Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Multiple cues on the physiochemical, mesenchymal, and intracellular trafficking interactions with nanocarriers to maximize tumor target efficiency
Received 4 March 2015
Accepted for publication 16 April 2015
Published 17 June 2015 Volume 2015:10(1) Pages 3989—4008
DOI https://doi.org/10.2147/IJN.S83951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Sang-Woo Kim, Dongwoo Khang
Nanomedicine Laboratory, Department of Molecular Medicine, School of Medicine, Gachon University, Incheon, South Korea
Abstract: Over the past 60 years, numerous medical strategies have been employed to overcome neoplasms. In fact, with the exception of lung, bronchial, and pancreatic cancers, the 5-year survival rate of most cancers currently exceeds 70%. However, the quality of life of patients during chemotherapy remains unsatisfactory despite the increase in survival rate. The side effects of current chemotherapies stem from poor target efficiency at tumor sites due to the uncontrolled biodistribution of anticancer agents (ie, conventional or current approved nanodrugs). This review discusses the effective physiochemical factors for determining biodistribution of nanocarriers and, ultimately, increasing tumor-targeting probability by avoiding the reticuloendothelial system. Second, stem cell-conjugated nanotherapeutics was addressed to maximize the tumor searching ability and to inhibit tumor growth. Lastly, physicochemical material properties of anticancer nanodrugs were discussed for targeting cellular organelles with modulation of drug-release time. A better understanding of suggested topics will increase the tumor-targeting ability of anticancer drugs and, ultimately, promote the quality of life of cancer patients during chemotherapy.
Keywords: cancer, anticancer nanodrugs, mesenchymal stem cell, intracellular trafficking
Introduction
After monumental remarks from Dr Richard Feynman at the APS (American Physical Society) meeting in 1956, advances in nanotechnology (NT) have catalyzed the growth and development of the semiconductor and electronics industry, where the average transistor density of a CPU inside our laptops has reached several billion.1 In spite of this, NT is rarely applied in the field of clinical medicine, even though medicine is a critical subject for human beings.
Today, cancer still remains a challenging and invincible disease. For example, the death rate due to tumors (or neoplasms) in the US has remained constant for the past 30 years, whereas the death rate due to heart diseases has significantly decreased, especially for individuals of age 65 years and over.2
As previously addressed by several insightful reviews, the major limitation of current cancer therapeutics is the poor targeting efficiency to tumor sites.3–5 For example, the target efficiency of most free and antibody-conjugated cancer drugs is still below a few percent.3,6 In other words, the majority of anticancer drugs spread not only to tumors, but also to normal tissues, organs, and even the brain. This is the primary reason for observed chemotherapy side effects.
Recently, alternative approaches have been introduced, such as stem cell therapy. Stem cells have innate homing abilities (migration through transendothelium) to cancer cells, and thus conjugation of nanodrugs can maximize the tumor target efficacy. Furthermore, stem cells are highly resistant to anticancer drugs compared with normal and cancer cells.7,8 However, excessive dosing with unrationalized drug design will eventually degrade the functions of stem cells and induce unexpected side effects (such as unexpected orphan diseases).
This review discusses three major issues to overcome current limitations (ie, low targeting percentage at tumor sites). The first is the physiochemical material-dependence of nanodrug biodistribution and clearance analysis. This will present a novel strategy for targeting specific organ and tumor sites, especially for improving the low rate of drug accumulation in tumors. Second, cell-conjugated nanodrug therapeutics are discussed for treating specific cancers with the aid of Trojan-mediating cells (ie, mesenchymal stem cells [MSCs] loaded with nanodrugs). This approach aims to increase both tumor searching and targeting abilities of nanodrug therapeutics. Finally, understanding optimal intracellular trafficking of conjugated anticancer drugs in cancer cells can further increase the efficacy of anticancer drugs. Endosome-assisted intracellular transport can bypass the tumor efflux and can thus deliver anticancer molecules into targeted organelles, such as mitochondria and nuclei, without experiencing drug efflux.
The increasing tumor-targeting efficiency of anticancer drugs can minimize the potential toxicity of anticancer drugs through optimized biodistribution and fast clearance and can, ultimately, significantly reduce the dose of anticancer drug.9
In conclusion, this review highlights the most influential physiochemical material cues for controlling the biodistribution of nanocarriers to increase tumor target efficacy. The possibility of stem cell-based cancer NT and intracellular–molecular interactions of nanocarriers (including organelles targeting) is introduced. The discussion highlights the latest advances in cancer nanomedicine and provides novel strategies to increase the tumor target efficiency that could replace current classical clinical chemotherapy in the near future.
Role of physiochemical and biomolecular factors in regulating biodistribution
Although the tumor-targeting efficacy of antibody-conjugated receptors is two to a few times better than conventional chemotherapy drugs, this is valid only for overexpressed tumor receptors at membrane.6,10–16 Specifically, epidermal growth factor receptor (EGFR) is the most well-known cancer receptor, and EGFR–antibody interactions of cancer drugs were previously reported.6,10,17 However, a more revolutionary approach than current antibody receptor targeting is necessary to further increase targeting efficacy. For example, the utilization of selective organ accumulation with respect to shape and size dimensions of nanomaterials would be advantageous in targeting specific organs (ie, in the case of source of tumor tissues) (Figure 1). Unfortunately, there has been limited information hitherto on targeting of specific organs and tissues by controlling the chemistry, shape, and size of nanoparticles (NPs).18 The prospects and limitations of the physiochemical properties of NP and associated biodistribution will be discussed in this section (Table 1).
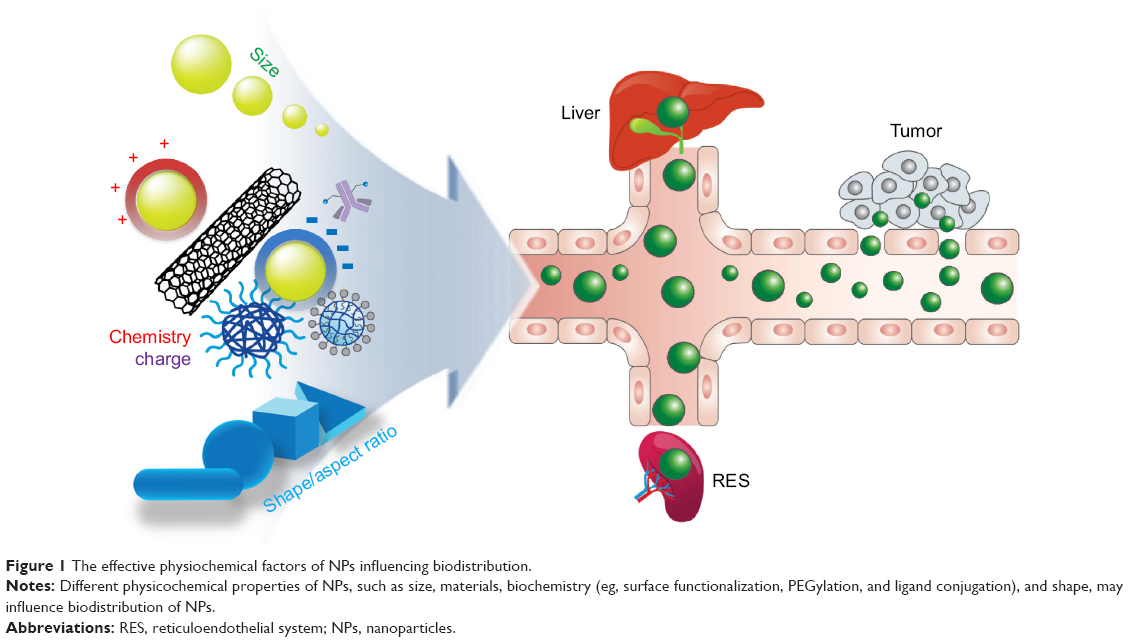 | Figure 1 The effective physiochemical factors of NPs influencing biodistribution. |
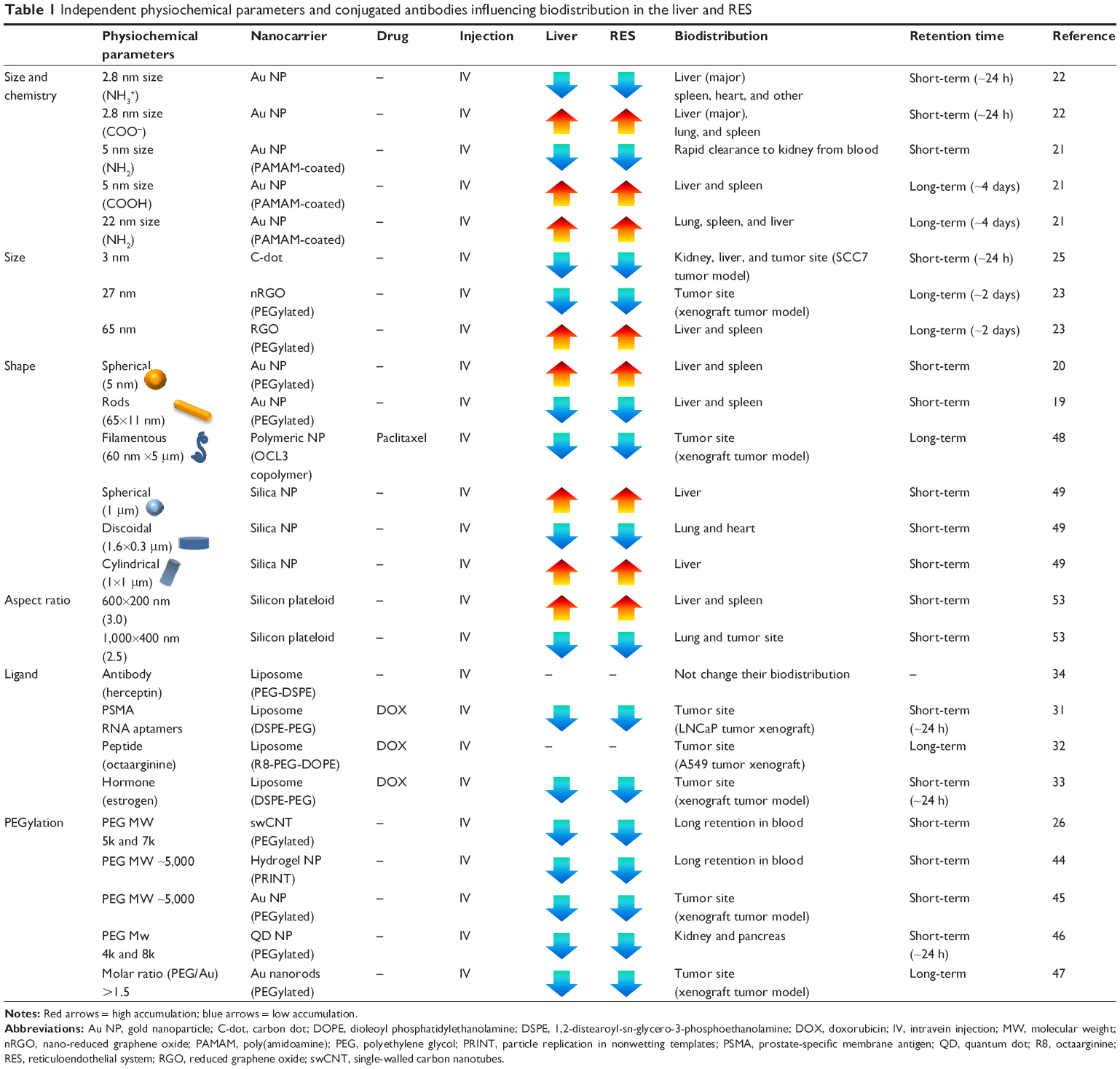 | Table 1 Independent physiochemical parameters and conjugated antibodies influencing biodistribution in the liver and RES |
Biodistribution and clearance of major nanoparticles
Gold nanoparticles
Gold (Au) nanorods (65×11 nm) identified a clear shift of biodistribution.19 Biodistribution of polyethylene glycol (PEG)-coated Au nanorods after intravenous injection (IV) showed a clear shift in the biodistribution from blood to liver, with increasing accumulation from 30 minutes to 72 hours. Although the authors coated PEG onto the Au NPs to neutralize surface charge and to reduce the toxicity, a drastic shift in biodistribution stalled at the liver raised concerns about liver toxicity. Another study identified more accumulation of PEG-coated Au nanosphere in the liver and spleen (57%) than Au nanorods (15%) after 24 hours of IV injection.20
A separate study further demonstrated a shift in biodistribution and clearance by changing size and chemistry. In this study, Au NP (5 nm, functionalized with NH2) exhibited rapid clearance to the kidney from the blood.21 However, higher accumulation in the liver and spleen by increasing size (ie, 22 nm with NH2) and altering surface chemistry (ie, 5 nm with COOH) was observed.21 Urinary excretion confirmed rapid clearance of 5 nm of Au NP (NH2), whereas larger size (ie, 22 nm with NH2) and alternative surface chemistries (5 nm with COOH) exhibited slower urinary excretion.21 A similar study also demonstrated that size and surface chemistry of Au NP strongly influence their biodistribution.22 Interestingly, the positively charged Au NP (2.8 nm, functionalized with NH3+) exhibited more accumulation in the spleen and other organs than the negatively charged Au NP (2.8 nm, functionalized with COOH–) with identical urinary excretion time.
Carbon nanomaterials (graphene, dot, nanotube)
PEGylated reduced graphene oxide (RGO-PEG, sheet diameters of 65 nm) significantly accumulated in the liver and spleen.23 However, PEGylated nano-reduced GO (nRGO-PEG) with smaller sizes (eg, sheet diameters of 27 nm) reduced liver and spleen accumulation, but increased accumulation at the tumor sites (skin xenograft tumor model).23 Moreover, nRGO-PEG shows significantly prolonged blood circulation, which allows a greater chance of passive tumor targeting. Although both materials (ie, RGO-PEG, nRGO-PEG) accumulated in the liver, liver toxicology tests indicated normal ranges up to 90 days.24 Oral administration of both RGO-PEG (65 nm) and nRGO-PEG (27 nm) recorded the highest accumulation in stomach, but cleared after a day. In contrast, intraperitoneal injection (in the abdominal cavity) sustained liver and spleen accumulation even after 7 days.24 The accumulation of GO NPs by intraperitoneal administration showed longer liver and spleen accumulation (ie, 1–7 days) than IV or PO administration (ie, 4 hours to 1 day).24 The additional clinical advantage of the studied nRGO-PEG is their implementation in photothermal therapy using 808 nm-wavelength laser ablation (but requires a greater injection dose of 20 mg/kg).
Due to the ultrasmall dimensions (ie, 3 nm), most carbon dots were promptly cleared by the urinary system (ie, from the kidney to the bladder) after 24 hours, despite initial accumulation in the liver, spleen, heart, and lungs (Table 1).25 Biodistribution of carbon dots by IV injection recorded a fast clearance rate, compared with other injection routes (eg, intramuscular and subcutaneous).
For single-walled carbon nanotubes (swCNTs), short-term (or transient) and long-term accumulation analysis revealed time-dependent variation of biodistribution. Long-term biodistribution of PEG-coated swCNTs showed selective accumulation in the liver and spleen after a day.26 Although the half-clearance time of swCNTs from the liver and spleen was approximately 20–30 days, molecular weight (MW) of PEG on the surface of the particles shortens the clearance time.26 In particular, higher PEG MW (eg, both 5k and 7k of PEG) showed significantly decreased half-clearance time (ie, approximately 1 day) in the liver and spleen, whereas 2k PEG showed high retention time (ie, 15 days). Instant observations of transient biodistribution analysis showed that the lungs were the first organ with notable accumulation (3 seconds after IV) and sustained for several minutes (Figure 2A and B).27 Liver accumulation was apparent after 17 seconds. Biodistribution of most organs reached steady state after 20 seconds. However, the spleen showed incremental real-time Raman intensity, which corresponds to accumulation, from 20 seconds to several minutes after IV. It was observed that there were significant transient fluctuations (during the initial 10 seconds) for nondominant organs, such as the brain, muscle, kidney, and lungs, which, however, stabilized after 20 seconds (Figure 2A and B). Thus, both long-term and short-term evaluation of nanomaterial biodistribution must be considered simultaneously.
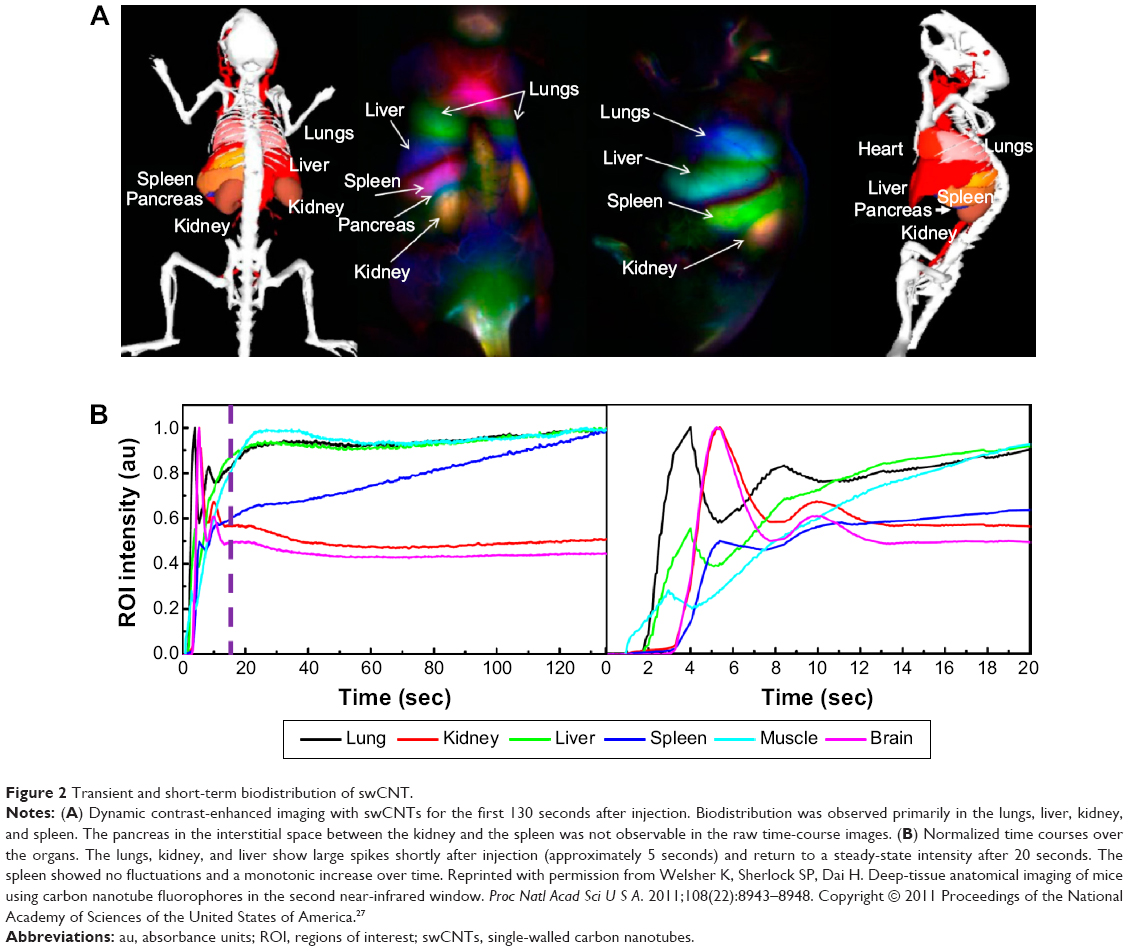 | Figure 2 Transient and short-term biodistribution of swCNT. |
Liposome nanoparticle
Liposomal encapsulation of doxorubicin (minimum size of 90±5 nm) has the advantage of prolonging blood circulation.28,29 Owing to limited size, bioconjugation with antibodies or ligands is an available strategy for liposome NP to promote targeting efficiency to tumor sites. In this regard, accessing tumor target sites by antibody conjugations was suggested by other studies.30 To improve the tumor-specific targeting of liposome, doxorubicin (DOX)-encapsulated liposome (similar to DOXIL) conjugated with an RNA aptamer (cell-penetrating peptide: DOX-aptamosomes) was designed to target the prostate-specific membrane antigen, and the results demonstrated selective delivery to the tumor sites in vivo (ie, skin xenograft tumor model).31 The accumulation of DOX-aptamosomes was observed in tumor sites, whereas unmodified DOX liposomes were mostly distributed to the liver and kidney (after 24 hours of IV). In addition, cell-penetrating peptide, octaarginine (R8)-conjugated liposomal DOX showed greater accumulation in A549 non-small lung cancer cells than normal liposomal DOX.32 In another study, DOX-encapsulating liposomes conjugated with estrogen (ES)-conjugated liposome (ES-liposome) were selectively delivered to breast carcinoma cells in vivo.33 The concentration of ES-liposome was significantly higher at the tumor sites than free DOX (ie, skin xenograft tumor model), whereas the concentration of ES-liposome was significantly lower than free DOX in the liver and kidney (after IV). Thus, ES-anchored liposome has increased tumor-targeting efficacy by avoiding liver accumulation.
Although antibodies conjugation increases cancer intracellular uptake (eg, via endocytosis), the antibody conjugation cannot significantly influence in vivo biodistribution.34,35 For instance, the conjugation of the Herceptin antibody (anti-Her2) with liposomes did not result in any alteration in the biodistribution (ie, accumulation pattern) and was mainly filtered in the reticuloendothelial system (RES) organs (eg, spleen and lymph node), compared with nonconjugated liposomes at 24 hours after IV.34
Avoiding liver and RES accumulation and increasing tumor targeting
NPs are most likely to interact with immune cells (neutrophil and macrophages) in the RES system (liver and spleen) through blood circulation, and finally get excreted by urinary clearance.36,37 To minimize the RES accumulation, prolonging the blood circulation is necessary. In contrast, extended circulation time of NPs in the blood system increases the chance of immune interactions by the RES system. As such, maximized drug delivery targeting to the tumor site or various organs (in case of tumor sources) through optimal blood circulation time is a critical point. In this section, various physiochemical factors (ie, chemistry, surface charge, and shape) of nanodrugs effectively influencing RES accumulation will be discussed. Specifically, smaller sized particles (d<5 nm) quickly accumulate in the liver and spleen, but are promptly cleared via blood circulation thereafter.21 However, longer clearance times of liver and spleen accumulation have been observed with particle sizes larger than 10 nm. For a toxicological perspective, this accumulation in the liver and spleen is somewhat dangerous.38–41 The benefit of achieving a two- to threefold increase in tumor-targeting efficacy using nanomaterials does not outweigh the cost of elevated drug accumulation in the liver and spleen. Consequently, it cannot justify the clinical replacement of current chemotherapeutics (with/without antibody-assisted therapeutics). Thus, alternative strategies are required for nanocarriers that dually promote tumor targeting and minimize nanocarrier deposition in the liver and the RES. In this section, distinctive strategies will be discussed for improved targeting of tumor tissues with less accumulation in the liver and spleen. Specifically, the role of chemistry, surface charge, and shape of nanocarriers will be discussed, along with the use of polymeric coatings, to either independently or simultaneously influence in vivo bioaccumulation.
Role of molecular weight of coating polymers for biodistribution
PEG layers on the surface of NPs (spherical NPs of size less than 200 nm) can prolong the blood circulation time along with avoiding RES accumulation.42,43 In particular, MW (5k) of surface PEG chain efficiently reduced liver uptake by avoiding plasma protein adsorption.44,45 However, when the PEG chain’s MW was increased beyond 5k, no considerable avoidance of RES accumulation was observed.42 A recent study demonstrated that the surface MW of PEG is strongly correlated with PEG chain. Specific weight of PEG chain (5k) led to an optimal dense “brush conformation” that allowed reduced plasma protein adsorption.44 As such, MW of PEG-coating polymer is a significant factor in facilitating in vivo biodistribution. For quantum dots (~3.2 nm), altering PEG MW on quantum dots can induce different organ biodistribution.46 Specifically, intermediate MW (4k–8k) exhibited organ distribution at the kidneys and pancreas, while small MW (2k) resulted in rapid accumulation at the liver.
In addition, PEG grafting level (PEG:Au molar ratio) on Au nanorods also can control their biodistribution. In this study, surface grafting with PEG:Au molar ratio of 1.5 showed the most prolonged blood circulation, and thus demonstrated increased tumor-targeting ability with decreased liver and spleen accumulation.47
Surface charge and size contribution for biodistribution
When Au NPs were functionalized with positive amine groups (NH), most of the NPs accumulated in the kidney (5 nm radius) and cleared within 4 days. Liver and spleen showed relatively lower Au NP distribution at this dimension.21 However, when Au NPs were coated with the carboxyl (5 nm of COOH) group, they were mostly accumulated in the liver and spleen. Furthermore, COOH–Au NPs exhibited slow urinary excretion compared with NH-Au NPs (5 nm radius).21 Furthermore, the Au NPs (11 nm of COOH) accumulated in the spleen and heart with reduced clearance rate to the kidney after IV injection (1 hour).21 At larger sizes (ie, >22 nm), NH-modified Au NPs showed increased lung accumulation compared with the liver and spleen. The urinary excretion was faster for 5 and 11 nm than for 22 nm of Au NPs (Table 1).21
Shape and aspect ratio are critical factors for altering bioaccumulation
The filament-shaped polymeric NPs (60×5,000 nm) demonstrated significant long-term circulation in the blood and effective delivery of anticancer drug (ie, paclitaxel) to tumor sites compared with spherically shaped NPs (50 nm).48 More recent studies examined the biodistribution of various shapes (eg, discoidal, spherical, hemispherical, cylindrical shapes of silica-based NPs with identical volumes) and demonstrated a strong correlation between shape and biodistribution to specific organs (ie, the lung, liver, heart, and spleen, but not the brain and kidney).49 Cylindrical (1 μm in diameter) and spherical (1 μm in diameter) silica NPs significantly accumulate in the liver after IV injection. In the spleen, discoidal and hemispherical silica NPs (1.6 μm in diameter) showed the most increased accumulation. Specifically, micron-sized discoidal particles (1.6 μm in diameter) tend to accumulate more in the lungs and the heart than NPs of other shapes. As such, discoidal silica NPs selectively influenced specific organ biodistribution without RES accumulation, compared with other shapes of silica NPs (Table 1).
In another in vitro study, rod-shaped NPs (ie, higher aspect ratio particles) dramatically enhanced cancer uptake efficiency compared with alternative shapes of (spherical and disk-shaped) polystyrene nano and microparticles, especially coated with an anticancer drug targeting breast cancer (trastuzumab: Herceptin).50 This study demonstrated that shape-dependent interactions of NPs and molecules can maximize the uptake level of cancer cells and, ultimately, inhibit cancer cell growth (in vitro).50 Rod-shaped geometries, which feature higher surface area to volume ratio than spheres, adsorbed a greater amount of Her2 receptors (ie, 24% more antibody than sphere).
However, NPs with diameters of less than 100 nm exhibited the opposite trend. Higher uptake NPs of Au spherical geometry than of Au rods was observed in HeLa cells. Intracellular uptake rates of Au nanosphere (size: 50 nm) were 40 times higher than that of Au nanorods (size: 14×50 nm/cell).51,52
Different aspect ratios of plateloid particles (ie, of porous silicon) and corresponding tumor-targeting efficacy were also identified (in vivo).53 Plateloid dimensions of 1,000×400 nm (2.5 aspect ratio) exhibited more tumor-specific targeting than 600×200 nm (3.0 aspect ratio) plateloid; also, 600×200 nm plateloids accumulated more in the liver and the spleen. The biodistribution shift of plateloid particles from the liver and spleen moved to the lungs and tumor sites. As such, lung accumulation was concurrent with increased tumor accumulation for the examined aspect ratios (ie, 2.5>3.0) in this study.53,54 Attaching RGD antibodies enhanced tumor site targeting than uncoated plateloid disk controls. Biochemical interaction was increased by two- to threefold by changing the particle aspect ratio. Tumor vasculature studies also indicated greater circulation in tumor vessels for specific aspect ratios of plateloid particles (1,000×400 nm). As such, choosing the appropriate size and shape of NPs can increase tumor cell uptake about 5–10 times in tumor sites (from just 1%–2% ID/g to 10% of ID/g). Taken together, surface chemistry is a major parameter in influencing biodistribution when the size of the NP is less than 10 nm. Physical shape (aspect ratio), however, is a more significant factor when the size of NP is over 100 nm.
Mesenchymal stem cell-assisted nanodrug delivery to tumor sites
Cell-mediated nanodrug delivery demonstrated the most active drug delivery to tumor sites than any other drug-delivery strategies.55–59 This may be attributed to specific antibody receptor-based NP drug deliveries that possess limited functions for targeting tumors. In this regard, living cells such as red blood cells (or hematopoietic stem cells), lymphocytes (T cells), and MSCs were considered to be living vehicles for targeting cancer cells (ie, hijack and tumor tropic agents, respectively).60 Circulatory cells (ie, red blood, monocyte, and lymphocytes) were advantageous for actively delivering NPs to the vascularized tumor sites through blood circulation.61–63 As an alternative approach, stem cells were introduced in view of their homing ability. Specifically, stem cells are capable of transendothelial migration (ie, rather than localize within blood vessels) and can actively migrate to cancer cells (Figure 3).64 An additional advantage of human MSCs (hMSCs) is that they support a wide range of classification determinant (CD) expression, which includes positive for CD 44, CD 73, CD 90, CD 105, and CD 106, and negative for CD 14, 34, and 45.56,64 This wide set of CD allows multiple functional strategies for tumor-killing procedures. In conclusion, MSCs are suitable for conjugation with NPs and deliver loaded anticancer molecules to tumor tissues (Figure 3), which can ultimately increase targeting efficiency and reduce undesirable side effects from untargeted chemotherapeutics (Table 2).
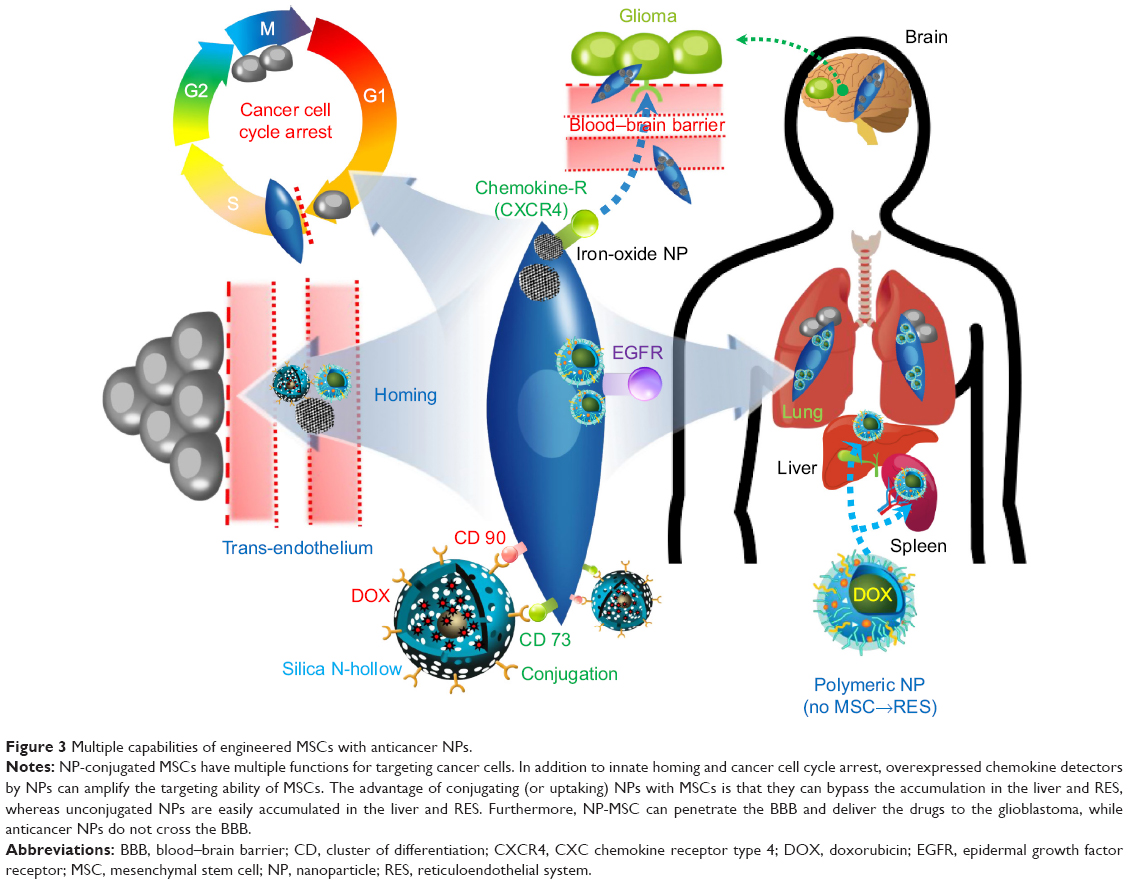 | Figure 3 Multiple capabilities of engineered MSCs with anticancer NPs. |
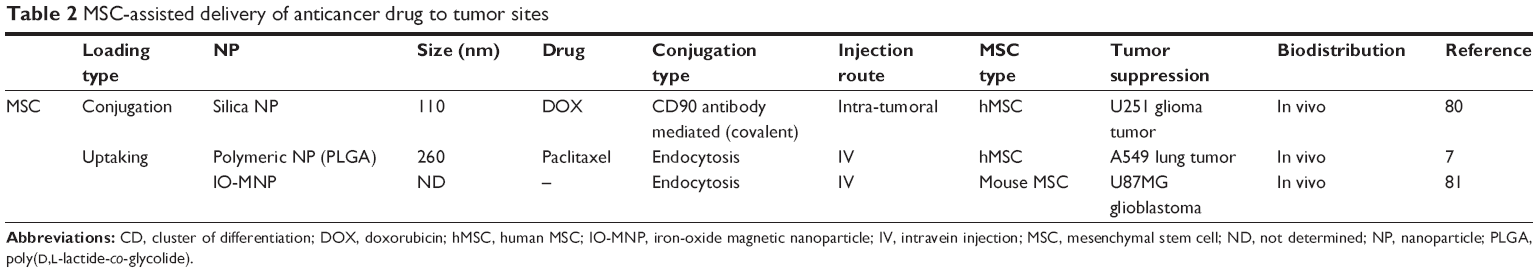 | Table 2 MSC-assisted delivery of anticancer drug to tumor sites |
Tumor-targeting ability and fate of mesenchymal stem cells
The hMSCs are ideal cells for targeting deep-seated tumor tissue in view of their homing ability (as discussed in the “Mesenchymal stem cell-assisted nanodrug delivery to tumor sites” section). Targeted delivery of anticancer drugs and cytokines to inhibit cancer growth corresponds to an improved therapeutic index (ie, toxic dose/dose of drug).57 Specifically, when interferon β (IFN-β) was loaded with NP on hMSC, the combinational strategy increased the anticancer effectiveness. Importantly, hMSCs are highly resistant to anticancer drugs because hMSCs have an active efflux pump system, such as ABC transporter.7,8
MSCs showed the ability to migrate to tumor sites such as glioma, melanoma, ovarian, breast, and colon cancers.65–67 The reason for this well-known tropism may be the detection ability (via various antigen receptors) of hMSCs of secreted growth factors and chemokines by cancer cells.65 Further, EGFRs displaying (via the transfection of cDNA-encoding EGFR) on MSCs can enhance migratory response against glioma or melanoma in vivo.68 Comprehensively, hMSCs have multisensory tumor detection systems, such as embedded chemokine detectors (stromal cell-derived factor 1 [SDF-1], transforming growth factor [TGF]), CXC motif receptor (CXCR4), and various tumor surface receptors.68 For nontumor-bearing animals, hMSCs initially resided in the lungs and migrated to the liver and spleen. For wounded mice, hMSCs migrated to the injured sites and remained for 3–5 days.66 Long-term biodistribution (ie, fate) of hMSCs after 4 weeks (IV injection) showed that most of the hMSCs had resided in the lymph node and spleen.69
Cancer inhibition or stimulation ability of stem cells
Stem cells are capable of inducing apoptosis and inhibiting proliferation and cell cycle arrest for cancer cells (Figure 3).70 In addition, they can stimulate immune cells such as natural killer cells, T-lymphocytes, and macrophages.71–74 In the case of lung carcinoma and colon cancer, MSCs showed antitumorigenic effects.75–77 The major mechanism of tumor growth inhibition was the inhibition of protein kinase Akt activation, which is well known for inhibiting both apoptosis and cell cycle arrest.56,78 In addition, IFN-β4 expressed by MSCs greatly inhibited MDA-MB-231 tumors.59
In contrast, it has also been reported that the fate of stem cells may influence metastatic potency when MSCs are cocultured with breast cancer cell lines, such as MCF-7 and MDA-MB-231 cell lines.79 This study found that specific chemokines (CCL5) produced by MSC can promote metastatic ability.69 As such, a thorough investigation of the cancer growth and metastatic potential of MSCs must precede their consideration ahead of clinical application.
Conjugation and uptake of nanoparticles with stem cells
Covalent conjugating silica NPs with MSC membrane proteins, such as CD 73 and CD 90, successfully load drugs on the membrane of hMSC.80 Uptake of some nonbioconjugated NPs (ie, without antibodies) through endocytic delivery by hMSCs was inevitable, but resulted in no significant adverse effect on hMSC proliferation and tropic ability toward human glioma cells (U-251).80 In addition, conjugating CD 90 antibodies to silica NPs at higher concentrations (ie, greater than 1 mg/ml of NPs) increased the conjugation of NPs on the surface of hMSCs. The loading rate of DOX on silica NPs was 18.2% and showed a pH-sensitive release of the attached drugs. After intratumoral injection, DOX-loaded silica NP-hMSC exhibits the greatest distribution (~7 days) at the U-251 glioma tumor tissue (in vivo) compared with DOX-silica NP and nonconjugated hMSC. Attaching NP and NP-DOX did not alter the ability to migrate to tumor cells. However, antibody attachment slightly reduced migration distance (or distance) compared with hMSCs without antibody attachment. Thus, attachment of antibody for supporting multiple functions may influence the migration speed (homing speed) of hMSCs and, thus, should be examined in advance.
Nanoengineered MSCs also boast longer retention times of cytotoxic drugs. These nanoengineered MSCs can actively accumulate in tumor sites and slowly release the cytotoxic drugs. This study examined hMSC uptake of biocompatible polymer-based NP (PLGA) (rather than conjugated NP on the extracellular membrane), but did not observe any alteration in MSC viability, differentiation, or migration ability.7 Cytotoxic drug (eg, paclitaxel) showed poor intracellular accumulation in hMSCs because MSCs have active drug efflux transporters, such as P-glycoprotein. Selective accumulation of hMSCs (with polymeric NP uptake) in the lungs was observed, while free polymer NPs (without hMSCs) mainly accumulated in the liver and spleen (Figure 3).7 As such, hMSCs can inhibit the accumulation of NP in the liver and RES systems when uptaken.
In addition, another study suggested that iron-oxide magnetic NP (IO-MNP), uptaken by mMSCs (mouse MSC), can bypass the blood–brain barrier (BBB).81 In this study, the uptaking of IO-MNP induced chemokine receptor CXCR4 (SDF-1 chemokine receptor) expression on MSCs and resulted in improved cell homing to the U87MG glioblastoma brain tumor, which showed a high expression of the chemokine SDF-1 alpha (Figure 3). These IO-MNPs could be efficiently uptaken in mMSCs without changes in cell viability and differentiation. Interesting features of this study were the multiple abilities of mMSCs for glioblastoma searching and repairing nerve tissue (regeneration) toward traumatic brain injury (across BBB from IV) (Figure 4A–C).
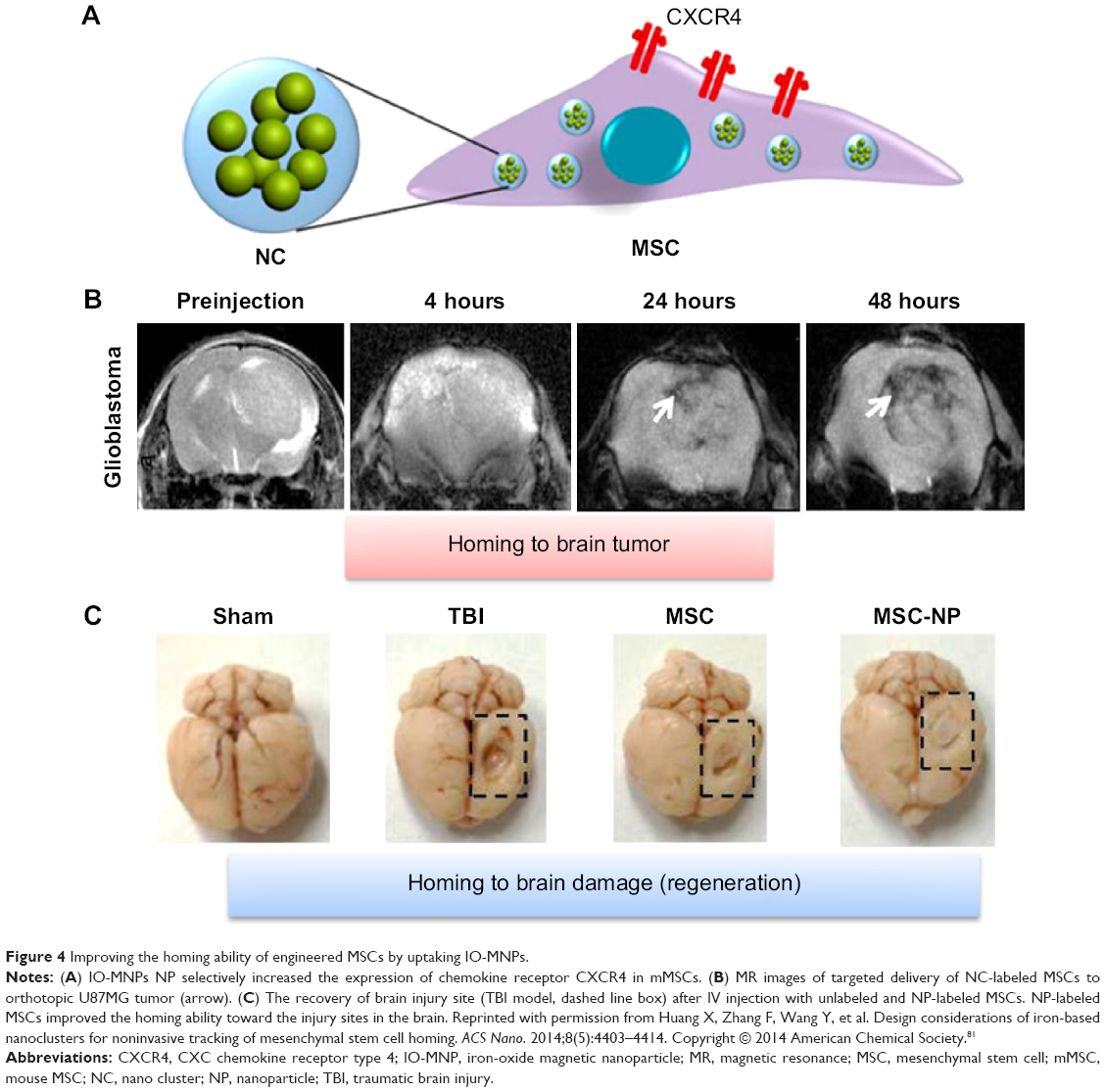 | Figure 4 Improving the homing ability of engineered MSCs by uptaking IO-MNPs. |
Transportation of anticancer molecules to cancer organelles
It has been reported that intracellular uptake efficiency of 100 nm-sized NPs estimated a 10- to 250-fold increase over 1–10 μm-sized microparticles.82 In addition, efficient intracellular delivery of anticancer drugs closely correlates with elusive cancer resistive response. Specifically, uptake via endosome intracellular pathways can overcome cancer cell efflux.83 Thus, optimized intracellular drug delivery can reduce the dosing of anticancer drugs and thus minimize the side effects of chemotherapy. The previous sections have emphasized how tumor-targeting efficacy can be enhanced with controlled design of physiochemical material properties in drug-delivery systems. In addition, living hMSCs were introduced for enhancing targeting efficiency (ie, homing) to tumor cells. However, it is important to deliver drugs not only to the tumor tissues, but also into the desired organelles within cancer cells. Intracellular targeting represents the delivery of therapeutic agents to specific organelles.84 As previous studies and reviews have described NPs uptake in detail, this review will evaluate the effective parameters to optimize intracellular transport of nanocarriers (Table 3 and Figure 5).85–89
 | Figure 5 Intracellular targeting to organelles using physiochemical parameters of anticancer nanodrugs. |
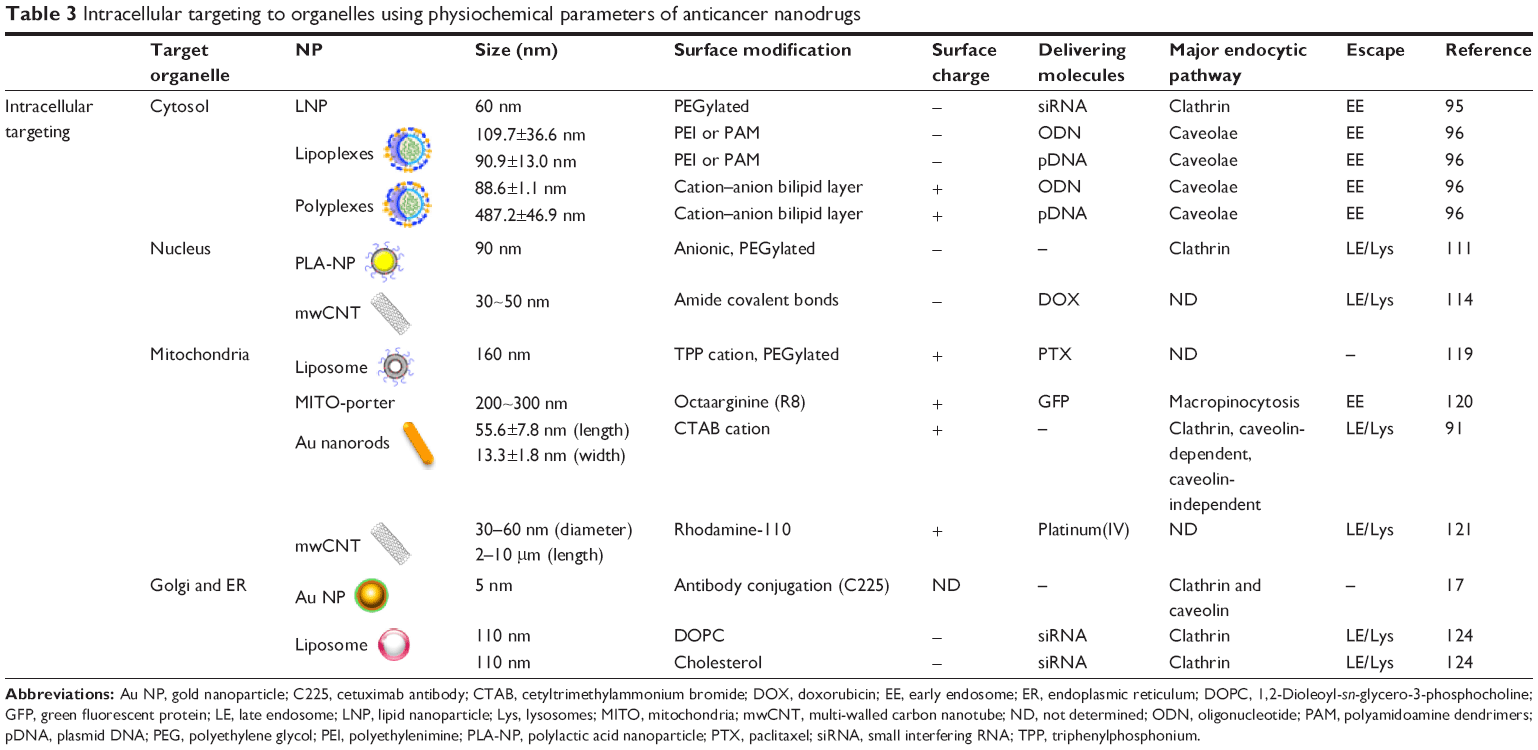 | Table 3 Intracellular targeting to organelles using physiochemical parameters of anticancer nanodrugs |
Intracellular trafficking of nanoparticles
Intracellular localization is the key factor in determining optimal drug delivery into specific organelles (eg, nucleus, Golgi apparatus, endoplasmic reticulum [ER], or mitochondria).89 Extensive studies have been conducted to elucidate the type of uptake and corresponding intracellular pathways of drug delivery.90 However, clear identification of intracellular pathways following specific uptake remains unclear. For example, it was reported that clathrin-mediated endocytosis can bypass fusion with lysosomes (Lys) to avoid lysosomal degradation.89 In contrast, clathrin- and caveolin-dependent uptakes are highly associated with lysosomal fusion and membrane leakage.91 Furthermore, some NPs (eg, cross-linked micelles) can bypass early endosomes (EEs), but reach the Lys within 30 minutes.92 However, it was generally concluded that most of the early-stage endosome delivery merges with primary Lys and evolves into secondary Lys (which exhibit larger sizes).90,93 These opposing interpretations of nearly identical intracellular trafficking reveal that the endocytic pathway is a dynamic and transient phenomenon.
Endosomal escape
Endosomal escape is considered to be an early stage of membrane destabilized by pH variations.94 Because the lysosomal stage is considered to be a stage of degradation by highly active enzymes (ie, acidic hydrolase proteins), endosomal escape of weak drug molecules is desirable prior to degradation for drug release. The biogenesis and maturation of NP delivery organelles with respect to time in cytosols was recently identified.95,96 In this study, small molecules (such as siRNA [siRNA, small interfering RNA]) escaped from the endosome to the cytosol with extremely low efficacy (ie, 1%–2% of total siRNA) in HeLa cells and showed differential release amount at each endosomal stage (vs time), such as EEs (0% until 1.5 hours), late endosomes (LE) (80% until 5 hours), and Lys (very few % after 5 hours) (Figure 6A–C).95 In vivo analysis of intracellular trafficking also showed identical trends, but rapid changes in the fate of EE, LE, and Lys, compared with in vitro (Figure 6B).95 Initial time of cytosolic escape for NPs depended on cell type (about 30 minutes), but again showed nearly identical trends.95
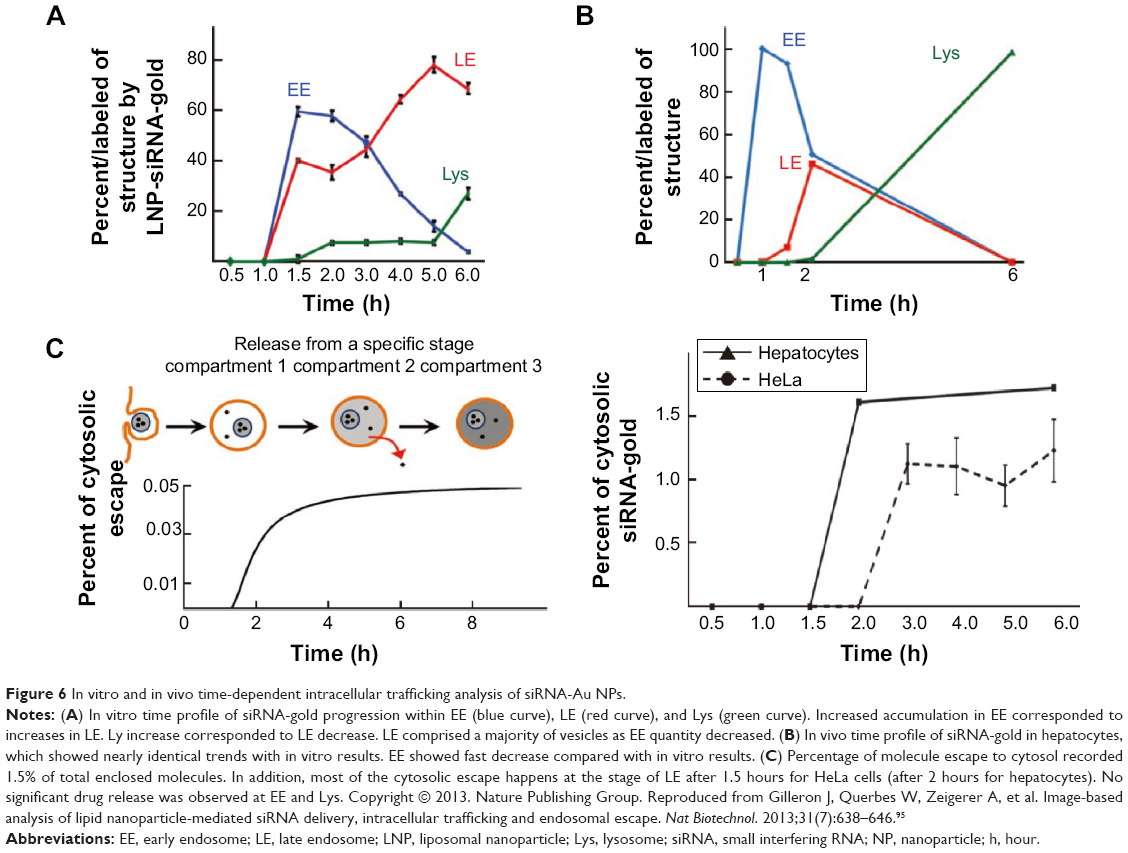 | Figure 6 In vitro and in vivo time-dependent intracellular trafficking analysis of siRNA-Au NPs. |
Furthermore, time-dependent transport of drug molecules from the cytosol to the nuclei (HeLa cells) was clearly identified using real-time observation (Figure 7A–C). Abrupt endosomal release of active molecules in the cytosol was followed by rapid nucleus internalization.96 The time frame between sudden burst from the endosome (ie, endosomal escape) and complete cell nucleus internalization was approximately 20 minutes. Increased number of endosome bursts within a given cell promoted cell nucleus internalization.
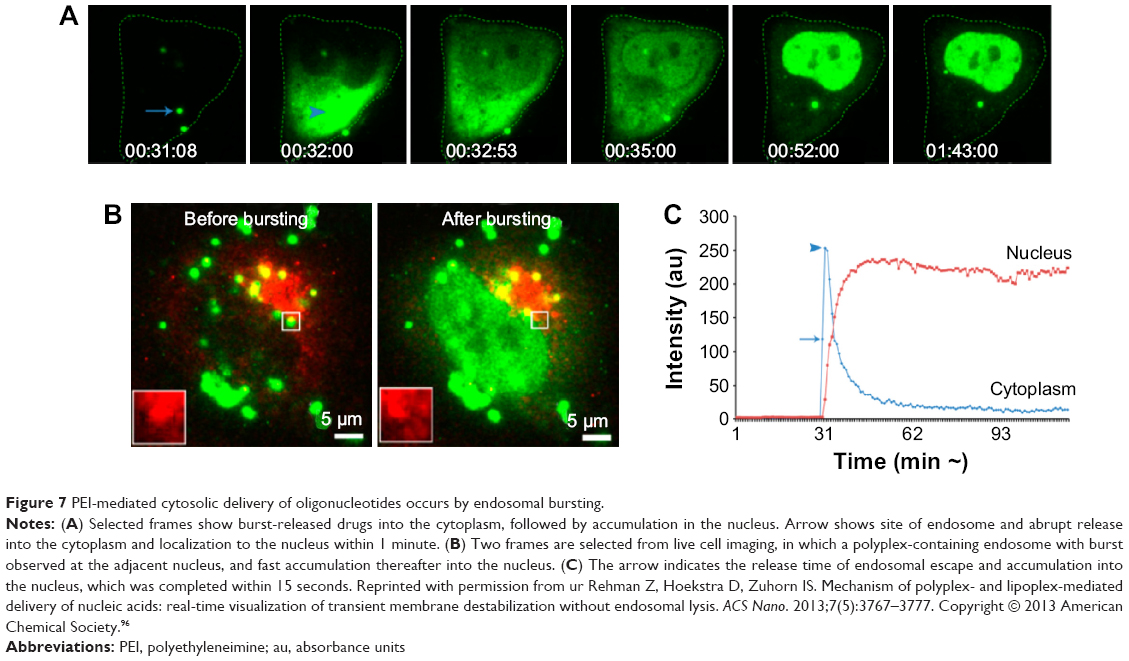 | Figure 7 PEI-mediated cytosolic delivery of oligonucleotides occurs by endosomal bursting. |
Weakness of PEG for drug delivery
(PEG)ylation is the most common method for conveying nucleic acids and active molecules with controlled retention time in the blood.26 Although the high density of PEG layer on NPs can increase their blood concentration by avoiding plasma protein adsorption, the efficiency of intracellular drug targeting significantly diminished by PEG degradation.97 This PEG dilemma underscored the practical limitations of PEG use in drug-delivery systems. PEGylation strongly inhibited cell uptake and endosomal escape, which present critical disadvantages for drug-delivery applications.97 Specifically, it was interpreted that lysosomal environments featuring low pH and greater acidic enzymes can degrade the activity of embedded molecules (eg, siRNA, pDNA [plasmid DNA], ODN [oligonucleotide], and proteins) in coated (or encapsulated) PEG (Figure 5).98–108 PEGylation may not be advantageous at the degradation stage (Ly), and, consequently, burst release at the EE stage is recommended.
Endolysosomal escape, advantageous targeting to nucleus
Intracellular conditions in cancer cells are quite complicated, and thus more than one stimulus can precisely control the drug-release time.109 For example, burst release of drugs on nanocarriers at costimulatory environments, such as pH gradients and protease enzyme conditions, could maximize therapeutic efficacy, especially drug delivery to the cell nucleus. Thus, a wide range of stimuli can trigger the drug release to a specific intracellular target at a controlled time inside.109
The endolysosomal escape of drugs from NPs is of great importance for the efficacy of drug-delivery system when the nanocarriers (including materials and active molecules) are susceptible to lysosomal degradation.110 The physiochemical factors that determine endosomal escape or lysosomal degradation have not yet been explicitly elucidated. However, previous research demonstrated that surface charge can influence the fate of endosomal escape or lysosomal degradation (Figure 5). Specifically, cationic and anionic NPs showed different routes of endocytosis. Although cationic NPs are highly internalized compared with anionic NPs, only anionic NPs experienced lysosomal degradation after clathrin-associated uptake.111 A recent study also demonstrated that the bond strength between NPs and anticancer drug is a significant factor in reaching the endolysosomal stage. This is because covalent amide bonds induced release of cancer drugs under conditions of high lysozyme density (Ly stage) and reduced pH in the vicinity of cancer nucleus.112,113 As such, choosing stable biomaterials, conjugation types (eg, covalent bonds), and chemically stable molecules is advantageous for increasing the delivery of anticancer drug into the nucleus through the endolysosomal escape, without experiencing degradation (eg, PEG with small molecules) at the lysosomal (or late endosomal) stages.114
Targeting to mitochondria
The primary role of mitochondria is to produce adenosine triphosphate in the cell, through metabolic reactions and processes. Moreover, mitochondria are critically involved in deciding cell apoptosis. In cancer, the apoptosis pathway of mitochondria is suppressed by overexpression of antiapoptotic proteins, such as Bcl-2 family.115,116 Thus, the current strategy for mitochondria-targeting nanocarriers is to trigger apoptosis in cancer cells.117 To date, several NPs (eg, liposomes, Au nanorods) have been reported to selectively target mitochondria. Because of the greater negative membrane potential (−130 to 150 mV) of mitochondria, targeting mitochondria by attractive electrostatic surface charge (ie, positive surface charge) was possible (Figure 5).117,118 Specifically, lipophilic cations (+) with triphenylphosphonium effectively delivered a proapoptotic chemotherapeutic paclitaxel to the mitochondria in vitro and in vivo.119 In addition, mitochondria-porter (a liposome-based carrier system for delivery of macromolecules into mitochondria) was surface modified with a high density of octaarginine (R8), which can highly accumulate in the mitochondria via membrane fusion.120 Au nanorods that exhibit a positive surface charge with cetrimonium bromide (CTAB) cations would be an ideal vehicle for drug delivery to negatively charged mitochondria.91 In this study, the cetrimonium bromide-capped Au nanorods showed that significantly increased accumulation in the mitochondria of A459 lung cancer cells resulted in the killing of cancer cells without affecting the functioning of normal cells. In the same manner, conjugation of rhodamine-110, a lipophilic cation, to mwCNTs significantly promoted internalization in the mitochondria of MCF-7 breast tumor cells by charge attractions and thus efficiently delivered cisplatin (platinum [IV]).121 In summary, mitochondrial targeting by cationic NPs can enhance the therapeutic efficacy of anticancer molecules and, ultimately, initiate signal cascades by activating Bax and Bak (proteins in mitochondria that regulate apoptotic cell death).122
Fate of endocytic nanoparticles
Elucidating the intracellular mechanism through endocytosis and exocytosis of various NPs is essential for anticipating the fate of nanocarriers. It is well established that uptaken NPs experience intracellular pathways (eg, EE, LE, and Lys) before reaching cell organelles (eg, cytosol, nucleus, mitochondria).88,123 However, few transport mechanisms were demonstrated for conveying NP to Golgi or ER sites. In this regard, nanoconjugation (eg, antibody) can enhance localization to the Golgi and Ly through EE.17 Specifically, EGFRs induced greater endocytosis when conjugated with Au NP and highlighted the importance of conjugation for cellular transport.17 In addition, cholesterol and phospholipid liposome NPs were localized into the ER and Golgi apparatus after endosome/Ly stages, and were eventually effluxed by permeability glycoprotein and ABCG1 efflux pumps (Figure 5).124 A recent review also discussed intracellular transport pathways of nanosized particles (toxin) to Golgi or ER sites. In particular, several toxins (eg, ricin, shiga, and anthrax) were translocated into the Golgi or ER after endolysosomal trafficking (eg, EE, LE, and Lys), and then secreted outside by secretory pathway (Figure 5).125–127
The exocytosis mechanism of various NPs has not yet been fully understood, in contrast to endocytosis mechanisms.88,128,129 Recent reviews reported the exocytosis of internalized NPs with respect to various physiochemical factors such as size, shape, and surface chemistry. For example, smaller Au NPs (14 nm) showed a faster rate of exocytosis than larger Au NPs (both 50 and 74 nm).130 In addition to NP size, the shape is another physiochemical factor influencing exocytosis of NP. In particular, rod-shaped Au NPs (ie, 20×30 nm, 14×50 nm, and 7×42 nm [diameter × length]) experience accelerated rates of exocytosis (ie, approximately fourfold faster) than spherically shaped Au NPs (ie, 14, 50 nm).51 Furthermore, cationic Au NPs (serum-coated NPs with a size of 15 nm) exhibited long retention times in cells by intracellular agglomeration, whereas PEGylated Au NPs (10 nm in diameter with negative charge) were rapidly excreted by cancer cells.131
Discussion and perspectives
Unfortunately, most nanodrugs that are approved by the US Food and Drug Administration or that are in clinical trials are currently confined to liposome-based drug encapsulation.132,133 Most inorganic nanodrugs are not considered safe drug-delivery materials owing to their accumulation in the liver and RES system, even though they have higher antitumor efficacy.134 As such, the biodegradation and clearance of inorganic nanodrugs are critical issues that must be addressed in order to achieve clinical approval.
In this line, increasing target efficacy without elevating accumulation in the RES and liver is a mandatory criterion in terms of immune toxicity.135 Specifically, future studies should evaluate the immune activity (ie, toxicity) by examining the accumulation of NPs (conjugated with drugs). In this regard, several reviews discussed the in vivo toxicity and anticipated immunological response of NPs.135–137
As discussed in the “Abstract” and “Introduction”, current chemotherapies, including antibody-based approaches, are difficult for supporting a patient’s quality of life due to the negative side effects of chemotherapy drugs.138 This is not an issue of drug efficacy, but rather the result of poor tumor-targeting efficacy. For this reason, this review introduced a broad set of strategies for overcoming current limitations to tumor target efficiency by highlighting current core technologies. Specifically, this review emphasized the application of nanoscale surface science, engineering, and the prospects of controlled interaction between active molecules and target cells.
One important element in reducing the side effects of drug therapeutics that was not fully discussed in this review is cancer drug efflux. Efflux pump proteins (eg, ABC transporters) are commonly overexpressed in tumor cells, which promote efflux of the cancer drug from the cytosol to outside the cell membrane.139–142 Endosomal drug delivery can bypass this drug efflux activity, generate greater amounts of endosomes that encapsulate the nanodrugs, and, ultimately, maximize therapeutic efficacy by safely delivering uptaken drugs to the targeted organelles (ie, the nucleus or mitochondria). Previous reviews have discussed cellular uptake of nanosized vectors via dynamin-dependent or -independent pathways, where the uptaken agents mostly experienced endosomal and lysosomal degradation.123,128,143 As such, nanosized drug agents are one of the most efficient materials for evading drug efflux from cancer cells without cotreatments (eg, verapamil and cyclosporine A).
In addition, physiochemical characteristics of specific organelles can be utilized for intracellular targeting. Specifically, mitochondrial apoptosis-inducible strategies by modifying the surface charge (ie, positive charge) of various NPs (eg, Au, liposomal, and mwCNT) are promising examples of targeting specific organelles using physiochemical materials–intracellular interactions.91,117–121
Although the safety of stem cell-based therapeutics remains controversial, engineered stem cell therapies have shown promising potential for clinical application due to their ability to search and communicate with tumor cells.55,56,66 For example, autologous (ie, harvested and cultured from the patient) MSCs can be conjugated with specially designed nanomaterials for personalized drug therapy to effectively destroy tumor cells. As discussed, NPs cannot easily penetrate the BBB, and present a challenge for drug delivery to brain tumors (ie, gliomas). However, nanoengineered MSCs (ie, through uptaken IO-MNP, as discussed earlier) bypassed the BBB and delivered anticancer molecules to the brain tumor site.
Furthermore, the time-dependent release of active molecules from endosome and Lys was recently analyzed.111,114 Design of the encapsulation strategy and choosing appropriate conjugation bonds with reference to the types of nanomaterial and anticancer agent are both critical factors for controlling drug release (ie, burst release at the vicinity of targeted organelles versus nonspecific release in the cytosol). The associated intracellular effects of physiochemical properties of nanomaterials are summarized in Table 3.
In conclusion, this review featured a diverse set of strategies currently being explored for minimizing anticancer drug dosing (of both small and macromolecules) by maximizing target efficacy. The combinatorial therapies introduced in this review provided multiple cues on the physiochemical, mesenchymal, and intracellular trafficking interactions with nanocarriers to maximize tumor-targeting efficiency, and supported guidelines in the future design of cancer therapeutics with controlled physiochemical-engineered cellular therapies.
Acknowledgments
This research was supported by grants from the National Research Foundation of Korea (2014R1A2A1A11052615) and the Korea Health Technology R&D Project through the KHIDI, funded by the Ministry of Health and Welfare (HI14C1802).
Disclosure
The authors report no conflict of interest in this work.
References
Jackson FI. Richard Feynman. J Roy Soc Med. 1997;90(6):360. | ||
Kochanek KD, Xu J, Murphy SL, Minino AM, Kung HC. Deaths: final data for 2009. Natl Vital Stat Rep. 2011;60(3):1–116. | ||
Park K. Facing the truth about nanotechnology in drug delivery. ACS Nano. 2013;7(9):7442–7447. | ||
Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12(11):958–962. | ||
Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: a review of current status and future prospects. Nano Today. 2014;9(2):223–243. | ||
Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65(24):11631–11638. | ||
Sadhukha T, O’Brien TD, Prabha S. Nano-engineered mesenchymal stem cells as targeted therapeutic carriers. J Control Release. 2014; 196:243–251. | ||
Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther. 2011;89(4):491–502. | ||
Wong CC, Cheng KW, Rigas B. Preclinical predictors of anticancer drug efficacy: critical assessment with emphasis on whether nanomolar potency should be required of candidate agents. J Pharmacol Exp Ther. 2012;341(3):572–578. | ||
Yang L, Mao H, Wang YA, et al. Single chain epidermal growth factor receptor antibody conjugated nanoparticles for in vivo tumor targeting and imaging. Small. 2009;5(2):235–243. | ||
El-Sayed IH, Huang X, El-Sayed MA. Selective laser photo-thermal therapy of epithelial carcinoma using anti-EGFR antibody conjugated gold nanoparticles. Cancer Lett. 2006;239(1):129–135. | ||
Owen SC, Patel N, Logie J, et al. Targeting HER2+ breast cancer cells: lysosomal accumulation of anti-HER2 antibodies is influenced by antibody binding site and conjugation to polymeric nanoparticles. J Control Release. 2013;172(2):395–404. | ||
Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120(1–2):18–26. | ||
Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188(6):759–768. | ||
Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5(2):147–159. | ||
Johnson JR, Ford CH, Newman CE, Woodhouse CS, Rowland GF, Simmonds RG. A vindesine-anti-CEA conjugate cytotoxic for human cancer cells in vitro. Br J Cancer. 1981;44(3):472–475. | ||
Bhattacharyya S, Bhattacharya R, Curley S, McNiven MA, Mukherjee P. Nanoconjugation modulates the trafficking and mechanism of antibody induced receptor endocytosis. Proc Natl Acad Sci U S A. 2010;107(33): 14541–14546. | ||
Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. | ||
Niidome T, Yamagata M, Okamoto Y, et al. PEG-modified gold nanorods with a stealth character for in vivo applications. J Control Release. 2006;114(3):343–347. | ||
Lipka J, Semmler-Behnke M, Sperling RA, et al. Biodistribution of PEG-modified gold nanoparticles following intratracheal instillation and intravenous injection. Biomaterials. 31(25):6574–6581. | ||
Balogh L, Nigavekar SS, Nair BM, et al. Significant effect of size on the in vivo biodistribution of gold composite nanodevices in mouse tumor models. Nanomedicine. 2007;3(4):281–296. | ||
Hirn S, Semmler-Behnke M, Schleh C, et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur J Pharm Biopharm. 2011; 77(3):407–416. | ||
Yang K, Wan J, Zhang S, Tian B, Zhang Y, Liu Z. The influence of surface chemistry and size of nanoscale graphene oxide on photothermal therapy of cancer using ultra-low laser power. Biomaterials. 2012; 33(7):2206–2214. | ||
Yang K, Gong H, Shi X, Wan J, Zhang Y, Liu Z. In vivo biodistribution and toxicology of functionalized nano-graphene oxide in mice after oral and intraperitoneal administration. Biomaterials. 2013; 34(11):2787–2795. | ||
Huang X, Zhang F, Zhu L, et al. Effect of injection routes on the biodistribution, clearance, and tumor uptake of carbon dots. ACS Nano. 2013;7(7):5684–5693. | ||
Liu Z, Davis C, Cai W, He L, Chen X, Dai H. Circulation and long-term fate of functionalized, biocompatible single-walled carbon nanotubes in mice probed by Raman spectroscopy. Proc Natl Acad Sci U S A. 2008; 105(5):1410–1415. | ||
Welsher K, Sherlock SP, Dai H. Deep-tissue anatomical imaging of mice using carbon nanotube fluorophores in the second near-infrared window. Proc Natl Acad Sci U S A. 2011;108(22):8943–8948. | ||
Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85(18):6949–6953. | ||
Allen TM. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol Sci. 1994;15(7):215–220. | ||
Maruyama K, Ishida O, Takizawa T, Moribe K. Possibility of active targeting to tumor tissues with liposomes. Adv Drug Deliv Rev. 1999; 40(1–2):89–102. | ||
Baek SE, Lee KH, Park YS, et al. RNA aptamer-conjugated liposome as an efficient anticancer drug delivery vehicle targeting cancer cells in vivo. J Control Release. 2014;196:234–242. | ||
Biswas S, Deshpande PP, Perche F, Dodwadkar NS, Sane SD, Torchilin VP. Octa-arginine-modified pegylated liposomal doxorubicin: an effective treatment strategy for non-small cell lung cancer. Cancer Lett. 2013;335(1):191–200. | ||
Paliwal SR, Paliwal R, Pal HC, et al. Estrogen-anchored pH-sensitive liposomes as nanomodule designed for site-specific delivery of doxorubicin in breast cancer therapy. Mol Pharm. 2012;9(1):176–186. | ||
Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–16740. | ||
Hatakeyama H, Akita H, Ishida E, et al. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int J Pharm. 2007;342(1–2):194–200. | ||
Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–1603. | ||
Frank MM, Fries LF. The role of complement in inflammation and phagocytosis. Immunol Today. 1991;12(9):322–326. | ||
Lasagna-Reeves C, Gonzalez-Romero D, Barria MA, et al. Bioaccumulation and toxicity of gold nanoparticles after repeated administration in mice. Biochem Biophys Res Commun. 2010;393(4):649–655. | ||
Balasubramanian SK, Jittiwat J, Manikandan J, Ong CN, Yu LE, Ong WY. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials. 2010;31(8):2034–2042. | ||
Cho WS, Cho M, Jeong J, et al. Acute toxicity and pharmacokinetics of 13 nm-sized PEG-coated gold nanoparticles. Toxicol Appl Pharmacol. 2009;236(1):16–24. | ||
Kim JS, Yoon TJ, Yu KN, et al. Toxicity and tissue distribution of magnetic nanoparticles in mice. Toxicol Sci. 2006;89(1):338–347. | ||
Gref R, Luck M, Quellec P, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18(3–4):301–313. | ||
Prencipe G, Tabakman SM, Welsher K, et al. PEG branched polymer for functionalization of nanomaterials with ultralong blood circulation. J Am Chem Soc. 2009;131(13):4783–4787. | ||
Perry JL, Reuter KG, Kai MP, et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 2012; 12(10):5304–5310. | ||
Zhang G, Yang Z, Lu W, et al. Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice. Biomaterials. 2009;30(10):1928–1936. | ||
Choi HS, Ipe BI, Misra P, Lee JH, Bawendi MG, Frangioni JV. Tissue- and organ-selective biodistribution of NIR fluorescent quantum dots. Nano Lett. 2009;9(6):2354–2359. | ||
Akiyama Y, Mori T, Katayama Y, Niidome T. The effects of PEG grafting level and injection dose on gold nanorod biodistribution in the tumor-bearing mice. J Control Release. 2009;139(1):81–84. | ||
Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007; 2(4):249–255. | ||
Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141(3):320–327. | ||
Barua S, Yoo JW, Kolhar P, Wakankar A, Gokarn YR, Mitragotri S. Particle shape enhances specificity of antibody-displaying nanoparticles. Proc Natl Acad Sci U S A. 2013;110(9):3270–3275. | ||
Chithrani BD, Chan WC. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7(6):1542–1550. | ||
Jin H, Heller DA, Sharma R, Strano MS. Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: single particle tracking and a generic uptake model for nanoparticles. ACS Nano. 2009;3(1):149–158. | ||
van de Ven AL, Kim P, Haley O, et al. Rapid tumoritropic accumulation of systemically injected plateloid particles and their biodistribution. J Control Release. 2012;158(1):148–155. | ||
Adeli M, Soleyman R, Beiranvand Z, Madani F. Carbon nanotubes in cancer therapy: a more precise look at the role of carbon nanotube-polymer interactions. Chem Soc Rev. 2013;42(12):5231–5256. | ||
Gao Z, Zhang L, Hu J, Sun Y. Mesenchymal stem cells: a potential targeted-delivery vehicle for anti-cancer drug, loaded nanoparticles. Nanomedicine. 2013;9(2):174–184. | ||
Fritz V, Jorgensen C. Mesenchymal stem cells: an emerging tool for cancer targeting and therapy. Curr Stem Cell Res Ther. 2008;3(1):32–42. | ||
Hu YL, Fu YH, Tabata Y, Gao JQ. Mesenchymal stem cells: a promising targeted-delivery vehicle in cancer gene therapy. J Control Release. 2010;147(2):154–162. | ||
Roger M, Clavreul A, Venier-Julienne MC, et al. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials. 2010;31(32):8393–8401. | ||
Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96(21):1593–1603. | ||
Choi MR, Stanton-Maxey KJ, Stanley JK, et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007; 7(12):3759–3765. | ||
Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531–541. | ||
Anselmo AC, Gupta V, Zern BJ, et al. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano. 2013;7(12):11129–11137. | ||
Smith BR, Ghosn EE, Rallapalli H, et al. Selective uptake of single-walled carbon nanotubes by circulating monocytes for enhanced tumour delivery. Nat Nanotechnol. 2014;9(6):481–487. | ||
Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–216. | ||
Menon LG, Picinich S, Koneru R, et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007;25(2):520–528. | ||
Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–2623. | ||
Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. | ||
Sato H, Kuwashima N, Sakaida T, et al. Epidermal growth factor receptor-transfected bone marrow stromal cells exhibit enhanced migratory response and therapeutic potential against murine brain tumors. Cancer Gene Ther. 2005;12(9):757–768. | ||
Albarenque SM, Zwacka RM, Mohr A. Both human and mouse mesenchymal stem cells promote breast cancer metastasis. Stem Cell Res. 2011;7:163–171. | ||
Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat Rev Cancer. 2014;14(10):683–691. | ||
Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726–736. | ||
Maccario R, Podesta M, Moretta A, et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of CD4+ T-cell subsets expressing a regulatory/suppressive phenotype. Haematologica. 2005;90(4):516–525. | ||
Nemeth K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15(1):42–49. | ||
Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105(4):1815–1822. | ||
Ohlsson LB, Varas L, Kjellman C, Edvardsen K, Lindvall M. Mesenchymal progenitor cell-mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol Pathol. 2003;75(3):248–255. | ||
Maestroni GJ, Hertens E, Galli P. Factor(s) from nonmacrophage bone marrow stromal cells inhibit Lewis lung carcinoma and B16 melanoma growth in mice. Cell Mol Life Sci. 1999;55(4):663–667. | ||
Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. 2007;21(2):304–310. | ||
Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26(12):3027–3036. | ||
Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. | ||
Li L, Guan Y, Liu H, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano. 2011; 5(9):7462–7470. | ||
Huang X, Zhang F, Wang Y, et al. Design considerations of iron-based nanoclusters for noninvasive tracking of mesenchymal stem cell homing. ACS Nano. 2014;8(5):4403–4414. | ||
Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13(12):1838–1845. | ||
Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6(9):688–701. | ||
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3): 329–347. | ||
Kam NW, Liu Z, Dai H. Carbon nanotubes as intracellular transporters for proteins and DNA: an investigation of the uptake mechanism and pathway. Angew Chem. 2006;45(4):577–581. | ||
Verma A, Uzun O, Hu Y, et al. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat Mater. 2008;7(7):588–595. | ||
Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell interactions. Small. 2010;6(1):12–21. | ||
Iversen T, Skotland T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: present knowledge and need for future studies. Nano Today. 2011;6:176–185. | ||
Biswas S, Torchilin VP. Nanopreparations for organelle-specific delivery in cancer. Adv Drug Deliv Rev. 2014;66:26–41. | ||
Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7(10):1322–1337. | ||
Wang L, Liu Y, Li W, et al. Selective targeting of gold nanorods at the mitochondria of cancer cells: implications for cancer therapy. Nano Lett. 2011;11(2):772–780. | ||
Sahay G, Kim JO, Kabanov AV, Bronich TK. The exploitation of differential endocytic pathways in normal and tumor cells in the selective targeting of nanoparticulate chemotherapeutic agents. Biomaterials. 2010;31(5):923–933. | ||
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 6th ed. New York, NY: Garland Science; 2014. | ||
Martens TF, Remaut K, Demeester J, De Smedt SC, Braeckmans K. Intracellular delivery of nanomaterials: how to catch endosomal escape in the act. Nano Today. 2014;9:344–364. | ||
Gilleron J, Querbes W, Zeigerer A, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31(7):638–646. | ||
ur Rehman Z, Hoekstra D, Zuhorn IS. Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano. 2013;7(5):3767–3777. | ||
Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 2011;63(3):152–160. | ||
Kogure K, Moriguchi R, Sasaki K, Ueno M, Futaki S, Harashima H. Development of a non-viral multifunctional envelope-type nano device by a novel lipid film hydration method. J Control Release. 2004;98(2):317–323. | ||
Yamada Y, Nomura T, Harashima H, Yamashita A, Katoono R, Yui N. Intranuclear DNA release is a determinant of transfection activity for a non-viral vector: biocleavable polyrotaxane as a supramolecularly dissociative condenser for efficient intranuclear DNA release. Biol Pharm Bull. 2010;33(7):1218–1222. | ||
Khalil IA, Kogure K, Futaki S, et al. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther. 2007;14(8):682–689. | ||
Masuda T, Akita H, Nishio T, et al. Development of lipid particles targeted via sugar-lipid conjugates as novel nuclear gene delivery system. Biomaterials. 2008;29(6):709–723. | ||
Hatakeyama H, Akita H, Kogure K, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14(1):68–77. | ||
Nakamura Y, Kogure K, Futaki S, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119(3):360–367. | ||
Mudhakir D, Akita H, Tan E, Harashima H. A novel IRQ ligand-modified nano-carrier targeted to a unique pathway of caveolar endocytic pathway. J Control Release. 2008;125(2):164–173. | ||
Hatakeyama H, Ito E, Akita H, et al. A pH-sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA-containing nanoparticles in vitro and in vivo. J Control Release. 2009;139(2):127–132. | ||
Nakamura Y, Kogure K, Yamada Y, Futaki S, Harashima H. Significant and prolonged antisense effect of a multifunctional envelope-type nano device encapsulating antisense oligodeoxynucleotide. J Pharm Pharmacol. 2006;58(4):431–437. | ||
Nakamura T, Moriguchi R, Kogure K, Shastri N, Harashima H. Efficient MHC class I presentation by controlled intracellular trafficking of antigens in octaarginine-modified liposomes. Mol Ther. 2008;16(8):1507–1514. | ||
Suzuki R, Yamada Y, Harashima H. Efficient cytoplasmic protein delivery by means of a multifunctional envelope-type nano device. Biol Pharm Bull. 2007;30(4):758–762. | ||
Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12(11):991–1003. | ||
Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16(10):1217–1226. | ||
Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443. | ||
Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205(2):275–292. | ||
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. | ||
Lee YK, Choi J, Wang W, et al. Nullifying tumor efflux by prolonged endolysosome vesicles: development of low dose anticancer-carbon nanotube drug. ACS Nano. 2013;7(10):8484–8497. | ||
Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27(50):6398–6406. | ||
Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75(2):241–251. | ||
Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 2000;41(2):235–250. | ||
Boddapati SV, D’Souza GG, Erdogan S, Torchilin VP, Weissig V. Organelle-targeted nanocarriers: specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008;8(8):2559–2563. | ||
Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J Control Release. 2012;159(3):393–402. | ||
Yamada Y, Akita H, Kamiya H, et al. MITO-Porter: a liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778(2):423–432. | ||
Yoong SL, Wong BS, Zhou QL, et al. Enhanced cytotoxicity to cancer cells by mitochondria-targeting MWCNTs containing platinum(IV) prodrug of cisplatin. Biomaterials. 2014;35(2):748–759. | ||
Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. | ||
Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758. | ||
Un K, Sakai-Kato K, Oshima Y, Kawanishi T, Okuda H. Intracellular trafficking mechanism, from intracellular uptake to extracellular efflux, for phospholipid/cholesterol liposomes. Biomaterials. 2012;33(32):8131–8141. | ||
Sandvig K, Torgersen ML, Engedal N, Skotland T, Iversen TG. Protein toxins from plants and bacteria: probes for intracellular transport and tools in medicine. FEBS Lett. 2010;584(12):2626–2634. | ||
Zornetta I, Brandi L, Janowiak B, et al. Imaging the cell entry of the anthrax oedema and lethal toxins with fluorescent protein chimeras. Cell Microbiol. 2010;12(10):1435–1445. | ||
Tarrago-Trani MT, Storrie B. Alternate routes for drug delivery to the cell interior: pathways to the Golgi apparatus and endoplasmic reticulum. Adv Drug Deliv Rev. 2007;59(8):782–797. | ||
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. | ||
Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58(1):32–45. | ||
Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomed. 2014;9(Suppl 1):51–63. | ||
Oh N, Park JH. Surface chemistry of gold nanoparticles mediates their exocytosis in macrophages. ACS Nano. 2014;8(6):6232–6241. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–680. | ||
Bhattacharyya S, Kudgus RA, Bhattacharya R, Mukherjee P. Inorganic nanoparticles in cancer therapy. Pharm Res. 2011;28(2):237–259. | ||
Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5(4):487–495. | ||
Fadel TR, Fahmy TM. Immunotherapy applications of carbon nanotubes: from design to safe applications. Trends Biotechnol. 2014;32(4):198–209. | ||
Magdolenova Z, Collins A, Kumar A, Dhawan A, Stone V, Dusinska M. Mechanisms of genotoxicity. A review of in vitro and in vivo studies with engineered nanoparticles. Nanotoxicology. 2014;8(3):233–278. | ||
Burish TG, Carey MP, Krozely MG, Greco FA. Conditioned side effects induced by cancer chemotherapy: prevention through behavioral treatment. J Consult Clin Psychol. 1987;55(1):42–48. | ||
Xue X, Liang XJ. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin J Cancer. 2012;31(2):100–109. | ||
Dong X, Mumper RJ. Nanomedicinal strategies to treat multidrug-resistant tumors: current progress. Nanomedicine. 2010;5(4):597–615. | ||
Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Nanomedicine for targeted cancer therapy: towards the overcoming of drug resistance. Drug Resist Updat. 2011;14(3):150–163. | ||
Chen CJ, Chin JE, Ueda K, et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47(3):381–389. | ||
Rajendran L, Knolker HJ, Simons K. Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov. 2010;9(1):29–42. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.