Back to Journals » Journal of Blood Medicine » Volume 12
Murine Leukemia-Derived Extracellular Vesicles Elicit Antitumor Immune Response
Authors Pando A, Fast L, Dubielecka PM, Chorzalska A, Wen S ![]() , Reagan J
, Reagan J
Received 28 February 2021
Accepted for publication 20 April 2021
Published 17 May 2021 Volume 2021:12 Pages 277—285
DOI https://doi.org/10.2147/JBM.S308861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Alejandro Pando, Loren Fast, Patrycja M Dubielecka, Anna Chorzalska, Sicheng Wen, John Reagan
Division of Hematology/Oncology, Department of Medicine, Rhode Island Hospital and the Warren Alpert School of Medicine at Brown University, Providence, RI, USA
Correspondence: Loren Fast
Division of Hematology/Oncology, Department of Medicine, Rhode Island Hospital and the Warren Alpert School of Medicine at Brown University, 31 S Killingly Road, Foster, RI, 02825, USA
Tel +1 401 533-8658
Email [email protected]
Background: Extracellular vesicles (EVs) are heterogeneous lipid bilayer particles secreted by cells. EVs contain proteins, RNA, DNA and other cargo that can have immunomodulatory effects. Cancer-derived EVs have been described as having immunomodulating effects in vivo with immunosuppressive and pro-tumor growth capabilities. However, cancer-derived EVs have also been harnessed and utilized for anti-cancer potential.
Methods: To assess the immunomodulatory effect of EVs produced by acute myeloid leukemia (AML) cells, we isolated vesicles secreted by the murine AML cell line, C1498, and investigated their effect on in vitro and in vivo immune responses.
Results: These leukemia-derived EVs were found to induce increased proliferation of CD3+ cells and enhanced cytolytic activity of CD3+ cells directed toward leukemic cells in vitro. Injection of leukemia-derived EVs into syngeneic naïve mice induced T cell responses in vivo and resulted in enhanced immune responses upon T cell re-stimulation in vitro.
Conclusion: These findings indicate that C1498-derived EVs have immunomodulatory effects on cell-mediated immune responses that could potentially be utilized to facilitate anti-leukemia immune responses.
Keywords: extracellular vesicles, acute myeloid leukemia, immune responses
Introduction
There is an increasing emphasis on harnessing the patient’s immune system to achieve anti-cancer responses. A variety of approaches have been tested including activating the patient’s immune system directly using various stimuli. Alternative adoptive immunotherapy approaches involve removing the patient’s lymphocytes and then expanding, activating and modifying the lymphocytes in vitro prior to reinfusing them back into the patient. Our group has developed a cellular immunotherapy approach in which a large number of G-CSF mobilized haploidentical donor CD3+ cells (1 −2 x 108 CD3+ cells/kg) are infused into patients who had received 100 cGy of total body irradiation prior to the infusion.1 This approach was shown to induce responses in about half of patients with refractory hematological malignancies (14/26, 5 complete remissions). The course of the treatment following infusion included rapid development (a median time of 14 hours) of a cytokine storm characterized by very high levels of IL-6 and high fevers. This response could be controlled by administration of corticosteroids, if needed. In almost all patients, no remaining donor cells could be detected in the patient at two weeks.
While effective, the mechanism(s) responsible for the anti-cancer responses in this therapeutic protocol have not been determined. One potential mechanism is that the activation of the alloreactive effector cells responsible for eliminating the haploidentical donor cells cross-react with tumor cells. A number of studies have shown that alloreactive effector cells cross-react with cancer cells.2–7 To explore this possibility further, we obtained blood from newly diagnosed leukemic patients, isolated the CD3+ cells, stimulated them with allogeneic stimulator cells and then tested the ability of alloreactive effector cells that were generated to lyse cells in a syngeneic population of cells containing leukemic cells.8 It was observed that about half of the patient’s CD3+ cells were able to lyse syngeneic leukemic cell containing populations when stimulated with allogeneic cells. In trying to identify characteristics of the responders and nonresponders, we found that lack of response was associated with increased expression of cytolytic CD4+ cells, increased expression of CD39 by CD8+ cells and an increased number of γδ T cells.
There has been an increased understanding of the extracellular vesicles (EVs) and the functional implications of the cargo that is carried by them. EVs are nano-sized heterogeneous lipid bilayer particles released by almost all cells and secreted at higher numbers in cancer cells.9 Initially thought to be cellular debris, over the last decade EVs have been shown to be involved in numerous biological processes such as intercellular communication, antigen presentation, protein secretion, and RNA shuttling.10 EVs carry DNA, proteins, RNA, bioactive lipids, and other cargo that is typically representative of the parent cell.11,12 Depending on the cell of origin, EVs can modulate physiological and pathological processes including proliferation, differentiation, inhibition, quiescence, and/or cellular death. EVs produced by cancer cells have been shown to enhance malignancy by transmission of regulatory factors to normal cells. EVs have also been shown to enhance anti-tumor immune responses by inducing immunity to antigens that are carried by tumor EVs. This raised the question of whether EVs were contributing to responses or lack of responses in the cellular immunotherapy protocol. To begin to test the role of EVs in this protocol, we isolated EVs from C1498, a murine AML cell line, and examined their effect on in vivo and in vitro immune responses directed toward C1498 cells. The results showed that the EVs derived from the C1498 cell line enhanced in vitro and in vivo anti-tumor responses.
Materials and Methods
Mice
C57BL/6J female mice (#000664 Jackson Laboratory, Bar Harbor, ME), 7 to 9 weeks of age (20–25g) were housed one week before the experiments. Mice were injected intraperitoneally with C1498-derived EVs or C1498 cells. Control mice were injected with PBS. All studies were approved by the Lifespan Institutional Animal Care and Use Committee.
Cell Lines and Reagents
The murine AML cell line (C1498) was obtained from ATCC. C1498 arose spontaneously in a C57BL/6J mouse. C1498 cells grow aggressively in C57BL/6J mice, when injected intravenously into syngeneic mice resulting in the development of acute leukemia.13 C1498 cells were cultured at 37°C in 5% CO2 in DMEM (ATCC) containing 10%fetal calf serum (FCS, Atlanta Biologicals) that had been depleted of extracellular vesicles by 2 centrifugation steps at 100,000 x×g for 70 minutes plus 100 U/mL penicillin/streptomycin (Gibco).
Isolation and Characterization of EVs
EVs were isolated from C1498 culture medium as previously described.14 Conditioned culture medium was obtained by expanding the C1498 cells until sixteen 175 cm2 flasks were obtained each containing 120 mL of medium at a cell density of approximately 2×106 cells/mL. Briefly, C1498 conditioned cell culture medium was subjected to a series of centrifugation steps starting at 3300 g for 5 minutes and 2000 g for 10 minutes at room temperature followed by 10,000 g for 1 hour at 4 °C and 100,000 g at 4 °C for 70 minutes with collection of the 1100,000 g pellet. The higher speed centrifugation steps were done using a Surespin 630 rotor and a Thermo WX ULTRA ultracentrifuge. The EVs pellet was then resuspended in PBS and centrifuged at 100,000 ×g for 70 minutes at 4 °C. The freshly prepared C1498 EVs were directly used for experiments.
Characterization of mouse C1498 derived EVs was carried out by electron microscopy as previously described.14 The representative images of C1498-derived EVs are shown in Supplemental Figure S.4A. Number and size distribution of vesicles was determined on a NanoSight NS500 (Malvern Instruments, Malvern, UK) (Supplemental Figure S.4B).
Proliferation and Cytolytic Analyses
C57BL/6J mice were injected ip with 200 uL PBS (i), or PBS containing C1498-derived EVs (ii), or C1498 cells (iii). At 7- or 14-days post injection, spleens were obtained from these mice and a ssingle-cellsuspension was prepared. The cells were washed twice with MLC medium (RPMI 1640 containing 4% EV depleted FCS, 2 mg/mL glucose, 2 mM glutamine, Pen/Strep and 10μM 2-mercaptoethanol). The splenocytes were resuspended in MLC medium and counted, and then stimulated with C1498-derived EVs or mitomycin C treated C1498 cells. C1498 cells (25 × 106 cells/mL MLC medium) and 25ug mitomycin C/mL were incubated for 30 minutes at 37°C. Following three washes with MLC medium, the treated cells were resuspended in MLC medium and counted. 1×105 responder splenocytes were co-incubated with mitomycin treated C1498 cells (1 × 104 cells), C1498 derived EVs, or PBS in triplicate wells in a 96 well flat bottom plate in a final volume of 200 μL MLC medium. High EV in vitro conditions (C1) was defined as 15 µg of EV protein added per 1×106 responder cells, and low EV condition (C2) was defined as adding 3 µg of EV protein per 1×106 responder cells. On the fourth day, 1 μCi of 3H thymidine (Perkin Elmer Health Sciences, Shelton, CT, USA) was added to each well, incubated for 4 hours at 37° and the DNA was collected on filter paper to measure thymidine incorporation and assess the proliferation of stimulated splenocytes.
To measure cytolytic activity, cultures were set up by adding 4×106 responder spleen cells to 1×106 mitomycin C treated C1498 cells, or C1498 derived-EVs in a final volume of 2 mL of MLC medium per well in a 24 well plate. On day 5 of culture, the cells were collected, centrifuged, and aliquots of the supernatants were stored at −20°C. The cells were then resuspended in RHG medium (RPMI 1640, 20 mM HEPES, 4% FCS and pen/strep). The cells were counted and used as effectors against 51Cr labeled target cells (C1498 cells). 51Cr labeled cells were prepared by incubating 5×106 target cells with 15 μCi 51Cr (Perkin Elmer Health Sciences, Shelton, CT, USA) in 200 µL of medium for 45 minutes at 37℃ with frequent agitation. After three washes with RHG medium the cells were counted. Each set of effector cells was added in duplicate to wells in 96 well V-bottom plates. Responder cell numbers ranged from 1×106 cells per well in 100 µL RHG medium in 2-fold serial dilutions to 1.25×105 cells per well. 51Cr-labeled target cells at 1×104 cells/100 µL were added to all wells. Control wells contained 100 µL RHG medium or 100 µL 1N HCl in addition to the target cells. After four hours incubation at 37℃, 100 μL of the supernatant was collected from each well and the 51Cr present in each sample measured using a gamma counter. Percent specific lysis was determined by subtracting the counts obtained from the medium containing wells from the experimental value divided by the counts obtained in the wells containing 1 N HCl minus the counts obtained from the medium containing wells, and then multiplied by 100. The results are presented as lytic units/106 cells in which 1 lytic unit (LU) is defined as the number of cells required to achieve 30% lysis of 1×104 target cells. All experiments were replicated four times.
Cytokine Measurements
Aliquots of the supernatants collected on day 5 of the mixed lymphocyte cultures were thawed on the same day and tested for cytokine levels using a multiplex CBA assay measuring IL-2, IL-4, IL-6, TNF-α, IFN-γ, IL-10, and IL-17A levels according to manufacturer’s instructions (BD Biosciences, San Jose, CA) using the LSRII flow cytometer. FACSARRAY software was used for analysis.
Immunophenotypic Analysis
Cell phenotypes were evaluated by multiparameter flow cytometry. Cells were incubated with a panel of labeled monoclonal antibodies (mAbs) anti-CD3 AF700, CD4 BV605, CD8 APC-H7, CD25 BV786, PD-1 APC, TIGIT BV421, and LAG3 BV711 (BD Biosciences) in staining buffer on ice in the dark for 20 min. Cells were then washed in the staining buffer and re-suspended in staining buffer and analyzed on an LSRII flow cytometer. Isotype controls were used for each experiment. Analysis of the results used FlowJo 10 software.
CFSE staining was conducted according to manufacturer’s instructions (Thermo Fisher Scientific). Briefly, cells were labeled with CFSE by adding 1 mL of freshly prepared CFSE (2 μM in PBS containing 2% EV free FCS) to cells (up to 1×108 cells) in 1 mL of PBS 2% EV free FCS. The tube containing this mixture was covered with foil and incubated at 337°Cfor 5 minutes. Cells were pelleted, washed twice with 10 mL of PBS 2% EV free FCS, resuspended, counted and used for experiments.
Statistical Analysis
Statistical analysis was performed utilizing GraphPad Prism 7.0 (GraphPad Software). Student’s t-test, unpaired, two-tail testing was applied to populations to determine the p values indicated in the figures. p values <0.05 were determined to be statistically significant. All data are represented as mean ± SD.
Results
Leukemia-Derived EVs Induce Increased Splenocyte Proliferation in vitro
To investigate the immunomodulatory ability of leukemia-derived EVs, we assessed the impact of culturing splenocytes obtained from naïve C57BL/6J mice with cells from the C1498 AML cell line, with two different concentrations of purified C1498-derived-EVs or when C1498 cells and C1498-EVs were combined. Results showed that the addition of C1498 cells or C1498-derived EVs alone to splenocytes induced a significant proliferative response and that combining C1498 cells and C1498-derived EVs provided an additive effect (Figure 1A, Supplemental Fig. S.1A). Increased C1498-derived EVs concentrations (C1 vs C2) trended toward increased proliferative responses (Figure 1A, Supplemental Fig. S.1A).
 |
Figure 1 C1498 derived EVs induce splenocyte and CD3+ proliferation. Splenocytes (1 x 105 cells) were cultured with PBS, C1498 cells, different concentrations of C1498 derived EVs (C1 = high, C2 = low), or both C1498 cells and C1498 derived EVs. (A) Comparison of tritiated thymidine incorporation (mean ± SD) of combined splenocyte proliferation activity from splenocytes isolated from naïve mice (control) and splenocytes isolated from EV immunized mice (treated) (mean ± SD). (B) CD3+ splenocytes from PBS injected, C1498 derived EV immunized, or C1498 cell immunized mice were labeled with CFSE and co-cultured in vitro with medium, C1498 derived EVs, or C1498 cells. On day 4 the splenocytes were analyzed by flow cytometry and the number of CFSE low cells (proliferating cells) were measured as fold difference from PBS injected naïve mice cultured with medium (mean ± SD). (C) Representative flow cytometry plots of CFSE labeled splenocytes stained with anti-CD3 after four days of culture. CD3+ expression on the vertical axis and CFSE expression on the horizontal axis. Column 1: cells underwent in vivo injection of PBS, column 2 cells underwent in vivo injection of C1498 derived EVs, column 3: cells underwent in vivo injection of C1498 cells. Row 1: cells underwent in vitro culture with medium, row 2: cells underwent in vitro culture with C1498 derived EVs, row 3: cells underwent in vitro culture with C1498 cells. Left upper quadrant represents CD3+ CFSElo population. Left bottom quadrant represents CD3- CFSElo population. Asterisks indicate significant differences (*p < 0.05, **p<0.01). |
Immunizing Naïve Mice of Leukemia-Derived EVs Enhances Immune Responses
To assess the effect of immunization on splenocyte responses, C57BL/6J mice were injected with, C1498-derived EVs, or PBS (Supplemental Fig. S.1B). Splenocytes were obtained at 7 days or 14 days post injection and cultured with C1498 cells, EVs from C1498 cells, or a combination of the two. Results demonstrated overall significantly increased proliferation responses from splenocytes isolated from EV immunized mice over splenocytes isolated from the naïve mice (Figure 1A, Supplemental Fig. S.1).
To investigate which cell sub-populations were undergoing proliferation, splenocytes obtained from mice injected with PBS, C1498-derived EVs, or C1498 AML cells were labeled with CFSE prior to culture, and CFSE positive CD3+ cells were assessed on day 4 of culture. Gating strategy is found in Supplemental Figure S.2. For all three in vitro stimuli, the proliferating splenocytes (CFSElo) from PBS injected mice exhibited a preferential proliferation of CD3– cells(35.5± 8.6% CD3+ cells) while splenocytes from EV immunized mice (59.1 ± 1.1% CD3+ cells) or AML cell immunized mice (77.1 ± 14.3% CD3+ cell) exhibited preferential proliferation of CD3+ cells (Figure 1B and C). To investigate which subpopulations of CD3+ cells proliferated in response to EVs, the frequencies of CD4+ and CD8+ cells in the CFSElo CD3+ cells were determined. CD8+ T cells from all three groups of mice comprised a significantly greater fraction of the proliferating cells than CD4+ T cells (Figure 2A and B).
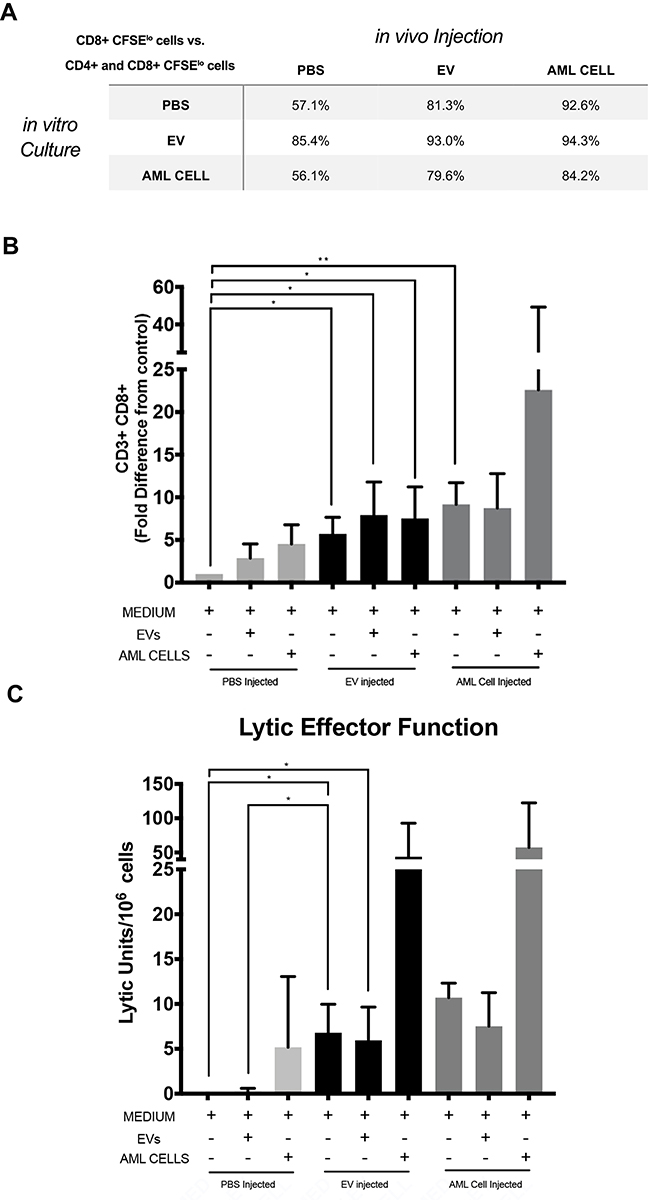 |
Figure 2 C1498 derived EVs preferentially activate CD8+ T cells and C1498 derived EV immunization increases lytic effector function. CFSE labeled splenocytes from in vivo PBS injected, C1498 derived EV immunized, or C1498 cell immunized mice were co-cultured in vitro with medium, C1498 derived EVs, or C1498 cells. On day 4 the cultures were harvested and stained with a panel of antibodies including anti-CD3, anti-CD8, anti-CD4, and underwent immunophenotypic analysis. (A) Table shows the fraction of CD8+ and CD4+ CFSElo population that are CD8+ cells. (B) Flow cytometry bar graphs of measured CD3+ CD8+ proliferation of each condition. Experiment repeated at least 3 times. Data is presented as fold difference in CFSE low cells (proliferating cells) from PBS injected naïve mice cultured with medium (mean ± SD). (C) Splenocytes isolated from C57BL/6J mice 7- and 14-days following injection with PBS, C1498 derived EVs, or AML cells were co-cultured in vitro with PBS, C1498 derived EVs, or AML cells for five days and the responding cells were tested for their ability to lyse 51Cr labeled C1498 cells. Data is presented as LU/106 effector cells and represents replicates from 4 experiments. Asterisks indicate significant differences (*p < 0.05, **p<0.01). |
To investigate the effect of exposure to leukemia-derived EVs on cytolytic activity, immune splenocytes were co-cultured with either PBS, leukemia-derived EVs, or C1498 cells for 5 days and then tested for their ability to lyse C1498 target cells. These immune splenocytes cultured for 5 days in medium exhibited the ability to lyse C1498 cells and this lytic activity was enhanced by co-culturing the immune splenocytes with leukemia-derived EVs or mitomycin C treated C1498 cells (Figure 2C). Increased levels of the inflammatory cytokines IL-6, IL-10, TNF-α, and IFN-γ were observed in the supernatants of immune splenocytes cultured in medium (Figure 3A–D). An additional increase in production of TNF-α and IFN-γ was seen when the splenocytes from the immunized mice were cultured with leukemia derived EVs and/or C1498 cells.
 |
Figure 3 C1498 derived EVs increase cytokine production. Splenocytes obtained from PBS injected, C1498 derived EV immunized, or C1498 cell immunized mice co-cultured in vitro with medium, C1498 derived EVs, or with C1498 cells were incubated for five days at 37°C. Supernatants were then collected and stored at −20°C. These supernatants were tested for cytokine levels using the mouse Th1/Th2/Th17A CBA assay. The cytokines shown are as follows: Panel (A), TNFα; Panel (B), IFNγ; Panel (C) IL6, Panel (D), IL10. Error bars indicate standard deviation of of triplicate samples from a representative of three independent experiments. Asterisks indicate significant differences (*p < 0.05, **p<0.01, ***p<0.001). |
CD3+ cells from immune mice upregulated the expression of activation induced inhibitory receptors PD-1, TIGIT, and LAG3 when analyzed after collection (Supplemental Fig. S.3). The increased expression of these markers suggests that EVs can induce activation in CD3+ cells but the increased expression of these inhibitory receptors by these activated CD3+ cells could result in inhibited responses if the ligands for these inhibitory receptors are present in the environment (Supplemental Fig. S.3).
Discussion and Conclusion
Tumor cells secrete increased numbers of extracellular vesicles and have the ability to both suppress and enhance immune activity depending on the underlying pathologic state, parental cell derivation, and absolute EV number. These two seemingly juxtaposed notions likely depend on the microenvironment context and complexity of the interactions amongst the components within that environment. In this study, we tested the impact of EVs obtained from a murine acute myeloid leukemic cell line, C1498, on in vivo and in vitro immune responses. Immune responses that were enhanced by immunization compared to responses by spleen cells from control unimmunized mice included increased proliferation, increased lytic effector function, a bias toward enhanced CD8+ cell proliferation and proinflammatory cytokine production. In addition, T lymphocytes from these immunized mice exhibited increased expression of activation markers including the inhibitory receptors PD-1, TIGIT, and Lag3. These findings suggest that EVs may be used instead of cells to induce immune responses and raises the possibility that EVs could be used as a component of immunotherapy protocols.15 Notably, observed increased expression of inhibitory receptors could possibly represent an inhibitory hurdle for the responses of these cells if the ligands for these receptors are present in the environment.
The possibility also exists that the EVs could directly present antigen. The ability of EVs to present antigen was supported with the findings that EVs express multiple different H-2 class I histocompatibility antigen chains and MHC class 1b proteins, indicating that EVs are able to present antigens to the adaptive immune system. These antigens can be recognized by immune cells and elicit a proliferative immune response. EVs also expressed the antigen CD160 on their plasma membrane which has broad specificity for binding to classical and nonclassical MHC class I molecules.16 CD160 is a ligand for Herpesvirus entry mediator (HVEM), that is considered a proposed immune checkpoint inhibitor with anti-tumor activity, further suggesting that EVs may harbor antigenic presentation potential.17 This may be important if patients are infused with haploidentical CD3+ cells. These CD3+ cells could respond in a restricted or alloreactive manner to EVs circulating in the cancer patient and the responses to the EVs could influence whether the hematological malignant cells are attacked.
Eliciting effective T cell responses has been a major component in producing anti-leukemia responses. Therefore, inducing the effective antigen-specific cytolytic CD8+ T cell is vital to producing anti-leukemia responses. Others demonstrated that leukemia-derived exosomes can induce proliferation of CD4+ T cells and suggested that tumor-derived EVs can partially suppress T cell activation.18,19 One of the components found in EVs produced by AML cells are microRNAs (miRNA).20,21 Characterization of these EVs have identified a selected subset of these miRNAs21 Some of these miRNAs have been found to modulate immune responses. MiR21 has been shown to inhibit immune function by causing premature apoptosis of T lymphocytes while miR145 and miR448 have been shown to enhance anti-tumor responses.21,22 In our study, we found that leukemia-derived EVs produced significant proliferation and activation of CD8+ T cells in mice immunized with EVs. Perhaps this is due to preferential expression of specific miRNA. Another possibility is the preferential expression of MHC class I molecules on EVs relative to the expression of MHC class II molecules. Interestingly, splenocytes from mice injected with leukemia derived EVs responded to leukemia derived EV re-exposure at a significantly higher level than splenocytes exposed to EVs for the first time, suggesting that EVs are able to prime murine splenocytes.
The finding that EVs isolated from a leukemic cell line are able to induce immune responses could suggest a potential role of leukemic cell derived EVs for generating anti-leukemic responses in the cellular immunotherapy protocol. A study by Lulla et al showed that activating and expanding leukemia antigen-specific T cells from stem cell donor lymphocytes enhanced anti-leukemic responses and minimized graft-versus-host disease when infused into recipients following a hematopoietic stem cell transplant.23 Because CD3+ cells from a non-tumor bearing haploidentical donor are infused into the patient with refractory hematological malignancy, one possibility is that the circulating EVs isolated from the patient could be used to activate leukemia-specific donor CD3+ cells prior to infusion. The EVs could induce effector cells restricted to peptides presented by syngeneic MHC molecules or alloreactive effector cells able to recognize and lyse the patient’s leukemic cells. The EVs could also potentially be used to test the responses of potential donors to identify which donor would generate the strongest anti-leukemic responses.
Our study suggests that tumor-derived EVs should be considered a component of possible anti-leukemia vaccines and that modification of them could be a possible path to a reproducible personalized medicine. Tumor-derived EVs have the advantage of containing unique tumor-associated antigens from the parental cell which they derive from and also contain immune stimulatory proteins. Tumor-derived EVs also have the advantage of not containing cell organelles and are highly reproducible and storable. Tumor-derived EV effects on target cells involve complex interactions, but harnessing these vesicles for anti-leukemia purposes is an important possibility warranting further studies.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
These studies were approved by the Lifespan Hospital Institutional Review Board. The studies followed the “Guide for the Care and Use of Laboratory Animals. 8th edition.”
Acknowledgments
The authors would like to thank the Brown University Molecular Pathology Core for conducting the electron microscopy studies.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Funding
These studies were supported by 5P30GM110759-04 and by funds provided by the Division of Hematology/Oncology, Rhode Island Hospital.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Colvin GA, Berz D, Ramanathan M, et al. Nonengraftment haploidentical cellular immunotherapy for refractory malignancies: tumor responses without chimerism. Biol Blood Marrow Transplant. 2009;15(4):421–431. doi:10.1016/j.bbmt.2008.12.503
2. Burdach S, Kolb HJ. The vigor of defense against non-self: potential superiority of allorestricted T cells in immunotherapy of cancer? Front Oncol. 2013;3:100. doi:10.3389/fonc.2013.00100
3. Kumari S, Walchli S, Fallang LE, et al. Alloreactive cytotoxic T cells provide means to decipher the immunopeptidome and reveal a plethora of tumor-associated self-epitopes. Proc Natl Acad Sci U S A. 2014;111(1):403–408. doi:10.1073/pnas.1306549111
4. Paciucci PA, Macphail S, Zarling JM, Bach FH. Lysis of syngeneic solid tumor cells by alloantigen stimulated mouse T and non-T cells. J Immunol. 1980;124(1):370–375.
5. Schirrmacher V, Hubsch D, Garrido F. Syngeneic tumor cells can induce alloreactive T killer cells: a biological role for transplantation antigens. Proc Natl Acad Sci U S A. 1980;77(9):5409–5413. doi:10.1073/pnas.77.9.5409
6. Sensi ML, Parenza M, Parmiani G. Alloreactivity and tumor antigens: generation of syngeneic antilymphoma killer lymphocytes by alloimmunization of mice with normal cells. J Natl Cancer Inst. 1983;70(2):291–297.
7. Stauss HJ, Xue S, Gillmore R, et al. Exploiting alloreactivity for tumour immunotherapy. Vox Sang. 2004;87(Suppl 2):227–229. doi:10.1111/j.1741-6892.2004.00493.x
8. Pando A, Reagan JL, Nevola M, Fast LD. Induction of anti-leukemic responses by stimulation of leukemic CD3+ cells with allogeneic stimulator cells. Exp Hematol Oncol. 2018;7:25. doi:10.1186/s40164-018-0118-5
9. Pando A, Reagan JL, Quesenberry P, Fast LD. Extracellular vesicles in leukemia. Leuk Res. 2018;64:52–60. doi:10.1016/j.leukres.2017.11.011
10. Mahaweni NM, Kaijen-Lambers ME, Dekkers J, Aerts JG, Hegmans JP. Tumour-derived exosomes as antigen delivery carriers in dendritic cell-based immunotherapy for malignant mesothelioma. J Extracell Vesicles. 2013;2. doi:10.3402/jev.v2i0.22492
11. Simons M, Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21(4):575–581. doi:10.1016/j.ceb.2009.03.007
12. Quesenberry PJ, Aliotta J, Deregibus MC, Camussi G. Role of extracellular RNA-carrying vesicles in cell differentiation and reprogramming. Stem Cell Res Ther. 2015;6:153. doi:10.1186/s13287-015-0150-x
13. Mopin A, Driss V, Brinster C, Detailed A. Protocol for characterizing the murine C1498 cell line and its associated leukemia mouse model. J Vis Exp. 2016;116.
14. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi:10.1038/nri855
15. Yao Y, Wang C, Wei W, et al. Dendritic cells pulsed with leukemia cell-derived exosomes more efficiently induce antileukemic immunities. PLoS One. 2014;9(3):e91463. doi:10.1371/journal.pone.0091463
16. Tu TC, Brown NK, Kim TJ, et al. CD160 is essential for NK-mediated IFN-gamma production. J Exp Med. 2015;212(3):415–429. doi:10.1084/jem.20131601
17. Vigano S, Banga R, Bellanger F, et al. CD160-associated CD8 T-cell functional impairment is independent of PD-1 expression. PLoS Pathog. 2014;10(9):e1004380. doi:10.1371/journal.ppat.1004380
18. Whiteside TL. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem Soc Trans. 2013;41(1):245–251. doi:10.1042/BST20120265
19. Huang F, Wan J, Hao S, Deng X, Chen L, Ma L. TGF-beta1-silenced leukemia cell-derived exosomes target dendritic cells to induce potent anti-leukemic immunity in a mouse model. Cancer Immunol Immunother. 2017;66(10):1321–1331. doi:10.1007/s00262-017-2028-5
20. Wallace JA, O’Connell RM. MicroRNAs and acute myeloid leukemia: therapeutic implications and emerging concepts. Blood. 2017;130:11. doi:10.1182/blood-2016-10-697698
21. Agha DM, Rouas R, Najar M, et al. Identification of acute myeloid leukemia bone marrow circulating MicroRNAs. Int J Mol Sci. 2020;21(19).
22. Agha DM, Rouas R, Najar M, et al. Impact of bone marrow miR-21 expression on acute myeloid leukemia T lymphocyte fragility and dysfunction. Cells. 2020;9:9. doi:10.3390/cells9071640
23. Lulla P, Naik S, Vasileiou S, et al. Clinical effects of administering leukemia-specific donor T cells to patients with AML/MDS post-allogeneic transplant. Blood. 2020. doi:10.1182/blood.2020009471
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.