closure, a typical progeroid appearance, and varying degrees of growth and developmental delay, caused by biallelic mutations of the PYCR1 gene, coding for the mitochondrial protein pyrroline-5-carboxylate reductase 1 that catalyzes the last step in proline biosynthesis.91
Additional progeroid syndromes resulting from mutations in genes not involved in nuclear envelope architecture, DNA repair or mitochondrial pathways are also described.92–94 Examples include marfanoid-progeroid-lipodystrophy syndrome, Penttinen syndrome, SHORT syndrome, and others.92–94 Marfanoid-progeroid-lipodystrophy syndrome is characterized by a neonatal progeroid appearance associated with facial dysmorphism, congenital lack of subcutaneous fat and incomplete signs of Marfan syndrome, resulting from mutations in the FBN1 gene coding for fibrillin-1, a component of the extracellular matrix of connective tissue.92 Penttinen syndrome is characterized by a prematurely aged appearance with lipoatrophy, epidermal and dermal atrophy along with hypertrophic lesions that resemble scars, thin hair, proptosis, underdeveloped cheekbones, and marked acro-osteolysis, resulting from mutations in the PDGFRB gene, which encodes the platelet-derived growth factor receptor β.93 SHORT syndrome is characterized by short stature, hyperextensibility of joints and/or inguinal hernia, ocular depression, Rieger anomaly (a defective development of the eye that often leads to glaucoma), and teething delay, and results from mutations of the PIK3R1 gene that encodes a regulatory subunit of phosphatidyl inositol-3 kinase, involved in cell proliferation and survival.94
Patient Management and Therapeutic Approaches
Unfortunately, for most SPS, no cure is yet available, and the primary strategy for patient management is to prevent or treat signs and symptoms as early as possible in order to improve survival and quality of life.2 For example, a complete management guide for HGPS is available from the Progeria Research Foundation, and includes recommendations for both families and health care professionals.6,24 HGPS children are at high risk for heart attack and stroke at any age, so that annual visit with a pediatric cardiologist is recommended, and low-dose aspirin should be considered for all children with HGPS for both stroke and heart attack prevention. It is also recommended that HGPS children undergo magnetic resonance imaging (MRI) of the brain, and MR angiography (MRA) of the major arteries in the brain and neck to determine the presence of arterial narrowing and stroke annually.24 In addition, high-calorie diet, exercise and hydrotherapy are recommended to counteract atherosclerosis, body fat reduction and muscular atrophy, and use of sunscreens, eye care, shoe pads, and hearing aids are recommended for daily activities.6,24 Similarly, symptomatic treatments for WS include cholesterol-lowering drugs in case of abnormal lipid profile, treatment of skin ulcers, surgical treatment of cataracts, control of type 2 diabetes mellitus, and standard treatment of tumors. Moreover, dietary/physical regimens and smoking avoidance are recommended to reduce atherosclerosis risk.5,62 Also, the treatment of atypical progeroid syndromes is directed toward the specific symptoms that are treated with standard therapies, including treatment of diabetes, if present, and use of cholesterol-lowering drugs, and may require the coordinated efforts of different specialists.95 Therefore, an early molecular diagnosis in specialized centers prevents a long diagnostic journey and allows to establish a proper management.2
The discovery of the genetic defects underlying SPS, such as HGPS, WS and progeroid laminopathies, and the molecular characterization of the patients have led to the availability and/or development of several cell culture and animal models of these diseases, that have been used to investigate their pathogenetic mechanisms as well as for testing several therapeutic compounds.23 Among the various therapeutic options investigated, the farnesyltransferase inhibitor (FTI) lonafarnib is the first and only known drug treatment receiving US Food and Drug Administration (FDA) approval (November, 2020) to extend lifespan in children with HGPS and progeroid laminopathies.96 Another trial tested whether the combination of lonafarnib with two drugs that inhibit the formation of the farnesyl group, namely pravastatin and zoledronic acid, was more effective than lonafarnib alone.97 However, the trial results showed that the three-drug combination was equally effective than giving lonafarnib on its own.97 Therefore, the three-drug combination is not currently recommended as a replacement therapy for lonafarnib.24 Treatment of HGPS fibroblasts with the mTOR inhibitor rapamycin, or with the rapamycin analog everolimus, reversed the cellular phenotype of HGPS fibroblasts, showing that the mutant protein progerin was cleared through autophagic mechanisms.98 Similarly, everolimus treatment increased proliferative ability, reduced nuclear blebbing and delayed senescence in several cell lines from patients with HGPS, atypical WS and other laminopathies.99 A clinical trial that administers everolimus plus lonafarnib to children with progeria is ongoing, and results of this trial are expected in 2023.24
Gene Editing Approaches
Gene editing approaches based on CRISPR/Cas9-based technologies showed promising results in HGPS cells and animal models, suggesting that in the future these approaches could represent novel options for the treatment of progeroid diseases.100,101 Specifically, gene editing is performed using a Cas9 endonuclease and a guide RNA that directs Cas9 to the target DNA region of interest, where it introduces cuts into the DNA strands allowing for the removal of the existing DNA and the insertion of replacement DNA.100,101 Using this technology, it was shown that CRISPR/Cas9-mediated lamin A/progerin reduction, attenuated weight loss, extended lifespan, and improved health status in HGPS mice.101 Similarly, Santiago-Fernández et al developed a CRISPR/Cas9-based approach to introduce frameshift mutations in the LMNA gene and showed that it led to a reduction in progerin and lamin A protein levels in HGPS fibroblasts, and reduced progerin expression, extended lifespan, and improved health status in HGPS mice.100 These studies showed the preclinical efficacy of gene editing in animal models of progeria, suggesting a possible use of this technology in humans in the near future.100,101 However, the putative off-target events and adverse effects of the Cas9 nuclease, including the generation of random mutations, the induction of severe immune responses against the CRISPR–Cas9 system in the treated patients, and the risk of also disrupting the wild-type LMNA allele, pose challenges to the clinical translation of gene disruption strategies for the treatment of progeria, suggesting that further investigation is required to ensure the safety of these approaches.100,101
More recently, a base editing approach has been proposed as a possible treatment for HGPS.102 Base editing is a CRISPR-Cas9-based genome editing technology that allows the introduction of precise point mutations in the DNA without generating double-strand breaks.103 DNA base editors comprise fusions between a catalytically impaired Cas nuclease and a base-modification enzyme that operates on single-stranded DNA.103 Two classes of DNA base editors have been described: cytosine base editors (CBEs) that convert a C•G base pair into a T•A base pair, and adenine base editors (ABEs) that convert an A•T base pair to a G•C base pair.103 ABE-mediated correction of the LMNA c.1824C>T mutation in fibroblasts derived from children with HGPS reduced the abundance of progerin protein and restored normal nuclear morphology.102 Furthermore, when delivered into a mouse model of human progeria, the ABE corrected the LMNA c.1824C>T allele in various tissues, reduced the vascular pathology, and increased the median lifespan of the animals.102 However, albeit base editing approaches hold the potential as possible treatment options for HGPS and other SPS, further investigation of potential immune responses to treatment and optimization of delivery strategies are required prior to translating these findings into the clinic.102
Discussion
The advent of rapid DNA sequencing methods has greatly accelerated the capability to uncover the genetic basis of several progeroid syndromes, leading to a rapid increase in the number of identified causative genes in recent years, and revealing a highly clinical and genetic heterogeneity underlying these conditions that often show overlapping signs and symptoms2–5 Therefore, an expert clinical evaluation coupled to NGS of panels of genes involved in premature-ageing disorders, rather than single-gene screening, is currently recommended for molecular diagnosis in specialized centers, and requires an interdisciplinary collaboration among several specialists in order to establish as early as possible an accurate clinical and molecular diagnosis for a proper patient management. More comprehensive genomic testing including whole-exome sequencing and whole-genome sequencing may be considered if single-gene testing or use of a multi-gene panel fails to confirm a diagnosis in an individual with progeroid features.2–4 For example, a large panel of 82 genes involved in these syndromes has been recently applied for the screening of 66 patients with a clinical suspicion of premature-ageing disorders, allowing to confirm or establish a final diagnosis in 26% of the cases, so that authors have suggested that it could represent a valid first-level analysis before whole-genome sequencing approaches.4
In addition, the identification of the genetic cause of several progeroid syndromes has led to the development of animal models of these disorders that have been increasingly used to investigate potential therapeutic strategies.23 Moreover, the characterization of the mutations underlying these conditions poses the basis for future gene editing and base editing approaches.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Coppedè F. Premature aging syndrome. Adv Exp Med Biol. 2012;724:317–331.
2. Lessel D, Kubisch C. Hereditary syndromes with signs of premature aging. Dtsch Arztebl Int. 2019;116(29–30):489–496. doi:10.3238/arztebl.2019.0489
3. Schnabel F, Kornak U, Wollnik B. Premature aging disorders: a clinical and genetic compendium. Clin Genet. 2021;99(1):3–28. doi:10.1111/cge.13837
4. Grelet M, Blanck V, Sigaudy S, et al. Outcomes of 4 years of molecular genetic diagnosis on a panel of genes involved in premature aging syndromes, including laminopathies and related disorders. Orphanet J Rare Dis. 2019;14(1):288. doi:10.1186/s13023-019-1189-z
5. Coppedè F. The epidemiology of premature aging and associated comorbidities. Clin Interv Aging. 2013;8:1023–1032. doi:10.2147/CIA.S37213
6. Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford progeria syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® (Internet). Seattle: University of Washington; 2019. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK1121/#top.
7. Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi:10.1038/nature01629
8. De Sandre-giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. doi:10.1126/science.1084125
9. Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci USA. 2004;101(24):8963–8968. doi:10.1073/pnas.0402943101
10. Arancio W, Pizzolanti G, Genovese SI, Pitrone M, Giordano C. Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review. Gerontology. 2014;60(3):197–203. doi:10.1159/000357206
11. Köhler F, Bormann F, Raddatz G, et al. Epigenetic deregulation of lamina-associated domains in Hutchinson-Gilford progeria syndrome. Genome Med. 2020;12(1):46. doi:10.1186/s13073-020-00749-y
12. Gonzalo S, Kreienkamp R. DNA repair defects and genome instability in Hutchinson-Gilford progeria syndrome. Curr Opin Cell Biol. 2015;34:75–83. doi:10.1016/j.ceb.2015.05.007
13. The UMD-LMNA mutations database; January 31, 2020. Available from: https://www.umd.be/LMNA/.
14. Barrowman J, Wiley PA, Hudon-Miller SE, Hrycyna CA, Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum Mol Genet. 2012;21(18):4084–4093. doi:10.1093/hmg/dds233
15. Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12(16):1995–2001. doi:10.1093/hmg/ddg213
16. Navarro CL, De Sandre-giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13(20):2493–2503. doi:10.1093/hmg/ddh265
17. Navarro CL, Esteves-Vieira V, Courrier S, et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur J Hum Genet. 2014;22(8):1002–1011. doi:10.1038/ejhg.2013.258
18. Puente XS, Quesada V, Osorio FG, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88(5):650–656. doi:10.1016/j.ajhg.2011.04.010
19. Cabanillas R, Cadiñanos J, Villameytide JA, et al. Néstor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A(11):2617–2625. doi:10.1002/ajmg.a.34249
20. Torras-Llort M, Medina-Giró S, Escudero-Ferruz P, et al. A fraction of barrier-to-autointegration factor (BAF) associates with centromeres and controls mitosis progression. Commun Biol. 2020;3(1):454. doi:10.1038/s42003-020-01182-y
21. Romero-Bueno R, de la Cruz Ruiz P, Artal-Sanz M, Askjaer P, Dobrzynska A. Nuclear organization in stress and aging. Cells. 2019;8(7):664. doi:10.3390/cells8070664
22. Ullrich NJ, Gordon LB. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol. 2015;132:249–264.
23. Coppedè F. Premature aging syndrome. In: Ram JL, Conn M, editors. Conn’s Handbook of Models for Human Aging.
24. The progeria research foundation. MA, USA: Peabody. Available from: https://www.progeriaresearch.org/.
25. Kandhaya-Pillai R, Hisama FM, Bucks SA, et al. Novel LMNA mutations in Greek and Myanmar patients with progeroid features and cardiac manifestations. Aging Pathobiol Ther. 2020;2(2):101–105. doi:10.31491/APT.2020.06.021
26. Starke S, Meinke P, Camozzi D, et al. Progeroid laminopathy with restrictive dermopathy-like features caused by an isodisomic LMNA mutation p.R435C. Aging (Albany NY). 2013;5(6):445–459. doi:10.18632/aging.100566
27. Navarro CL, Cadiñanos J, De Sandre-giovannoli A, et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005;14(11):1503–1513. doi:10.1093/hmg/ddi159
28. Moulson CL, Go G, Gardner JM, et al. Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J Invest Dermatol. 2005;125(5):913–919. doi:10.1111/j.0022-202X.2005.23846.x
29. Denecke J, Brune T, Feldhaus T, et al. A homozygous ZMPSTE24 null mutation in combination with a heterozygous mutation in the LMNA gene causes Hutchinson-Gilford progeria syndrome (HGPS): insights into the pathophysiology of HGPS. Hum Mutat. 2006;27(6):524–531. doi:10.1002/humu.20315
30. Cenni V, D’Apice MR, Garagnani P, et al. Mandibuloacral dysplasia: a premature ageing disease with aspects of physiological ageing. Ageing Res Rev. 2018;42:1–13. doi:10.1016/j.arr.2017.12.001
31. Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426–431. doi:10.1086/341908
32. Ben Yaou R, Navarro C, Quijano-Roy S, et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur J Hum Genet. 2011;19(6):647–654. doi:10.1038/ejhg.2010.256
33. Hitzert MM, van der Crabben SN, Baldewsingh G, et al. Mandibuloacral dysplasia type B (MADB): a cohort of eight patients from suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines. Orphanet J Rare Dis. 2019;14(1):294. doi:10.1186/s13023-019-1269-0
34. Spear ED, Hsu ET, Nie L, Carpenter EP, Hrycyna CA, Michaelis S. ZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin A cleavage activity and/or protein stability. Dis Model Mech. 2018;11(7):dmm033670. doi:10.1242/dmm.033670
35. Filesi I, Gullotta F, Lattanzi G, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23(2):150–158. doi:10.1152/physiolgenomics.00060.2005
36. Fisher HG, Patni N, Scheuerle AE. An additional case of Nestor-Guillermo progeria syndrome diagnosed in early childhood. Am J Med Genet A. 2020;182(10):2399–2402. doi:10.1002/ajmg.a.61777
37. Paquet N, Box JK, Ashton NW, et al. Nestor-Guillermo progeria syndrome: a biochemical insight into barrier-to-autointegration factor 1, alanine 12 threonine mutation. BMC Mol Biol. 2014;15(1):27. doi:10.1186/s12867-014-0027-z
38. Bolderson E, Burgess JT, Li J, et al. Barrier-to-autointegration factor 1 (Banf1) regulates poly [ADP-ribose] polymerase 1 (PARP1) activity following oxidative DNA damage. Nat Commun. 2019;10(1):5501. doi:10.1038/s41467-019-13167-5
39. Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947–950. doi:10.1038/ng.2670
40. Elouej S, Harhouri K, Le Mao M, et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat Commun. 2020;11(1):4589. doi:10.1038/s41467-020-18146-9
41. Shastry S, Simha V, Godbole K, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95(10):E192–197. doi:10.1210/jc.2010-0419
42. Pelosini C, Martinelli S, Ceccarini G, et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism. 2014;63(11):1385–1389. doi:10.1016/j.metabol.2014.07.010
43. Reinier F, Zoledziewska M, Hanna D. Mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome in the context of inherited lipodystrophies. Metabolism. 2015;64(11):1530–1540. doi:10.1016/j.metabol.2015.07.022
44. Lessel D, Hisama FM, Szakszon K, et al. POLD1 germline mutations in patients initially diagnosed with Werner syndrome. Hum Mutat. 2015;36(11):1070–1079. doi:10.1002/humu.22833
45. Okada A, Kohmoto T, Naruto T, et al. The first Japanese patient with mandibular hypoplasia, deafness, progeroid features and lipodystrophy diagnosed via POLD1 mutation detection. Hum Genome Var. 2017;4(1):17031. doi:10.1038/hgv.2017.31
46. Ajluni N, Meral R, Neidert AH, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf). 2017;86(5):698–707. doi:10.1111/cen.13311
47. Elouej S, Beleza-Meireles A, Caswell R, et al. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (MDPL). Metabolism. 2017;71:213–225. doi:10.1016/j.metabol.2017.03.011
48. Sasaki H, Yanagi K, Ugi S, et al. Definitive diagnosis of mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome caused by a recurrent de novo mutation in the POLD1 gene. Endocr J. 2018;65(2):227–238. doi:10.1507/endocrj.EJ17-0287
49. Wang LR, Radonjic A, Dilliott AA, McIntyre AD, Hegele RA. A de novo POLD1 mutation associated with mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome in a family with Werner syndrome. J Investig Med High Impact Case Rep. 2018;6:2324709618786770. doi:10.1177/2324709618786770
50. Wang XW, Lu LY, Xie Y, Yu XJ. A Chinese girl with mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) diagnosed via POLD1 mutation detection. Chin Med J (Engl). 2020;133(16):2009–2011. doi:10.1097/CM9.0000000000000986
51. Yu PT, Luk HM, Mok MT, Lo FI. Evolving clinical manifestations of mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome: from infancy to adulthood in a 31-year-old woman. Am J Med Genet A. 2021;185(3):995–998. doi:10.1002/ajmg.a.62035
52. Murdocca M, Spitalieri P, De Masi C, et al. Functional analysis of POLD1 p.ser605del variant: the aging phenotype of MDPL syndrome is associated with an impaired DNA repair capacity. Aging (Albany NY). 2021;13(4):4926–4945. doi:10.18632/aging.202680
53. Ono K, Wang X, Kim SO, Armstrong LC, Bornstein P, Han J. Metaxin deficiency alters mitochondrial membrane permeability and leads to resistance to TNF-induced cell killing. Protein Cell. 2010;1(2):161–173. doi:10.1007/s13238-010-0017-y
54. Rivera-Torres J, Acín-Perez R, Cabezas-Sánchez P, et al. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics. 2013;91:466–477. doi:10.1016/j.jprot.2013.08.008
55. Sieprath T, Corne TD, Nooteboom M, et al. Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus. 2015;6(3):236–246. doi:10.1080/19491034.2015.1050568
56. Coppedè F, Migliore L. DNA repair in premature aging disorders and neurodegeneration. Curr Aging Sci. 2010;3(1):3–19. doi:10.2174/1874609811003010003
57. Petr MA, Tulika T, Carmona-Marin LM, Scheibye-Knudsen M. Protecting the aging genome. Trends Cell Biol. 2020;30(2):117–132. doi:10.1016/j.tcb.2019.12.001
58. Hisama FM, Oshima J, Martin GM. How research on human progeroid and antigeroid syndromes can contribute to the longevity dividend initiative. Cold Spring Harb Perspect Med. 2016;6(4):a025882. doi:10.1101/cshperspect.a025882
59. Yu CE, Oshima J, Fu YH, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272(5259):258–262. doi:10.1126/science.272.5259.258
60. Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008;124(4):369–377. doi:10.1007/s00439-008-0562-0
61. Oshima J, Sidorova JM, Monnat RJ
62. Oshima J, Martin GM, Hisama FM. Werner syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® (Internet). Seattle: University of Washington; 2016. Available from: www.ncbi.nlm.nih.gov/books/NBK1514/.
63. Goto M, Ishikawa Y, Sugimoto M, Furuichi Y. Werner syndrome: a changing pattern of clinical manifestations in Japan (1917~2008). Biosci Trends. 2013;7(1):13–22.
64. Yokote K, Chanprasert S, Lee L. WRN mutation update: mutation spectrum, patient registries, and translational prospects. Hum Mutat. 2017;38(1):7–15. doi:10.1002/humu.23128
65. Satoh M, Imai M, Sugimoto M, Goto M, Furuichi Y. Prevalence of Werner’s syndrome heterozygotes in Japan. Lancet. 1999;353(9166):1766. doi:10.1016/S0140-6736(98)05869-3
66. Masala MV, Scapaticci S, Olivieri C, et al. Epidemiology and clinical aspects of Werner’s syndrome in North Sardinia: description of a cluster. Eur J Dermatol. 2007;17(3):213–216. doi:10.1684/ejd.2007.0155
67. Friedrich K, Lee L, Leistritz DF, et al. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet. 2010;128(1):103–111. doi:10.1007/s00439-010-0832-5
68. Saha B, Lessel D, Nampoothiri S, et al. Ethnic-specific WRN mutations in South Asian Werner syndrome patients: potential founder effect in patients with Indian or Pakistani ancestry. Mol Genet Genomic Med. 2013;1(1):7–14. doi:10.1002/mgg3.1
69. Uhrhammer NA, Lafarge L, Dos Santos L, et al. Werner syndrome and mutations of the WRN and LMNA genes in France. Hum Mutat. 2006;27(7):718–719. doi:10.1002/humu.9435
70. Huang S, Lee L, Hanson NB, et al. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27(6):558–567. doi:10.1002/humu.20337
71. The Werner syndrome mutational database. Seattle, WA, USA: Department of Pathology, University of Washington. Available from: http://www.pathology.washington.edu/research/werner/database/.
72. Melcher R, von Golitschek R, Steinlein C, et al. Spectral karyotyping of Werner syndrome fibroblast cultures. Cytogenet Cell Genet. 2000;91(1–4):180–185. doi:10.1159/000056841
73. The international registry of Werner syndrome. Seattle, WA, USA: Department of Pathology, University of Washington; January 4, 2019. Available from: http://www.pathology.washington.edu/research/werner/registry/gene.html.
74. Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;362(9382):440–445. doi:10.1016/S0140-6736(03)14069-X
75. Lessel D, Vaz B, Halder S, et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet. 2014;46(11):1239–1244. doi:10.1038/ng.3103
76. Lopez-Mosqueda J, Maddi K, Prgomet S, et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. Elife. 2016;5:e21491. doi:10.7554/eLife.21491
77. Lessel D, Saha B, Hisama F, et al. Atypical Aicardi-Goutieres syndrome: is the WRN locus a modifier? Am J Med Genet A. 2014;164A(10):2510–2513. doi:10.1002/ajmg.a.36664
78. Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83(4):655–666. doi:10.1016/0092-8674(95)90105-1
79. Kaneko H, Kondo N. Clinical features of Bloom syndrome and function of the causative gene, BLM helicase. Expert Rev Mol Diagn. 2004;4(3):393–401. doi:10.1586/14737159.4.3.393
80. Kitao S, Shimamoto A, Goto M, et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22(1):82–84. doi:10.1038/8788
81. Colombo EA, Locatelli A, Cubells Sánchez L, et al. Rothmund-Thomson syndrome: insights from new patients on the genetic variability underpinning clinical presentation and cancer outcome. Int J Mol Sci. 2018;19(4):1103. doi:10.3390/ijms19041103
82. Karikkineth AC, Scheibye-Knudsen M, Fivenson E, Croteau DL, Bohr VA. Cockayne syndrome: clinical features, model systems and pathways. Ageing Res Rev. 2017;33:3–17. doi:10.1016/j.arr.2016.08.002
83. Lehmann J, Seebode C, Martens MC, Emmert S. Xeroderma pigmentosum - facts and perspectives. Anticancer Res. 2018;38(2):1159–1164. doi:10.21873/anticanres.12335
84. Rizza ERH, DiGiovanna JJ, Khan SG, Tamura D, Jeskey JD, Kraemer KH. Xeroderma pigmentosum: a model for human premature aging. J Invest Dermatol. 2021;141(4S):
85. Ferri D, Orioli D, Botta E. Heterogeneity and overlaps in nucleotide excision repair disorders. Clin Genet. 2020;97(1):12–24. doi:10.1111/cge.13545
86. Wambach JA, Wegner DJ, Patni N, et al. Bi-allelic POLR3A loss-of function variants cause autosomal-recessive Wiedemann-Rautenstrauch syndrome. Am J Hum Genet. 2018;103(6):968–975. doi:10.1016/j.ajhg.2018.10.010
87. Gatti RA, Berkel I, Boder E, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336(6199):577–580. doi:10.1038/336577a0
88. Sandoval N, Platzer M, Rosenthal A, et al. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum Mol Genet. 1999;8(1):69–79. doi:10.1093/hmg/8.1.69
89. AlSabbagh MM. Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges. 2020;18(9):943–967.
90. Ehmke N, Graul-Neumann L, Smorag L, et al. De novo mutations in SLC25A24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am J Hum Genet. 2017;101(5):833–843. doi:10.1016/j.ajhg.2017.09.016
91. Reversade B, Escande-Beillard N, Dimopoulou A, et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet. 2009;41(9):1016–1021. doi:10.1038/ng.413
92. Passarge E, Robinson PN, Graul-Neumann LM. Marfanoid–progeroid–lipodystrophy syndrome: a newly recognized fibrillinopathy. Eur J Hum Genet. 2016;24(9):1244–1247. doi:10.1038/ejhg.2016.6
93. Johnston JJ, Sanchez-Contreras MY, Keppler-Noreuil KM, et al. A point mutation in PDGFRB causes autosomal-dominant Penttinen syndrome. Am J Hum Genet. 2015;97(3):465–474. doi:10.1016/j.ajhg.2015.07.009
94. Dyment DA, Smith AC, Alcantara D, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158–166. doi:10.1016/j.ajhg.2013.06.005
95. National organization for rare disorders. Danbury, CT, USA. Available from: https://rarediseases.org/.
96. Dhillon S. Lonafarnib: first approval. Drugs. 2021;81(2):283–289. doi:10.1007/s40265-020-01464-z
97. Gordon LB, Kleinman ME, Massaro J, et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford Progeria syndrome. Circulation. 2016;134(2):114–125. doi:10.1161/CIRCULATIONAHA.116.022188
98. Cao K, Graziotto JJ, Blair CD. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3(89):89ra58. doi:10.1126/scitranslmed.3002346
99. DuBose AJ, Lichtenstein ST, Petrash NM, et al. Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc Natl Acad Sci U S A. 2018;115(16):4206–4211. doi:10.1073/pnas.1802811115
100. Santiago-Fernández O, Osorio FG, Quesada V, et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat Med. 2019;25(3):423–426. doi:10.1038/s41591-018-0338-6
101. Beyret E, Liao HK, Yamamoto M, et al. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat Med. 2019;25(3):419–422. doi:10.1038/s41591-019-0343-4
102. Koblan LW, Erdos MR, Wilson C, et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature. 2021;589(7843):608–614. doi:10.1038/s41586-020-03086-7
103. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–788. doi:10.1038/s41576-018-0059-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.