Back to Journals » Biologics: Targets and Therapy » Volume 8
Multikinase inhibitors use in differentiated thyroid carcinoma
Authors Jasim S, Ozsari L, Habra M
Received 27 August 2014
Accepted for publication 21 October 2014
Published 4 December 2014 Volume 2014:8 Pages 281—291
DOI https://doi.org/10.2147/BTT.S57619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Doris Benbrook
Sina Jasim,1,* Levent Ozsari,2,* Mouhammed Amir Habra2
1Division of Endocrinology, Diabetes, Metabolism, and Nutrition, Mayo Clinic, Rochester, MN, USA; 2Department of Endocrine Neoplasia and Hormonal Disorders, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
*These authors contributed equally in this work
Abstract: Thyroid cancer is the most common endocrine malignancy, and its incidence is increasing. Standard therapy for most patients with localized differentiated thyroid cancer (DTC) includes surgery, radioactive iodine, and thyroid hormone replacement. A minority of thyroid cancer patients requires systemic therapy for metastatic disease. Patients with metastatic DTC do not usually benefit from traditional cytotoxic chemotherapy. In this review, we describe newly developed small-molecule tyrosine kinase inhibitors (TKIs) that are being actively tested and used in the management of advanced thyroid cancer. The use of TKIs as a form of molecular targeted therapy is evolving based on understanding of the pathways involved in DTC. Disrupting tumor vascular supply by targeting vascular endothelial growth factor receptor signaling is the most commonly used approach to treat advanced/metastatic DTC. Other mechanisms include targeting BRAF, MAPK/ERK kinase, or mammalian target of rapamycin signaling. Although TKIs appear to have superior efficacy compared to cytotoxic chemotherapy, they can cause substantial adverse effects; symptomatic management of adverse effects, dose adjustment, or cessation of therapy may be required.
Keywords: differentiated thyroid cancer, progression-free survival, adverse effects, targeted therapy, sorafenib, lenvatinib
Introduction
Thyroid cancer is the most common endocrine malignancy and the fifth most common cancer in women, with an estimated 62,980 new cases expected in the United States in 2014.1 Thyroid cancers are divided into four major histological types: papillary (85%), follicular (11%), medullary (3%), and anaplastic (1%).2,3 Papillary and follicular thyroid cancers (PTC and FTC) are referred to as differentiated thyroid cancers (DTCs).
In the past few decades, the incidence of thyroid carcinoma has increased, but thyroid cancer mortality has remained stable at 0.5 cases per 100,000 persons.4 The reasons for the worldwide increase in thyroid cancer are unclear; some authors suspect that the rise in thyroid cancer incidence reflects the increasing use of imaging studies leading to incidental identification, while others suggest that there is a true increase even in large tumors possibly related to environmental changes.5,6 The increase rate among thyroid cancers is mostly due to an increase in PTC diagnosis.7
Prognostic factors in DTC include age at diagnosis, sex, family history, preoperative thyroid-stimulating hormone levels, and postoperative stimulated thyroglobulin values.8 The mortality rate is higher in patients diagnosed with DTC after the age of 45 years than in younger patients.9,10 Older patients are also more likely to have aggressive histological variants, extensive neck disease, and distant metastases at diagnosis. Sex is another important prognostic factor, as DTC is more frequent in women than men while the relative mortality is much higher in men compared to women.1,11
The histologic findings of malignant thyroid nodules provide important prognostic information. The oncocytic (Hurthle cell) variant of FTC is associated with worse prognosis and it is also less responsive to radioiodine therapy. Furthermore, tall-cell, columnar-cell, and diffuse sclerosing variants of PTC carry worse prognosis compared to conventional PTC.12
BRAF gene mutation (p.V600E) is the most prevalent (29%–69%) point mutation in PTC.13,14 It has been associated with aggressive behavior, extrathyroidal invasion, lymph node metastasis, and advanced stage in PTC.15–18 Moreover, BRAF mutation was shown to have a low positive predictive value of 28% and a high negative predictive value of 87% for PTC recurrence,19 suggesting that its use in thyroid cancer prognostic evaluation should be exercised with caution. The use of BRAF mutation for PTC prognosis is still controversial since it is found in about half of PTCs,16 with <10%–15% of the tumors displaying aggressive behavior.17
RAS mutations are associated with follicular thyroid neoplasia and almost half of cases of FTC,13 but the sensitivity and specificity are not sufficient to support these mutations’ use as a molecular marker for the prediction of prognosis.20,21
RAS is another protein in the inner surface of the cell membrane and essential part of the mitogen-activated protein kinase (MAPK) signaling pathway. Activating RAS mutations can be seen in about 10%–20% of PTCs but are more common in FTC (40%–50%) of follicular carcinomas and 20%–40% of poorly differentiated and anaplastic carcinomas.22
RET/PTC rearrangements are seen in about 10%–20% of PTCs, especially in young adults and occasionally in benign thyroid nodules.23–25 PAX8/PPAR-γ rearrangement is detected in about one-third of FTC and in a follicular variant of PTC but not in classic PTC and it is associated with tumor multifocality and vascular invasion.26
Distant metastases can be seen at the time of initial diagnosis in 2%–12% of DTC patients27–29 and can be detected during subsequent follow-up in about 5%–30% of patients.27,29,30 The 10-year overall survival rate for DTC patients with distant metastases ranges from 20% to 51%. The remarkable variation in metastatic burden estimation and its effect on survival is likely to be explained by the various methods reported in the literature for assessing metastatic sites, as well as the retrospective nature of published data.29,30
Traditionally, metastatic DTC has been considered a chemotherapy-resistant malignancy, and our ability to effectively treat these patients was very limited. In the past decade, remarkable advances have been achieved through better characterization of molecular pathways involved in DTC. These molecular pathways play an important role for the cancer cell survival and aggressiveness mostly via the activation of multiple intracellular receptors and tyrosine kinases. In particular, small-molecule tyrosine kinase inhibitors (TKIs), which block key signaling pathways involved in tumor growth, are showing efficacy in thyroid cancer. In this review, we describe the current treatment of advanced DTC, with a focus on the TKIs that are being actively tested and used in this disease.
Traditional treatment of DTC
Surgery
Surgery is the treatment of choice for localized or locally invasive DTC. Total thyroidectomy is the gold-standard treatment when the size of the primary tumor is more than 1 cm.2 Total thyroidectomy is also recommended for multifocal disease. A limited surgical approach (lobectomy) may be justified in patients with unifocal tumor <1 cm in diameter or multifocal papillary microcarcinomas. If there is evidence of lymph node involvement in patients with papillary cancer, central neck (level IV) dissection in addition to total thyroidectomy is recommended.2,31 Even in the absence of obvious nodal involvement preoperatively, central dissection is recommended for patients with a primary tumor >4 cm in diameter.2
Radioactive iodine
In DTC patients, radioactive iodine (RAI) is used to ablate residual thyroid tissue and small-volume cancer deposits. RAI therapy is recommended after thyroidectomy in patients with tumor size >4 cm, extrathyroidal extension, lymph node involvement, worrisome histologic subtypes, or known distant metastasis.2
Radioiodine-refractory DTC is defined by documenting disease progression after receiving a cumulative RAI dose of >600 mCi, disease progression within a few months (usually within 12 months) after receiving therapeutic dose of RAI (usually >100 mCi), or the absence of RAI uptake in known metastases seen by other cross-body imaging techniques.32,33
Cytotoxic chemotherapy
Patients with progressive disease are candidates for cytotoxic chemotherapy. Major drugs in this group include doxorubicin, paclitaxel, and cisplatin. Systemic chemotherapy is highly toxic and usually associated with very low response rates that are usually partial and short lived.34,35 Prior to the development of targeted therapies, cytotoxic chemotherapy was used in patients with rapidly progressive, locally advanced or metastatic disease who had limited treatment options.
Tyrosine kinase inhibitors
TKIs were introduced in thyroid cancer management based on the expanding knowledge of molecular pathways involved in thyroid carcinoma. In RAI-resistant DTC that is progressive and not amenable to surgical resection, these drugs are often offered after careful consideration of potential side effects and possible benefits from disease control. While TKIs improved partial response rates, progression-free survival (PFS), and stable disease rates, the impact of TKIs on overall survival has not been well documented.36
Most TKIs studied in DTC target signaling by vascular endothelial growth factor (VEGF) and its receptors (VEGFRs). Anti-VEGF TKIs are ATP-mimetic agents that bind to the ATP-binding site of the intracellular tyrosine kinase domain of VEGFRs (Figure 1).37,38 Most agents in this class of drugs have been approved to treat other malignancies, and recently the US Food and Drug administration (FDA) and the European Medicine Agency (EMA) approved sorafenib for the management of progressive DTC.39,40 We here summarize the clinical experience reported for the important TKIs that are being tested or have potential for use in DTC (Table 1).
 | Figure 1 A simplified illustration about the intracellular pathways and mechanisms of action for select small-molecule kinase inhibitors in differentiated thyroid carcinoma. |
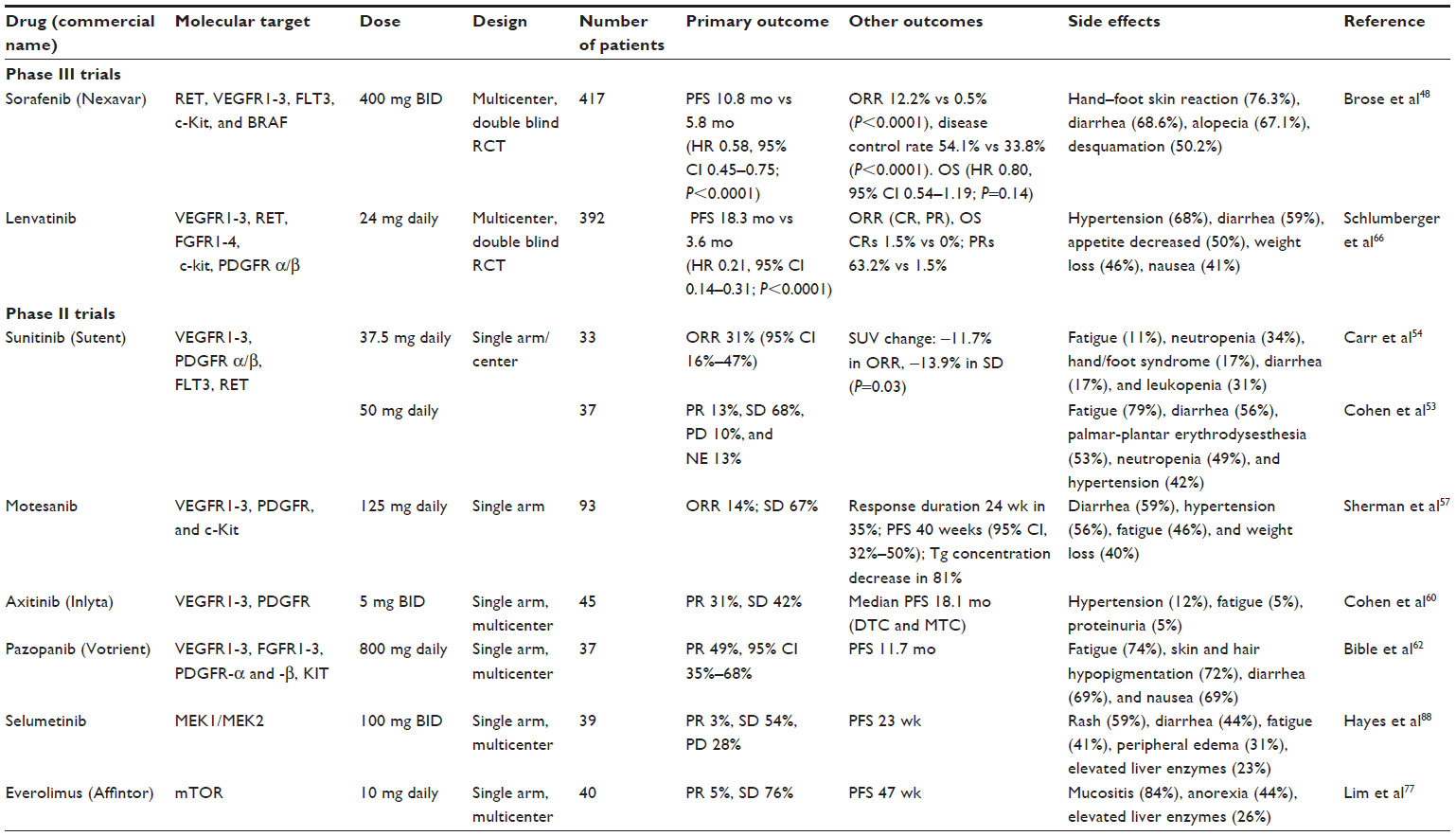 | Table 1 Summary of main Phase II and III clinical trials of tyrosine kinase inhibitors in differentiated thyroid cancer |
Sorafenib
Sorafenib is an oral TKI that targets rearranged during transfection (RET), VEGFR1-3, FMS-like tyrosine kinase 3 (FLT3), c-Kit, and BRAF.41 Inhibition of MAPK activation after sorafenib treatment was shown in colon, pancreas, and breast cancer cell lines. In colon, breast, and non-small-cell lung cancer xenograft models, sorafenib demonstrated a broad spectrum of antitumor activity.42
A number of trials, mainly Phase II, have examined sorafenib’s efficacy in metastatic thyroid cancer.41,43–46 In a meta-analysis that included seven small Phase II studies using sorafenib in DTC patients, the overall partial response, stable disease, and progressive disease rates were 21%, 60%, and 20%, respectively. Sorafenib had been discontinued in 16% of patients because of toxicities or intolerance. The dose of sorafenib had been reduced in 56% of patients. Sorafenib toxicities include hypertension, fatigue, hand–foot skin reaction, squamous cell carcinoma, nausea, vomiting, diarrhea, elevated levels of liver enzymes, weight loss, proteinuria, arthralgia, and bone marrow suppression.40
A recently published multicenter, randomized, double-blind, placebo-controlled, Phase III trial (DECISION) enrolled 417 patients with progressive and RAI-resistant DTC; a 1:1 randomization scheme was used. This study only included patients who did not use any other TKIs or chemotherapy earlier. Sorafenib (starting at a maximal dose of 400 mg twice daily) use (n=207 patients) was associated with a PFS duration of 10.8 months compared to 5.8 months for the placebo-treated group (n=210 patients), P<0.0001. Overall survival rate between sorafenib and placebo group was not statistically significant. The reason for similar overall survival rate could be drug crossover between groups.47
The DECISION study has suggested that BRAF and RAS mutations were not predictive markers for sorafenib effect on PFS. The most frequent treatment-emergent adverse events in the sorafenib group were hand–foot skin reaction (76.3%), diarrhea (68.6%), alopecia (67.1%), and other skin reactions (50.2%).48 Cardiovascular effects of sorafenib include hypertension, prolonged QT interval, cardiac ischemia, and congestive heart failure.49 Gastrointestinal perforation is an unusual complication of anti-VEGF therapy, including sorafenib; it requires drug discontinuation.
In the DECISION trial, dose reductions were required in 64% of patients receiving sorafenib and nearly 19% of patients receiving placebo.48 Lower doses of sorafenib were also used in some studies for patients with RAI-resistant DTC, pulmonary metastases from PTC, and brain metastasis from FTC, with the same apparent therapeutic effect and better tolerance.50,51
Sorafenib is currently approved in the US and the European Union (EU) to treat renal cell carcinoma, hepatocellular carcinoma, and now DTC as well, based on data from the DECISION trial.48
Sunitinib
Sunitinib is another oral multikinase inhibitor that inhibits VEGFR1-3, platelet-derived growth factor receptor (PDGFR)-α and -β, stem cell factor receptor (c-Kit), FLT3, and RET.52
The role of sunitinib in the treatment of metastatic DTC has been evaluated with several studies. In 2008, a Phase II study showed partial response and stable disease rates of 13% and 68%, respectively, in patients with DTC refractory to curative treatment.53 A more recent Phase II study of sunitinib in patients with advanced DTC had 7% complete response, 25% partial response, and 48% stable disease rates.54 Adverse effects of sunitinib have significant overlap with those of sorafenib, including hypertension, hand–foot syndrome, mucositis, and fatigue. Sunitinib is currently approved in the US and EU for treating advanced renal cell carcinoma, gastrointestinal stromal tumor (GIST), and advanced pancreatic neuroendocrine tumors.
Motesanib
Motesanib is an oral small-molecule multikinase inhibitor that inhibits VEGFR1-3, PDGFR, and c-Kit. In vivo studies have shown that motesanib selectively targets neovascularization by inhibiting VEGF-induced vascular permeability and cellular proliferation.55 Antitumor activity of motesanib was demonstrated in a Phase I study that included five patients with DTC.56 In an open-label Phase II study of 93 patients with progressive and RAI-resistant DTC, motesanib use resulted in objective response in 14% of subjects and stable disease in 67%, with an estimated median PFS of 40 weeks (95% confidence interval 32%–50%). Similar to sorafenib and sunitinib, motesanib’s most common adverse effects included diarrhea, hypertension, weight loss, and fatigue.57 Motesanib is not available commercially.
Axitinib
Axitinib is an orally available and potent selective small-molecule inhibitor of VEGFR1-3. An in vitro study showed that this drug blocked endothelial cell proliferation and decreased tumor vascularization.58 In an early trial, axitinib showed activity in patients with some tumor types, including thyroid cancer.59 During a Phase II trial, axitinib was associated with a response rate of 30% in patients with advanced RAI-resistant DTC, and 38% of patients had stable disease lasting ≥16 weeks.60 The most common side effects were diarrhea, hypertension, fatigue, and decreased appetite and weight.61 Axitinib is approved in the US and EU to treat advanced renal cell carcinoma that has not responded or has recurred after one prior systemic therapy.
Pazopanib
Pazopanib is an orally active small-molecule inhibitor of VEGFR1-3, fibroblast growth factor receptors (FGFR) 1–3, PDGFR-α and -β, interleukin-2 receptor inducible T-cell kinase (Itk), leukocyte-specific protein tyrosine kinase (Lck), c-Kit, and transmembrane glycoprotein receptor tyrosine kinase (c-Fms). The partial response rate was 49% and lasted more than 1 year for patients with RAI-refractory and/or metastatic DTC.62 Dose reduction was required because of adverse effects such as fatigue, skin and hair hypopigmentation, diarrhea, and nausea.62 Hepatotoxicity is a known toxicity of pazopanib, which could be fatal and has generated a black-box warning. Liver enzymes must be monitored before and during therapy. Other side effects include cardiac dysfunction and QT prolongation, thromboembolic events, gastrointestinal perforation, and hypertension. A Phase II trial is ongoing to compare pazopanib treatment interruption with continuous therapy in RAI-refractory progressive DTC (PAZOTHYR 2012-003162-41, NCT01813136). Pazopanib is approved in the US and EU for treating patients with advanced renal cell carcinoma and soft-tissue sarcoma.
Lenvatinib
Lenvatinib is an orally available multikinase inhibitor that targets VEGFR1-3, FGFR1-4, c-Kit, and PDGFR-β. The inhibition of FGFRs offers an opportunity to overcome resistance to VEGF/VEGFR inhibitors. In vitro, lenvatinib decreased lymphatic vessel density in human breast cancer xenografts.63 In Phase I trials, the most common drug-related adverse effects were hypertension, diarrhea, nausea, stomatitis, proteinuria, and vomiting.64 In a Phase II study, 59% of RAI-refractory DTC patients had a partial response. Stable disease was observed in 36% of patients, with a median PFS of 13.3 months.65 In a Phase III trial (SELECT), the median PFS was 18.3 months in patients with progressive RAI-resistant DTC who received lenvatinib (n=261), compared with 3.6 months for placebo-treated matched patients (n=131).66 This study compared lenvatinib (starting dose of 24 mg) with placebo in a 2:1 double-blind randomized allocation. The complete response rate was 1.5% in the lenvatinib group and 0% in the placebo group. The median overall survival time was not reached because the study allowed crossover between the two study arms. Efficacy was seen in different prespecified subgroups, including patients who received one prior anti-VEGF therapy. In this study, no unexpected toxicities were reported. The most common side effects of lenvatinib included hypertension, diarrhea, reduced appetite, weight loss, and nausea. Dose reduction was needed in about 80% of the subjects who received lenvatinib, and drug discontinuation secondary to side effects was needed in about 14% of patients.66 Lenvatinib is not yet available commercially.
BRAF inhibitors
BRAF is a proto-oncogene that produces the B-Raf protein. Raf proteins are serine/threonine kinases that function as oncoproteins.67 There are three kinds of Raf kinase proteins: A-Raf, B-Raf, and C-Raf. B-Raf has a role in signaling that leads to cell growth.
BRAF mutations result in signals for cellular proliferation being triggered without an external stimulus, ie, uncontrolled cell proliferation. So far, 65 different BRAF mutations have been reported.68 The most frequent is p.V600E, in which valine (V) is substituted with glutamic acid (E). This mutation is found especially in thyroid cancer, colon cancer, and melanoma.67
Because deregulation of BRAF kinase can cause cancer, BRAF kinase is a potential target for cancer treatment. Two types of small-molecule BRAF kinase inhibitors have been developed. Type 1 inhibitors bind to the active conformation, and type 2 inhibitors bind to an inactive site of BRAF kinase.67 Well-known BRAF inhibitors are vemurafenib and dabrafenib, described as follows.
Vemurafenib
Vemurafenib is an orally administered type 1 BRAF inhibitor. This agent inhibits not only BRAF V600E but also V600D, V600K, and V600R.67 At higher concentrations, vemurafenib inhibits V600E-mutant BRAF, CRAF, wild-type BRAF, and other kinases.69
In xenograft models, it showed activity in BRAF-mutated melanoma.70 Clinical response was established in three patients with BRAF (V600E)-mutated PTC in a Phase I trial.71 Progressive RAI-refractory BRAF V600-mutant PTC patients had a 35% partial response rate to vemurafenib, and the 6-month disease control rate was 58%. The most frequent side effects were rash, fatigue, weight loss, taste alteration, squamous cell carcinoma, and alopecia.67 This drug can be considered in patients with radiologically progressive and RAI-resistant V600-mutant PTC in whom antiangiogenic therapy is contraindicated.67 A pilot study is in progress to test vemurafenib with radioiodine in patients with DTC and poor DTC (14-031NCT02145143). The FDA and EMA have approved this drug for patients with unresectable or metastatic malignant melanoma with BRAF V600E mutation.
Dabrafenib
Dabrafenib is an oral type 1 BRAF kinase inhibitor. Dabrafenib is selective for mutant BRAF.72 At higher concentrations, dabrafenib also inhibits wild-type BRAF and CRAF kinases and other kinases such as salt-inducible kinase 1 (SIK1), NIMA-related kinase 11 (NEK11), and LIM domain kinase 1 (LIMK1).
In a Phase I trial, patients with BRAF-mutant metastatic PTC had a 33% response rate to dabrafenib.72 A Phase II trial comparing dabrafenib and dabrafenib plus trametinib is ongoing in patients with BRAF-mutant metastatic PTC (OSU-12064 NCI-2012-01700, NCCNGSK20014, NCT01723202). A Phase I study of combination therapy with dabrafenib and lapatinib ditosylate in patients with unresectable refractory thyroid cancer is also under way (NCI-2013-01748 9354, 13-061, P30CA008748, U01CA069856, NCT01947023). General side effects of dabrafenib are pyrexia, fatigue, headache, neutropenia, thrombocytopenia, leukopenia, astenia, squamous cell carcinoma, basal cell carcinoma, rash, and skin papillomas.67 This drug was approved by the FDA and EMA for BRAF V600E mutation-positive unresectable or metastatic melanoma.
MEK inhibitors
The nucleus of a cell communicates with the surface receptors through the MAPK/extracellular signal-regulated kinase (ERK) (MEK) cascade. Activation of this cascade results in cell proliferation, which leads to tumors. Aberrant activation of MAPK not only promotes cell division but also inhibits sodium-iodide symporter and thyroid peroxidase, which can lead to RAI-resistant PTC. Activation starts with certain mutations on oncogenes such as KRAS, NRAS, HRAS, and BRAF.73 The MEK cascade has become a therapeutic target for anticancer drugs.
Selumetinib
Selumetinib is an oral MEK1/MEK2 inhibitor that was recently reported to increase RAI uptake and retention in 60% of RAI-refractory DTC patients. The efficacy of selumetinib was more pronounced in patients harboring RAS mutations; however, these findings should be validated in a larger cohort of DTC patients.74 A Phase III trial comparing the efficacy of selumetinib vs placebo in patients with DTC is ongoing (NCT01843062). Selumetinib is not available commercially.
Trametinib
Trametinib is another orally active drug that reversibly inhibits MEK1 and 2. A Phase I clinical trial showed activity in patients with BRAF-mutant melanoma. The most common adverse effects were papulopustular rash, diarrhea, peripheral edema, fatigue, and acneiform dermatitis.75 There are two ongoing Phase II studies of trametinib in patients with DTC. The first is determining the effect of dabrafenib with or without trametinib in patients with radioiodine-refractory, BRAF-mutated DTC (OSU-12064 NCI-2012-01700, NCCNGSK20014, NCT01723202). The second trial is evaluating whether trametinib enhances the effects of RAI therapy (NCI-2014-01106 13-157, 9446, U01CA069856, P30CA008748, NCT02152995). Trametinib is commercially available both in the US and EU for the indication of unresectable or metastatic melanoma with BRAF V600 mutation.
mTOR inhibitors
Mammalian target of rapamycin (mTOR) is a protein that affects the phosphatidylinositol-3 kinase (PI3K)/Akt pathway. Activation of this pathway results in cell proliferation and reduction in iodide uptake in thyroid cells.76 Antagonizing mTOR is a reasonable strategy against proliferating cancer cells and RAI-resistant thyroid cancer.
The rapamycin analog, everolimus, is the major drug of this group. Everolimus blocks cell proliferation. This drug has an antiproliferative effect and antiangiogenic properties. Phase II study results showed a low response rate in thyroid cancer patients who are resistant to or not appropriate for RAI.77 Even with the low response rate, the median PFS was 43 weeks. The most common side effects were mucositis, anorexia, and hepatic enzyme elevation. Everolimus is approved to treat hormone-receptor positive breast cancer, unresectable or metastatic pancreatic neuroendocrine tumors, and advanced renal-cell carcinoma after failure with sunitinib or sorafenib.
TKI combination therapies
The variety of targets on the kinase pathway has increased interest in combination therapies. Some of these therapies increase RAI uptake in thyroid cancer. These data bring new approaches for combination therapies especially for RAI-resistant thyroid cancer.
VEGFR and mTOR inhibitors
Inhibiting angiogenesis by blocking VEGFRs is a valuable strategy for cancer therapy. However, antiangiogenic regimens can cause increased circulating angiogenic factors such as VEGF.78 Some preclinical reports showed that exposure to some VEGFR inhibitors may increase metastasis.79 This complication may be reduced by combining a TKI with an agent that has a complementary underlying biological mechanism. mTOR inhibitors are potent antiangiogenic drugs. They decrease VEGF serum levels by inhibiting hypoxia-inducible factor 1 formation in vitro.80 In a xenograft model, mTOR inhibitor everolimus counterbalanced the VEGF elevation created by sunitinib, and a significant reduction in tumor size was seen.79 Combination therapy with everolimus and sorafenib showed promising results in Hurthle cell and medullary thyroid cancer patients.81 The major adverse effects were liver transaminase elevation, hyperglycemia, and pancreatitis.
BRAF and MEK inhibitors
The use of BRAF inhibitors is limited because of side effects and tumor resistance. Molecular studies have shown that inhibition of the MAPK pathway with BRAF and MEK inhibitors is useful in overcoming resistance.82 A Phase I/II trial of a selective BRAF inhibitor (dabrafenib) combined with an MEK inhibitor (trametinib) in patients with metastatic melanoma and BRAF V600 mutations resulted in improved PFS compared with that for dabrafenib alone. Although the risk of pyrexia increased, the rate of adverse skin lesions was significantly reduced.83 A Phase II trial comparing these treatments is underway in patients with BRAF-mutant metastatic PTC (OSU-12064NCI-2012-01700, NCCNGSK20014, NCT01723202).
TKI-related side effects
Because of the serious adverse effects of TKIs, physicians should perform a detailed examination before prescribing this group of drugs. The medical history and laboratory data should be focused on cardiovascular, hepatic, and renal status. Patients with uncontrolled hypertension or disease in areas at high risk of developing fistulas or bleeding cannot be candidates.49 Patient performance status should be assessed by using a validated scale such as Eastern Cooperative Oncology Group (ECOG) scoring. Most clinical trials include patients with ECOG scores >2. We do not have enough evidence about the tolerance of TKIs in patients with poor performance status.52
The most common side effect of antiangiogenic drugs is hypertension. Although hypertension is a side effect, it is also considered a biomarker to predict response to therapy.49 QT prolongation is one of the cardiovascular side effects of TKIs. Patients with prolonged QT or who are using antiarrhythmic drugs are not candidates for TKIs. Before starting TKI treatment, patients should have baseline electrocardiogram (ECG) and possibly echocardiogram. Although uncommon, congestive heart failure is another cardiovascular side effect that can be seen with TKIs. This side effect is the result of cardiomyotoxicity and is exacerbated with hypertension. Standard treatment for heart failure may be required for some patients.
Proteinuria can be caused by disturbed microvascular permeability as a result of TKI treatment. Baseline renal function and periodic urinalysis are recommended. There are limited data about TKI usage in patients with end-stage renal disease.
Hepatic transaminase and bilirubin elevation are seen generally with pazopanib and sunitinib treatment. Hepatotoxicity can be fatal. Liver function testing is recommended at baseline and monthly for 4 months.49
Hematological side effects of TKIs are bone marrow suppression, thrombosis, and tumor-related hemorrhage. These side effects can be fatal. TKI treatment should be discontinued at least 7 days before the surgery and should not be started until wound healing is complete. There are not enough data on TKIs used in patients with cerebral or gastrointestinal hemorrhage in the previous 6 months. Serial complete blood counts are recommended at baseline and each clinical visit.
The most common dermatological side effects of TKIs, especially with sorafenib, are hand–foot skin reaction and rash. In the case of severe lesions, treatment can be ceased permanently. Before starting sorafenib, evaluation for squamous cell carcinomas should be performed.
TKIs may exacerbate hypothyroidism in patients who are using levothyroxine therapy. Increasing levothyroxine dose and close monitoring are required.
When compared to cytotoxic chemotherapy, TKIs are well-tolerated cancer treatment drugs. Most of their side effects can resolve spontaneously or after dose reduction or drug discontinuation.84 Although patients are less likely to discontinue TKIs than cytotoxic chemotherapy, some challenging and fatal side effects can nevertheless be seen under TKI therapy.
Patients using multiple drugs for different reasons should be careful before starting TKI therapy. Most TKIs are metabolized in the liver by the cytochrome P450 system.85 Drugs that inhibit or induce this system can change TKIs’ plasma concentration.86 Patients should be aware of drug interactions with TKIs.
Following are the recommendations for patients who are considering or already using TKIs:
- Preexisting hypertension should be adequately managed and controlled before starting TKIs.87 The development of severe hypertension requires TKI dose modification with initiating antihypertensive therapy. Patients who already have hypertension before TKI treatment may need to intensify antihypertensive regimen as well as close follow-up.
- Close monitoring and correction of electrolyte abnormality are warranted against QT prolongation and Torsade de pointes. Attention should be paid to avoid combining TKIs with other drugs known to prolong QT interval.49
- Before starting TKI therapy, clinical assessment, baseline ECG, and occasionally echocardiography are recommended. Congestive heart failure is not common with TKIs; however, if it occurs, it can be fatal.49
- Proteinuria is another side effect of TKIs. Patients should also have a baseline urinalysis and possibly protein-to-creatinine ratio assessment before starting TKI therapy especially with some VEGFR inhibitors known to induce proteinuria.86
- The most common dermatologic reaction of TKIs, namely, hand–foot syndrome, affects the quality of life. Drug interruption can reduce the skin toxicity. Supportive measures and skin care can help to ameliorate this adverse effect. Squamous cell carcinomas can also develop.
- A rare but life-threatening complication of TKIs is gastrointestinal perforation or fistula development. Major risk factors for this complication are recent sigmoidoscopy or colonoscopy, history of external beam irradiation, underlying tumor, diverticulitis, and bowel obstruction.
- Although asymptomatic pancreatic enzyme elevation can be seen with TKIs, acute pancreatitis is rare.
- TKIs can reduce wound healing, cause thrombosis, and cause mucosal bleeding.
Conclusion
TKIs are promising targeted therapies under investigation for RAI-resistant DTC. The FDA has approved sorafenib in these patients, and lenvatinib has shown remarkable efficacy in DTC in therapy-naïve as well as after failing one prior TKI therapy. The clinical role of other TKIs including BRAF, MEK, and mTOR inhibitors is emerging. Current research efforts are focused on refining combination treatment strategies and determining which patients will obtain the most benefit from TKIs with the least toxicity.
Disclosure
MAH received research support from Eisai Inc. The authors declare no other conflicts of interest.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer, Cooper DS, Doherty GM, Haugen BR, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167–1214. | |
Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the US, 1985–1995 [see comments]. Cancer. 1998;83(12):2638–2648. | |
Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006;295(18):2164–2167. | |
Youserm DM, Huang T, Loevner LA, Langlotz CP. Clinical and economic impact of incidental thyroid lesions found with CT and MR. Am J Neuroradiol. 1997;18(8):1423–1428. | |
Cronan JJ. Thyroid nodules: is it time to turn off the US machines? Radiology. 2008;247(3):602–604. | |
Aschebrook-Kilfoy B, Grogan RH, Ward MH, Kaplan E, Devesa SS. Follicular thyroid cancer incidence patterns in the United States, 1980–2009. Thyroid. 2013;23(8):1015–1021. | |
Soares P, Celestino R, Melo M, Fonseca E, Sobrinho-Simoes M. Prognostic biomarkers in thyroid cancer. Virchows Arch. 2014;464(3):333–346. | |
Russell MA, Gilbert EF, Jaeschke WF. Prognostic features of thyroid cancer. A long-term followup of 68 cases. Cancer. 1975;36(2):553–559. | |
Hay ID, Thompson GB, Grant CS, et al. Papillary thyroid carcinoma managed at the Mayo Clinic during six decades (1940–1999): temporal trends in initial therapy and long-term outcome in 2444 consecutively treated patients. World J Surg. 2002;26(8):879–885. | |
Gilliland FD, Hunt WC, Morris DM, Key CR. Prognostic factors for thyroid carcinoma. A population-based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) program 1973–1991. Cancer. 1997;79(3):564–573. | |
DeLellis RA. Pathology and Genetics of Tumours of Endocrine Organs. Lyon: IARC Press; 2004. | |
Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6(4):292–306. | |
Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12(2):245–262. | |
Kim TH, Park YJ, Lim JA, et al. The association of the BRAF(V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: a meta-analysis. Cancer. 2012;118(7):1764–1773. | |
Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13(3):184–199. | |
Soares P, Sobrinho-Simoes M. Cancer: small papillary thyroid cancers – is BRAF of prognostic value? Nat Rev Endocrinol. 2011;7(1):9–10. | |
Patel KN. Genetic mutations, molecular markers and future directions in research. Oral Oncol. 2013;49(7):711–721. | |
Xing M. Prognostic utility of BRAF mutation in papillary thyroid cancer. Mol Cell Endocrinol. 2010;321(1):86–93. | |
Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20(7):697–706. | |
Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TW, Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol. 1999;50(4):529–535. | |
Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol. 2011;7(10):569–580. | |
Bongarzone I, Fugazzola L, Vigneri P, et al. Age-related activation of the tyrosine kinase receptor protooncogenes RET and NTRK1 in papillary thyroid carcinoma. J Clin Endocrinol Metab. 1996;81(5):2006–2009. | |
Fenton CL, Lukes Y, Nicholson D, Dinauer CA, Francis GL, Tuttle RM. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J Clin Endocrinol Metab. 2000;85(3):1170–1175. | |
Guerra A, Sapio MR, Marotta V, et al. Prevalence of RET/PTC rearrangement in benign and malignant thyroid nodules and its clinical application. Endocr J. 2011;58(1):31–38. | |
Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am J Surg Pathol. 2002;26(8):1016–1023. | |
Mazzaferri EL, Jhiang SM. Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med. 1994;97(5):418–428. | |
Nixon IJ, Whitcher MM, Palmer FL, et al. The impact of distant metastases at presentation on prognosis in patients with differentiated carcinoma of the thyroid gland. Thyroid. 2012;22(9):884–889. | |
Mihailovic J, Stefanovic L, Malesevic M. Differentiated thyroid carcinoma with distant metastases: probability of survival and its predicting factors. Cancer Biother Radiopharm. 2007;22(2):250–255. | |
Dinneen SF, Valimaki MJ, Bergstralh EJ, Goellner JR, Gorman CA, Hay ID. Distant metastases in papillary thyroid carcinoma: 100 cases observed at one institution during 5 decades. J Clin Endocrinol Metab. 1995;80(7):2041–2045. | |
Caplan RH, Strutt PJ, Wickus GG. Subclinical hormone secretion by incidentally discovered adrenal masses. Arch Surg. 1994;129(3):291–296. | |
Schlumberger M, Brose M, Elisei R, et al. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014;2(5):356–358. | |
Sacks W, Braunstein GD. Evolving approaches in managing radioactive iodine-refractory differentiated thyroid cancer. Endocr Pract. 2014;20(3):263–275. | |
Busaidy NL, Cabanillas ME. Differentiated thyroid cancer: management of patients with radioiodine nonresponsive disease. J Thyroid Res. 2012;2012:618985. | |
Haugen BR. Management of the patient with progressive radioiodine non-responsive disease. Semin Surg Oncol. 1999;16(1):34–41. | |
Haugen BR, Sherman SI. Evolving approaches to patients with advanced differentiated thyroid cancer. Endocr Rev. 2013;34(3):439–455. | |
Morabito A, De Maio E, Di Maio M, Normanno N, Perrone F. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist. 2006;11(7):753–764. | |
Korpanty G, Smyth E. Anti-VEGF strategies – from antibodies to tyrosine kinase inhibitors: background and clinical development in human cancer. Curr Pharm Des. 2012;18(19):2680–2701. | |
Thomas L, Lai SY, Dong W, et al. Sorafenib in metastatic thyroid cancer: a systematic review. Oncologist. 2014;19(3):251–258. | |
Dadu R, Waguespack SG, Sherman SI, et al. Efficacy and tolerability of different starting doses of sorafenib in patients with differentiated thyroid cancer. Oncologist. 2014;19(5):477–482. | |
Hoftijzer H, Heemstra KA, Morreau H, et al. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol. 2009; 161(6):923–931. | |
Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr Pharm Des. 2002;8(25):2255–2257. | |
Kloos RT, Ringel MD, Knopp MV, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol. 2009;27(10):1675–1684. | |
Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol. 2008;26(29):4714–4719. | |
Schneider TC, Abdulrahman RM, Corssmit EP, Morreau H, Smit JW, Kapiteijn E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: final results of a phase II trial. Eur J Endocrinol. 2012;167(5):643–650. | |
Ahmed M, Barbachano Y, Riddell A, et al. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol. 2011;165(2):315–322. | |
Brose MS, Nutting C, Jarzab B, et al. Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer: the phase 3 DECISION trial. Oncol Res Treat. 2014;37:130–131. | |
Brose MS, Nutting CM, Jarzab B, et al; DECISION Investigators. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319–328. | |
Carhill AA, Cabanillas ME, Jimenez C, et al. The noninvestigational use of tyrosine kinase inhibitors in thyroid cancer: establishing a standard for patient safety and monitoring. J Clin Endocrinol Metab. 2013;98(1):31–42. | |
Chen L, Shen Y, Luo Q, Yu Y, Lu H, Zhu R. Response to sorafenib at a low dose in patients with radioiodine-refractory pulmonary metastases from papillary thyroid carcinoma. Thyroid. 2011;21(2):119–124. | |
Shen Y, Ruan M, Luo Q, et al. Brain metastasis from follicular thyroid carcinoma: treatment with sorafenib. Thyroid. 2012;22(8):856–860. | |
Houk BE, Bello CL, Kang D, Amantea M. A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin Cancer Res. 2009;15(7):2497–2506. | |
Cohen EE, Needles BM, Cullen KJ, et al. Phase 2 study of sunitinib in refractory thyroid cancer. J Clin Oncol. 2008;26(15):6025. | |
Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16(21):5260–5268. | |
Polverino A, Coxon A, Starnes C, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and Kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res. 2006;66(17):8715–8721. | |
Rosen LS, Kurzrock R, Mulay M, et al. Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25(17):2369–2376. | |
Sherman SI, Wirth LJ, Droz JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med. 2008;359(1):31–42. | |
Inai T, Mancuso M, Hashizume H, et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165(1):35–52. | |
Rugo HS, Herbst RS, Liu G, et al. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol. 2005;23(24):5474–5483. | |
Cohen EEW, Rosen LS, Vokes EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26(29):4708–4713. | |
Locati LD, Licitra L, Agate L, et al. Treatment of advanced thyroid cancer with axitinib: phase 2 study with pharmacokinetic/pharmacodynamic and quality-of-life assessments. Cancer. 2014;120(17):2694–2703. | |
Bible KC, Suman VJ, Molina JR, et al. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11(10):962–972. | |
Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459–5465. | |
Boss DS, Glen H, Beijnen JH, et al. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours. Br J Cancer. 2012;106(10):1598–1604. | |
Sherman SI, Jarzab B, Cabanillas ME, et al. A phase II trial of the multitargeted kinase inhibitor E7080 in advanced radioiodine (RAI)-refractory differentiated thyroid cancer (DTC). J Clin Oncol. 2011; 29(15):abstr5503. | |
Schlumberger M, Tahara M, Wirth LJ, et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib (E7080) in patients with 131 I-refractory differentiated thyroid cancer (SELECT). J Clin Oncol. 2014;32(5s):abstrLBA6008. | |
Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: from the laboratory to clinical trials. Crit Rev Oncol Hematol. 2014;90(3):220–232. | |
Roskoski R. RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun. 2010;399(3):313–317. | |
Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. | |
Yang H, Higgins B, Kolinsky K, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70(13):5518–5527. | |
Kim KB, Cabanillas ME, Lazar AJ, et al. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid. 2013;23(10):1277–1283. | |
Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–1901. | |
Trujillo JI. MEK inhibitors: a patent review 2008–2010. Expert Opin Ther Pat. 2011;21(7):1045–1069. | |
Ho AL, Grewal RK, Leboeuf R, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623–632. | |
Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | |
de Souza ECL, Ferreira ACF, de Carvalho DP. The mTOR protein as a target in thyroid cancer. Expert Opin Ther Targets. 2011;15(9):1099–1112. | |
Lim SM, Chang H, Yoon MJ, et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann Oncol. 2013;24(12):3089–3094. | |
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–239. | |
Fuereder T, Jaeger-Lansky A, Hoeflmayer D, et al. mTOR inhibition by everolimus counteracts VEGF induction by sunitinib and improves anti-tumor activity against gastric cancer in vivo. Cancer Lett. 2010;296(2):249–256. | |
Lane HA, Wood JM, McSheehy PM, et al. mTOR Inhibitor RAD001 (Everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin Cancer Res. 2009;15(5):1612–1622. | |
Sherman EJ, Ho AL, Fury MG, et al. Phase II study of everolimus and sorafenib for the treatment of metastatic thyroid cancer. J Clin Oncol. 2013;31(15):abstr6024. | |
Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364(8):772–774. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | |
Eckstein N, Röper L, Haas B, et al. Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. J Exp Clin Cancer Res. 2014;33:15. | |
Josephs DH, Fisher DS, Spicer J, Flanagan RJ. Clinical pharmacokinetics of tyrosine kinase inhibitors: implications for therapeutic drug monitoring. Ther Drug Monit. 2013;35(5):562–587. | |
Cabanillas ME, Hu MI, Durand JB, Busaidy NL. Challenges associated with tyrosine kinase inhibitor therapy for metastatic thyroid cancer. J Thyroid Res. 2011;2011:985780. | |
Kandula P, Agarwal R. Proteinuria and hypertension with tyrosine kinase inhibitors. Kidney Int. 2011;80(12):1271–1277. | |
Hayes DN, Lucas AS, Tanvetyanon T, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18(7):2056–2065. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.