Back to Journals » Drug Design, Development and Therapy » Volume 15
Multiple Administrations of Itraconazole Increase Plasma Exposure to Pyrotinib in Chinese Healthy Adults
Authors Liu Y, Zhang Q, Lu C, Hu W
Received 24 March 2021
Accepted for publication 26 May 2021
Published 10 June 2021 Volume 2021:15 Pages 2485—2493
DOI https://doi.org/10.2147/DDDT.S312310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Yueyue Liu, Qian Zhang, Chao Lu, Wei Hu
Department of Clinical Pharmacology, The Second Hospital of Anhui Medical University, Hefei, 230601, People’s Republic of China
Correspondence: Chao Lu; Wei Hu
Department of Clinical Pharmacology, The Second Hospital of Anhui Medical University, Hefei, 230601, People’s Republic of China
Tel/Fax + 86-551 63806057
Email [email protected]; [email protected]
Purpose: Pyrotinib, an irreversible human epidermal growth factor receptor 2 (HER2), is a epidermal growth factor receptor double-target tyrosine kinase inhibitor used for treating HER2-positive breast cancer. This study aimed to evaluate the impact of the strong CYP3A4 inhibitor itraconazole on the safety and pharmacokinetics of pyrotinib in Chinese healthy adults.
Patients and Methods: This was an open-label, randomized, self-control study. Eighteen healthy adults were included in this trial. They received a single 80 mg dose of pyrotinib orally on days 1 and 9, and a 200 mg once-daily dose of itraconazole on days 6 through 22. Blood samples were obtained, and the drug concentration was detected using liquid chromatography/tandem mass spectrometry.
Results: Compared with pyrotinib alone, the exposure to pyrotinib co-administered with itraconazole substantially increased, and the Cmax and AUC0-t increased by 2.78- and 10.8-fold, respectively. No serious adverse events were reported in this trial, and no participant dropped out of the trial because of adverse events.
Conclusion: The exposure to pyrotinib was substantially affected by the action of itraconazole. The concomitant use of pyrotinib with itraconazole might require dose modification of pyrotinib. All treatments were well tolerated in healthy participants.
Clinical Trial Registry: http://www.chinadrugtrials.org.cn/clinicaltrials.prosearch.dhtml, CTR20191866.
Keywords: drug–drug interaction, itraconazole, pharmacokinetics, pyrotinib, safety
Introduction
Pyrotinib is a new type of irreversible epidermal growth factor receptor/human epidermal growth factor receptor 2 (double tyrosine kinase inhibitor mainly used to treat HER2-positive breast cancer.1 The first-in-patient study suggested that pyrotinib was well tolerated and showed encouraging efficacy in HER2-positive patients with metastatic breast cancer.2 Previous randomized Phase II clinical trial showed that pyrotinib alone or in combination with capecitabine was beneficial to patients with HER2-positive breast cancer.3 Several studies have also shown the safety and efficacy of pyrotinib in treating patients with HER-2-positive breast cancer.4–8 In addition, pyrotinib has also been developed for treating HER-2-positive gastric cancer, non small cell lung cancer, and other HER2-positive solid tumors, with clinical trials currently underway.1,9–12 Considering the important clinical value of pyrotinib, it was given priority review and approved by the China Food and Drug Administration in 2018.1
The pharmacokinetic study of pyrotinib showed that, after the first dose of pyrotinib 80–400 mg, the median time to peak concentration (Tmax) was 3–5 h and the mean terminal half-life (T1/2) was 11.4–15.9 h. No major accumulation of pyrotinib was noted following repeated daily administration. A linear relationship was observed between the peak concentration (Cmax) and area under the concentration–time curve at the steady state (AUCss) of pyrotinib and the dose.2 Pyrotinib was mainly metabolized by CYP3A4 to produce O-despicoline, carbonylation O-despicoline, dehydro-pyrrolidine, carbonyl-N-methylpyrrole alkylmethylpyrrolidine, hydroxymethyl pyrrolidine and hydroxymethyl pyrrolidine, and double oxidation and dehydrogenation metabolites.13 Data reported that the amino acid residue Lys190 of human serum albumin was covalently bound to pyrotinib through Michael addition, and the covalently bound pyrotinib could be separated from human plasma protein, oxidized and metabolized, and excreted through feces.14 Diarrhea was the most common adverse event (AE) of pyrotinib. The incidence and severity of diarrhea were dose dependent.2
The in vitro enzyme phenotype test showed that CYP3A4 was the principal enzyme in the metabolism of pyrotinib.13 It was suggested that the effects of combined CYP3A4 inhibitors on the pharmacokinetics and metabolism of pyrotinib should be monitored clinically. Most of the drugs used in many patients with cancer are metabolized by CYP3A4, and the changes in the plasma concentration or metabolic behavior caused by drug interaction are directly linked to the safety and efficacy of the drug. It was suggested that when pyrotinib was co-administered with CYP3A4 inhibitors, changes in the exposure of pyrotinib might increase patients’ risk of medication, leading to treatment failure or toxicity. However, no reports were available to show the effect of strong CYP3A4 inhibitors on the pharmacokinetics of pyrotinib. Itraconazole, a strong CYP3A4 inhibitor, is recommended by regulatory authorities as a representative inhibitor in drug–drug interaction studies.15,16 Therefore, itraconazole was used to explore whether the pharmacokinetic parameters of pyrotinib were affected by CYP3A4 inhibitors.
Methods
Drug and Clinical Trial Information
The pyrotinib maleate tablets used in this study were produced and provided by Jiangsu Hengrui Pharmaceutical Co., Ltd (Dalian, China) with a specification of 80 mg. Itraconazole capsule was produced by Xi’an Janssen Pharmaceutical Co., Ltd. (Xian, China) with a specification of 0.1 g. The trial was launched on September 14, 2019. The first participant signed the informed consent form on September 16, 2019. All participants were enrolled on September 18, 2019. On September 19, 2019, the participant completed the first dose. The observation for adverse reactions in all participants ended on November 30, 2019.
Participants
Healthy male and female participants, aged 18–45 years (including both ends), had a body mass index of 19.0–26.0 kg/m2 and weighed no less than 45 kg (female) and 50 kg (male). All participants were confirmed to be in good health by the researchers, based on comprehensive physical examination (vital signs and physical examination), routine laboratory examination (blood routine, blood biochemistry, and urine routine), serum virological examination (hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antigen/antibody), coagulation function (plasma prothrombin time, activated partial thromboplastin time, international standardized ratio),12-lead electrocardiogram, chest x-ray, heart color ultrasound, human chorionic gonadotropin serum test, drug screening (morphine, methamphetamine, and ketamine), and alcohol tests within 1 week before administration. Participants who recently participated in clinical trials of other drugs or took drugs (including vitamins, supplements, and herbal medicines); were allergic to drugs, especially pyrotinib tablets, itraconazole, or other excipients; were pregnant; were involved in drug use and alcohol abuse; and could not tolerate venipuncture blood collection were excluded. In addition, all participants signed the informed consent form before the trial; fully understood the content, process, and possible adverse reactions of the study; and were able to complete the study in accordance with the requirements of the trial.
Study Design
The study was a single-center, open, single-dose, self-control clinical trial. A total of 18 healthy participants were scheduled to be enrolled in the group. They were examined during the screening period within 7 days prior to administration, and eligible participants were admitted to the Phase I clinical trial ward the day before the initial dose. Healthy adults received a single dose of pyrotinib 80 mg orally on days 1 and 9, and a 200 mg once-daily dose of itraconazole on days 6 through 22 (Figure 1). All treatments were given after overnight fasting for at least 10 h. Safety assessment was carried out on day 5. In terms of drug administration, the participants began to eat the standard meal 30 min before administration in the morning, finished the meal within 30 min, and then took orally 80 mg of pyrotinib and/or 200 mg of itraconazole with 240 mL of warm water 30 min after meal. The participants were required to abstain from water within 1 h before administration and following fasting within 4 h after administration. The participants were discharged from the hospital after completing the group examination on the morning of day 23 and were hospitalized for 24 days. In addition, participants were followed up telephonically from 28 to 32 days after the initial administration.
 |
Figure 1 Study design. |
Blood Sample Collection and Bioanalytical Methods
The blood samples for pyrotinib were collected 0 (predose), 1, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, and 96 h after administration on day 1 and 0 (predose),1, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 96, 168, and 336 h after administration on day 9. About 4 mL of blood was obtained from each blood collection point. The blood samples were centrifuged, and the plasma was stored in a −70 °C refrigerator for detection.
The concentration of pyrotinib was determined by validated liquid chromatography/tandem mass spectrometry (LC–MS/MS). LC–MS/MS analysis was carried out using a UPLC 30A LC system (Shimadzu, Japan) coupled with a QTRAP 5500 triple quadrupole MS (AB Sciex, Canada) equipped with an ESI ion spray source.
Chromatographic separation was performed using 0.1% formic acid aqueous solution and 10mM ammonium acetate in water (A) and acetonitrile (B) as the mobile phase on BEH C18 column (50 × 2.1 mm, 1.7 μm, Waters). Gradient elution was used as follows: 10% B at 0–0.3 min, 10–95% B at 0.3–0.8 min, and then maintained for 1.1 min; 95–10% B at 1.9–2.2 min; and 10% B at 2.2–2.9 min with a flow rate of 0.55 mL/min. The injection volume was 1 μL, and the total run time for a single sample analysis was 2.9 min. Mass spectrometric analysis was performed using positive ESI in a multiple reaction-monitoring mode with both Q1 and Q3 resolution sets to unity. Ion pairs of m/z 583.4→ 138.2 for pyrotinib and m/z 392.2→161.9 for labetalol were selected. Pyrotinib displayed good linearity in the range of 1.00–500 ng/mL.
Pharmacokinetic Evaluations and Statistical Analyses
The pharmacokinetic parameters of pyrotinib were calculated using the non compartment model of Phoenix WinNonlin 8.1 (Pharsight Corporation, CA, USA) according to the actual sampling time, including the main outcomes: the maximum observed concentration (Cmax), the area under the blood concentration–time curve from time 0 to the time of last measurable concentration collection (AUC0-t), and extrapolated to infinite time (AUC0-∞). The time to Cmax (Tmax), elimination half-life (T1/2), apparent clearance (CL/F), and apparent volume of distribution (Vz/F) were also calculated.
Plasma pyrotinib pharmacokinetic parameters AUC0-∞, AUC0-t, and Cmax after natural logarithm transformation were fitted to obtain the corrected least square mean difference between the combination drug and pyrotinib single drug by fitting a mixed-effects model and its estimate of 90% confidence interval. When using mixed-effects model analysis with SAS 9.4 software (SAS Institute Inc., NC, USA) medication (itraconazole combined with pyrotinib or pyrotinib alone) was used as a fixed effect, and participants participated in the model fitting as a random effect. The corrected least-squares mean difference and its 90% confidence interval obtained from this analysis were then converted into the corrected geometric mean ratio and its 90% confidence interval by picking up the antilog.
Safety Assessments
In this study, safety evaluation ran through all the time, and the safety of the drug was evaluated by adverse event recording, laboratory examination, vital signs, physical examination, and 12-lead ECG. Also, the participants were followed up by telephone at the end of the trial. The researchers collected all observed or actively reported adverse events to determine and record the severity of AEs (classified according to Common Terminology Criteria for Adverse Events version 5.0) and the relationship between AE and research drugs.17 AEs included adverse drug reactions, new diseases in the course of the study, and aggravation of previous diseases. In addition, the researchers could record significant clinical changes, including any unexpected symptoms, signs, abnormal physical examination results, and objective test results as AEs. If an adverse event occurred during the trial, the participants were required to undergo safety follow-up until all AEs disappeared or reached a stable state, or a reasonable explanation was obtained.
Results
Study Participants
A total of 18 participants (14 male and 4 female, with an average age of 29.9 years) were included in this study. All the participants passed the screening test to prove that they were in good health. Except one case who was Mongolian, the other participants were of Han nationality. The demographic characteristics of the participants are shown in Table 1.
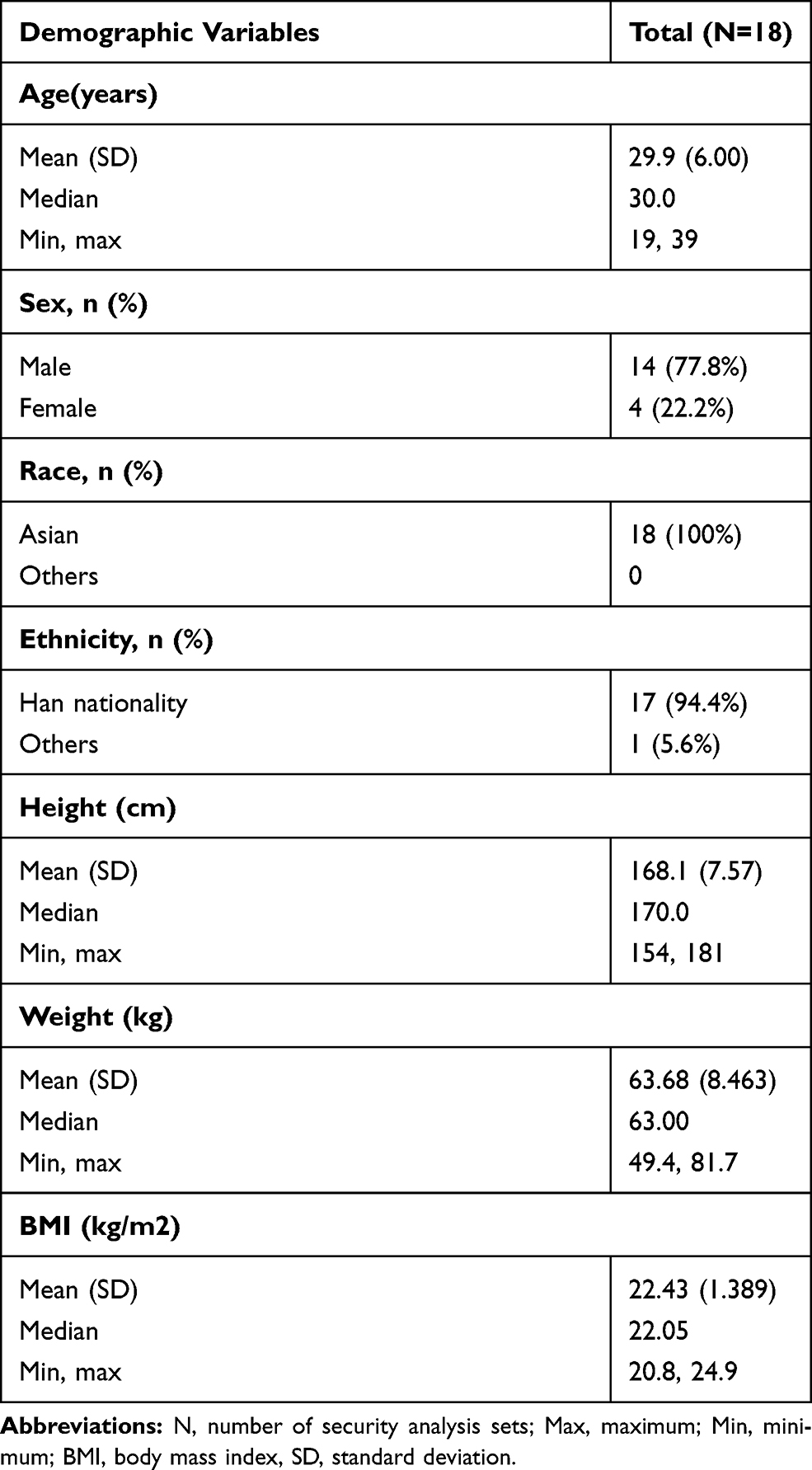 |
Table 1 Demographic Characteristics |
Plasma Concentrations of Pyrotinib
In different administration stages (pyrotinib alone or itraconazole combined with pyrotinib), the mean plasma concentration–time curves of pyrotinib in plasma after the oral administration of pyrotinib 80 mg are shown in Figure 2. The results of blood concentration analysis showed that pyrotinib could be detected in most of the participants 1 h after administration and reached the Cmax 5–6 h after administration of a single oral administration of 80 mg pyrotinib at different stages of administration (pyrotinib alone or itraconazole combined with pyrotinib). The mean plasma concentration–time curves and individual plasma concentration–time curves were observed. The findings were as follows: (1) The blood concentrations in 16 participants were lower than those of LLOQ 72 h after administration of pyrotinib. Also, the plasma concentrations of all participants were lower than those of LLOQ 96 h after the administration of pyrotinib. (2) In the combined administration stage, the plasma concentration of pyrotinib could still be detected in all participants 96 h after administration. Also, the plasma concentration could still be detected in eight participants, except that the plasma concentration of ten participants was found to be lower than that of LLOQ by the analytical method 336 h after the administration of pyrotinib.
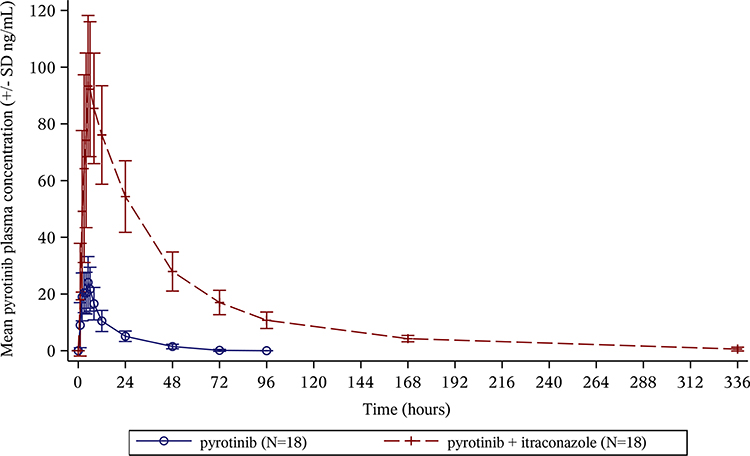 |
Figure 2 Mean plasma concentration of pyrotinib during the study period. The error bars represent the standard deviations. |
Pharmacokinetic Parameters
The PK parameters of pyrotinib with and without itraconazole are presented in Table 2, while the comparisons of PK parameters of pyrotinib are shown in Table 3. After administering pyrotinib alone or co-administering pyrotinib and itraconazole, the Tmax of pyrotinib was comparable, with a median of about 5 h. The elimination half-life of pyrotinib was prolonged, the CL/F rate decreased, and the exposure increased after co-administration with itraconazole compared with a single administration. The least-squares geometric mean ratio (pyrotinib combined with itraconazole/pyrotinib alone) of Cmax, AUC0-t, and AUC0-∞, and their 90% confidence interval, were 3.78 (3.479–4.117), 11.8 (10.78–12.89), and 11.4 (10.48–12.35), respectively. It indicated that after administering itraconazole combined with pyrotinib, the Cmax, AUC0-t, and AUC0-∞ of pyrotinib increased by 2.78-, 10.8- and 10.4-fold, respectively.
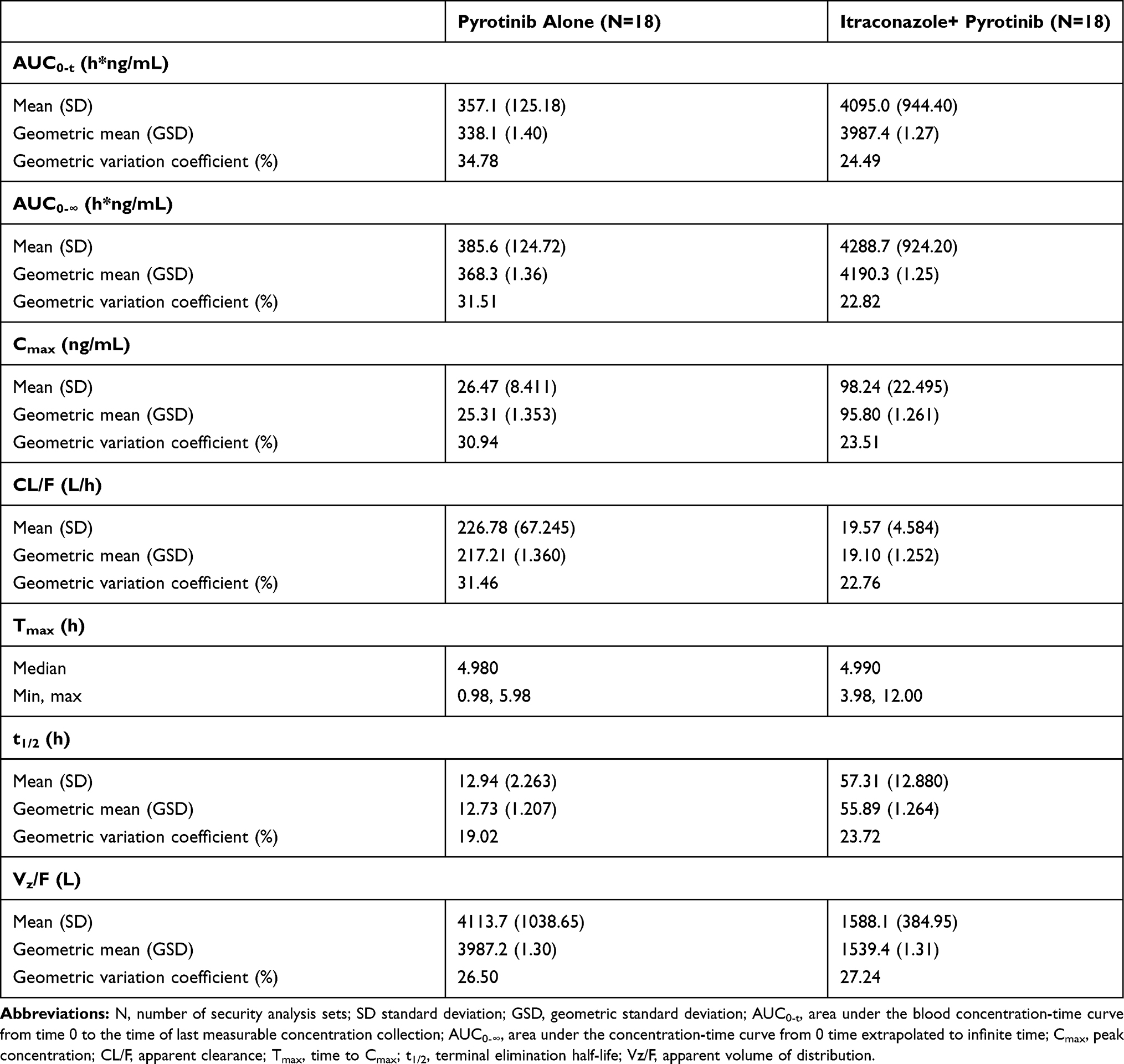 |
Table 2 Pharmacokinetic Parameters of Pyrotinib in Plasma During the Study Period (Analysis Set of Pharmacokinetic Parameters) |
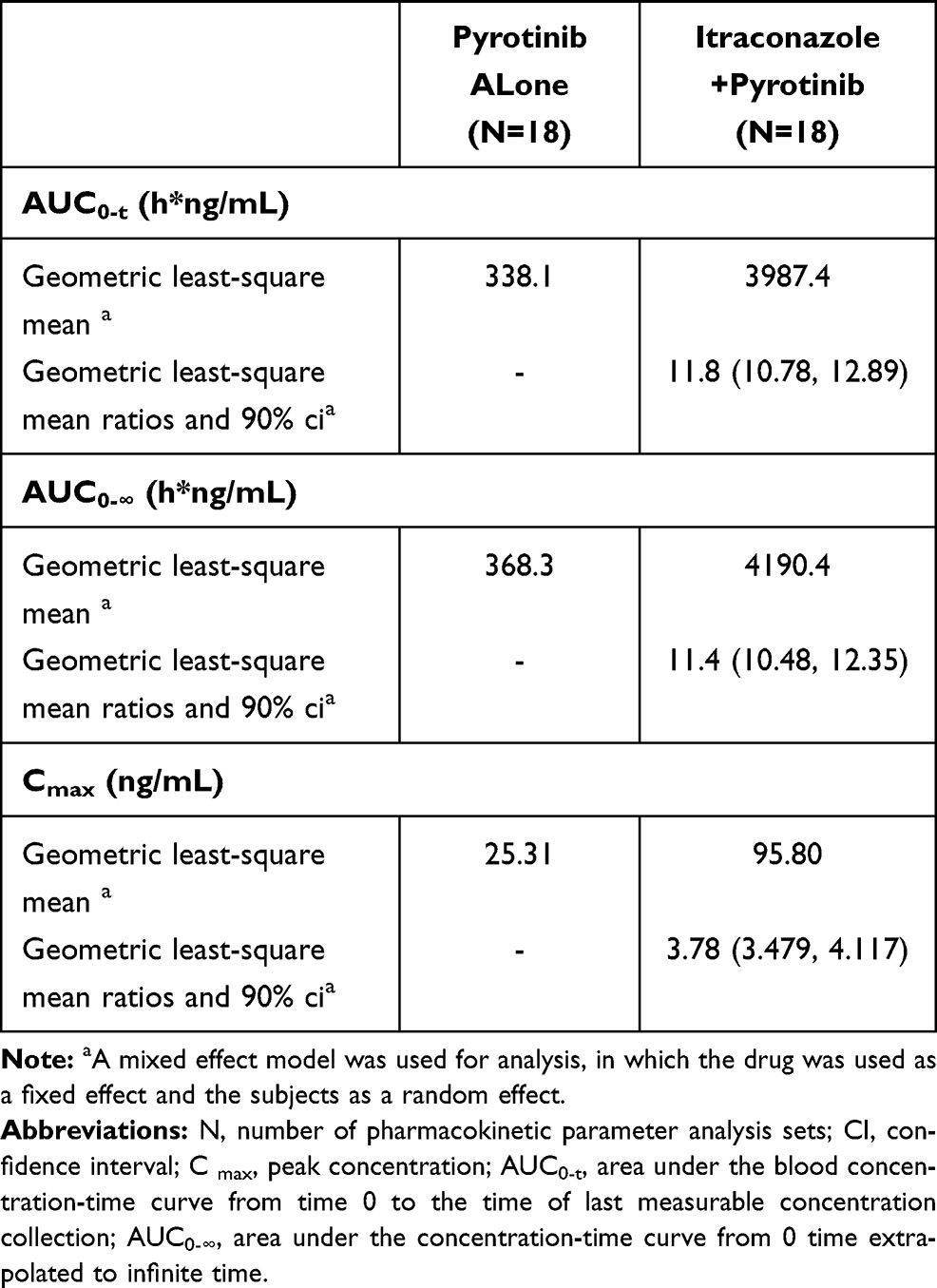 |
Table 3 Comparison of Pharmacokinetic Parameters of Pyrotinib Alone and Itraconazole Combined with Pyrotinib During the Study Period (Analysis Set of Pharmacokinetic Parameters) |
Safety Assessment
Common (incidence ≥ 10%) pyrotinib adverse reactions in this study included hypertriglyceridemia (27.8%), decreased white blood cell count (11.1%), decreased neutrophil count (11.1%), abdominal pain (11.1%), and diarrhea (11.1%). Other side effects of pyrotinib included increased blood bilirubin levels (5.6%) and upper respiratory tract infection (5.6%). The common adverse reactions of (incidence ≥ 10%) itraconazole included diarrhea (16.7%) and headache (11.1%). Other adverse reactions of itraconazole included abdominal distension (5.6%), oropharyngeal pain (5.6%), skin rash (5.6%), and eyelid edema (5.6%). The summary of all AEs during treatment is shown in Tables 4 and 5. The severity of all AEs in this study was grade 1 or 2. No AEs of grade 3 and above occurred. All AEs were self-resolving without special treatment.
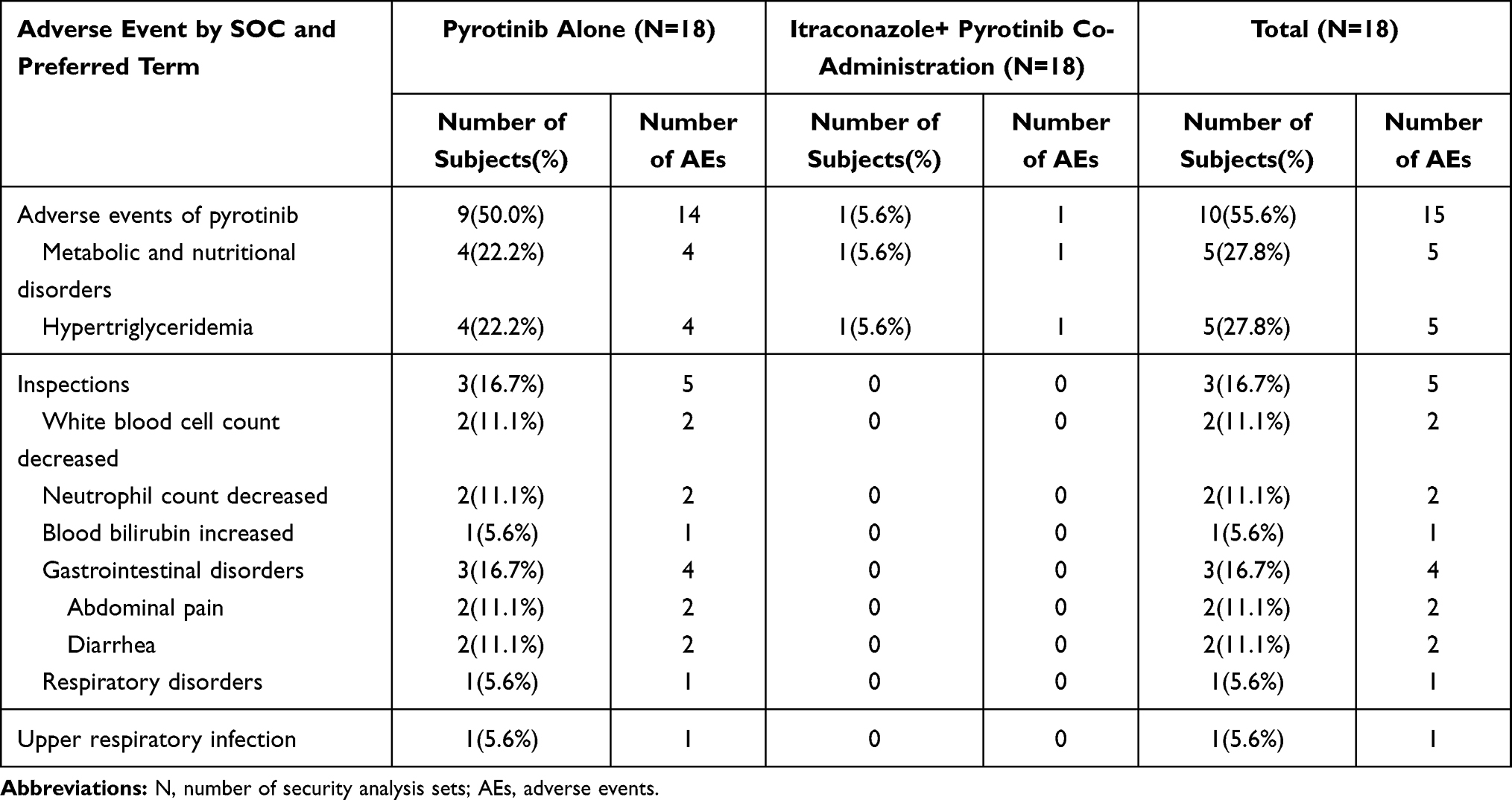 |
Table 4 Summary of Adverse Events to Pyrotinib |
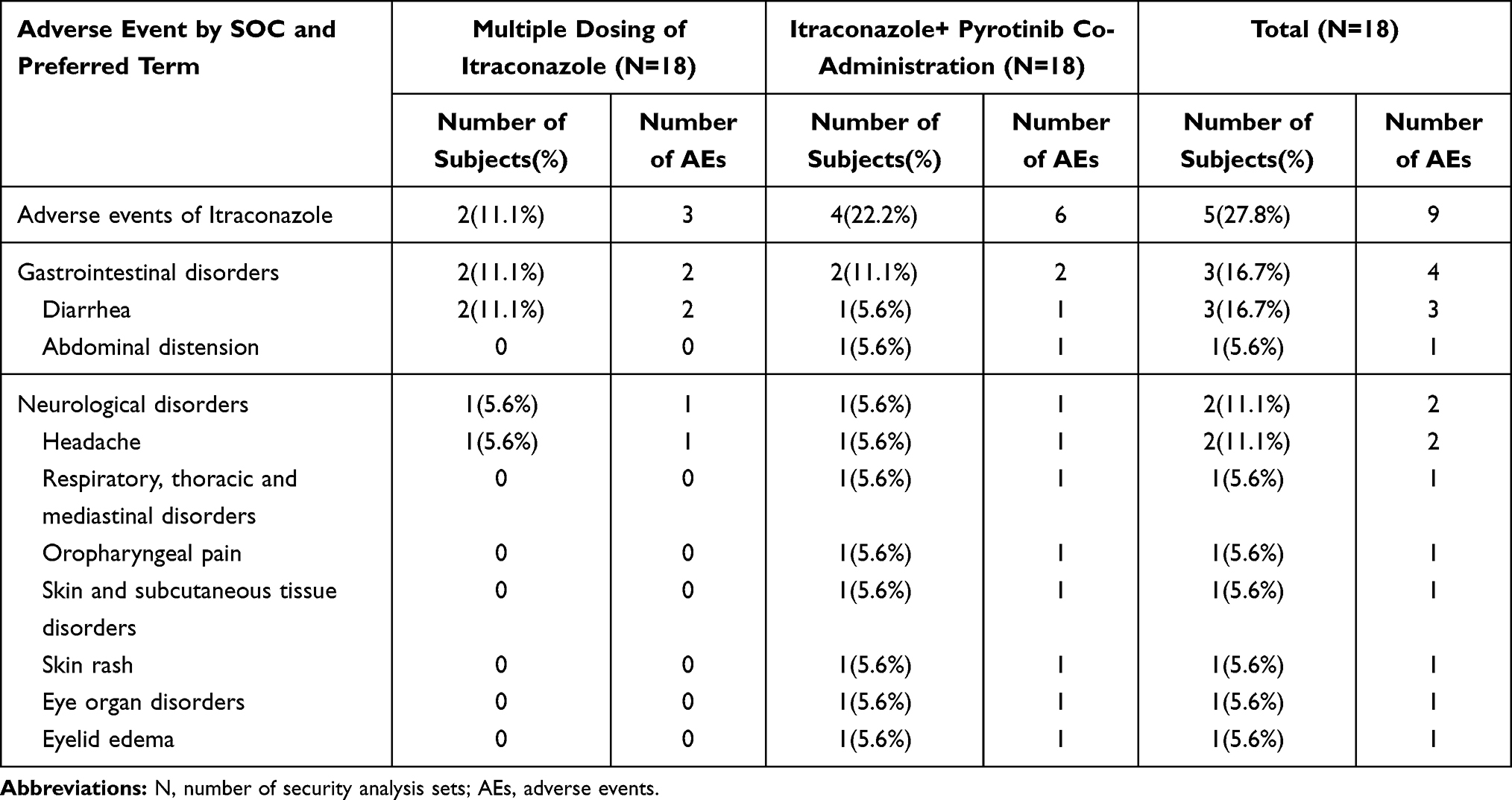 |
Table 5 Summary of Adverse Events to Itraconazole |
Discussion
The study was the first clinical drug–drug interaction research on the use of pyrotinib. This trial, conducted on healthy participants, demonstrated that pyrotinib co-administered with itraconazole was safe and well tolerated. Itraconazole could also increase the exposure, prolong the elimination period, and decrease the clearance rate of pyrotinib. In previous studies, pyrotinib was safe and well tolerated in healthy participants.14 Therefore, in this trial, we chose healthy participants as the target population.
According to the guidelines for drug–drug interactions, this study chose itraconazole as a potent inhibitor of CYP3A4 to investigate drug–drug interactions. The commonly used clinical dose of itraconazole is 200–400 mg per day. Compared with a single dose of 200 mg of itraconazole, a single dose of 400 mg leads to a limited increase in the inhibitory effect of CYP3A.18 Therefore, itraconazole was given at a dose of 200 mg once a day in this study. Preclinical studies on the CYP450 enzyme phenotype of pyrotinib showed that pyrotinib was mainly metabolized by CYP3A4.13,14 Therefore, when pyrotinib was used in combination with CYP3A4 inhibitors, it reduced the metabolism of pyrotinib and increased its exposure in the body. Considering that the recommended dose of pyrotinib in the Phase III clinical trial of pyrotinib was 400 mg, the exposure of pyrotinib might increase during concomitant use with itraconazole. Consequently, this study chose 80 mg as the single dose of pyrotinib for safety reasons.
The T1/2 of a single dose of pyrotinib in healthy participants was about 15–20 h. Therefore, the cleaning period of pyrotinib was designed to be 5 days to ensure that pyrotinib was basically eliminated from the body before itraconazole was taken. In addition, a 3-day introduction period of itraconazole was designed before the combination of pyrotinib and itraconazole, to maximize the extent of inhibition of the interacting drugs under steady-state conditions. It was predicted that the elimination of pyrotinib would be slower after combined administration. Therefore, the pharmacokinetic blood collection time was prolonged to 336 h, during which itraconazole was given continuously to maintain its inhibition of the CYP3A4 enzyme.
The results demonstrated that the elimination T1/2 of pyrotinib was prolonged, the CL/F decreased, and Cmax, AUC0-t, and AUC0-∞ increased by 2.78-, 10.8-, and 10.4-fold, respectively, after co-administration with itraconazole. An increase in pyrotinib AUC combined with an increase in its elimination T1/2 and a decrease in pyrotinib CL/F showed the key role of CYP3A4 in pyrotinib elimination. Neratinib is an analog of pyrotinib. On co-administering with the potent CYP3A4 inhibitor, ketoconazole significantly increased the exposure of neratinib in humans, with the Cmax and AUC increased by 3.2- and 4.8-fold, respectively. It was thus recommended to adopt dose adjustment for neratinib when administered with CYP3A4 inhibitors.19 The effects of CYP3A4 inhibitors on pyrotinib pharmacokinetics were more profound in magnitude compared with the effects observed on neratinib. Therefore, dose adjustment/reduction was recommended to be adopted when patients taking pyrotinib were administered with CYP3A4 inhibitors. In addition, pyrotinib was a weak inhibitor of CYP2C19 and also a substrate for P-glycoprotein.13 Therefore, pharmacokinetic interactions might occur during use with CYP2C9-metabolized drugs or P-glycoprotein inhibitors, but it requires further investigation.
Previous studies on pyrotinib showed that gastrointestinal diseases and adverse reactions on the skin and subcutaneous tissue were common AEs, especially diarrhea, nausea, vomiting, and palmoplantar redness syndrome.1 Diarrhea was the most common AE and also the most common ≥ III AE in all studies. The incidence, severity, duration, and frequency of diarrhea in individual participants were dose dependent. Grade III diarrhea mainly occurred in the 400 mg dose group.2 The most common hematological AEs included decreased white blood cell count, decreased neutrophil count, decreased hemoglobin level, and elevated aspartate aminotransferase, alanine aminotransferase, serum creatinine, and serum bilirubin levels.1 In the present study, the common adverse reactions (incidence ≥ 10%) of pyrotinib included hypertriglyceridemia (27.8%), decreased white blood cell count (11.1%), decreased neutrophil count (11.1%), abdominal pain (11.1%), and diarrhea (11.1%), which were roughly consistent with previous findings. The present study did not report any serious AEs, including AEs leading to withdrawal from the trial and AEs leading to death. During the study, no participants had abnormal results of vital signs and 12-lead ECG after baseline, with clinical significance. The physical examination indexes of all participants were normal before and after administration, with no abnormal change. Except for one participant who used Kaisailu on days 13 and 14 due to abdominal distension, another participant did not use the combination medication during the study period.
Conclusions
Compared with pyrotinib alone, the exposure to pyrotinib co-administered with itraconazole substantially increased, while the Cmax and AUC0-t increased by 2.78- and 10.8-fold, respectively. The concomitant use of pyrotinib with itraconazole might require dose modification of pyrotinib. A single oral dose of 80 mg pyrotinib and a combination with 200 mg itraconazole were well tolerated in Chinese healthy volunteers.
Data Sharing Statement
The original data of this study will not be shared due to confidentiality.
Ethical Approval
The study was approved by the ethics committee of The Second Hospital of Anhui Medical University (Hefei, China) and performed according to the ethical principles formulated in the Helsinki Declaration of the World Medical Congress and its amendments.
Informed Consent
All participants included in this study signed informed consent.
Acknowledgments
The authors thank all study participants. This study was sponsored by Jiangsu Hengrui Pharmaceutical Co., Ltd. and supported by the National Major Scientific and Technological Special Project (2020ZX09201014).
Disclosure
The authors are accountable for the content and writing of this manuscript and report no conflicts of interest related to this study.
References
1. Blair HA. Pyrotinib: first global approval. Drugs. 2018;78(16):1751–1755. doi:10.1007/s40265-018-0997-0
2. Ma F, Li Q, Chen S, et al. Phase I study and biomarker analysis of pyrotinib, a novel irreversible pan-erbb receptor tyrosine kinase inhibitor, in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol. 2017;35(27):3105–3112. doi:10.1200/JCO.2016.69.6179
3. Ma F, Ouyang Q, Li W, et al. Pyrotinib or lapatinib combined with capecitabine in HER2-positive metastatic breast cancer with prior taxanes, anthracyclines, and/or trastuzumab: a randomized, phase II study. J Clin Oncol. 2019;37(29):2610–2619. doi:10.1200/JCO.19.00108
4. Song GH, Li HP, Di LJ, et al. [Efficacy and safety of oral pyrotinib in HER2 positive metastatic breast cancer: real-world practice]. Beijing Da Xue Xue Bao Yi Xue Ban. 2020;52(2):254–260. [Chinese].
5. Lin Y, Lin M, Zhang J, et al. Real-world data of pyrotinib-based therapy in metastatic HER2-positive breast cancer: promising efficacy in lapatinib-treated patients and in brain metastasis. Cancer Res Treat. 2020. doi:10.4143/crt.2019.633
6. Li X, Yang C, Wan H, et al. Discovery and development of pyrotinib: a novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur J Pharm Sci. 2017;110:51–61. doi:10.1016/j.ejps.2017.01.021
7. Li Q, Guan X, Chen S, et al. Safety, efficacy, and biomarker analysis of pyrotinib in combination with capecitabine in HER2-positive metastatic breast cancer patients: a phase I clinical trial. Clin Cancer Res. 2019;25(17):5212–5220. doi:10.1158/1078-0432.CCR-18-4173
8. Gourd E. Pyrotinib shows activity in metastatic breast cancer. Lancet Oncol. 2017;18(11):e643. doi:10.1016/S1470-2045(17)30755-6
9. Zhou C, Li X, Wang Q, et al. Pyrotinib in HER2-mutant advanced lung adenocarcinoma after platinum-based chemotherapy: a multicenter, open-label, single-arm, phase II study. J Clin Oncol. 2020;38:JCO2000297. doi:10.1200/JCO.20.00297
10. Wu Y, Ni J, Chang X, Zhang X, Zhang L. Successful treatment of pyrotinib for bone marrow metastasis induced pancytopenia in a patient with non-small-cell lung cancer and ERBB2 mutation. Thorac Cancer. 2020;11(7):2051–2055. doi:10.1111/1759-7714.13480
11. Wang Y, Jiang T, Qin Z, et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann Oncol. 2019;30(3):447–455. doi:10.1093/annonc/mdy542
12. Lian X, Zhu C, Lin H, et al. Radiosensitization of HER2-positive esophageal cancer cells by pyrotinib. Biosci Rep. 2020;40(2). doi:10.1042/BSR20194167.
13. Zhu Y, Li L, Zhang G, et al. Metabolic characterization of pyrotinib in humans by ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J Chromatogr B. 2016;1033:117–127. doi:10.1016/j.jchromb.2016.08.009
14. Meng J, Liu XY, Ma S, et al. Metabolism and disposition of pyrotinib in healthy male volunteers: covalent binding with human plasma protein. Acta Pharmacol Sin. 2019;40(7):980–988. doi:10.1038/s41401-018-0176-6
15. US FDA. Guidance for industry drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendation, 2012. Draft guidance. 2013.
16. European Medicines Agency. Guideline on the investigation of drug interactions. 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientificguideline/2012/07/WC500129606.pdf.
17. U.S. Department of Health and Human Service. Common terminology criteria for adverse events. November 27, 2017. Available from: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5.
18. Liu L, Bello A, Dresser MJ, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug-drug interaction studies. J Clin Pharmacol. 2016;56(2):143–151. doi:10.1002/jcph.562
19. Abbas R, Hug BA, Leister C, Burns J, Sonnichsen D. Pharmacokinetics of oral neratinib during co-administration of ketoconazole in healthy subjects. Br J Clin Pharmacol. 2011;71(4):522–527. doi:10.1111/j.1365-2125.2010.03845.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.