Back to Journals » The Application of Clinical Genetics » Volume 12
Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype–phenotype correlations
Authors Oliveira JP ![]() , Ferreira S
, Ferreira S ![]()
Received 3 October 2018
Accepted for publication 14 January 2019
Published 5 March 2019 Volume 2019:12 Pages 35—50
DOI https://doi.org/10.2147/TACG.S146022
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Martin Maurer
João Paulo Oliveira,1,2,3 Susana Ferreira1,3
1Department of Genetics, Faculty of Medicine, University of Porto, Alameda Hernâni Monteiro, 4200-319 Porto, Portugal; 2Service of Medical Genetics, São João University Hospital Centre, Alameda Hernâni Monteiro, 4200-319 Porto, Portugal; 3i3S – Institute for Research and Innovation in Health / [Instituto de Investigação e Inovação em Saúde], University of Porto, 4200-135 Porto, Portugal
Abstract: Fabry disease (FD) is a rare X-linked glycosphingolipidosis resulting from deficient α-galactosidase A (AGAL) activity, caused by pathogenic mutations in the GLA gene. In males, the multisystemic involvement and the severity of tissue injury are critically dependent on the level of AGAL residual enzyme activity (REA) and on the metabolic load of the disease, but organ susceptibility to damage varies widely, with heart appearing as the most vulnerable to storage pathology, even with relatively high REA. The expression of FD can be conceived as a multidomain phenotype, where each of the component domains is the laboratory or clinical expression of the causative GLA mutation along a complex pathophysiologic cascade pathway. The AGAL enzyme activity is the most clinically useful marker of the protein phenotype. The metabolic phenotype and the pathologic phenotype are diverse expressions of the storage pathology, respectively, assessed by biochemical and histological/ultrastructural methods. The storage phenotypes are the direct consequences of enzyme deficiency and hence, together with the enzymatic phenotype, constitute the more specific diagnostic markers of FD. In the pathophysiology cascade, the clinical phenotypes are most distantly linked to the underlying genetic causation, being critically influenced by the patients’ gender and age, and modulated by the effects of variation in other genetic loci, of polygenic inheritance and of environmental risk factors. A major challenge in the clinical phenotyping of patients with FD is the differential diagnosis between its nonspecific, later-onset complications, particularly the cerebrovascular, cardiac and renal, and similar chronic illnesses that are common in the general population. Comprehensive phenotyping, whenever possible performed in hemizygous males, is therefore crucial for grading the severity of pathogenic GLA variants, to clarify the phenotypic correlations of hypomorphic alleles, to define benign polymorphisms, as well as to establish the pathogenicity of variants of uncertain significance.
Keywords: Fabry disease phenotypic variants, α-galactosidase, GLA gene, biomarkers, pathophysiology cascade, phenocopies
Introduction
Fabry disease (FD, OMIM#301500) is a rare X-linked glycosphingolipidosis resulting from deficient α-galactosidase A (AGAL; EC 3.2.1.22) activity, caused by inherited or de novo pathogenic mutations in its gene (GLA; OMIM*300644).1–3 AGAL is a lysosomal hydrolase that catalyzes the removal of terminal nonreducing α-d-galactose residues in polysaccharides, glycolipids, and glycopeptides.4 In humans, impairment of AGAL activity leads to widespread intralysosomal accumulation of globotriaosylceramide (Gb3: Galα1→4Galβ1→4Glcβ1→Cer), starting early in life, as well as of other minor compounds, including galabiosylceramide (Ga2: Galα1→4Galβ1→Cer), which has a more restricted organ distribution,2 namely to the kidneys. The major physiological source of Gb3 is globoside, an abundant glycolipid of cell membranes in different tissues and in erythrocytes.5
The storage pathology of FD can be morphologically demonstrated by electron microscopy (EM) examination of affected tissues, and specific in situ immunodetection of Gb3 is possible with anti-Gb3 monoclonal antibodies.2,6 Although Gb3 accumulation is regarded as the primary pathogenic event in FD, the secondary pathophysiological derangements arising therefrom, at cell, tissue, and organ levels, which ultimately give rise to the clinical manifestations, are still unclear.7–13 Progressive organ damage is thought to be critically linked to the involvement of vascular endothelial and smooth muscle cells, particularly in the microcirculation, as well as of pericytes; of cardiomyocytes, including those forming the cardiac conduction system; of different epithelial cells in the kidney, including podocytes, the parietal layer of the glomerular capsule, and the tubular epithelium; and of a variety of neuronal cell types in the central and peripheral nervous systems, particularly peripheral neurons in the autonomic and posterior root ganglia, and cells of the perineurium along the entire length of the peripheral nerve fibers.1,2,6 However, the extension of organ involvement and the severity of tissue injury observed in individual FD patients are critically dependent on the level of residual enzyme activity (REA) and the metabolic load of the disease, here defined as an estimator of the relative amount of AGAL substrates accumulated in individual FD patients. Furthermore, despite the systemic nature of the AGAL deficiency, susceptibility to clinically apparent dysfunction brought about by the abnormal glycosphingolipid catabolism varies extensively among organs,6 from fully resistant (eg, the liver) to highly susceptible (eg, the heart). Globotriaosylsphingosine (lysoGb3), a deacylated, amphiphilic Gb3 metabolite that accumulates in the plasma14 and is excreted in the urine15 of FD patients to much greater relative degrees than its parent compound, has recently been identified as a possible contributor to the metabolic load of the disease, likely playing a major pathogenic role in the cardiovascular, kidney, and neuronal injury.14,16,17
Overall, FD can be conceived as a multidomain disease phenotype, where each of the component domains is the laboratory or clinical expression of the causative GLA mutation along a complex pathophysiologic cascade pathway (Figure 1).18 The phenotypic domains that more closely mirror the genotypic effect are the enzymatic and the storage phenotypes, while the clinical phenotype is most influenced by additional genetic systems and environmental modulators and is critically dependent on age and gender.1–3 Because of the modulating effect of X-chromosome inactivation in the expression of X-linked diseases in females,19 the clinical phenotype and the natural history of FD should be defined in affected males, who have homogeneous tissue expression of the AGAL deficiency. For all the stated reasons, the establishment of genotype–phenotype correlations in FD requires robust evidence of altered AGAL-dependent glycosphingolipid homeostasis, particularly in patients with atypical clinical phenotypes or carrying a GLA variant of unknown significance (VUS).20
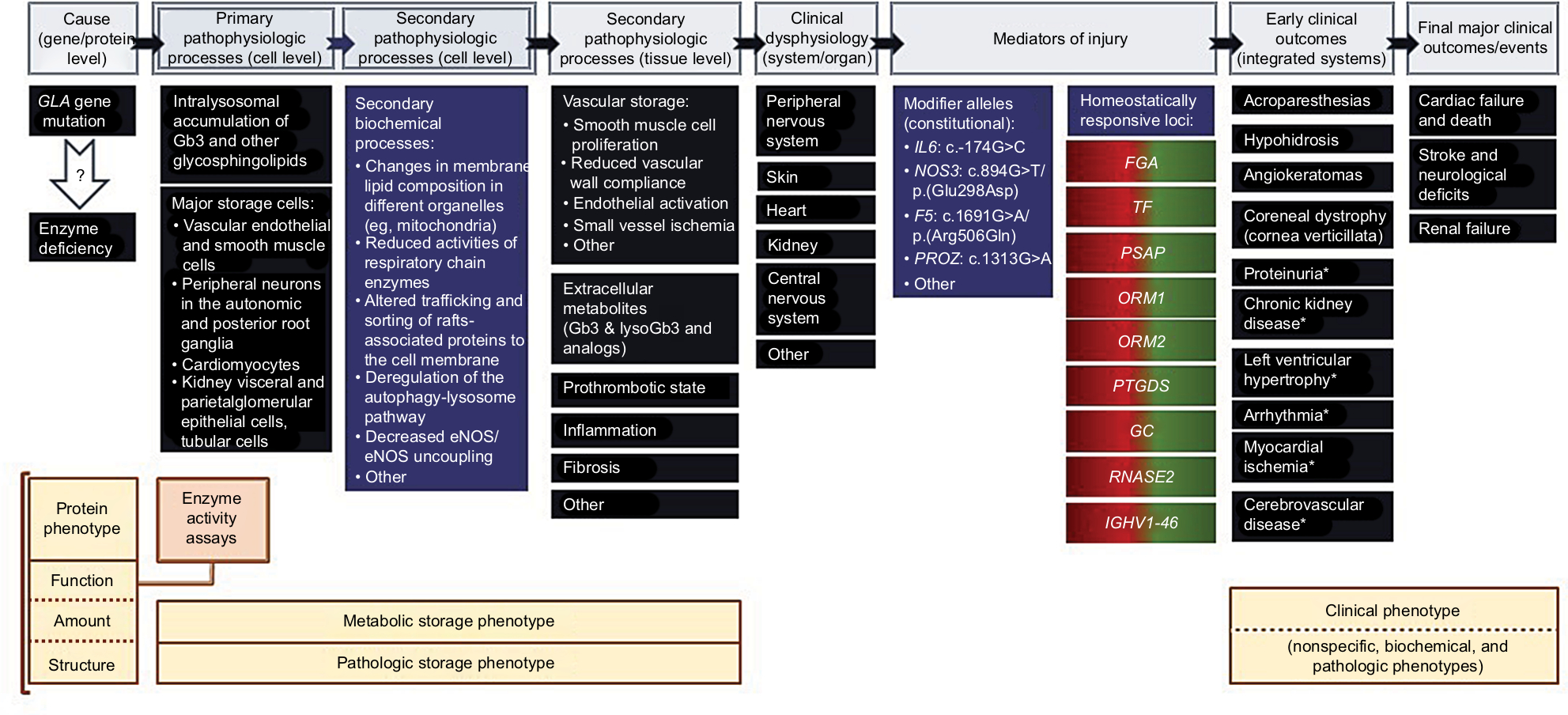 | Figure 1 The pathophysiology cascade of FD and its relevance for understanding the genotype–phenotype correlations. Notes: The pathophysiologic pathways linking a GLA gene mutation to a clinical phenotype of FD are represented at the top of the figure. Selected examples of related molecular and biochemical mechanisms; cellular, tissue, and organ pathology; modifier genes; and early and late clinical outcomes are presented below each stage in the pathophysiology cascade. The critical issue regarding the causality of FD is whether a specific GLA mutation causes a severe enough AGAL deficiency to drive the pathophysiologic cascade all the way down to the development of either a full-blown or an incomplete clinical phenotype of FD. Such GLA mutations have a major gene effect and, by themselves, are enough to cause FD in hemizygous males. The secondary pathophysiologic processes at the cell level are not immediately related to the AGAL deficiency but rather to the deleterious consequences of the lysosomal storage pathology upon the homeostasis of other subcellular compartments and the chemical composition of cell membranes. The secondary pathophysiologic processes at tissue level are derangements of general mechanisms of disease, brought about by the AGAL deficiency. Mediators of injury are genetic loci other than GLA that contribute to modulate the severity of the clinical phenotype of AGAL deficiency. Genetic loci that have minor alleles associated with increased risk of pathology in patients with FD are classified as “constitutional.” For example, IL6:c.-174G>C, NOS3:c.894G>T, FV:c.1691G>A (factor V Leiden), and PROZ:c.-13A>G polymorphisms have all been reported to be significantly associated with the presence of cerebral WMLs, whereas NOS3:c.894G>T polymorphism has additionally been positively associated with LVH.7 “Homeostatically responsive loci” are genetic loci that show dynamic adaption to the AGAL deficiency status. The selected examples of such loci are of genes that exhibit an opposite regulation trend in ERT-naïve FD patients (overexpression) compared to FD patients on ERT (under-expression), using a systems biology approach;18 the color gradient on the gene labels illustrate the homeostatic up- (red) and down-regulation (green). Biological processes in which those genes are involved include platelet degranulation (FGA, PSAP, TF); cellular response to reactive oxygen species (PSAP); acute inflammatory response (ORM1, ORM2); acute phase response (ORM1, ORM2); prostaglandin synthesis and regulation (PTGDS); metabolism of fat-soluble vitamins (GC); chemotaxis (RNASE2); and immunoglobulin receptor binding (IGHV1-46). Other major biological processes that are activated in ERT-naïve FD patients are wound healing, extracellular matrix remodeling, and peptidase activity. The AGAL protein phenotype is the closest to the genetic defect and can be described by the enzyme activity measured in a variety of assays; by the amount of protein produced; and by the molecular structure of the mutated protein. The enzyme activity is the most commonly used, clinically useful marker of the protein phenotype. The metabolic storage phenotype and the pathologic storage phenotype are different expressions of the underlying storage pathology of FD, respectively assessed by biochemical and histological/ultrastructural methods. The storage phenotypes are direct consequences of the enzyme deficiency and, together with the enzymatic phenotype, constitute the more specific diagnostic evidence of FD. In the FD pathophysiology cascade, the clinical phenotype is the most distantly linked to the genetic cause of the disease and critically influenced by gender and age. The clinical expression of FD depends not only on the major gene effect of a pathogenic GLA mutation but also on the constitutional modifier alleles present in each patient and on the intrinsic responsiveness of his/her homeostatically responsive loci; it is additionally modulated by the the subject’s multifactorial susceptibility background, resulting from polygenic additive minor gene effects and the complex interactions of this constitutional genetic susceptibility with both modifiable (eg, tobacco smoking) and nonmodifiable (eg, age, gender) environmental risk factors. (* indicates conditions that have multifactorial susceptibility in the general population). The early-onset neuropathic, dermatological, and ophthalmological manifestations of FD are the most diagnostically specific but are not observed in patients with significant residual AGAL activity. The late cerebrovascular, cardiac, and renal complications of FD are not FD-specific and their expression in different patients is modulated by the individual’s multifactorial susceptibility background. The clinical phenotype associated with a GLA hypomorphic allele may appear more severe than expected in such a patient. In the limit, a patient carrying a benign GLA variant may present with stroke, LVH, myocardial infarction, or CKD, exclusively due to his/her multifactorial background, and be erroneously diagnosed with FD, which is a major pitfall in establishing genotype-phenotype correlations in FD. Abbreviations: AGAL, α-galactosidase; CKD, chronic kidney disease; eNOS, endothelial nitric oxide synthase; ERT, enzyme replacement therapy; FD, Fabry disease; Gb3, globotriaosylceramide; LVH, left ventricular hypertrophy; lysoGb3, globotriaosylsphingosine; WMLs, white matter lesions. |
Enzymatic phenotype and the other ancillary protein phenotypes
The AGAL enzymatic phenotype is most directly correlated to the genotype in the pathophysiologic pathway of FD and the most accurate predictor of the clinical severity of FD.21 Still, the minimum level of in vivo AGAL enzyme activity required for maintaining normal lysosomal homeostasis and to keep substrate turnover within physiological ranges is not known and seems to differ between different cell types and tissues.20 Experimental data obtained in cultured skin fibroblasts suggest that lysosomal hydrolases involved in glycosphingolipid metabolism have a significant functional reserve and that substrate storage occurs only when the REA decreases below the critical threshold of 10%–15% of normal.22 For clinical purposes, AGAL activity is routinely measured in the protein extract of isolated peripheral blood leukocytes, which represents the gold standard method, as well as in whole blood, whole blood spots dried on filter paper (DBS), or in plasma,1,2,23,24 but can also be assayed in cultured skin fibroblasts,2,23 in intact leukocytes or leukocyte subpopulations,25 as well as in other biologic fluids, like urine and tears.2 Standard laboratory protocols for assaying AGAL activity in vitro are based upon the rate of cleavage of the synthetic fluorigenic substrate 4-methylumbelliferyl-α-d-galactopyranoside, usually in the presence of α-N-acetylgalactosamine, which is added to the reaction mixture to inhibit cross-reactivity with α-N-acetylgalactosaminidase (NAGAL; also known as α-galactosidase B). NAGAL activity accounts for a minor fraction of the total AGAL activity measured in healthy individuals (reportedly ranging from none up to ≈8% on leukocytes and to ≈20% on plasma),26,27 but for >40% of the total AGAL activity measured on leukocytes of FD patients with some residual AGAL activity,27 and for all the AGAL activity measured on fibroblasts of FD patients without residual AGAL activity.26 For quality control, it is recommended that another lysosomal enzyme (eg, β-galactosidase) be assayed on the same sample.23 Normal reference ranges for AGAL activity differ widely depending on the biological material used as the enzyme source as well as on the biochemical conditions of the assay, including the substrate concentration and whether an inhibitor of NAGAL is employed.28 When the enzyme assay is not available onsite, attention should be paid to proper sample processing, storage, shipping, and handling conditions.15,23,29 Furthermore, the AGAL activity measured on DBS is significantly influenced also by the blood collection technique.30 All these factors are potential confounders of the characterization of the AGAL enzymatic phenotype in individual subjects, including FD patients, and preclude direct comparisons of AGAL activity assayed in different biological samples or carried out in different laboratories. Relative AGAL activity – ie, the ratio between the enzyme activity measured in a study subject to the mean normal level, expressed as a percentage – is commonly used to stratify the severity of the enzyme deficiency in patients with FD, but cannot be accurately calculated when the enzyme testing laboratory only reports the upper and lower limits of the normal reference interval. By extrapolating data from in vitro clinical assays, the relative AGAL activity cutoff for diagnosing FD has been estimated as low as 10%31 or 15%,32 and as high as 20%1 or 30%–35%,20 whereas severe AGAL deficiency has been variably defined as <1%–2%1,31 to <5%23 of the mean normal level. Yet, these cutoffs should be interpreted with caution since it is uncertain whether the AGAL activity measured in vitro using a synthetic substrate truly reflects the in vivo activity in the affected tissues.
Since males with FD are hemizygous for a pathogenic GLA variant, with all their nucleated cells expressing the mutated protein, they can be reliably diagnosed by any of the standard AGAL enzyme assays,1,2 when the REA is unequivocally low. However, in affected females, who most typically carry a pathogenic GLA variant in heterozygosity, the AGAL enzyme phenotype ascertained in mixed or non-clonal cell populations, in tissues, or in biologic fluids, reflects the genetic mosaicism that results from X-chromosome inactivation;1,2,33 for this reason, the standard AGAL activity assays have a low diagnostic sensitivity for FD in females, with many patients exhibiting normal AGAL activities in leukocytes, DBS, or plasma,1,2,28,34,35 and the diagnosis of FD requires the identification of a pathogenic GLA variant.1,2 Nevertheless, the heterozygous status for pathogenic GLA variants can also be unequivocally diagnosed in affected females by demonstration of both normal and AGAL-deficient populations in clonal cell cultures (eg, skin fibroblasts).2
Besides by assaying the AGAL catalytic activity, the AGAL protein phenotype can be further characterized in terms of the amount of enzyme produced by a given GLA variant and by the molecular structure of the variant protein.
The immunocaptured AGAL protein concentration and enzyme activity were shown to be highly correlated in DBS and plasma samples from healthy newborns and adults, as well as in a relatively small cohort of males with FD.29 When overexpressed in vitro, no correlation between AGAL protein level and catalytic activity was observed for GLA mutations natively showing <6% REA, while for mutations associated with REA >6% the amount of protein was highly correlated with the enzyme activity.21 These data suggest that GLA mutations possessing <6% REA most likely interfere with the catalytic core of the enzyme, whereas GLA mutations possessing >6% REA most likely affect protein trafficking. Because many of the GLA mutations retaining significant REA are responsive in vitro to pharmacologic chaperones, like 1-deoxygalactonojirimycin (DGJ),21,36 their pharmacogenetic characterization has important therapeutic implications.36 Some GLA mutations that completely abolish AGAL enzyme activity despite being associated with significant AGAL protein levels are amenable to the stabilizing effect of DGJ only in terms of the amount of AGAL protein produced, without any recognizable increase of the enzyme activity.21 The rate of GLA gene transcription is modulated at the 5’UTR and two common single-nucleotide polymorphisms (SNPs) in that location, respectively, c.-30G>A and c.-10C>T, have been associated with increased plasma37,38 or decreased leukocyte AGAL enzyme activity.39–41 Coinheritance of these GLA 5’UTR SNPs with AGAL variants possessing residual catalytic activity may alter the expected enzymatic phenotype assayed in the plasma or in leukocytes.42,43 The pathogenicity of GLA mutations involving canonical and noncanonical splice sites is also critically dependent on the relative amount of aberrantly spliced AGAL mRNA in affected cells.44–46
Molecular modeling of GLA missense mutations has shown that variants compromising the active site of the enzyme; leading to broken disulfide bonds; or disrupting the hydrophobic core of the protein are associated with absent or severely deficient AGAL activity.4 With the major exception of active site residues, the amino acid residues affected in AGAL variants associated with significant REA tend to be more solvent-accessible than those associated with severe enzyme deficiency,4,21 and the mean number of main- and side-chain atoms influenced by any GLA missense mutation is also significantly higher in the latter variants.47 Knowledge of these AGAL molecular structural correlations allow clinicians to make general predictions on the severity of a given GLA mutation4 and may be particularly helpful for estimating the pathogenicity of variants of uncertain significance.
Pathology storage phenotype
Accumulation of undegraded AGAL substrates in primarily affected organs constitute the major evidence of FD. The abnormal intracellular storage of Gb3 can be either confirmed or suspected from histological, immunohistological, or ultrastructural examination of relevant tissue samples, with kidney,32,48 skin,49 and heart50 biopsies having been the most frequently used in the clinics. Whatever the tissue sample, the presence of extensive Gb3 deposits in vascular endothelial cells is considered a marker of severe AGAL deficiency.2,9,49
Since lysosomal Gb3 deposits are almost entirely extracted by tissue processing for paraffin embedding, the characteristic appearance of affected cells in tissues examined by light microscopy (LM), using standard histological preparations, is cytoplasmic vacuolization, which may be missed on routine histologic examination.48,51 However, some residual Gb3 remains in the vacuoles, which can be specifically identified by immunohistochemical staining with anti-Gb3 antibodies,6,52 even in archival paraffin-embedded tissue specimens.53 When using immunohistological methods to make the diagnosis of FD, physiological expression of Gb3 should not be mistaken as evidence of pathological Gb3 storage. This is most relevant for the diagnosis of FD nephropathy,53 because the human kidney has one of the highest contents of Gb3 in the body, with particularly high physiological expressions at the cell membranes of podocytes, cortical tubular epithelial cells, and vascular endothelial cells.54 Immunocytochemical detection of Gb3 inclusions in peripheral blood mononuclear cells,55 or in desquamated kidney tubular epithelial cells in the urinary sediment,56 as well as the morphologic identification of the tubular epithelial storage cells on examination of the urinary sediment under phase-contrast microscopy using a polarized light filter,56 has also been proposed as useful cytopathology diagnostic tests for FD.
Because the lysosomal glycosphingolipid deposits are preserved by tissue embedding for EM study, LM examination of methylene blue- or toluidine blue-stained semi-thin EM scout sections enables direct visualization of Gb3 inclusions within the affected cells of FD patients. On ultrastructural examination by transmission EM, the most distinctive pathologic feature in the affected organs of FD patients is the presence of intralysosomal osmiophilic inclusions consisting of alternating dark- and light-staining bands of variable periodicity, which are described either as “myelin figures,” when the laminated material shows a concentric appearance, or as “zebra bodies,” when they exhibit a parallel arrangement.6 Although these inclusions are not pathognomonic of FD,20 their observation in relevant tissue specimens obtained in appropriate clinical contexts confirms the clinical diagnosis of FD and has been recommended as the gold standard for the differential diagnosis of FD in adult subjects presenting with left ventricular hypertrophy (LVH) or chronic kidney disease (CKD) and a VUS in the GLA gene.57,58 Stored Gb3 in affected cells can also be specifically detected by immuno-EM methods,59 but these are not commonly available.
Metabolic storage phenotype
Biochemical evidence of increased levels of AGAL substrates in affected organs, as well as (arguably)20 in the plasma, urine, and other body fluids, can also be used as a diagnostic criterion of FD.2,14,15,20,21,23,60–65 To this end, a number of validated methods, based on different analytical techniques, including ELISA, chromatography (eg, HPLC), mass spectrometry, as well as combinations of the latter two (eg, HPLC–tandem mass spectrometry), are available for measuring Gb3, lysoGb3, and Ga2 in tissues and body fluids. Unfortunately, differences in the sensitivity of distinct assays, in the internal standards used for mass spectrometry-based quantification, in the units employed for reporting laboratory results, and in the collection method and sample processing for urine testing, hinder direct comparisons of the results provided by different laboratories.
Although Gb3 concentration in the primarily affected organs exceeds the normal level from ten to several hundred times,5 tissue biopsies are rarely obtained for Gb3 quantification. Accordingly, the plasma level and the urinary excretion of Gb3 and lysoGb3 have been the most commonly assayed FD storage products in the clinical practice. Plasma Gb3 concentration is elevated three- to fourfold within the micromolar range in most males with severe AGAL deficiency, but persists within normal levels in a substantial proportion of affected males with REA, and in the majority of females, particularly those carrying pathogenic GLA variants associated with REA.23,58,60,61,66 Urinary Gb3 excretion is consistently high in the hemizygous males and in nearly all of the heterozygous females for severe GLA mutations, but remains within normal range in variable proportions of males and in most females carrying less severe GLA mutations.60 Although the plasma and urinary Gb3 levels are poorly predictive of FD manifestations in individual patients,23,60 the urinary Gb3/creatinine ratio has been shown to correlate with the GLA genotype in both children and adults.67
In contrast to Gb3, lysoGb3 is detectable only at trace levels in the plasma of healthy individuals and is not at all detectable in normal urine samples, even when employing ultrasensitive mass spectroscopy analytical techniques.68 For this reason, lysoGb3 is better than Gb3 at discriminating between FD patients and healthy individuals. Plasma lysoGb3 is consistently elevated to high nanomolar concentrations in affected males with severe AGAL deficiency,58,62,65,66,69 and in all58,65 or most69 of the heterozygote females for severe GLA mutations; however, females carrying GLA variants associated with REA, particularly if asymptomatic, may exhibit plasma lysoGb3 concentrations within the normal range.58,66 No58 or only minimal65,69 overlapping was observed in plasma lysoGb3 values between men and women carrying severe GLA mutations, and only a small overlap exists between the values measured in males with severe AGAL deficiency and those who have REA.58,62,65,66,69,70 An empirically determined plasma lysoGb3 cutoff level has proved useful to discriminate between GLA mutations causing severe FD phenotypes, with multiple major organ involvement, from mutations associated with attenuated, later-onset clinical phenotypes, often with single-organ involvement, irrespective of patient gender and current clinical condition.62
Although the urinary excretion of Gb3 and lysoGb3 varies in parallel among FD patients,15 males with REA may have detectable lysoGb3 in the urine with Gb3 remaining within normal range.71 Since urinary Gb3 is derived primarily from desquamated tubular epithelial cells, it could be a more specific biomarker of the kidney involvement in FD, while urinary lysoGb3, which is likely filtered from the plasma by the kidneys,68 more closely reflects the systemic metabolic load of the disease.
Only two clinical studies published so far32,72 have reported data permitting to correlate the various phenotypic domains that are pertinent to either confirm the diagnosis or make the differential diagnosis of FD, including tissue Gb3 concentration in affected organs.
On the baseline characterization of male patients with FD who were enrolled in a trial of enzyme therapy, evidence of Gb3 storage was observed in toluidine blue-stained semi-thin plastic sections of the kidney biopsy of a 43-year-old proteinuric male carrying a mild GLA mutation, while his Gb3 levels in plasma, urine, and kidney tissues remained within normal range.32 This observation suggests that, at least in the kidneys, the histopathological phenotype might be the most sensitive to demonstrate the abnormal Gb3 storage in FD.
In two related males aged 41 and 47 years, and one unrelated female aged 60 years, with history of unexplained LVH, arrhythmias, and progressive heart failure (HF), which had initially manifested in early adulthood, myocardial Gb3 concentration in tissue samples obtained at the time of heart transplantation was >100-fold increased, as compared with healthy individuals and patients with obstructive hypertrophic cardiomyopathy (HCM).72 Massive vacuolization of cardiomyocytes was observed on LM examination of endomyocardial biopsies, while the EM study showed intralysosomal lamellar deposits resembling myelin bodies in cardiomyocytes, as well as vacuoles containing granular material in interstitial cells. On EM examination of diagnostic kidney biopsies obtained in one patient from each family, despite the normal renal function and absence of proteinuria, vacuoles containing lamellar and/or granular inclusions were present in parietal epithelial, tubular, and interstitial cells, but not in the podocytes. The granular material observed within vacuoles of heart and kidney cells and the absence of podocyte involvement are not features of the pathology storage phenotype of FD. Notably, all three patients had normal AGAL activity in leukocytes, as well as normal Gb3 levels in plasma and urine sediment, and the GLA gene sequencing did not reveal any pathogenic variant, which excluded the diagnosis of FD. These data suggest that the ultrastructural heart and kidney pathology storage phenotypes, together with the plasma and urine Gb3 levels, are more specific than the Gb3 tissue concentration for the differential diagnosis of FD and this rare familial Gb3-associated HCM, at least in patients with advanced HF.
Finally, the pharmacodynamic response of plasma and urinary Gb3 and lysoGb3 levels to enzyme replacement therapy,73–77 as well as to oral pharmacological chaperone therapy,78,79 is useful for assessing efficacy and monitoring treatment responses, and their reduction to (near-)normal concentrations has been proposed as a therapeutic goal for FD.80
Clinical phenotypes and their pathology correlates
Males diagnosed with FD may exhibit a wide spectrum of partial overlapping phenotypes, with several patterns of organ involvement ranging from multisystemic to heart-limited.1–3 In general, the lower the intrinsic enzyme activity of the causative AGAL mutation, the more severe is the overall clinical phenotype, the earlier is the onset of clinical manifestations, and the more extensive is the organ involvement.69 However, the wide phenotypic variability observed among patients with the same GLA mutation, even when they are related males, illustrates the complexity of the genotype-to-phenotype correlations in FD and the need to consider additional genetic and environmental modulating factors for their proper understanding.7,18
The most evocative, presenting classic features of FD, summarized in Table 1,1,2 are neuropathic pain, manifesting as either chronic intermittent acroparesthesias or episodic crises of extreme severity; multiple skin angiokeratomas; and cornea verticillata, a characteristic whorled corneal opacity that does not affect vision. These signs and symptoms typically become apparent in childhood or adolescence, but the corneal dystrophy may be initially overlooked since its diagnosis requires a high index of suspicion. Nonspecific gastrointestinal symptoms, more often abdominal pain and diarrhea, as well as impaired sweating, heat and/or cold sensitivity, exercise intolerance and easy fatigability, are additional commonly reported early-onset manifestations of classic FD. Albuminuria, usually first detected during late childhood or adolescence, is the earliest standard laboratory marker of the kidney involvement; however, protocol kidney biopsies have demonstrated the presence of Gb3 inclusions in all glomerular cell types (podocytes and endothelial and mesangial cells), as well as progressive podocyte foot process effacement, even in normoalbuminuric children.81,82 While the neuropathic pain symptoms tend to wane in adulthood, all the other early-onset signs and symptoms of classic FD persist in the long term. The natural course of classic FD is complicated by progressive CKD, ultimately leading to end-stage renal disease (ESRD), as well as by cardiac (eg, LVH, arrhythmias, ischemic heart disease) and cerebrovascular disease (eg, transient ischemic attacks, stroke), which are currently the major causes of premature morbidity and mortality.83 White matter lesions (WMLs) – ie, localized hyperintensities on brain magnetic resonance imaging T2-weighted and fluid-attenuated inversion-recovery sequences, which are considered a surrogate marker of cerebral small vessel disease84 – are present in almost half of the patients with FD, and their prevalence increases with age,69,85 with classically affected men exhibiting the highest risk.69 Since asymptomatic WMLs have been identified in school-age children and adolescents with classic FD,86 the natural history of FD-related brain microvascular disease may start early in life and follow a protracted, asymptomatic course for many years. The AGAL activity measured in classically affected males is typically null or severely reduced, but patients with REA <1% have considerably faster CKD progression rates than patients with higher REA levels.32
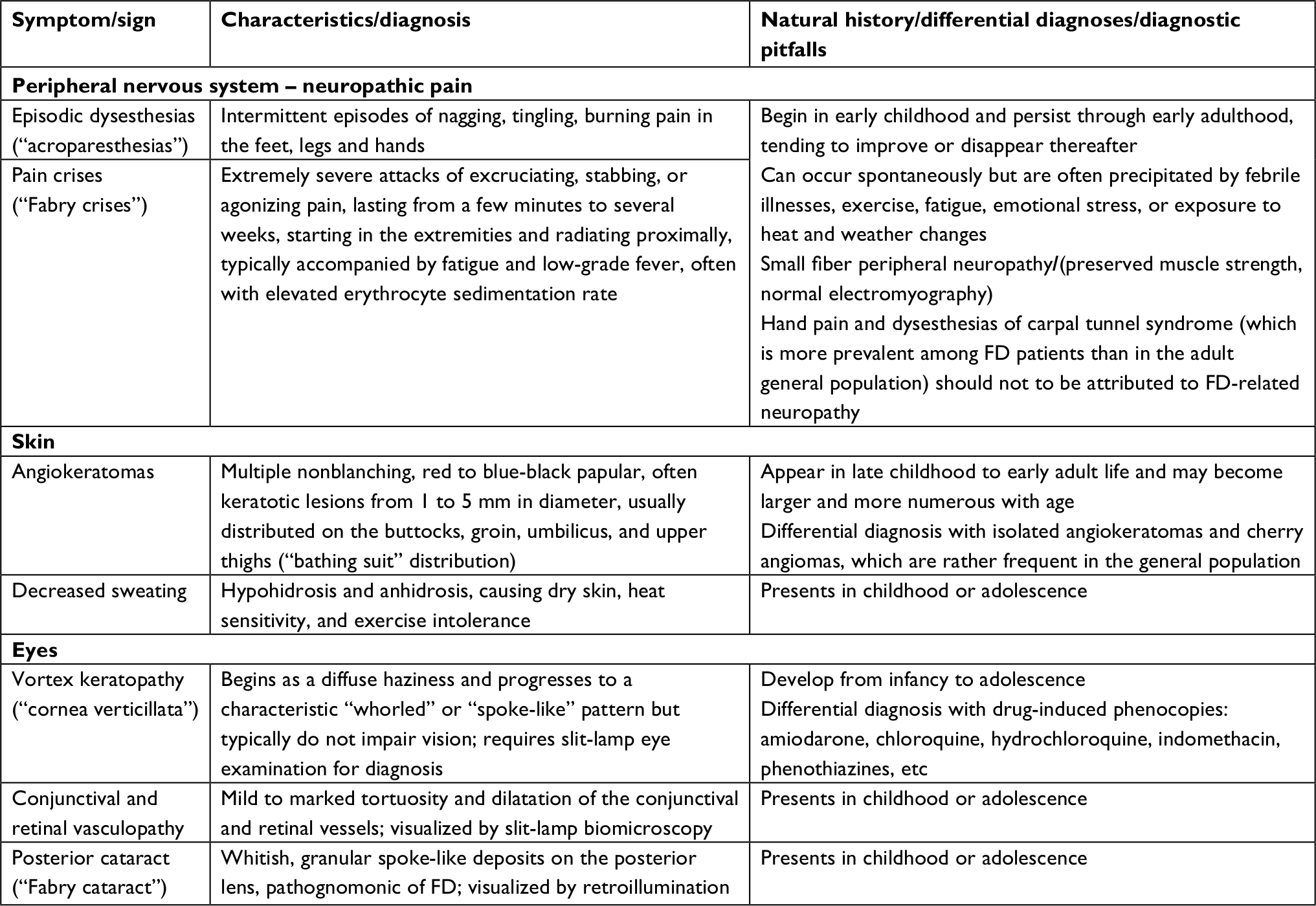 | Table 1 Cardinal presenting features of classic Fabry disease (FD) in children and adolescents |
With an estimated prevalence of 1:40,000–1:60,000 among newborn males,2 classic FD qualifies as an “ultra-rare” disorder. However, it has been more recently realized that clinically milder, later-onset phenotypic variants of FD due to GLA mutations associated with substantial REA have about ≈5–10 times higher prevalence in the general population than the classic phenotype.1,83 Such affected males usually lack the manifestations of early neuropathic, cutaneous, and ocular involvement that characterize classic FD, but over time develop cardiac complications, which becomes clinically noticeable only in the fifth to eighth decades of life, when patients with classic FD would be severely affected or would have died. Some of these patients have coexisting evidence of kidney involvement, usually mild-to-moderate proteinuria which, in some cases, is associated with slowly deteriorating renal function; the observation of significantly increased levels of urinary biomarkers of glomerular (especially type IV collagen) and tubular (especially N-acetyl-beta-glucosaminidase) dysfunction in adult patients with normal renal function and normoalbuminuria suggests that they may be biochemical flags of an incipient stage in the natural history of the kidney involvement in FD.87 Tissue biopsies or autopsy studies of patients presenting with this primarily cardiac phenotype (“cardiac variant”) showed extensive Gb3 storage in cardiomyocytes and, in the subset of patients with kidney involvement, also in podocytes, but with no or only minimal Gb3 accumulation in other kidney cell types; notably, the vascular endothelium was spared in both organs.2 The leukocyte AGAL activity measured in males with FD cardiac variants is most often in the range of ≈3%–10% of the normal mean. Still other patients, who had not experienced any of the early-onset symptoms and signs of classic FD, develop a predominantly renal phenotype (“renal variant”), with progressive CKD in early adulthood, culminating in ESRD in the fourth to sixth decades of life.1,2 Since the severe renal complications precede clinically overt cardiac disease in these patients, the observation of mild-to-moderate LVH in young adults presenting with CKD of uncertain etiology may be a diagnostic clue to FD. The leukocyte AGAL activity measured in males with FD renal variants is usually in the range of ≈1%–5% of the normal mean, and their glomerular kidney pathology is similar to that described in classic FD nephropathy,88 with Gb3 accumulation in all types of glomerular cells, including in the capillary endothelium.51 Yet, the dichotomous classification of the phenotypic spectrum of FD into “classic” and “atypical” or “nonclassic” forms has also been advocated.62,69
Taken together, these data suggest that different cells have different cutoffs of AGAL activity sufficiency, with cardiomyocytes and podocytes becoming AGAL-insufficient at relatively higher REA levels than the vascular endothelial cells. It is also possible that higher cell turnover rates are a protective factor against the build-up of Gb3 in susceptible cells, contributing to the lower susceptibility of vascular endothelia compared to that of terminally differentiated cells, like cardiomyocytes, podocytes, and neurons.
Although many heterozygous females may remain asymptomatic throughout their lives, a significant percentage develops vital organ complications, with only a minority exhibiting multisystemic clinical phenotypes as severe as those observed in classically affected males.1–3 Affected females from families with classic FD tend to have more organ-limited disease, with later clinical onset than their male counterparts, but >70% develop cornea verticillata,2,80 regardless of whether they have additional FD manifestations. Overall, women with classic FD have a higher risk of developing complications compared with women with nonclassic FD.69 The prevalence of WMLs in females with FD is comparable to that in males.85 In a large FD registry study, about 25% of the participant females have had a severe cerebrovascular, cardiac, or renal event before the seventh decade of life.89 Although the affected females experienced first stroke or significant cardiac complications in the early fifth to mid-sixth decades of life (ie, about 5–10 years later than males), ESRD was reached at the median age of 38 years in both genders. Overall, the life-expectancy of untreated males and females with FD is respectively reduced to ~50 and 70 years.1
Other common later-onset, nondiagnostic manifestations of FD
Besides the cardiac, cerebrovascular, and renal complications that constitute the major prognostic determinants of life expectancy in patients with FD, other nonspecific symptoms are commonly observed in affected adults which are not life-threatening but may significantly contribute to their considerably lower health-related quality of life, as compared to the general population.90
Aside from the small fiber neuropathy and cerebrovascular disease, FD is associated with neuropsychological handicap in several domains, including executive functioning, information processing speed, and attention; with mild impairment of motor performance, which gets worse with increasing age; with affective disorders, including depression; with daytime sleepiness; and with decreased health-related quality of life.91,92 Prevalence rates of clinically significant depression in adults with FD have ranged from 15% to 62%, with males reporting severe depression more often than females (36% vs 22%).93 Depressive symptoms within the clinical range have also been reported by teenagers with FD.94 Currently available data suggest that depression in FD patients, rather than organic in nature, is a reflection of the degree to which clinical symptoms interfere with daily life: in keeping with this concept, the most common factor associated with depression is neuropathic pain.91
Hearing loss, tinnitus, and vertigo are more prevalent in patients with FD than in the age-matched general population,1,2,95 but the underlying etiology remains unclear in many patients.96 The age onset of FD-related neuro-otologic symptoms is earlier, and their clinical severity is higher in hemizygous males than in heterozygous females. In a large registry-based study, clinical evidence of cochlear or vestibular dysfunction was present in more than half of the males since 40 years of age, and in more than half of the females after the sixth decade of life.95 Tinnitus and hearing loss may be early symptoms in children with classic FD1,2 and are also more prevalent among patients with later-onset FD variants than in the age-matched general population.97 In most cases, hearing loss is sensorineural and slowly progressive, but the risk of sudden hearing loss is also increased in FD patients.
The overall prevalence of gastrointestinal symptoms in patients with FD, including abdominal pain, bloating, nausea, constipation and diarrhea, is higher in children than in adults,98 and abdominal pain and/or diarrhea may be the initial manifestation of classic FD in children (reported by 23.2% of the boys and 11.4% of the girls in a registry-based study).99 However, up to 28% of adult participants in a FD registry complained of significant abdominal pain, and up to 19.2% complained of diarrhea.98 Major pathophysiological mechanisms thought to contribute to the gastrointestinal symptoms of FD are the dysfunction of the autonomic nervous system responsible for gut motility; the vasculopathy affecting the gastrointestinal circulation; and secondary tissue inflammation.100,101 Glycosphingolipid storage in enteric neurons controlling gut motility and in ganglion cells of the submucosal and the myenteric plexuses, as well as in endothelial and smooth muscle cells of the mesenteric microvasculature, including vessels supplying the enteric nervous system, ultimately leading to ischemic damage, are the relevant pathology correlates.100,101
A major clinical dilemma posed by the nonspecific manifestations of FD is whether they are indeed causally related to FD or, instead, are comorbid conditions arising by chance in FD patients. When the underlying diagnosis is not suspected, true FD-related complications are likely to be overlooked as diagnostic clues, while the erroneous interpretation of concurrent common complex disorders as FD-related is more likely to occur in patients with either a confirmed or suspected diagnosis of FD.
Genetic pathology of FD and clinical approach to establishing genotype–phenotype correlations
The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GLA) currently reports more than 900 GLA gene variants, of which ≈75% are point mutations, most of them classified as pathogenic. Nonsense point mutations (comprising ≈14% of the known GLA point mutations) and point mutations affecting the GLA mRNA splicing (comprising ≈6% of all known GLA point mutations), as well as all the non-point mutations – ie, small and gross insertions, deletions, and duplications, as well as complex rearrangements – are invariably pathogenic and should not pose major problems in the characterization of their phenotypic correlations. The consequences of GLA missense mutations are very much dependent on their location and impact on the tridimensional structure of the AGAL protein,4 with those affecting the enzyme active site causing particularly severe AGAL deficiency. Although most of the known GLA missense mutations are classified as pathogenic, some behave as hypomorphic alleles (perhaps contributing additively to the multifactorial risk of cerebrovascular disease as metabolic modulators),42,43 and others are entirely benign polymorphisms.20
Comprehensive phenotyping is, therefore, crucial for grading the severity of pathogenic GLA variants, to clarify the phenotypic correlations of hypomorphic alleles, to define benign polymorphism, as well as to establish the pathogenicity of allelic VUS. Whenever possible, this phenotypic characterization should be carried out in hemizygous males, if necessary identified prospectively on cascade family screening. Diffuse angiokeratomas102 and typical cornea verticillata80,103 in adults are highly predictive clinical signs for the diagnosis of classic FD, but have also been observed in patients with later-onset FD variants103 or carrying hypomorphic GLA variants.43 Contrastingly, quantitative sensory testing and intraepidermal nerve fiber density cannot reliably distinguish patients with FD from those without FD.104 Isolated or localized angiokeratomas and cherry haemangiomas,102 as well as carpal tunnel syndrome-related hand pain and dysesthesias,80 which are common conditions in the general adult population, should not be misdiagnosed as clinical evidence of FD: this can be minimized by taking into account the childhood- or adolescent-onset of the dermatological and neuropathic manifestations of classic FD. Same caution should be applied to the differential diagnosis of drug-induced phenocopies of cornea verticillata,105 which can be ruled out by a thorough medication history. In some cases, definitive diagnosis of FD will require biochemical20 or histopathological evidence57,58 of abnormal Gb3 accumulation in an affected organ.
A major pitfall in the clinical phenotyping of patients with FD is the differential diagnosis between its major but nonspecific later-onset cerebrovascular, cardiac, and renal disease complications and similar chronic illnesses that are common in the general population and may be superimposed on FD (see Table 2 for epidemiologic data on common complex disorders that are potential confounder phenocopies of FD manifestatons).106–114 In contrast to FD, atherosclerotic and hypertensive cerebrovascular106 and cardiac diseases,115,116 as well as CKD,108 which are highly prevalent conditions among adults and seniors, have multifactorial causation determined by polygenic inheritance,117–120 together with the complex interactions of the constitutional genetic susceptibility with both modifiable and nonmodifiable environmental risk factors. The same holds true for depression;121 hearing loss;122 functional gastrointestinal disorders,123 especially irritable bowel syndrome,124 which have many overlapping symptoms with FD-related gastrointestinal manifestations; and for WMLs,125 whose prevalence increases with aging, particularly in individuals with cardiovascular risk factors, predicting a higher risk of stroke, vascular cognitive impairment, dementia, and death.84 In younger patients, the differential diagnosis between the early clinical stages of multiple sclerosis and FD may be clinically challenging because of the similarities of the respective neuroradiological features.126,127
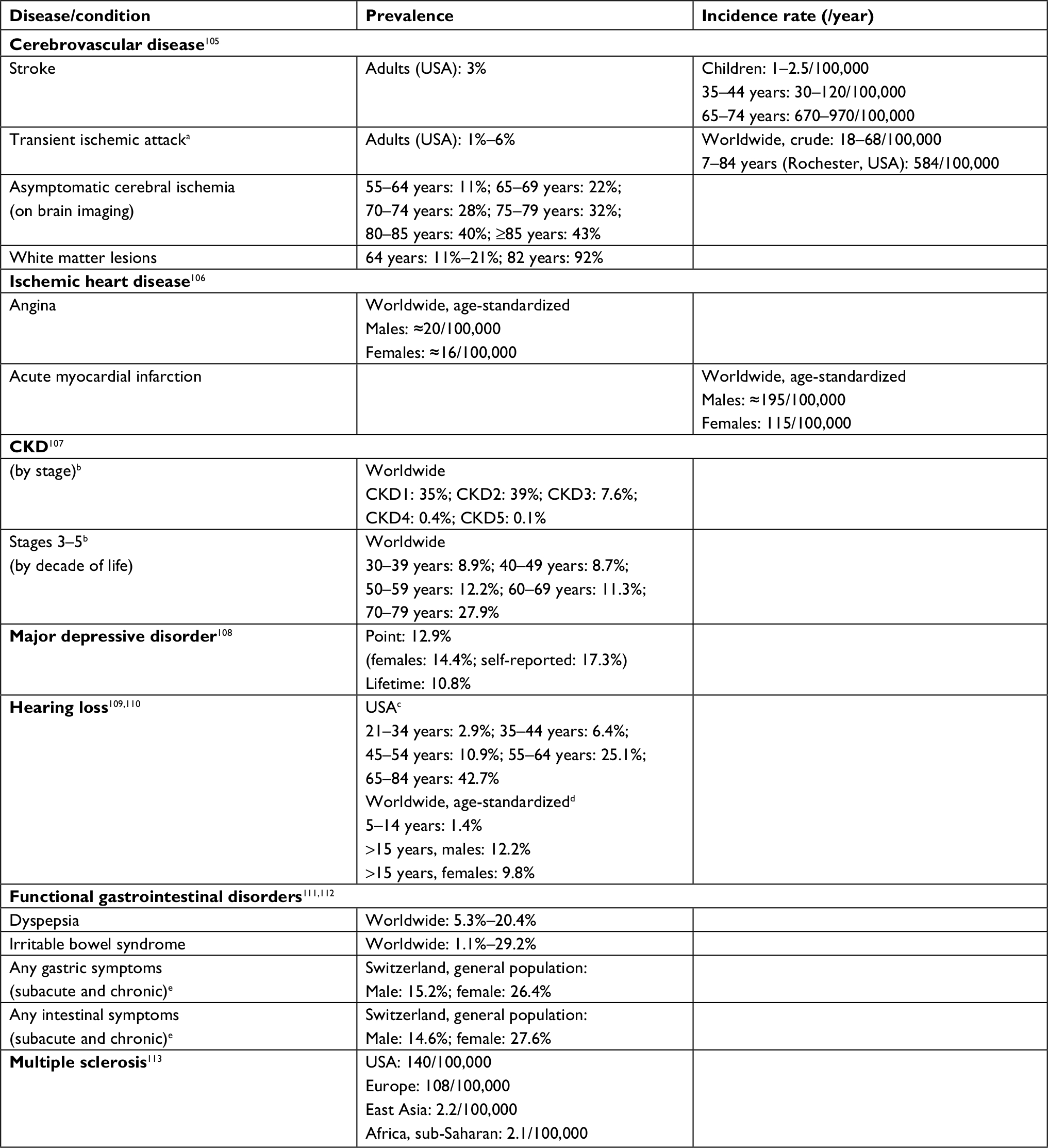 | Table 2 Epidemiologic data on the common complex diseases that can present as phenocopies of nonspecific Fabry disease complications Notes: aTIA classically defined as “a temporary abrupt-onset neurological deficit due to brain or retinal ischemia lasting less than 24 hours in duration;” applying the revised tissue-based definition of “transient episode of neurological dysfunction caused by focal brain, spinal cord, or retinal ischemia, without acute infarction,” the TIA decreased by 33%, increasing the rates of stroke by this amount. bCKD stages were defined on the basis of eGFR (expressed in mL/min/1.73m2) and UACR (expressed as mg albumin/g creatinine) as follows: stage 1, eGFR >90 + UACR >30; stage 2, eGFR 60–89 + UACR >30; stage 3, eGFR 30–59; stage 4, eGFR 29–15; and stage 5, eGFR <15. Hearing level was calculated as the better ear hearing threshold in decibels averaged over frequencies 0.5, 1.0, 2.0, and 4.0 kHz; cHearing impairment defined as a pure-tone average hearing level >25 dB in the worse ear; dHearing impairment defined as a pure-tone average hearing level ≥35 dB in the better ear. eDuration of gastrointestinal symptoms defined as subacute, when persisting for ≥1 week, and chronic, when persisting for ≥3 months. Abbreviations: CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; TIA, transient ischemic attack; UACR, urinary albumin/creatinine ratio. |
In males, inheritance of a pathogenic GLA variant is a sufficient condition for the development of a clinical phenotype of FD, including of one or more of its later-onset major complications. However, the individual polygenic heritage may have a major role in modulating the severity of the late clinical expression of FD among patients carrying the same pathogenic GLA variant. In addition, the patient’s inherited polygenic background and his interacting environmental risks may be the critical factor for the pathogenic expression of some hypomorphic GLA gene alleles, whose deleterious effects upon the AGAL-dependent glycosphingolipid catabolism are not severe enough to cause FD, unless in the additive context of a predisposing multifactorial risk.42 Furthermore, the cerebrovascular, cardiac, or renal disease coexisting in patients carrying a GLA gene variant may be entirely dependent on the subject’s multifactorial risk, and eventually be misdiagnosed as evidence of FD.
Besides LVH secondary to hypertensive heart disease and to other abnormal cardiac loading conditions, familial HCM is also a much more frequent cause of LVH than FD.128 Familial HCM has an estimated prevalence of at least 1:500 in the adult population, and in up to 60% of the cases the disease is an autosomal dominant trait caused by mutations in cardiac sarcomere protein genes. Hence, in patients presenting with isolated LVH, evidence for autosomal dominant inheritance in extended family history (particularly, male-to-male transmission) will exclude FD as the underlying diagnosis. Cardiac amyloidosis and mitochondrial cytopathies are rarer causes of LVH that, in appropriate clinical contexts, should be considered in the differential diagnosis of cardiac FD.129
The abovementioned epidemiologic data should be taken into due account in clinical practice and when interpreting pooled data from large observational FD registries, such as the “Fabry Outcome Survey” (https://clinicaltrials.gov/ct2/show/NCT03289065) and the “Fabry Registry” (https://clinicaltrials.gov/ct2/show/NCT00196742), but they are most relevant when assigning a pathogenic role to novel GLA variants or VUS, especially those identified in the context of case-finding studies among high-risk patient groups,83,130 as well as in newborn screening programs.83,131 Since testing for FD in high-risk patient populations is intrinsically biased with regard to the clinical manifestations, neglecting comprehensive phenotyping workup of probands and their affected relatives may lead to falsely attributing causality to hypomorphic alleles or benign GLA polymorphisms.42,43,71,132
Conclusion
Knowledge of the phenotypic correlations of the whole range of known GLA gene variants and elucidation of the pathophysiologic mechanisms linking the genetic mutations to their clinical manifestations are of paramount importance for all the stakeholders involved in the delivery of health care to FD patients and their families. These include the molecular genetics laboratories – that should incorporate on their clinical reports updated information on the genotype-phenotype correlations of any GLA gene variant they identify – as well as the pharmaceutical industry.
The flowcharts in Figure 2 summarize the key decision-making processes in FD diagnosis, according to the circumstances of patient ascertainment and the selected laboratory testing. Genotype-phenotype correlations provide clinicians with essential data on which to base major therapeutic decisions, the prognostic information to be discussed with patients and their at-risk relatives, and the preconceptional counseling. By ensuring the comprehensive characterization of their FD patients, keeping in mind the multiple phenotypic domains of the disease and the full spectrum of its clinical manifestations and their differential diagnoses, medical practitioners are uniquely qualified to contribute the necessary data to establish well-grounded genotype–phenotype correlations.
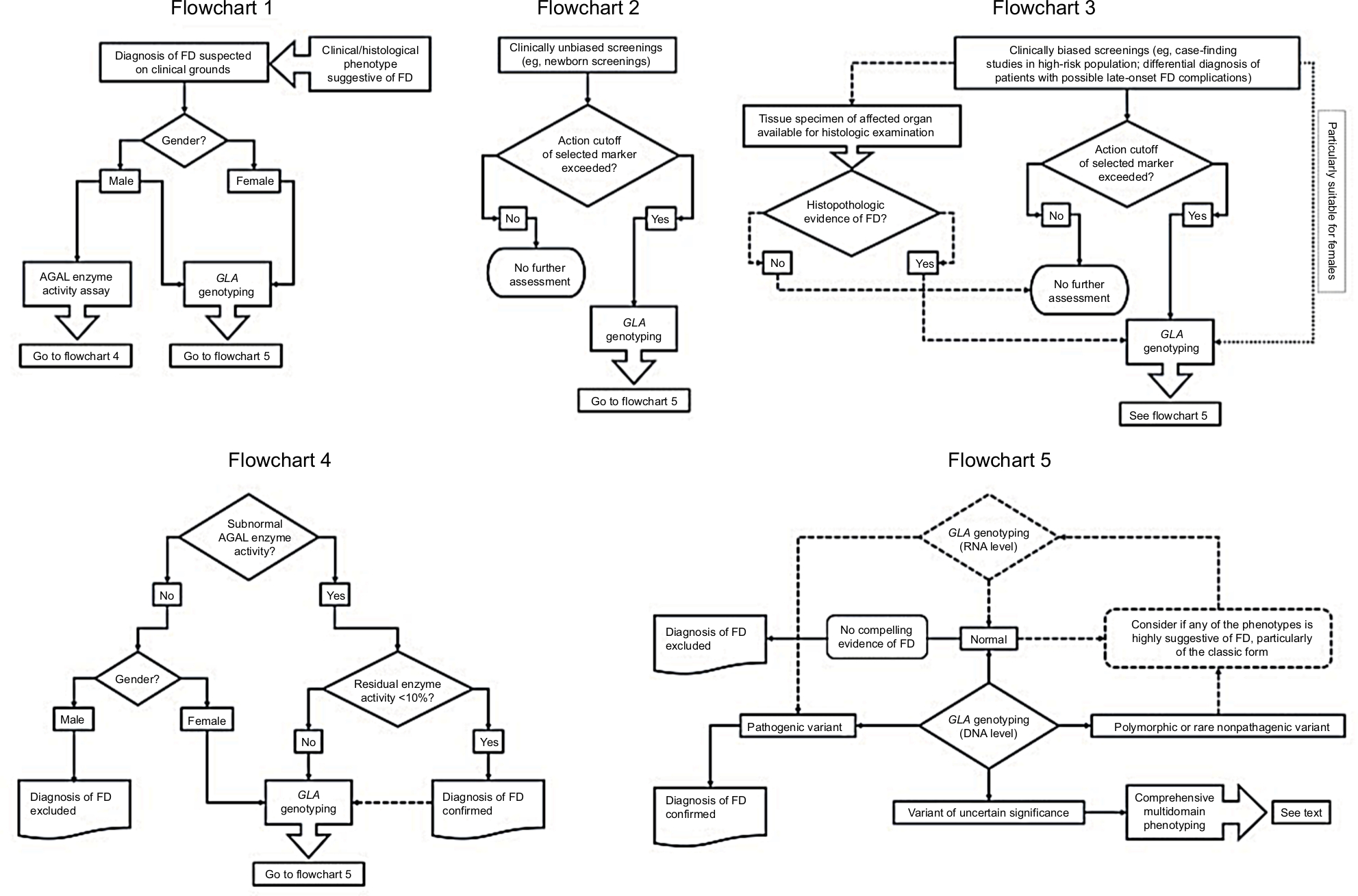 | Figure 2 Clinical decision-making processes in diagnosis of Fabry disease (FD) accounting for patient ascertainment and selected laboratory testing. Notes: Flowchart 1 – Standard algorithm for confirmation of the diagnosis of FD in patients with suggestive clinical or histological phenotypes (solid lines). The clinical or histological diagnosis of FD in males can be confirmed either by α-galactosidase (AGAL) activity assay or by GLA genotyping. Contrastingly, the identification of a pathogenic GLA variant is critical to confirm the diagnosis of FD in females, since the diagnostic sensitivity of routine AGAL assays is negatively influenced by the effect of random X-chromosome inactivation. Flowchart 2 – Standard algorithm for the diagnosis FD in clinically unbiased screenings (solid lines). If the predefined action cutoff value of the marker used for population screening (eg, AGAL activity assay or a stored glycosphingolipid) is not reached, no further assessment is required; however, positive results must always be confirmed by GLA genotyping. Flowchart 3 – Standard algorithm for the diagnosis of FD in clinically biased screenings (solid lines). Most of these studies have used AGAL activity assays as screening tool and only a few have used GLA genotyping as first-tier (dotted line); the latter approach is especially suitable for females. Availability of either archival or prospectively obtained tissue specimens for histopathological examination can be of great help to confirm or exclude the diagnosis of FD (dashed lines). Flowchart 4 – Standard algorithm for interpretation of AGAL enzyme activity results (solid lines). Normal AGAL enzyme activity excludes the diagnosis of FD in males but not in females, since heterozygotes for pathogenic GLA variants frequently exhibit normal AGAL activity. Residual AGAL activity less than about 10% of the normal mean confirms FD diagnosis in both genders, while subnormal results above that level call for GLA genotyping and detailed phenotypic profiling, particularly in women and individuals carrying GLA gene variants that modulate the enzymatic phenotype (eg, the GLA c.-10C>T single-nucleotide polymorphism in the 5’UTR). For establishing comprehensive genotype–phenotype correlations, GLA genotyping is recommended even in patients diagnosed by the enzyme assay (dashed line). Flowchart 5 – Standard algorithm for interpretation of GLA genotyping results (solid lines). GLA genotyping can be carried out in genomic DNA (gDNA) and/or in mRNA samples, which are represented in the flowchart by solid and dashed lozenges, respectively. Although rarely used as first-tier, the latter approach (dashed lines) is particularly helpful as a second-tier GLA genotyping method, when the patient’s phenotype is highly suggestive of FD but gDNA-based genotyping was nondiagnostic. In such cases, comprehensive gDNA-based genotyping should include the use of laboratory techniques (eg, multiplex ligation-dependent probe amplification – MLPA) enabling the detection of small and gross insertions, deletions, duplications, or other complex GLA variants, which are not identifiable by routine DNA sequencing methods. Detection of a pathogenic GLA variant, either in gDNA or mRNA assays, establishes the diagnosis of FD, irrespective of the patient’s gender. In patients without additional compelling evidence of FD (eg, significantly reduced AGAL activity; histopathological evidence of globotriaosylceramide storage), a normal GLA genotyping result at gDNA level excludes the diagnosis; this assumption also applies to a normal genotyping result at mRNA level in males. In females, however, a normal genotyping result at mRNA level may be misleading, due to the possibility of totally skewed X-chromosome inactivation in the source cells (eg, peripheral blood leukocytes), concealing the expression of the pathogenic GLA variant, and caution should be applied when using these data to exclude the diagnosis of FD. In such cases, GLA genotyping at gDNA level will be required. Detection of variants of uncertain significance requires a comprehensive multidomain phenotyping to ascertain their pathogenicity. |
Acknowledgments
We thank Dr Bojan Vujkovac (Slovenj Gradec, Slovenia) for his helpful review of and comments to the content of Figure 1 – the pathophysiologic pathways in FD. We thank the anonymous reviewers of the original draft of this manuscript for their contributions to the improvement of the final version.
Disclosure
João Paulo Oliveira is member of the European Advisory Board of the Fabry Registry, a global observational registry of patients with FD sponsored by Genzyme Corporation. He has received unrestricted research grants and funding for research projects from Genzyme Corporation; consulting honoraria and speaker’s fees from Genzyme Corporation; and conference registration fees and travel grants from Genzyme Corporation, Shire Human Genetic Therapies, and Amicus Therapeutics. Susana Ferreira has received unrestricted research grants and funding for research projects from Genzyme, A Sanofi Division; and conference registration fees and travel grants from Genzyme, A Sanofi Division, and Shire Human Genetic Therapies.
References
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5(1):30. | ||
Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Valle D, Beaudet AL, Vogelstein B, et al, editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2014. | ||
Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–427. | ||
Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337(2):319–335. | ||
Hřebíček M, Ledvinová J. Biochemistry of Fabry Disease. In: Elstein D, Altarescu G, Beck M, editors. Fabry Disease. 1st ed. Dordrecht: Springer Science+Business Media B.V.; 2010:81–104. | ||
Elleder M. Subcellular, cellular and organ pathology of Fabry disease. In: Elstein D, Altarescu G, Beck M, editors. Fabry Disease. 1st ed. Dordrecht: Springer Science+Business Media B.V.; 2010:39–79. | ||
Schiffmann R, Disease F. Fabry disease. Pharmacol Ther. 2009;122(1):65–77. | ||
Das AM, Naim HY. Biochemical basis of Fabry disease with emphasis on mitochondrial function and protein trafficking. Adv Clin Chem. 2009;49:57–71. | ||
Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010;99(2):99–108. | ||
Chévrier M, Brakch N, Céline L, et al. Autophagosome maturation is impaired in Fabry disease. Autophagy. 2010;6(5):589–599. | ||
Weidemann F, Sanchez-Niño MD, Politei J, et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013;8(1):116. | ||
Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122(3):19–27. | ||
Eikrem Ø, Skrunes R, Tøndel C, et al. Pathomechanisms of renal Fabry disease. Cell Tissue Res. 2017;369(1):53–62. | ||
Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci USA. 2008;105(8):2812–2817. | ||
Auray-Blais C, Ntwari A, Clarke JTR, et al. How well does urinary lyso-Gb3 function as a biomarker in Fabry disease? Clin Chim Acta. 2010;411(23–24):1906–1914. | ||
Sanchez-Niño MD, Carpio D, Sanz AB, Ruiz-Ortega M, Mezzano S, Ortiz A. Lyso-Gb3 activates Notch1 in human podocytes. Hum Mol Genet. 2015;24(20):5720–5732. | ||
Choi L, Vernon J, Kopach O, et al. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci Lett. 2015;594:163–168. | ||
Fernandes M, Husi H. Integrative systems biology investigation of Fabry disease. Diseases. 2016;4(4):35. | ||
Dobyns WB, Filauro A, Tomson BN, et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A. 2004;129A(2):136–143. | ||
Schiffmann R, Fuller M, Clarke LA, Aerts JM. Is it Fabry disease? Genet Med. 2016;18(12):1181–1185. | ||
Lukas J, Giese AK, Markoff A, et al. Functional characterisation of alpha-galactosidase A mutations as a basis for a new classification system in Fabry disease. PLoS Genet. 2013;9(8):e1003632. | ||
Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88(5):513–523. | ||
Gal A, Hughes DA, Winchester B. Toward a consensus in the laboratory diagnostics of Fabry disease: recommendations of a European expert group. J Inherit Metab Dis. 2011;34(2):509–514. | ||
Massaccesi L, Burlina A, Baquero CJ, Goi G, Burlina AP, Tettamanti G. Whole-blood alpha-D-galactosidase A activity for the identification of Fabry’s patients. Clin Biochem. 2011;44(10–11):916–921. | ||
Hölzl MA, Gärtner M, Kovarik JJ, et al. Quantification of α-galactosidase activity in intact leukocytes. Clin Chim Acta. 2010;411(21–22):1666–1670. | ||
Mayes JS, Scheerer JB, Sifers RN, Donaldson ML. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin Chim Acta. 1981;112(2):247–251. | ||
Andrade J, Waters PJ, Singh RS, et al. Screening for Fabry disease in patients with chronic kidney disease: limitations of plasma alpha-galactosidase assay as a screening test. Clin J Am Soc Nephrol. 2008;3(1):139–145. | ||
Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338–346. | ||
Fuller M, Lovejoy M, Brooks DA, Harkin ML, Hopwood JJ, Meikle PJ. Immunoquantification of alpha-galactosidase: evaluation for the diagnosis of Fabry disease. Clin Chem. 2004;50(11):1979–1985. | ||
Olivova P, der Veen Kvan, Cullen E, et al. Effect of sample collection on α-galactosidase A enzyme activity measurements in dried blood spots on filter paper. Clin Chim Acta. 2009;403(1–2):159–162. | ||
Desnick RJ, Banikazemi M, Wasserstein M. Enzyme replacement therapy for Fabry disease: an inherited nephropathy. Clin Nephrol. 2002;57(1):1–8. | ||
Branton MH, Schiffmann R, Sabnis SG, et al. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine. 2002;81(2):122–138. | ||
Echevarria L, Benistan K, Toussaint A, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89(1):44–54. | ||
Linthorst GE, Vedder AC, Aerts JMFG, Hollak CEM. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin Chim Acta. 2005;353(1–2):201–203. | ||
Pasqualim G, Simon L, Sperb-Ludwig F, et al. Fabry disease: a new approach for the screening of females in high-risk groups. Clin Biochem. 2014;47(7–8):657–662. | ||
Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430–438. | ||
Saifudeen Z, Desnick RJ, Ehrlich M. A mutation in the 5’ → untranslated region of the human alpha-galactosidase A gene in high-activity variants inhibits specific protein binding. FEBS Lett. 1995;371(2):181–184. | ||
Fitzmaurice TF, Desnick RJ, Bishop DF. Human alpha-galactosidase A: high plasma activity expressed by the -30G>A allele. J Inherit Metab Dis. 1997;20(5):643–657. | ||
Oliveira JP, Ferreira S, Reguenga C, Carvalho F, Månsson JE. The g.1170C>T polymorphism of the 5′ untranslated region of the human alpha-galactosidase gene is associated with decreased enzyme expression-evidence from a family study. J Inherit Metab Dis. 2008;31(Suppl 2):405–413. | ||
Oliveira JP, Ferreira S, Barceló J, et al. Effect of single-nucleotide polymorphisms of the 5′ untranslated region of the human α-galactosidase gene on enzyme activity, and their frequencies in Portuguese Caucasians. J Inherit Metab Dis. 2008;31(S2):247–253. | ||
Gervas-Arruga J, Cebolla JJ, Irun P, et al. Increased glycolipid storage produced by the inheritance of a complex intronic haplotype in the α-galactosidase A (GLA) gene. BMC Genet. 2015;16(1):109. | ||
Baptista MV, Ferreira S, Pinho-E-Melo T, et al. Mutations of the GLA gene in young patients with stroke: the PORTYSTROKE study-screening genetic conditions in Portuguese young stroke patients. Stroke. 2010;41(3):431–436. | ||
Ferreira S, Ortiz A, Germain DP, et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: data from individual patients and family studies. Mol Genet Metab. 2015;114(2):248–258. | ||
Ishii S, Nakao S, Minamikawa-Tachino R, Desnick RJ, Fan JQ. Alternative splicing in the alpha-galactosidase A gene: increased exon inclusion results in the Fabry cardiac phenotype. Am J Hum Genet. 2002;70(4):994–1002. | ||
Lai LW, Whitehair O, Wu MJ, O’Meara M, Lien YH. Analysis of splice-site mutations of the alpha-galactosidase A gene in Fabry disease. Clin Genet. 2003;63(6):476–482. | ||
Filoni C, Caciotti A, Carraresi L, et al. Unbalanced Gla mRNAs ratio quantified by real-time PCR in Fabry patients’ fibroblasts results in Fabry disease. Eur J Hum Genet. 2008;16(11):1311–1317. | ||
Matsuzawa F, Aikawa S, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in alpha-galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005;117(4):317–328. | ||
Fogo AB, Bostad L, Svarstad E, et al. Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry nephropathy (ISGFN). Nephrol Dial Transplant. 2010;25(7):2168–2177. | ||
Liguori R, Incensi A, de Pasqua S, et al. Skin globotriaosylceramide 3 deposits are specific to Fabry disease with classical mutations and associated with small fibre neuropathy. PLoS One. 2017;12(7): e0180581. | ||
Pieroni M, Chimenti C, De Cobelli F, et al. Fabry’s disease cardiomyopathy: echocardiographic detection of endomyocardial glycosphingolipid compartmentalization. J Am Coll Cardiol. 2006;47(8):1663–1671. | ||
Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64(3):801–807. | ||
Askari H, Kaneski CR, Semino-Mora C, et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007;451(4):823–834. | ||
Valbuena C, Leitão D, Carneiro F, Oliveira JP. Immunohistochemical diagnosis of Fabry nephropathy and localisation of globotriaosylceramide deposits in paraffin-embedded kidney tissue sections. Virchows Arch. 2012;460(2):211–221. | ||
Ergonul Z, Clayton F, Fogo AB, Kohan DE. Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr Nephrol. 2003;18(3):246–253. | ||
Üçeyler N, Böttger J, Henkel L, et al. Detection of blood Gb3 deposits as a new tool for diagnosis and therapy monitoring in patients with classic Fabry disease. J Intern Med. 2018;284(4):427–438. | ||
Selvarajah M, Nicholls K, Hewitson TD, Becker GJ. Targeted urine microscopy in Anderson-Fabry disease: a cheap, sensitive and specific diagnostic technique. Nephrol Dial Transplant. 2011;26(10):3195–3202. | ||
Smid BE, van der Tol L, Cecchi F, et al. Uncertain diagnosis of Fabry disease: consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol. 2014;177(2):400–408. | ||
Smid BE, van der Tol L, Biegstraaten M, Linthorst GE, Hollak CE, Poorthuis BJ. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet. 2015;52(4):262–268. | ||
Kanekura T, Fukushige T, Kanda A, et al. Immunoelectron-microscopic detection of globotriaosylceramide accumulated in the skin of patients with Fabry disease. Br J Dermatol. 2005;153(3):544–548. | ||
Winchester B, Young E. Laboratory diagnosis of Fabry disease. In: Elstein D, Altarescu G, Beck M, editors. Fabry Disease. 1st ed. Dordrecht: Springer Science+Business Media B.V.; 2010:111–132. | ||
Aerts JM, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J Inherit Metab Dis. 2011;34(3):605–619. | ||
Niemann M, Rolfs A, Störk S, et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet. 2014;7(1):8–16. | ||
Boutin M, Menkovic I, Martineau T, Vaillancourt-Lavigueur V, Toupin A, Auray-Blais C. Separation and analysis of lactosylceramide, galabiosylceramide, and globotriaosylceramide by LC-MS/MS in urine of Fabry disease patients. Anal Chem. 2017;89(24):13382–13390. | ||
Maruyama H, Miyata K, Mikame M, et al. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet Med. 2019;21(1):44–52. | ||
Duro G, Zizzo C, Cammarata G, et al. Mutations in the Gla gene and LysoGb3: is it really Anderson-Fabry disease? Int J Mol Sci. 2018;19(12):3726. | ||
Liao HC, Huang YH, Chen YJ, et al. Plasma globotriaosylsphingosine (lysoGb3) could be a biomarker for Fabry disease with a Chinese hotspot late-onset mutation (IVS4+919G>A). Clin Chim Acta. 2013;426:114–120. | ||
Auray-Blais C, Cyr D, Ntwari A, et al. Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. Mol Genet Metab. 2008;93(3):331–340. | ||
Gold H, Mirzaian M, Dekker N, et al. Quantification of globotriaosylsphingosine in plasma and urine of Fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin Chem. 2013;59(3):547–556. | ||
Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. 2017;28(5):1631–1641. | ||
Alharbi FJ, Baig S, Auray-Blais C, et al. Globotriaosylsphingosine (Lyso-Gb3) as a biomarker for cardiac variant (N215S) Fabry disease. J Inherit Metab Dis. 2018;41(2):239–247. | ||
Ferreira S, Auray-Blais C, Boutin M, et al. Variations in the Gla gene correlate with globotriaosylceramide and globotriaosylsphingosine analog levels in urine and plasma. Clin Chim Acta. 2015;447:96–104. | ||
Apelland T, Gude E, Strøm EH, et al. Familial globotriaosylceramide-associated cardiomyopathy mimicking Fabry disease. Heart. 2014;100(22):1793–1798. | ||
Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345(1):9–16. | ||
Schiffmann R, Kopp JB, Austin HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. | ||
van Breemen MJ, Sm R, Dekker N. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta. 1812;2011(1):70–76. | ||
Rombach SM, Aerts JM, Poorthuis BJ, et al. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One. 2012;7(10):e47805. | ||
Vedder AC, Linthorst GE, Houge G, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One. 2007;2(7):e598. | ||
Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III attract study. J Med Genet. 2017;54(4):288–296. | ||
Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry’s disease with the pharmacologic chaperone Migalastat. N Engl J Med. 2016;375(6):545–555. | ||
Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189–203. | ||
Tøndel C, Kanai T, Larsen KK, et al. Foot process effacement is an early marker of nephropathy in young classic Fabry patients without Albuminuria. Nephron. 2015;129(1):16–21. | ||
Najafian B, Svarstad E, Bostad L, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011;79(6):663–670. | ||
Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(2):284–293. | ||
Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666. | ||
Körver S, Vergouwe M, Hollak CEM, van Schaik IN, Langeveld M. Development and clinical consequences of white matter lesions in Fabry disease: a systematic review. Mol Genet Metab. 2018;125(3):205–216. | ||
Marchesoni C, Cisneros E, Pfister P, et al. Brain MRI findings in children and adolescents with Fabry disease. J Neurol Sci. 2018;395:131–134. | ||
Aguiar P, Azevedo O, Pinto R, et al. New biomarkers defining a novel early stage of Fabry nephropathy: a diagnostic test study. Mol Genet Metab. 2017;121(2):162–169. | ||
Warnock DG, Valbuena C, West M, Oliveira JP. Renal manifestations of Fabry disease. In: Elstein D, Altarescu G, Beck M, editors. Fabry Disease. 1st ed. Dordrecht: Springer Science+Business Media B.V.; 2010:211–243. | ||
Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry registry. Mol Genet Metab. 2008;93(2):112–128. | ||
Arends M, Hollak CE, Biegstraaten M. Quality of life in patients with Fabry disease: a systematic review of the literature. Orphanet J Rare Dis. 2015;10(1):77. | ||
Bolsover FE, Murphy E, Cipolotti L, Werring DJ, Lachmann RH. Cognitive dysfunction and depression in Fabry disease: a systematic review. J Inherit Metab Dis. 2014;37(2):177–187. | ||
Lohle M, Hughes D, Milligan A, et al. Clinical prodromes of neurodegeneration in Anderson-Fabry disease. Neurology. 2015;84(14):1454–1464. | ||
Cole AL, Lee PJ, Hughes DA, Deegan PB, Waldek S, Lachmann RH. Depression in adults with Fabry disease: a common and under-diagnosed problem. J Inherit Metab Dis. 2007;30(6):943–951. | ||
Bugescu N, Naylor PE, Hudson K, Aoki CD, Cordova MJ, Packman W. The psychosocial impact of Fabry disease on pediatric patients. J Pediatr Genet. 2016;5(3):141–149. | ||
Hegemann S, Hajioff D, Conti G, et al. Hearing loss in Fabry disease: data from the Fabry outcome survey. Eur J Clin Invest. 2006;36(9):654–662. | ||
Carmona S, Weinschelbaum R, Pardal A, et al. Neuro-otological and peripheral nerve involvement in Fabry disease. Audiol Res. 2017;7(2):176. | ||
Rodrigues J, Azevedo O, Sousa N, Cunha D, Mexedo A, Fonseca R. Inner ear involvement in Fabry disease: clinical and audiometric evaluation of a large cohort of patients followed in a reference centre. Eur J Med Genet. 2018;61(6):341–347. | ||
Hoffmann B, Schwarz M, Mehta A, Keshav S, Fabry Outcome Survey European Investigators. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007;5(12):1447–1453. | ||
Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res. 2008;64(5):550–555. | ||
Zar-Kessler C, Karaa A, Sims KB, Clarke V, Kuo B. Understanding the gastrointestinal manifestations of Fabry disease: promoting prompt diagnosis. Therap Adv Gastroenterol. 2016;9(4):626–634. | ||
Hilz MJ, Arbustini E, Dagna L, et al. Non-specific gastrointestinal features: could it be Fabry disease? Dig Liver Dis. 2018;50(5):429–437. | ||
Zampetti A, Orteu CH, Antuzzi D, et al. Angiokeratoma: decision-making aid for the diagnosis of Fabry disease. Br J Dermatol. 2012;166(4):712–720. | ||
van der Tol L, Sminia ML, Hollak CE, Biegstraaten M. Cornea verticillata supports a diagnosis of Fabry disease in non-classical phenotypes: results from the Dutch cohort and a systematic review. Br J Ophthalmol. 2016;100(1):3–8. | ||
van der Tol L, Verhamme C, van Schaik IN, van der Kooi AJ, Hollak CE, Biegstraaten M. In patients with an α-galactosidase A variant, small nerve fibre assessment cannot confirm a diagnosis of Fabry disease. JIMD Rep. 2016;28:95–103. | ||
Samiy N. Ocular features of Fabry disease: diagnosis of a treatable life-threatening disorder. Surv Ophthalmol. 2008;53(4):416–423. | ||
Ovbiagele B, Nguyen-Huynh MN. Stroke epidemiology: advancing our understanding of disease mechanism and therapy. Neurotherapeutics. 2011;8(3):319–329. | ||
Moran AE, Forouzanfar MH, Roth GA, et al. The global burden of ischemic heart disease in 1990 and 2010: the global burden of disease 2010 study. Circulation. 2014;129(14):1493–1501. | ||
Hill NR, Fatoba ST, Oke JL, et al. Global prevalence of chronic kidney disease: a systematic review and meta-analysis. PLoS One. 2016;11(7):e0158765. | ||
Lim GY, Tam WW, Lu Y, Ho CS, Zhang MW, Ho RC. Prevalence of depression in the Community from 30 countries between 1994 and 2014. Sci Rep. 2018;8(1):2861. | ||
Stevens G, Flaxman S, Brunskill E, et al. Global and regional hearing impairment prevalence: an analysis of 42 studies in 29 countries. Eur J Public Health. 2013;23(1):146–152. | ||
Nash SD, Cruickshanks KJ, Klein R, et al. The prevalence of hearing impairment and associated risk factors: the Beaver dam offspring study. Arch Otolaryngol Head Neck Surg. 2011;137(5):432–439. | ||
Canavan C, West J, Card T. The epidemiology of irritable bowel syndrome. Clin Epidemiol. 2014;6:71–80. | ||
Avramidou M, Angst F, Angst J, Aeschlimann A, Rössler W, Schnyder U. Epidemiology of gastrointestinal symptoms in young and middle-aged Swiss adults: prevalences and comorbidities in a longitudinal population cohort over 28 years. BMC Gastroenterol. 2018;18(1):21. | ||
Leray E, Moreau T, Fromont A, Edan G. Epidemiology of multiple sclerosis. Rev Neurol. 2016;172(1):3–13. | ||
Aronow WS. Hypertension and left ventricular hypertrophy. Ann Transl Med. 2017;5(15):310. | ||
Sanchis-Gomar F, Perez-Quilis C, Leischik R, Lucia A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann Transl Med. 2016;4(13):256. | ||
Dichgans M, Malik R, König IR, et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. Stroke. 2014;45(1):24–36. | ||
Orho-Melander M. Genetics of coronary heart disease: towards causal mechanisms, novel drug targets and more personalized prevention. J Intern Med. 2015;278(5):433–446. | ||
Piras D, Zoledziewska M, Cucca F, Pani A. Genome-wide analysis studies and chronic kidney disease. Kidney Dis. 2017;3(3):106–110. | ||
Malik R, Chauhan G, Traylor M, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524–537. | ||
Mullins N, Lewis CM. Genetics of depression: progress at last. Curr Psychiatry Rep. 2017;19(8):43. | ||
Tu NC, Friedman RA. Age-related hearing loss: unraveling the pieces. Laryngoscope Investig Otolaryngol. 2018;3(2):68–72. | ||
Adam B, Liebregts T, Holtmann G. Mechanisms of disease: genetics of functional gastrointestinal disorders-searching the genes that matter. Nat Clin Pract Gastroenterol Hepatol. 2007;4(2):102–110. | ||
Henström M, D’Amato M. Genetics of irritable bowel syndrome. Mol Cell Pediatr. 2016;3(1):7. | ||
Freudenberger P, Schmidt R, Schmidt H. Genetics of age-related white matter lesions from linkage to genome wide association studies. J Neurol Sci. 2012;322(1–2):82–86. | ||
Böttcher T, Rolfs A, Tanislav C, et al. Fabry disease: underestimated in the differential diagnosis of multiple sclerosis? PLoS One. 2013;8(8):e71894. | ||
Russo C, Riccio E, Pontillo G, et al. Multiple sclerosis and Fabry disease, two sides of the coin? The case of an Italian family. Mult Scler Relat Disord. 2018;26:164–167. | ||
Authors/Task Force members, Elliott PM, Anastasakis A, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task Force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of cardiology (ESC). Eur Heart J. 2014;35(39):2733–2779. | ||
Limongelli G, Masarone D, Verrengia M, et al. Diagnostic clues for the diagnosis of Nonsarcomeric hypertrophic cardiomyopathy (phenocopies): amyloidosis, Fabry disease, and mitochondrial disease. J Cardiovasc Echogr. 2018;28(2):120–123. | ||
Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. 2010;47(4):217–222. | ||
Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40. | ||
Rolfs A, Fazekas F, Grittner U, et al. Acute cerebrovascular disease in the young: the Stroke in Young Fabry patients study. Stroke. 2013;44(2):340–349. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.