Back to Journals » International Journal of Nanomedicine » Volume 14
Multifunctional PLGA-based nanoparticles as a controlled release drug delivery system for antioxidant and anticoagulant therapy
Authors Lee PC, Zan BS, Chen LT, Chung TW ![]()
Received 22 May 2018
Accepted for publication 19 December 2018
Published 26 February 2019 Volume 2019:14 Pages 1533—1549
DOI https://doi.org/10.2147/IJN.S174962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Pei-Chi Lee,1 Bo-Shen Zan,1 Li-Ting Chen,1 Tze-Wen Chung1,2
1Department of Biomedical Engineering, National Yang Ming University, Taipei 112, Taiwan; 2Drug Delivery Department, Center for Advanced Pharmaceutics and Drug Delivery Research, National Yang Ming University, Taipei 112, Taiwan
Background: Ischemia/reperfusion (I/R) injury causes the generation of many ROS such as H2O2 and leads to vascular thrombosis, which causes tissue damage.
Purpose: In this investigation, poly (lactideco-glycolide) (PLGA)-based nanoparticles are used for their anticoagulant and antioxidant properties in vascular therapy.
Methods: Both heparin and glutathione are entrapped on PLGA-stearylamine nanoparticles by layer-by-layer interactions.
Results: The drug release rate is successfully controlled with only 10.3% of the heparin released after 96 hours. An H2O2-responsive platform is also developed by combining silk fibroin and horse peroxidase to detect H2O2 in this drug delivery system. Besides, hyaluronic acid was decorated on the surface of nanoparticles to target the human bone marrow mesenchymal stem cells (hBMSCs) for cell therapy. The results of an in vitro study indicate that the nanoparticles could be taken up by hBMSCs within 2 hours and exocytosis occurred 6 hours after cellular uptake.
Conclusion: We propose that the multifunctional nanoparticles that are formed herein can be effectively delivered to the site of an I/R injury via the hBMSC homing effect. The proposed approach can potentially be used to treat vascular diseases, providing a platform for hBMSCs for the controlled delivery of a wide range of drugs.
Keywords: ischemia/reperfusion, I/R, heparin, glutathione, anticoagulant, antioxidant, human bone marrow mesenchymal stem cells, hBMSCs
Introduction
Ischemia/reperfusion (I/R) injury is tissue damage that is caused by the reperfusion of ischemic tissues. An I/R injury is associated with many physiological conditions such as myocardial infarction, vascular thromboembolic events, and transplant surgery.1–5 The reperfusion of blood flow generates a large amount of ROS and leads to cellular apoptosis by inducing the release of proinflammatory cytokines.3 ROS can cause permanent tissue damage and increase the morbidity and mortality rate after I/R injuries. Therefore, controlling oxidative stress during the reparation of ischemic tissues is important. Lee et al developed H2O2-responsive antioxidant nanoparticles (NPs) for treating I/R injury and H2O2-associated diseases.6 Hardy et al delivered the α-interaction domain peptide, combined with antioxidants curcumin or resveratrol, by using multifunctional poly (glycidyl methacrylate) NPs to reduce the tissue damage and oxidative stress that were caused by a cardiac I/R injury.7
Vascular dysfunction is a critical symptom of I/R injury.8 Endothelial cell activation and barrier disruption by hypoxia and loss of integrity of the vascular wall can easily induce thrombosis.9 Anticoagulant drugs are typically administered directly to patients with I/R injury, but the drug release rate is high and the short duration of circulation in the blood can lead to severe side effects following the rapid dissolution of clots in thrombo-occluded vessels.10 An anticoagulant drug such as heparin can easily cause system toxicity, allowing systemic coagulopathy and hemorrhage to occur.11 Also, the inappropriate delivery of anticoagulant drugs causes bleeding, patient incompliance, and the delivery of an inadequate amount of the drug to the injured area.12 Additionally, anticoagulant drugs fail to form a residue at the injury site owing to the plasma deactivation, preventing the effective therapeutic level from being reached. Therefore, drugs must be protected using multifunctional NPs to protect their stability in circulation.13
The development of controlled drug delivery systems has always been of interest in the field of biotechnology. Multifunctional drug carriers that are prepared from polymeric materials such as poly (lactide-co-glycolide) (PLGA), poly-(ethylene glycol) (PEG), poly(ε-caprolactone) (PCL), and poly (lactic acid) have been extensively used to carry drugs owing to their biocompatibility, biodegradability, and functionalizability.14–18 Recently, several drug carrier systems, which have used NPs, liposomes, micelles, and dendrimers, have had great influence on diagnosis and therapy. NPs are attractive nanocarriers with therapeutic applications in the early detection of biomarkers and the treatment of vascular diseases.19–22 Surface modification, such as by the layer-by-layer (LbL) technique, can be used to control the half-life of the encapsulated drug in the system. The fabrication of LbL NPs by alternately coating oppositely charged polymers or biomolecules on various colloids can produce a promising delivery system with high loading efficiency, easy functionalization, tunable multifunctionality, and responsiveness to stimuli.23,24 The main interactions of the layers depend on the electrostatic force between oppositely charged materials. Some researchers have also identified other interactions, including hydrogen bonding, biomolecule recognition, and sol–gel reactions in the preparation of LbL NPs.25,26 LbL NPs of appropriate size and surface functionalization have attracted the interest of materials engineers and bioscientists owing to their usefulness in drug delivery. Ramasamy et al showed that the assembly of liposomal NPs with PEGylated polyelectrolytes can be used to improve the systemic delivery of multiple anticancer drugs, which improves drug half-life and reduces burst release.27 Deng et al also demonstrated that the use of LbL NPs in the codelivery of doxorubicin and siRNA could achieve a high loading of therapeutics, intracellular delivery, an extended serum half-life, and tumor targeting.28
Many investigations have established that human bone marrow mesenchymal stem cells (hBMSCs) can repair vascular injuries.29,30 hBMSCs have been shown to home preferentially to areas of injury in response to signals of cellular damage, known as homing signals.31 The hBMSCs home to injured sites and secrete growth factors, which may play an important role in tissue regeneration and repair.32 Exploiting the abundant surface markers CD44 on hBMSCs, the hyaluronic acid (HA)-decorated NPs produced herein can readily target hBMSCs, and hBMSC-based NPs are then delivered to the injured vasculature.33
In this study, PLGA-based NPs were combined with heparin and glutathione (GSH) so that their anticoagulant and antioxidant properties could be exploited in treating I/R injury. The surfaces of the NPs are modified with HA to enable the mesenchymal stem cells (MSCs) to target for the purposes of cell therapy. An H2O2-responsive platform is developed by combining the silk fibroin (SF) and horse peroxidase (HRP) for use in our drug delivery system to detect the H2O2 under UV light irradiation, which we named SF/HRP/Optical (SHO) system.
Materials and methods
Aminolysis of PLGA sheets
PLGA (Lakeshore Biomaterials, Germany) sheets were prepared by the solvent casting method, as follows.34 PLGA was dissolved in tetrahydrofuran (THF). A 20% solution of PLGA in THF was prepared for casting onto a glass Petri dish using a pipette. Then, the samples were dried at room temperature for 1 hour to form the thin sheets. To incorporate amino groups onto the surface of the PLGA sheets, the sheets were immersed in a 6% (w/w) 1,6-hexanediamine (Sigma-Aldrich Co., St Louis, MO, USA)/isopropanol (DEA/IPA) solution at 50°C for 8 minutes and then rinsed in deionized water (DIW). Subsequently, the PLGA-NH2 sheets thus formed were dried at room temperature for 24 hours and immersed in a 0.1% FITC (Sigma) solution at 25°C for 24 hours. After the samples were labeled with FITC, the samples were washed in 95% ethanol.
Subsequently, the sheets were dried at room temperature to prepare NPs. The overall amino groups that were grafted onto the PLGA-NH2 sheets were quantified using the ninhydrin assay. In this assay, ninhydrin reacted with free amino groups on the PLGA surface to form a purple pigment, which was detected using a spectrophotometer in the range 550–570 nm. The ninhydrin powder (Sigma) was dissolved as a reagent in 95% ethanol (1 M). A PLGA-NH2 sheet with an area of 1 cm2 was immersed into the reagent at 70°C in a water bath. After 15 minutes, THF was added to terminate the reaction. The amino density per unit area of the sample was calculated by comparing the absorbance at 568 nm with a previously obtained calibration curve for DEA from 0.5 to 25 μg/mL (y=0.00891x−0.000529, R2=0.99658) using a microplate reader (Thermo Multiskan GO Microplate Spectrophotometer, Thermo Fisher Scientific, Waltham, MA, USA).
Synthesis of PLGA-SA NPs
PLGA-SA NPs were prepared as the core of the ultimate NPs and alternating oppositely charged layers of drugs were coated on them. Briefly, the PLGA-NH2 sheets (40 mg), SA (Alfa Aesar, Ward Hill, MA, USA) (5 mg), and polyvinyl alcohol (PVA; Tokyo Kasei Kogyo, Chuo-ku, Tokyo, Japan) (5 mg) were dissolved in a DMSO/acetone mixture at 50°C and stirred for 1 hour. Then, 0.3% PVA solution was (injected 100 mL/h) added to the mixture to control the size of the formed particles.17,35 After vigorous stirring for 5 minutes, the solution was subjected to ultracentrifugation (18,000 rpm, 1 hour) before being washed with DIW to remove the supernatant. The positively charged PLGA-SA cores were used in the LbL assembly of NPs.
Fabrication of PS/hpGHA NPs
To obtain antioxidant PLGA-SA (PS)/hpGHA NPs, which could target CD44, heparin (Sigma), GSH (Sigma), and HA (Lifecore Biomedical, Chaska, MN, USA) were coated on NPs by LbL assembly (Figure 1). First, PLGA-SA (+) NPs with positively charged layers of SA were suspended in DIW (10 mg/mL) in a well with sonicator tip for 1 minute. Negatively charged heparin (hp) (−) was then added to the PLGA-SA NP solution in a water bath with sonication at 4°C, causing it to be absorbed onto the PS owing to the electrostatic force, yielding PS/hp NPs. Thereafter, the solution of PS/hp NPs was subjected to ultracentrifugation (15,000 g, 0.5 hour) before being washed in DIW to remove the free heparin. The PS/hp NPs were collected and resuspended in the DIW to coat them with GSH. Polyethylenimine (PEI) (Sigma) was functionalized with GSH by reacting it with the carboxylic groups of GSH in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (Sigma) and N-hydroxysulfosuccinimide (NHS) (Sigma) to yield a positive charge on PS/hpG NPs. Thirty milligrams of GSH in Tris buffer (10 mM, pH =8.5) that contained 20 mg of EDC and 5.75 mg of NHS were vigorously stirred at room temperature. Then, 10 mg of PEI was added to the solution. After 4 hours, the formed GSH-conjugated PEI was washed three times using DIW. Purified GSH-PEI (G) (+) was added to the solution of PS/hp (−) NPs in a water bath with sonication at 4°C. Thereafter, the solution of PS/hpG NPs was subjected to ultracentrifugation (15,000 g, 0.5 hour), before being washed with DIW to remove the free GSH-PEI. The purified PS/hpG (−) NPs were further mixed with HA (+) at a concentration of 10 mg/mL in DIW following the procedure that was used to obtain the PS/hpGHA NPs.
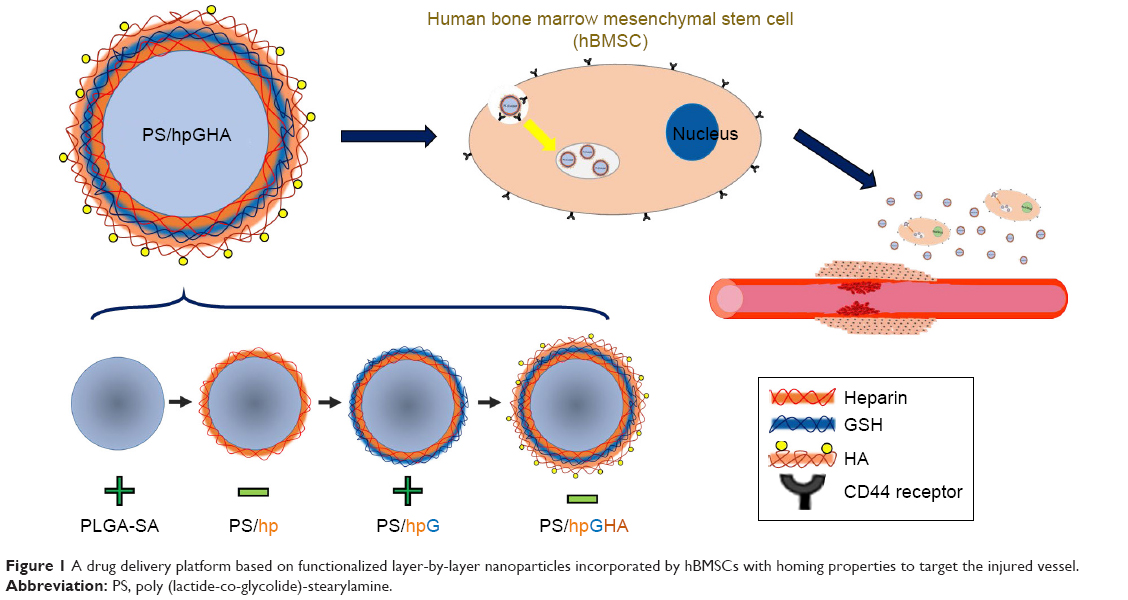 | Figure 1 A drug delivery platform based on functionalized layer-by-layer nanoparticles incorporated by hBMSCs with homing properties to target the injured vessel. |
The amounts of heparin on PS/hpGHA NPs were determined by toluidine blue O (TBO) (Sigma) assay.36 Each sample was added to 0.005% TBO solution (0.01 N HCl, 2% NaCl), which was mixed using a vortex mixer for 30 seconds at room temperature. Hexane (5 mL) was then added to the mixture, which was shaken vigorously for another 30 seconds to separate the heparin–dye complex formed in the mixture. The unreacted solution was removed, and the sample was diluted by adding absolute ethanol. The heparin was quantified by measuring the absorbance of the contents of each well at 610 nm using a microplate reader (Thermo Multiskan GO Microplate Spectrophotometer).
The amounts of GSH on PS/hpGHA NPs were determined using Ellman’s (DTNB) method.37 DTNB reagent at a concentration of 0.15 mg/mL in 0.1 M PBS (pH =8) was prepared. GSH at concentrations from 1 to 100 μg/mL or PS/hpGHA NPs in PBS were mixed with DTNB (5:1, V/V) for 20 minutes in the dark. The absorbance at 410 nm was measured using a microplate reader (Thermo Multiskan GO Microplate Spectrophotometer).
Size, zeta potential, and stability of NPs
The size and zeta potential of various NPs in DIW were evaluated at 25°C using a dynamic light scattering analyzer that was equipped with a zeta potential measurement device (Particulate Systems NanoPlus, Norcross, GA, USA). The core shell structures of the PLGA-SA and PS/hpGHA NPs were observed by transmission electron microscopy (TEM) (JEM-2000EXII, JOEL, Akishima, Tokyo, Japan). Briefly, an NP solution was placed on a 300 mesh copper grid that had been coated with carbon. The NPs on the copper grid were then stained with 0.2 wt% of phosphotungstic acid. After they were dried in air at room temperature, the morphology of the stained NPs was imaged by TEM. The PLGA-SA and PS/hpGHA NPs were added to PBS to evaluate the stability of multilayered structures. The sizes of the NPs were measured at 1, 12, 24, 48, and 96 hours.
In vitro release of heparin from PS/hpGHA NPs
Ten milligrams of PS/hpGHA NPs in 15 mL PBS (pH =7.4) was stirred at 200 rpm at 37°C. At 0.5, 1, 2, 4, 8, 24, 48, 72, and 96 hours, 1 mL of the sample was extracted and subjected to ultracentrifugation (15,000 g, 0.5 hour). The supernatant was collected for TBO testing to quantify the heparin. PS/hpGHA NP pellets were resuspended in 1 mL PBS in a well and then added to the original sample.
Coagulation properties of PS/hpGHA NPs
A kaolin clotting time test was carried out to determine the time to clot formation.38,39 The study protocol was approved by the Institutional Review Board at National Yang Ming University before the initiation of the study. Written informed consent was obtained from the healthy blood donors. Whole blood from healthy volunteers was collected in tubes that contained 3.2% sodium citrate, and the tubes were centrifuged at 2,000 g for 15 minutes to yield platelet poor plasma (PPP). PPP (100 μL) was mixed with 0.1% kaolin (100 μL) (First Chemical Products, Datong, Taipaei, Taiwan) in PBS for 3 minutes at 37°C. Twenty microliters of PBS, heparin (0.4 IU and 0.6 IU), and PS/hpGHA NPs (heparin =0.4 IU and 0.6 IU) were added at 37°C. After 1 hour, 0.2 M CaCl2 was added. After 10 seconds, 200 μL of each mixture was placed in 96-well plates to measure absorbance at 660 nm using a microplate reader (Thermo Multiskan GO Microplate Spectrophotometer). Absorbance was recorded every 15 seconds.
Detection of H2O2 using SHO system
Silk cocoons, Bombyx mori fibroin, were purchased from a silk center in Taiwan (Shih-Tan, Miao-Li, Taiwan). SF solutions were prepared as described elsewhere.40 Briefly, silk cocoons were boiled for 30 minutes in 0.02 M Na2CO3 and then rinsed thoroughly in distilled water to extract glue-like sericin proteins. The obtained SF was dissolved in 9.3 M LiBr solution with 20% (w/v) at 60°C for 4 hours and then dialyzed in DIW at room temperature for 48 hours. The final concentration of the SF solution was 8% (w/v), determined by weighing the residual solid in a fixed volume of solution after it was dried at 50°C for 24 hours. The 1% (w/v) SF solution, 10 U/mL HRP, and various concentrations of H2O2 (0, 0.40, 0.81, 1.62, 2.43, 3.24 mM) were added into 96-well plates at 37°C. After 1 hour, the 96-well plates were placed under a fluorescence microscope (Olympus IX71) using a UV filter (excitation filter: D350/50x, emission filter: D460/50 m). The fluorescence signal from SF was quantified using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA).
The H2O2-scavenging activity of PS/hpGHA NPs was quantified using an SHO system. Briefly, 1.62 mM H2O2 was added to various concentrations of GSH (0, 0.03, 0.08, 0.16, 0.33, 0.65 mM), PS/hpGHA (GSH =0.06 mM), or PLGA-SA NPs. After 30 minutes, the mixture was added to the SF (1%)/HRP (10 U/mL) solution at 37°C in 96-well plates. One hour later, the 96-well plates were placed under a fluorescence microscope (Olympus IX71) with a UV filter (excitation filter: D350, emission filter: D460/50 m). The fluorescence signal was quantified using Image-Pro Plus software (Media Cybernetics).
Cytotoxicity and antioxidant properties of PS/hpGHA NPs
L929 cells were purchased from Lonza. The cells were cultured in a 10 cm dish (Greiner Cellstar®, Frickenhausen, Baden-Württemberg, Germany) with minimum essential medium (Gibco) that had been supplemented with 10% (v/v) horse bovine serum at 37°C in 5% CO2 and a relative humidity of 90%; the medium was changed every 2 days. After 90% confluence, L929 cells were trypsinized using 0.25% trypsin/EDTA (Sigma-Aldrich) and transferred to 96-well plates (104 cell/well). After incubation, the cells were treated with PS/hpGHA NPs (0, 10, 50, 100, and 200 μg/mL) for 24 hours. The cells were then washed with PBS and the MTT solution (Sigma) was then added to each well and incubated for 3 hours in the dark. Formazan crystals thus formed were dissolved by adding 100 μL of DMSO. The absorbance of the formazan was measured at 570 nm using a microplate reader (Thermo Multiskan GO Microplate Spectrophotometer).
The antioxidant ability of PS/hpGHA NPs in L929 cells was also evaluated. H2O2 (10 μg/mL) was first mixed with PLGA-SA or PS/hpGHA NPs at 25°C (H2O2+PLGA-SA, H2O2+PS/hpGHA). After 3 hours, the L929 cells were treated with H2O2 (10 μg/mL), H2O2+PLGA-SA, or H2O2+PS/hpGHA. After another 24 hours, cell viability was evaluated by MTT assay. L929 cells were treated with lipopolysaccharide (LPS) to evaluate the anti-inflammatory activities of PS/hpGHA NPs. The cells were treated with PBS, PS/hpGHA NPs, LPS (100 ng/mL), or LPS (100 ng/mL) + PS/hpGHA NPs (50 μg/mL) for 24 hours. After 24 hours, cell viability was evaluated by MTT assay.
Superoxide detection and immunocytochemistry
Human umbilical vein endothelial cells (HUVECs) were purchased from Gibco. They were cultured in a 10 cm dish (Cellstar) with Medium 200 (Gibco) that had been supplemented with 2% (v/v) low serum growth supplement at 37°C and a relative humidity of 90% in 5% CO2; the medium was changed every other day. After 90% confluence, HUVECs were trypsinized using trypsin/EDTA solution (Gibco) with 0.025% trypsin and 0.01% EDTA in PBS and transferred to 96-well plates (5,000 cells/well). H2O2 (10 μg/mL) was first mixed with PLGA-SA or PS/hpGHA NPs at 25°C (H2O2+PLGA-SA, H2O2+PS/hpGHA). After 24 hours, the cells were treated with PBS (control), H2O2 (10 μg/mL), H2O2+PLGA-SA, or H2O2+PS/hpGHA. After 2 hours, the cells were washed with PBS and stained with mitochondrial superoxide (MitoSOX) indicator or hypoxia-inducible factor-1α (HIF-1α) antibody.
The intracellular ROS was detected using a MitoSOX Assay Kit (Abcam, Cambridgeshire, Cambridge, England), following the manufacturer’s instructions for labeling the MitoSOX and detecting red fluorescence. The cells were observed under a fluorescence microscope (Olympus IX71). The nuclei of the cells were observed using a DAPI filter (excitation filter: D350, emission filter: D460/50 m). The MitoSOX of the cells was observed using a TRITC filter (excitation filter: HQ535, emission filter: HQ610/75 m).
The cells were stained with HIF-1α antibody and then fixed in 0.25% glutaraldehyde for 15 minutes at 37°C. After permeabilization in 0.1% Triton X-100 and blocking by BSA for 5 minutes, the cells were immunostained. Rabbit anti-HIF-1α (dilution 1:500, GeneTex) and rabbit anti-ACTIN (dilution 1:400, Invitrogen) antibodies were used. Alexa Fluor 488-conjugated goat antirabbit secondary antibodies (1:500, Invitrogen) were used. The cells were observed under a fluorescence microscope (Olympus IX71). The nuclei of the cells were observed using OR through a DAPI filter (excitation filter: D350, emission filter: D460/50 m). HIF-1α in the cells was observed using an FITC filter (excitation filter: HQ480, emission filter: HQ535), and the F-actin in the cells was observed using a TRITC filter (excitation filter: HQ535, emission filter: HQ610/75 m).
Cellular uptake of PS/hpGHA NPs in hBMSCs
hBMSCs were purchased from Lonza. The cells were cultured in a 10 cm dish (Cellstar, Germany) with growth medium (PT-4105, Lonza) at 37°C in 5% CO2 and a relative humidity of 90%; the medium was changed every 2 days. After 90% confluence, hBMSCs were trypsinized using 0.25% trypsin/EDTA (Sigma-Aldrich) and transferred to 8-slide coverslips (104 cell/well). After incubation for 24 hours, the PS/hpGHA NPs or PLGA-SA NPs were used to treat hBMSCs for 15, 30, 60, and 120 minutes. Thereafter, the cells were washed three times in PBS and fixed with 4% paraformaldehyde for 15 minutes. Then, they were stained using the DAPI (Sigma) and observed using a confocal microscope (Zeiss, Oberkochen, Baden-Württemberg, Germany). The cells were irradiated with excitation wavelengths of 405 and 561 nm and their nuclei and the NPs were observed. The fluorescence signals from the NPs in the cells were quantified using Image-Pro Plus software (Media Cybernetics).
HA was used as an inhibitor for competitive inhibition studies. The hBMSCs were pretreated with HA (5 mg/mL) for 1 hour and then washed in PBS. The PS/hpGHA NPs were then incubated with hBMSCs for 2 hours. Thereafter, the cells were washed three times in PBS and fixed with 4% paraformaldehyde for 15 minutes. Another group of cells was treated only with PS/hpGHA NPs for comparison. All cells were stained with phalloidin-tetramethyl-rhodamine B isothiocyanate (Sigma) and DAPI to visualize F-actin and the nuclei of the cells, respectively. The cells were observed under a confocal microscope (Zeiss LSM880). They were irradiated with excitation wavelengths of 405, 488 and 561 nm. Their nuclei, F-actin and contained NPs were observed. The fluorescence signals from the NPs in the cells were quantified using Image-Pro Plus software (Media Cybernetics).
Retention time of NPs in hBMSCs
hBMSCs were seeded on 8-slide coverslip (104 cell/well). PLGA-SA or PS/hpGHA NPs were treated with hBMSCs for 6 hours and subsequently removed by washing three times using PBS. The cells were incubated by replacing the medium with a fresh medium at 0, 6, 12, and 24 hours. At the indicated times, the cells were washed three times using PBS and fixed with 4% paraformaldehyde for 15 minutes. Then, they were stained in DAPI and observed under a confocal microscope (Zeiss LSM880). Their ability to retain NPs was evaluated by quantifying the fluorescent intensity of the NPs in the cells using Image-Pro Plus software (Media Cybernetics).
Statistical analysis
All calculations were carried out in SigmaStat statistical software (Jandel Science, San Rafael, CA, USA). Statistical significance in Student’s t-test corresponded to a confidence level of 95%. Data are presented as mean±SD from triplicate measurements. Differences were considered statistically significant at P<0.05.
Results and discussion
Characterization of PS/hpGHA NPs
To develop a sustained controlled release system, PS/hpGHA NPs were prepared using an LbL technique. Briefly, the PLGA-SA was first made by nanoprecipitation. PLGA and SA were dissolved into the DMSO/acetone mixture at 50°C and stirred for 1 hour. Then, 0.3% PVA solution (100 mL/h) was injected to control the particle size. After vigorous stirring, the solution was subjected to ultracentrifugation and was then washed to remove the supernatant. PLGA-SA was used as the core of the NPs and then forming alternating oppositely charged coating layers of drugs. Various drugs and biomolecules were assembled directly on the PLGA-SA surface to obtain PS/hp, PS/hpG, and PS/hpGHA NPs, as schematically depicted in Figure 1. The main interaction of the layers depends on the electrostatic force between oppositely charged materials. Heparin, GSH, and HA can be easily entrapped by the LbL interaction.
In Figure 2A, the zeta potential of NPs is measured at each step of the coating process. Each layer caused a charge inversion on the surface, and the varying charge was consistent with the absorptions of biopolymers. Owing to the amine groups of SA, the zeta potential of PLGA-SA NPs became highly positive (+30.3 mV). In contrast, the absorption of heparin resulted in a negative zeta potential (−20.4 mV) because of the large amount of sulfite (SO3−2) on it. Then, PEI that was covalently bonded with GSH was used to provide the positive charge owing to its many amine groups, and the zeta potentials became +19.4 mV. Additionally, because HA has many carboxylic acid (COO−) groups, a negative zeta potential of PS/hpGHA NP was measured after the HA was absorbed (−17.6 mV). The absorption of heparin, GSH, and HA on the NPs was evidenced by the increase in hydrodynamic size to 223.9, 236.0, and 255.2 nm for the respective layers (Figure 2B). Electron microscopic images revealed the structure of the NPs before and after LbL assembly (Figure 2C). The PLGA-SA NPs had a solid and obvious boundary and sizes of about 20–35 nm (Figure 2C, PLGA-SA). After the final layers were added, the formation of a larger core–shell structure could be an evidence of the uniform coating of the PS/hpGHA NPs. The visible spread out dark shell in the image may be due to the longitudinal attachment of biopolymers (Figure 2C, PS/hpGHA). The particles in the TEM images were smaller (193.58 nm) than the average size that was obtained by DLS (255.20 nm), perhaps owing to the shrinkage of particles during the drying process. After HA was absorbed, the size of the PS/hpGHA NPs was more stable than that of the PLGA-SA NPs in PBS for up to 96 hours (Figure 2D). The stability of the size of the PS/hpGHA NPs with the highly negatively charged surfaced arose from their stable dispersion in PBS solution.
 | Figure 2 Physicochemical characterization of PS/hpGHA NPs. |
LbL NPs are a remarkable nanoplatform with sequentially deposited oppositely charged biopolymers enabling them to be loaded with multiple drugs. The drug release rate could be controlled because of the strong interaction between the layers, and the blood circulation time of drugs could be increased. Deng et al used LbL NPs to deliver both an anticancer drug and an siRNA for treating breast cancer. Their NPs were extremely stable, releasing <30% siRNA over the first 24 hours and providing an extended serum half-life of 28 hours.28 Ramasamy et al also used an LbL assembly of liposomal NPs to control the release of an anticancer drug. The amount of initial burst release rate of drug from NPs was reduced and followed by a more sustained release, greatly extending the systemic circulation time of the drug to achieve effective tumor drug delivery.27 Multiple drugs and biomaterials were incorporated into the LbL NPs herein. On the inner shells were heparin and GSH with anticoagulant and antioxidant properties. HA at the outer NPs supported CD44 targeting. The NPs also had a highly stable structure for 96 hours.
Anticoagulant property of PS/hpGHA NPs
In this investigation, the LbL technique was employed to control the release of heparin from NPs. Heparin is widely used for its anticoagulant activity. The anticoagulant mechanism demonstrated that heparin could enhance the ability of antithrombin III to form complexes with thrombin (factor Xa and factor IXa), thus inhibiting their activity.41 Figure 3A shows the release profile of heparin in vitro over 96 hours. A burst release of heparin was observed at 8 hours with 8.9% of the total heparin released. A relatively constant release rate was observed until 24 hours, owing to relatively strong electrostatic interactions between the heparin layer and the PLGA-SA NPs. The proportion of heparin released at 96 hours was only 10.3%.
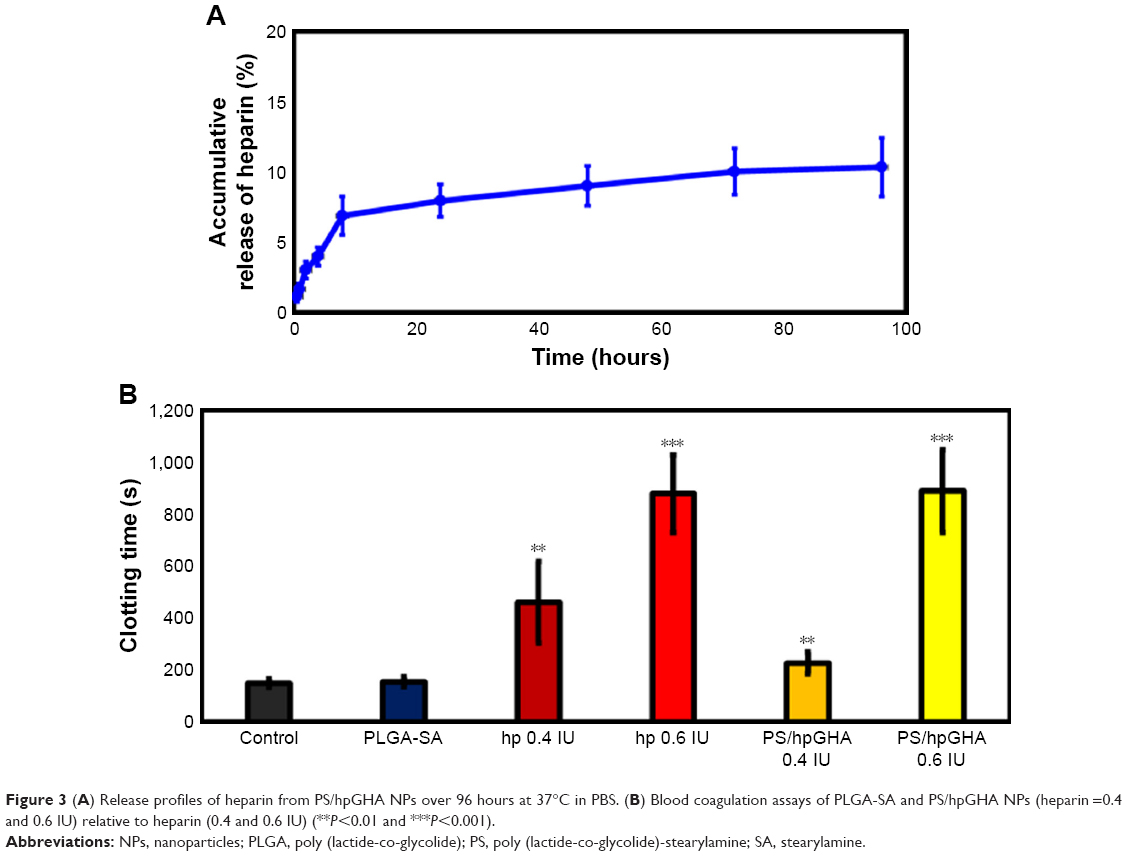 | Figure 3 (A) Release profiles of heparin from PS/hpGHA NPs over 96 hours at 37°C in PBS. (B) Blood coagulation assays of PLGA-SA and PS/hpGHA NPs (heparin =0.4 and 0.6 IU) relative to heparin (0.4 and 0.6 IU) (**P<0.01 and ***P<0.001). |
The antithrombogenic property of the PS/hpGHA NPs was evaluated by in vitro blood coagulation assay.42 In Figure 3B, the normal blood clotting times of the reference samples that contained PBS and PLGA-SA NPs were approximately 149.7–154.1 seconds. The addition of heparin to the blood sample prolonged its clotting time to 460.8 seconds (0.4 IU) and 878 seconds (0.6 IU) (Figure 3B), revealing that anticoagulant ability is closely related to heparin content. In an anticoagulation study in which PS/hpGHA NPs that contained 0.4 IU heparin were added to blood samples, the clotting time was increased to 227.0 seconds. Treating the blood sample with PS/hpGHA NPs with 0.6 IU heparin content yielded a clotting time of 888.8 seconds, which is similar to that achieved using pure heparin. These results suggest that the PS/hpGHA NPs that were developed in this study preserved the anticoagulation function of heparin.
Although heparin is an effective inhibitor of blood coagulation, it exhibits undesirable side effects, such as hemorrhagic complications, thrombocytopenia, and low bioavailability. The short period of circulation of heparin due to its negative charge and poor permeation causes repeated administrations to be required. In recent years, various studies have addressed the development of a heparin drug system using an LbL assembly, which enables both stable binding and sustained release. Yao et al developed a tri-layered PCL/chitosan (CS) vascular graft. Heparin was immobilized on the grafts through strong ionic bonding between heparin and CS fibers. The release rate of heparin was sustained for more than a month.43 Also, the interaction between heparin and NPs is very important because it determines the release profile and the biological function of heparin. Whitaker et al covalently immobilized heparin on tissue engineering scaffolds, which enabled stable binding, but made the release of heparin impossible.44 Therefore, we use ionic bonding instead of covalent bonding to achieve sustained release. Although only 10.3% of the heparin was released from PS/hpGHA NPs at 96 hours, it clearly functioned as an anticoagulant because of the heparin that was coated on the NPs providing enough free active sites for efficient contact with blood or plasma.45
Detection of H2O2 by SHO system
A new method that we named SHO system for detecting H2O2 that involves the addition of HRP to SF is introduced here. Mixing SF solution with HRP in the presence of H2O2 forms an SF transparent gel; HRP reacts with tyrosine in the SF to form tyrosine radicals in the presence of H2O2 in solution, forming in turn dityrosine bonds that cross-link the SF polymers to afford an SF gel, which emits blue fluorescent when it is irradiated by UV light;46 10 U/mL HRP was mixed with 1% SF solution, and various concentrations of H2O2 (0, 0.40, 0.81, 1.62, 2.43, and 3.24 mM) were added to the mixture at 37°C, which was left to stand for 1 hour. The images in Figure 4A reveal, under excitation at a wavelength of 325 nm, fluorescence of the formed transparent gel with an emission wavelength of 400 nm. As the concentration of H2O2 increased, the intensity of fluorescence increased (Figure 4B). However, increasing the concentration of H2O2 (to 2.43 and 3.24 mM) reduced the intensity, owing to the deactivation of HRP by H2O2.47
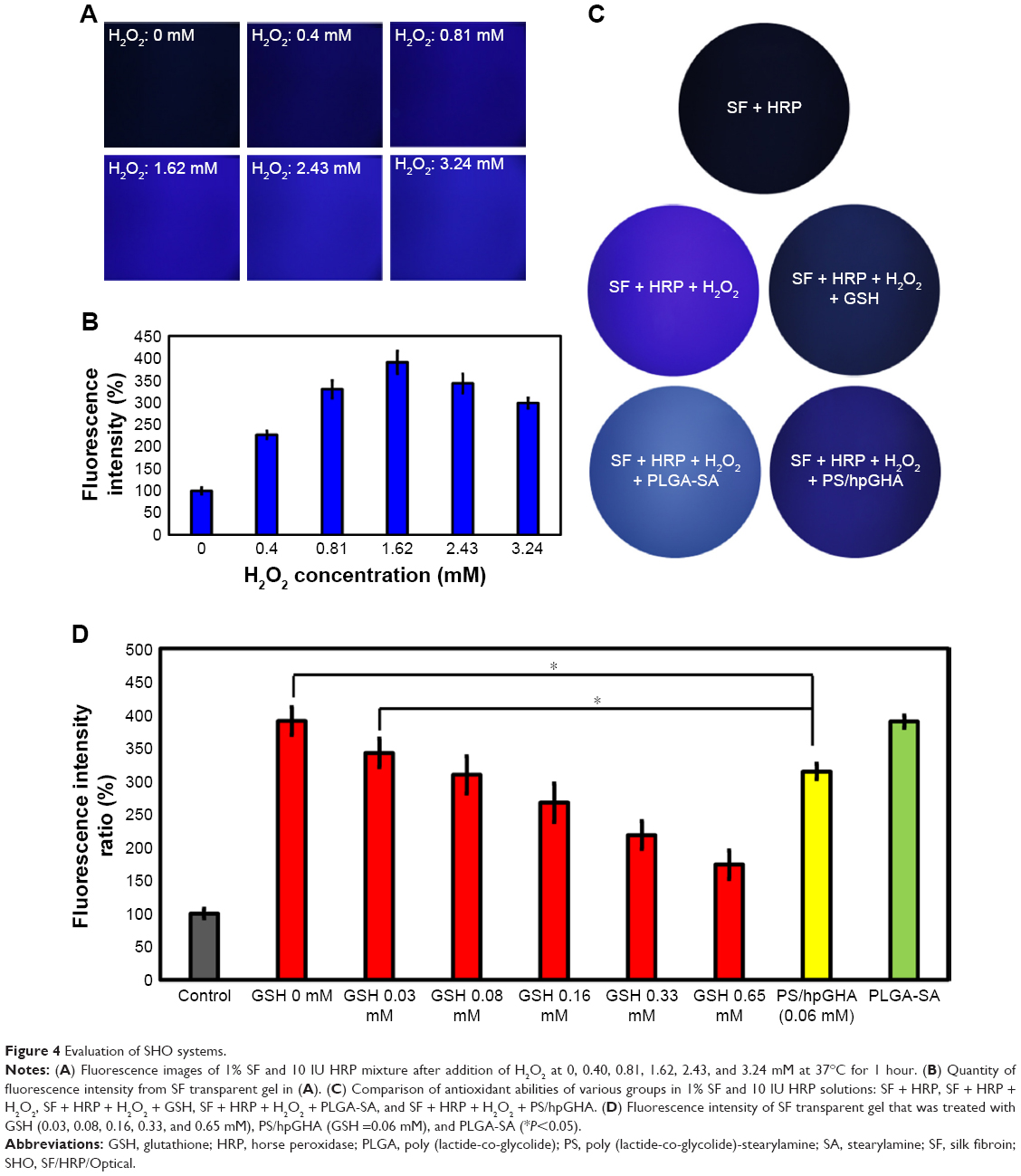 | Figure 4 Evaluation of SHO systems. |
The SHO system was used to evaluate the H2O2-scavenging ability of PS/hpGHA NPs. PS/hpGHA NPs contain GSH, which is a tripeptide that participates in antioxidant cellular defense.48,49 GSH was present mainly in its reduced form, which can be converted to GSSG (oxidized form) under oxidative stress and provides an effective defense against H2O2 as it can scavenge free radicals and reduce H2O2.50 GSH or PS/hpGHA NPs (GSH =0.06 mM) were mixed with H2O2 (1.62 mM) for 30 minutes, and then the mixture was added to SF/HRP solution at 37°C for 1 hour. In Figure 4C, the SF + HRP group without H2O2 showed no fluorescence, indicating that H2O2 is one of the main initiators of the cross-linking reaction that lead to the formation of a dityrosine bond. Adding H2O2 increased the fluorescence (SF + HRP + H2O2). Both SF + HRP + H2O2 + GSH and SF + HRP + H2O2 + PS/hpGHA groups exhibited considerably less fluorescence than the SF + HRP + H2O2 group because the proton that was donated by GSH neutralized the electrons at the center of H2O2. The thiol group of GSH is converted to a thyil radical (GS*) or a thiolate anion/radical (GS•). Then, the reduced H2O2 reduces the tyrosine radicals, inhibiting the cross-linking reaction. Results from the fluorescence images in Figure 4D were quantified; the fluorescence intensity of the PS/hpGHA group that contained 0.06 mM GSH was 313.91, which is close to that (309.14) of 0.08 mM free GSH.
Pichorner et al investigated the radical scavenging properties of GSH by generating phenoxyl radicals from tyrosine using HRP and H2O2.51 They showed that GSH efficiently inhibited the formation of dityrosine by measuring the intensity of dityrosine fluorescence as a result of the reaction. In our study, HRP was added to a solution of SF, which has phenol groups in tyrosine side chains. Subsequently, in the presence of H2O2, the SF transparent gel was formed so that we could detect the existence of H2O2 from the fluorescence intensity of the SF gel. Andersen found that silk solutions that were acted on by HRP and H2O2 formed gels were stable in water and were highly elastic.52 Aeschbach et al also referenced the enzymatic cross-linking of silks in a fundamental study of the formation of dityrosine cross-links by tyrosine oxidation.53
Antioxidant properties of PS/hpGHA NPs in vitro
After PS/hpGHA NPs were shown to reduce H2O2 effectively, the antioxidant ability of PS/hpGHA NPs in L929 cells was evaluated. H2O2 (10 μg/mL) was mixed with PLGA-SA or PS/hpGHA NPs for 3 hours at 25°C. Each mixture was used to treat cells for 24 hours, and the cell viability was then evaluated by MTT assay. Figure 5A shows the cell viability of L929 cells for 24 hours after treatment with PS/hpGHA at concentrations from 10 to 200 μg/mL. PS/hpGHA NPs exhibited excellent biocompatibility. The antioxidant activities of PS/hpGHA NPs in H2O2-stimulated cells were then investigated (Figure 5B). H2O2 was toxic to cells, as evidenced by a drop in cell viability (68.81%) (Figure 5B, H2O2 group). Also, PLGA-SA NPs did not protect cells from the H2O2-mediated oxidative stress, and cell viability was only 60.69%. In contrast, PS/hpGHA NPs had inhibitory effects on H2O2-induced toxicity, increasing the cell viability to 83.72%. Therefore, PS/hpGHA NPs had strong synergistic therapeutic effects as a highly potent H2O2-scavenging agent. We also used LPS as a stimulus and examined its effect on the viability of, and oxidative stress in, L929 cells. LPS can be used as a stimulus to induce oxidative stress damage through increased ROS generation. L929 cells were treated with PBS (control), PS/hpGHA NPs (50 μg/mL), LPS (100 ng/mL), and LPS (100 ng/mL) + PS/hpGHA NPs (50 μg/mL) for 24 hours. Figure 5C indicates that the viability of LPS-stimulated cells was 83.33%. In contrast, PS/hpGHA NPs inhibited LPS-induced toxicity, increasing the cell viability to 99.53%.
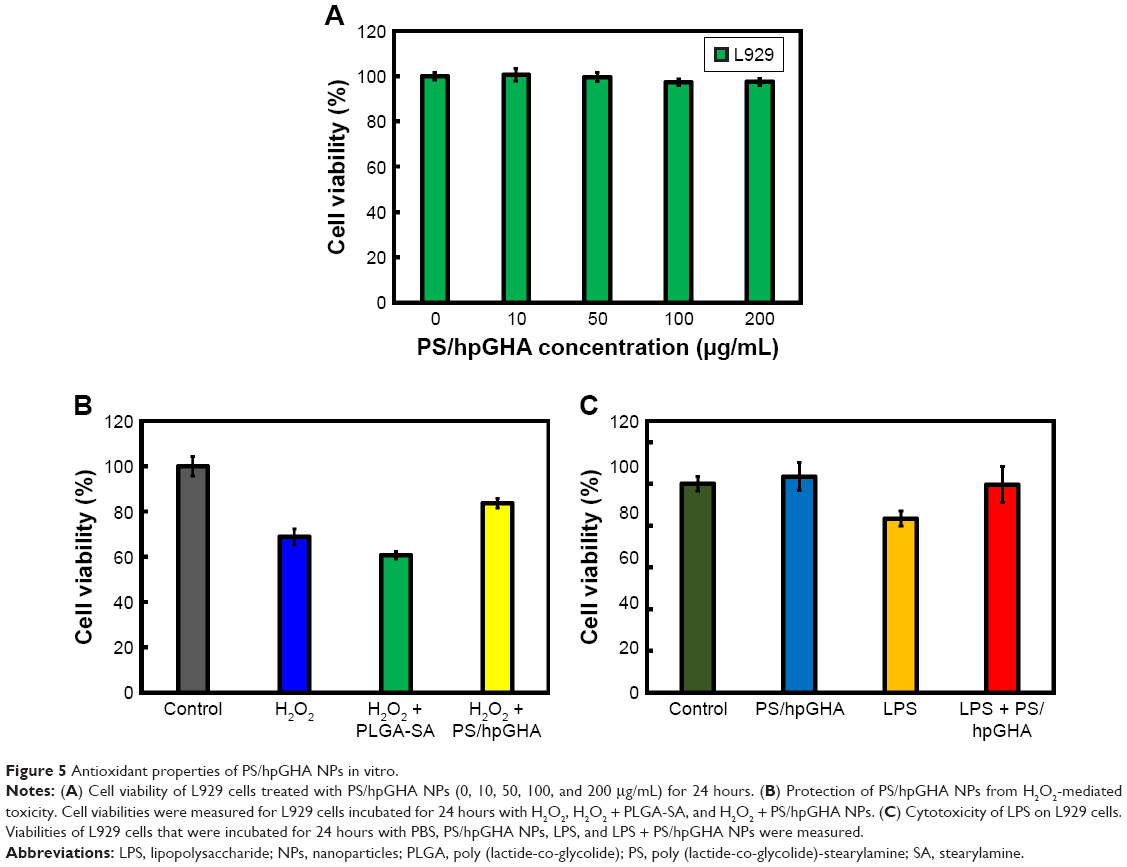 | Figure 5 Antioxidant properties of PS/hpGHA NPs in vitro. |
To determine whether H2O2 specifically induced oxidative stress, causing the death of HUVECs, and to provide evidence that the PS/hpGHA NPs effectively scavenged the generated ROS, MitoSOX Red was used. MitoSOX Red is a detection reagent for MitoSOX. It is oxidized by the superoxide, which is produced in mitochondria during H2O2-induced stress to emit red fluorescence. The cells were treated with PBS (control), H2O2, H2O2 + PLGA-SA, and H2O2 + PS/hpGHA NP for 2 hours. Figure 6 reveals an obvious red fluorescence from the cells that had been treated with H2O2 and H2O2 + PLGA-SA groups. The fluorescence in the cell decreased upon treatment with PS/hpGHA NPs, suggesting that GSH oxidation by ROS and the PS/hpGHA NPs could prevent the oxidative stress in HUVECs.
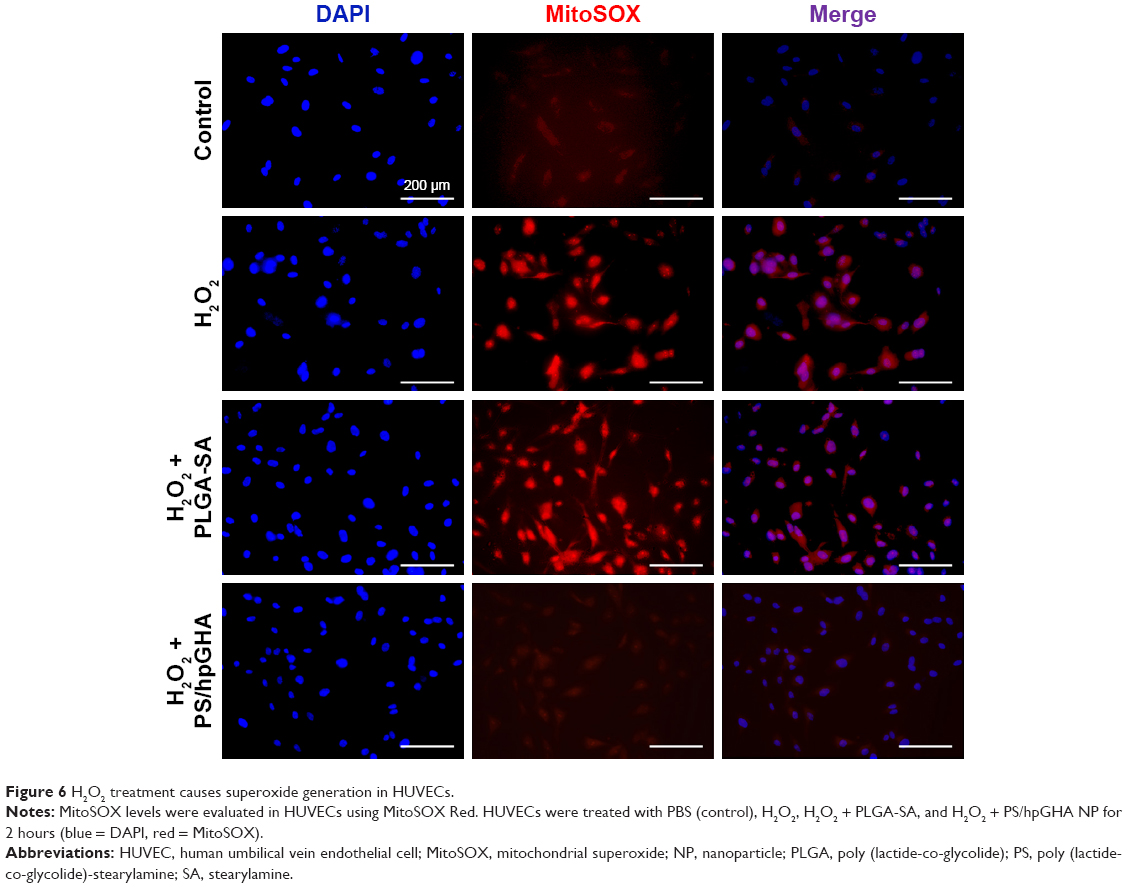 | Figure 6 H2O2 treatment causes superoxide generation in HUVECs. |
The effects of hypoxia on H2O2 treatment were examined. Hypoxia can be induced by H2O2, causing cell death. Hence, cells were stained with HIF-1α, which is a cytoplasmic protein that has a critical role in cells under reduced oxygen tension and responds to hypoxia cells following the treatment with PBS (control), H2O2, H2O2 + PLGA-SA, and H2O2 + PS/hpGHA NP for 2 hours. Figure 7 presents HIF-1α in cytoplasm upon treatment with PBS, representing the normal conditions. A comparison with control demonstrates that the fluorescence accumulates in the nuclei of H2O2-induced hypoxia cells (Figure 7, H2O2 and H2O2 + PLGA-SA), showing that the HIF-1α moved from the cytoplasm (Figure 7, white arrows) to become localized in the nuclei. In contrast, the cells with H2O2-induced hypoxia that were treated with H2O2 + PS/hpGHA NP (Figure 7, H2O2 + PS/hpGHA NP) exhibited no obvious changes under a microscope.
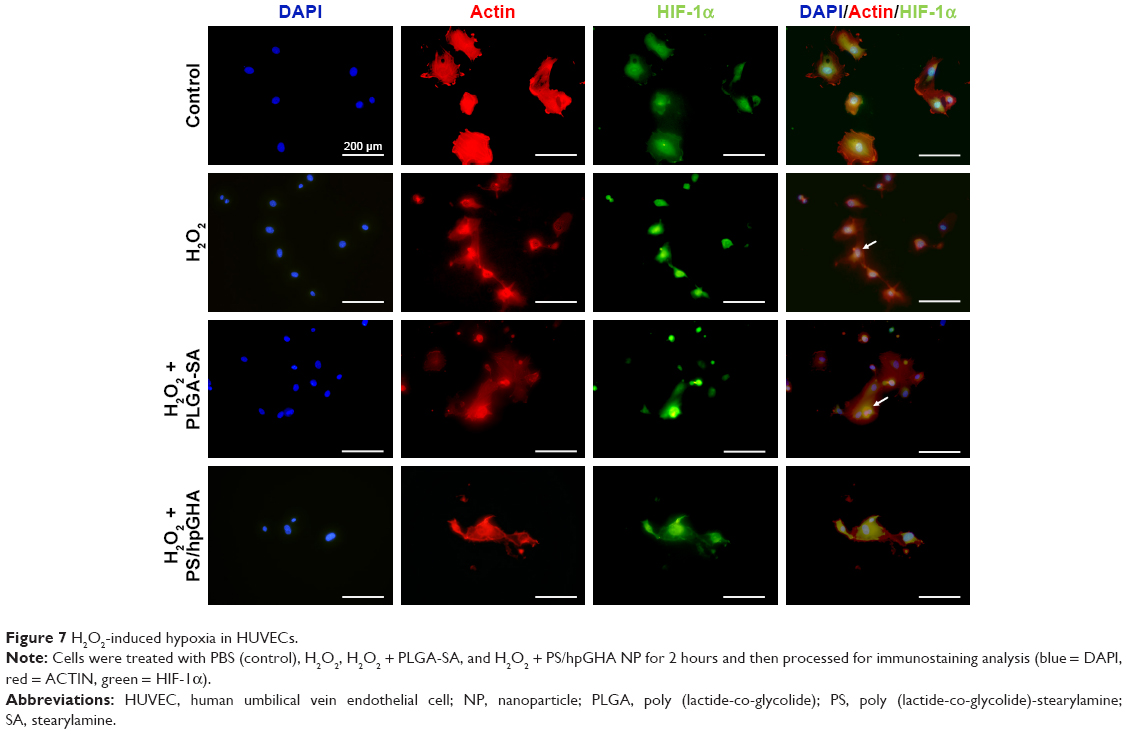 | Figure 7 H2O2-induced hypoxia in HUVECs. |
The production of a large amount of ROS is induced after blood flows following an ischemic period. Recent studies have demonstrated that oxidative stress may be primarily responsible for the pathogenesis of this injury. Therefore, the treatment of I/R injuries with antioxidant agents has tremendous potential. Kang et al examined a nanotheranostic agent that is based on the H2O2-triggered CO2-generating antioxidant poly (vanillin oxalate) (PVO).54 Intravenously administrated PVO NPs markedly strengthened the ultrasound signal at the site of a hepatic I/R injury and effectively suppressed liver damage by inhibiting inflammation and apoptosis. Xu et al used curcumin, which has antioxidant properties and is a promising protective agent against renal I/R injury.56 During myocardial I/R, a large amount of ROS is produced. Lee et al generated H2O2-responsive polymer NPs and examined their therapeutic effect on myocardial I/R injury by targeting them to the site of ROS overproduction to treat oxidative stress-associated diseases.6 Here, tri-functional NPs with antioxidant, anticoagulant, and targeting functions are developed.
Cellular uptake of PS/hpGHA NPs
Many studies have demonstrated the unique homing properties of MSCs for use in tissue repair. An MSC–based platform can effectively target in a drug delivery system.55 Therefore, NPs were based on hBMSCs herein in vascular therapy to exploit their anticoagulant and antioxidant effects. For the construction of hBMSCs as vehicles requires that the interactions of hBMSCs with NPs are understood. To investigate the enhancement of the cellular uptake as a result of CD44 selectivity, which is common marker of hBMSCs, hBMSCs were treated with PS/hpGHA NP-labeled FITC. Figure 8A shows the cell viability of hBMSCs 24 hours after treatment with various concentrations of PS/hpGHA from 10 to 200 μg/mL. The cell viability was 96.75%±7.04% even at a high concentration of 200 μg/mL, suggesting that PS/hpGHA was nontoxic to hBMSCs. Then, a confocal image was obtained to observe the cellular uptake of NPs during the short period. hBMSCs were treated with PLGA-SA or PS/hpGHA NPs (50 μg/mL) for 15, 60, 90, and 120 minutes to evaluate the selectivity of cellular uptake as a result of the interaction between CD44 and HA. In Figure 8B, the hBMSCs that had been treated with PLGA-SA showed no obvious signal at 15 minutes, but as the time passed, the signal in the hBMSCs became slightly stronger. The cells that were treated with PS/hpGHA fluoresced at 15 minutes, and as the time increased, the signal became stronger. At 120 minutes after treatment, the fluorescence of PS/hpGHA groups exceeded that of the PLGA-SA group, indicating that the cellular uptake of PS/hpGHA was higher than that of PLGA-SA. The amounts of PLGA-SA and PS/hpGHA that were taken up by the cells were obtained from the signal from the fluorescence image (PLGA-SA at 15 minutes =100%). As shown in Figure 8C, the fluorescence intensity in the PS/hpGHA group significantly exceeded that of the PLGA-SA-treated group (P<0.05) in hBMSCs at 15, 30, and 60 minutes. The intensity of PS/hpGHA NPs was nearly 1.32-fold higher than that of PLGA-SA in the hBMSCs. At 120 minutes, the increased cellular uptake could reach to 1.55-fold. Besides, the intensity of the signal from PS/hpGHA at 120 minutes in hBMSCs was 2.55-fold that that from PLGA-SA at 15 minutes in hBMSCs. These results indicate that the HA-coated NPs improved cellular uptake.
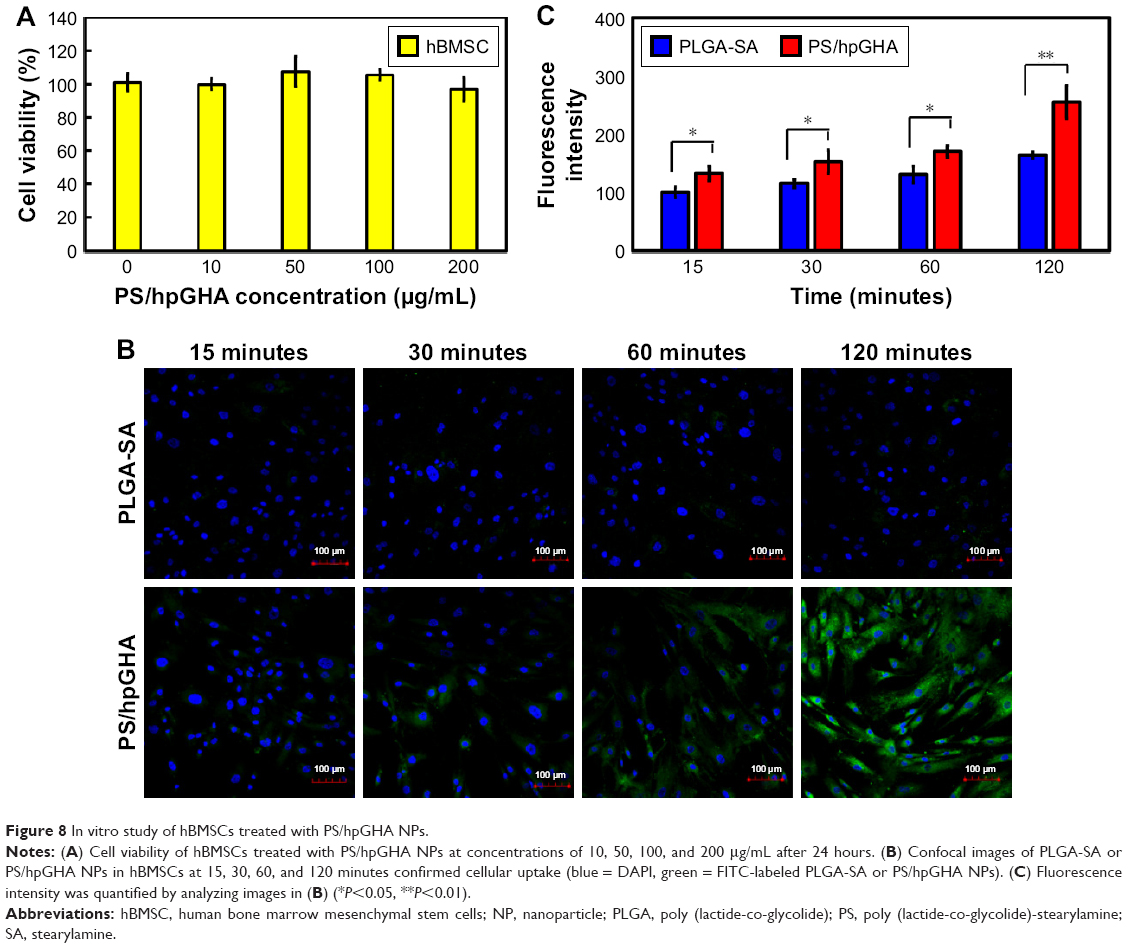 | Figure 8 In vitro study of hBMSCs treated with PS/hpGHA NPs. |
To verify the interaction between CD44 and HA, the mechanism of targeting was identified by pretreating the cells with HA to block the CD44 receptor. Cells were treated with free HA for 1 hour, and then HA was washed. The cells were treated with PLGA-SA or PS/hpGHA NPs for 2 hours. In Figure 9A, the fluorescence of PS/hpGHA in the pretreated HA group was reduced significantly in hBMSCs, and its intensity was 1/2.99 that of PS/hpGHA group (Figure 9B, PLGA-SA). The PLGA-SA group with HA pretreatment did not yield a significantly lower intensity than PS/hpGHA (Figure 9B). These results suggest that the enhanced uptake of PS/hpGHA NPs depended on the specific HA–CD44 interaction.
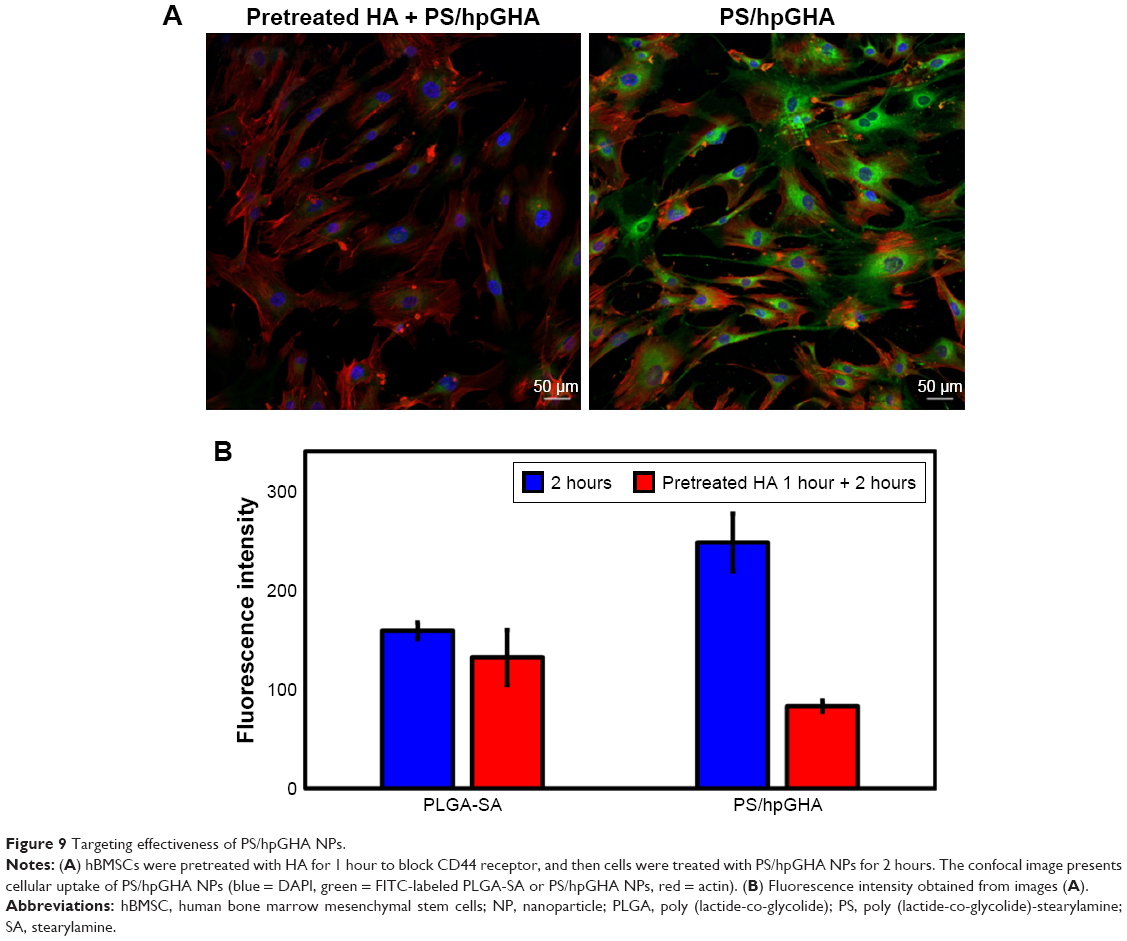 | Figure 9 Targeting effectiveness of PS/hpGHA NPs. |
Retention time of PS/hpGHA particles in hBMSCs
In this study, hBMSCs were used as an NP vector to deliver a drug to a target site. hBMSCs can home to the site of a vascular injury so the retention time of NPs in cells is important. This retention time is dominated by the dilution effect after cell division and efflux by exocytosis. We hypothesized that multifunctionality would be unaffected by exocytosis, so retention time in cells would also be unaffected. The cells were treated with PLGA-SA or PS/hpGHA for 6 hours (where retention time is indicated as 0 in Figure 10), and the cells were subsequently washed and cultured for up to 24 hours. In our result, the PLGA-SA signals in hBMSCs were significantly decreased over time, and no obvious signal was observed from them after 24 hours (Figure 10A, PLGA-SA). In contrast, PS/hpGHA signals were maintained for at least 24 hours (Figure 10A, PS/hpGHA). These results reveal the excellent stability of PS/hpGHA NPs in cells. More particles were retained in the PS/hpGHA group than the PLGA-SA group. To compare the retentions of NPs, the fluorescence images of hBMSCs were obtained and the fluorescence was quantified (Figure 10B). hBMSCs that had been treated with PS/hpGHA had a 1.9-fold stronger fluorescence signal after 6 hours of incubation than PLGA-SA-treated cells. The signal from PS/hpGHA was 1.7 times as strong as that from PLGA-SA after 24 hours of retention (*P<0.05). The improved retention effect of PS/hpGHA may be related to the ability of the particles to interact with CD44 receptors on the hBMSC membrane. The interaction between HA and CD44 receptors can improve the attachment of HA-coated particles.
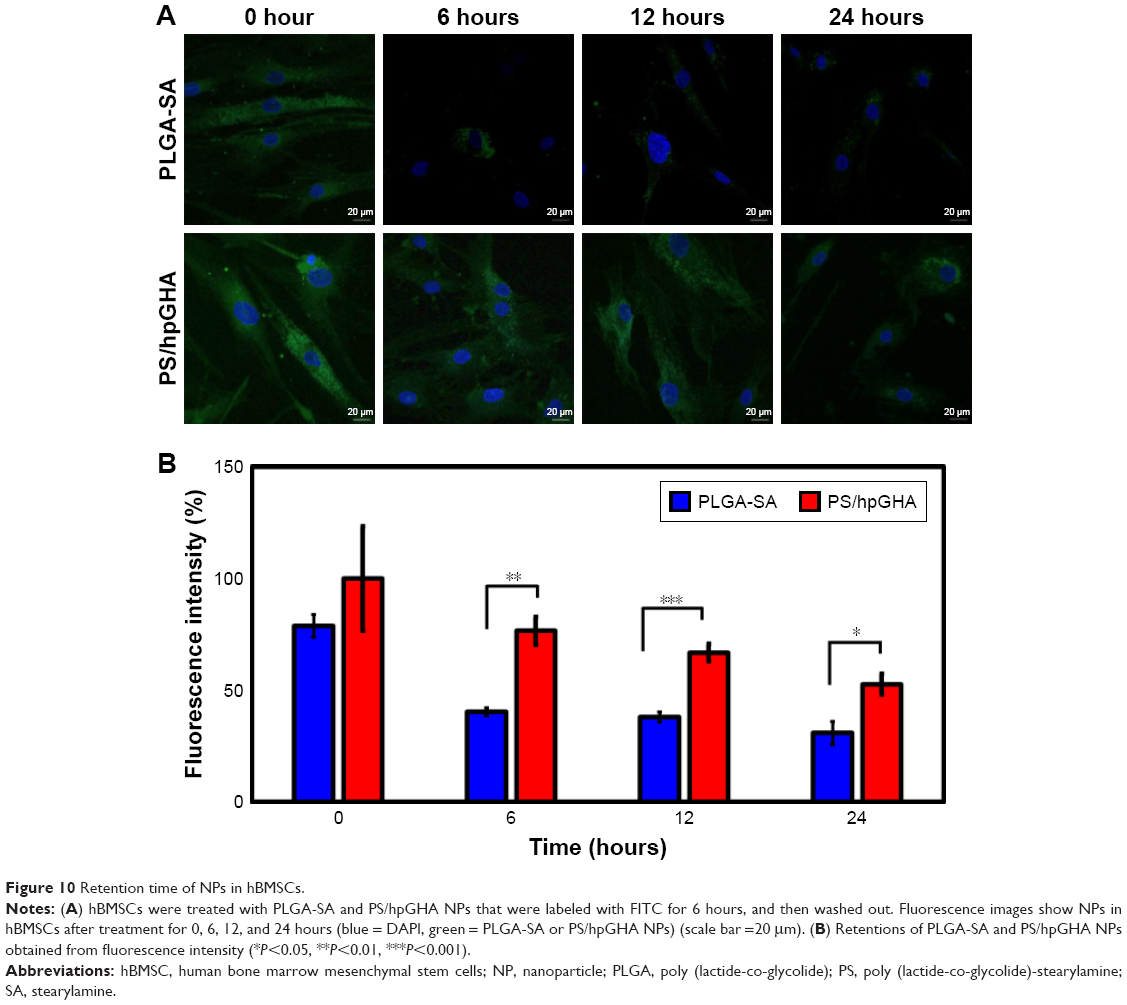 | Figure 10 Retention time of NPs in hBMSCs. |
Conclusion
In this study, we used PLGA-based NPs to achieve the anticoagulant and antioxidant ability for vascular therapy. Both heparin and GSH could easily be entrapped by the LbL interaction. We successfully controlled the drug release rate of heparin. The results suggest that the PS/hpGHA NPs developed in this study preserved the anticoagulation function of heparin. We also developed a new method (SHO system) to detect H2O2 for testing our drug delivery system. The PS/hpGHA NPs had inhibitory effects on H2O2-induced toxicity, increasing the cell viability to 83.72%. Therefore, PS/hpGHA NPs had strong synergistic therapeutic effects as a highly potent H2O2-scavenging agent. The results of in vitro study indicate that the HA-coated NPs improved cellular uptake, and the NPs could be taken up by hBMSCs and exocytosis occurred during short time period after cellular uptake. Therefore, we propose that the multifunctional NPs that are formed herein can be effectively delivered to the site of an I/R injury via the hBMSC homing effect. The proposed approach can potentially be used to treat vascular diseases, providing an hBMSC-based platform for the controlled delivery of a wide range of drugs and cell therapy.
Acknowledgment
The authors would like to thank the Ministry of Science and Technology, Taiwan, for financially supporting this research under contract no MOST-105-2811-E-010-001.
Disclosure
The authors report no conflicts of interest in this work.
References
Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100. | ||
Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. | ||
Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res. 2004;61(3):498–511. | ||
Weigand K, Brost S, Steinebrunner N, Büchler M, Schemmer P, Müller M. Ischemia/reperfusion injury in liver surgery and transplantation: pathophysiology. HPB Surg. 2012;2012:176723. | ||
Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation – from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10(2):79–89. | ||
Lee D, Bae S, Hong D, et al. H2O2-responsive molecularly engineered polymer nanoparticles as ischemia/reperfusion-targeted nanotherapeutic agents. Sci Rep. 2013;3(1):2233. | ||
Hardy N, Viola HM, Johnstone VP, et al. Nanoparticle-mediated dual delivery of an antioxidant and a peptide against the L-type Ca2+ channel enables simultaneous reduction of cardiac ischemia-reperfusion injury. ACS Nano. 2015;9(1):279–289. | ||
Yang Q, He GW, Underwood MJ, Yu CM. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am J Transl Res. 2016;8(2):765–777. | ||
Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78(6):539–552. | ||
Agyare E, Kandimalla K. Delivery of polymeric nanoparticles to target vascular diseases. J Biomol Res Ther. 2014;3(1):S1-001. | ||
Hossein HM, Hooman BM, Nasim Z. Intracranial hemorrhage in methanol toxicity: challenging the probable heparin effect during hemodialysis. J Res Phar Pract. 2017;6:186–189. | ||
Shoeb M, Fang MC. Assessing bleeding risk in patients taking anticoagulants. J Thromb Thrombolysis. 2013;35(3):312–319. | ||
Wo Y, Brisbois EJ, Bartlett RH, Meyerhoff ME. Recent advances in thromboresistant and antimicrobial polymers for biomedical applications: just say yes to nitric oxide (NO). Biomater Sci. 2016;4(8):1161–1183. | ||
Zhou S, Deng X, Yang H. Biodegradable poly(epsilon-caprolactone)-poly(ethylene glycol) block copolymers: characterization and their use as drug carriers for a controlled delivery system. Biomaterials. 2003;24(20):3563–3570. | ||
Italia JL, Bhatt DK, Bhardwaj V, Tikoo K, Kumar MN. PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J Control Release. 2007;119(2):197–206. | ||
Huang J, Zhang H, Yu Y, et al. Biodegradable self-assembled nanoparticles of poly (D,L-lactide-co-glycolide)/hyaluronic acid block copolymers for target delivery of docetaxel to breast cancer. Biomaterials. 2014;35(1):550–566. | ||
Tang L, Azzi J, Kwon M, et al. Immunosuppressive activity of size-controlled PEG-PLGA nanoparticles containing encapsulated cyclosporine a. J Transplant. 2012;2012(25):1–9. | ||
Moorthi A, Tyan YC, Chung TW. Surface-modified polymers for cardiac tissue engineering. Biomater Sci. 2017;5(10):1976–1987. | ||
Katsuki S, Matoba T, Koga JI. Anti-inflammatory nanomedicine for cardiovascular disease. Front Cardiovasc Med. 2017;22:87. | ||
Matoba T, Koga JI, Nakano K, Egashira K, Tsutsui H. Nanoparticle-mediated drug delivery system for atherosclerotic cardiovascular disease. J Cardiol. 2017;70(3):206–211. | ||
Liu M, Li M, Wang G. Heart-targeted nanoscale drug delivery systems. J Biomed Nanotechnol. 2014;10(9):2038–2062. | ||
Matoba T, Egashira K. Nanoparticle-mediated drug delivery system for cardiovascular disease. Int Heart J. 2014;55(4):281–286. | ||
Liu XQ, Picart C. Layer-by-layer assemblies for cancer treatment and diagnosis. Adv Mater. 2016;28(6):1295–1301. | ||
Becker AL, Johnston APR, Caruso F. Layer-by-layer-assembled capsules and films for therapeutic delivery. Small. 2010;6(17):1836–1852. | ||
Zhang X, Chen H, Zhang H. Layer-by-layer assembly: from conventional to unconventional methods. Chem Commun. 2007;21(14):1395–1405. | ||
Fu Y, Bai S, Cui S, Qiu D, Wang Z, Zhang X. Hydrogen-bonding-directed layer-by-layer multilayer assembly: reconformation yielding microporous films. Macromolecules. 2002;35(25):9451–9458. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano. 2013;7(11):9571–9584. | ||
Gu W, Hong X, Potter C, Qu A, Xu Q. Mesenchymal stem cells and vascular regeneration. Microcirculation. 2017;24(1):e12324. | ||
Kalinina NI, Sysoeva VY, Rubina KA, Parfenova YV, Tkachuk VA. Mesenchymal stem cells in tissue growth and repair. Acta Naturae. 2011;3(4):30–37. | ||
Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–216. | ||
Balaji S, Keswani SG, Crombleholme TM. The role of mesenchymal stem cells in the regenerative wound healing phenotype. Adv Wound Care. 2012;1(4):159–165. | ||
Bian XH, Zhou GY, Wang LN, et al. The role of CD44-hyaluronic acid interaction in exogenous mesenchymal stem cells homing to rat remnant kidney. Kidney Blood Press Res. 2013;38(1):11–20. | ||
Causa F, Battista E, della Moglie R, Guarnieri D, Iannone M, Netti PA. Surface investigation on biomimetic materials to control cell adhesion: the case of RGD conjugation on PCL. Langmuir. 2010;26(12):9875–9884. | ||
Fàbregas A, Sánchez-Hernández N, Ticó JR, et al. A new optimized formulation of cationic solid lipid nanoparticles intended for gene delivery: development, characterization and DNA binding efficiency of TCERG1 expression plasmid. Int J Pharm. 2014;473(1–2):270–279. | ||
Smith PK, Mallia AK, Hermanson GT. Colorimetric method for the assay of heparin content in immobilized heparin preparations. Anal Biochem. 1980;109(2):466–473. | ||
Güçlü K, Ozyürek M, Güngör N, Baki S, Apak R. Selective optical sensing of biothiols with Ellman’s reagent: 5,5′-Dithio-bis(2-nitrobenzoic acid)-modified gold nanoparticles. Anal Chim Acta. 2013;794:90–98. | ||
Chung TW, Lin PY, Wang SS, Chen YF. Adenosine diphosphate-decorated chitosan nanoparticles shorten blood clotting times, influencing the structures and varying the mechanical properties of the clots. Int J Nanomedicine. 2014;9:1655–1664. | ||
Bonar R, Favaloro E, Zebeljan D, et al. Evaluating laboratory approaches to the identification of lupus anticoagulants: a diagnostic challenge from the RCPA Haematology QAP. Pathology. 2012;44(3):240–247. | ||
Yang MC, Chi NH, Chou NK, et al. The influence of rat mesenchymal stem cell CD44 surface markers on cell growth, fibronectin expression, and cardiomyogenic differentiation on silk fibroin – hyaluronic acid cardiac patches. Biomaterials. 2010;31(5):854–862. | ||
Middeldorp S. Heparin: from animal organ extract to designer drug. Thromb Res. 2008;122(6):753–762. | ||
Bosch YP, Al Dieri R, Ten Cate H, et al. Measurement of thrombin generation intra-operatively and its association with bleeding tendency after cardiac surgery. Thromb Res. 2014;133(3):488–494. | ||
Yao Y, Wang J, Cui Y, et al. Effect of sustained heparin release from PCL/chitosan hybrid small-diameter vascular grafts on anti-thrombogenic property and endothelialization. Acta Biomater. 2014;10(6):2739–2749. | ||
Whitaker MJ, Quirk RA, Howdle SM, Shakesheff KM. Growth factor release from tissue engineering scaffolds. J Pharm Pharmacol. 2001;53(11):1427–1437. | ||
Argyo C, Cauda V, Engelke H, Rädler J, Bein G, Bein T. Heparin-coated colloidal mesoporous silica nanoparticles efficiently bind to antithrombin as an anticoagulant drug-delivery system. Chem Eur J. 2012;18(2):428–432. | ||
Partlow BP, Hanna CW, Rnjak-Kovacina J, et al. Highly tunable elastomeric silk biomaterials. Adv Funct Mater. 2014;24(29):4615–4624. | ||
Sakai S, Hirose K, Taguchi K, Ogushi Y, Kawakami K. An injectable, in situ enzymatically gellable, gelatin derivative for drug delivery and tissue engineering. Biomaterials. 2009;30(20):3371–3377. | ||
Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267(16):4928–4944. | ||
Fratelli M, Demol H, Puype M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99(6):3505–3510. | ||
Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clinica Chimica Acta. 2003;333(1):19–39. | ||
Pichorner H, Metodiewa D, Winterbourn CC. Generation of superoxide and tyrosine peroxide as a result of tyrosyl radical scavenging by glutathione. Arch Biochem Biophys. 1995;323(2):429–437. | ||
Andersen SO. The cross-links in resilin identified as dityrosine and trityrosine. Biochim Biophys Acta. 1964;93(1):213–215. | ||
Aeschbach R, Amadò R, Neukom H. Formation of dityrosine cross-links in proteins by oxidation of tyrosine residues. Biochim Biophys Acta. 1976;439(2):292–301. | ||
Kang C, Cho W, Park M, et al. H2O2-triggered bubble generating antioxidant polymeric nanoparticles as ischemia/reperfusion targeted nanotheranostics. Biomaterials. 2016;85:195–203. | ||
Gao Z, Zhang L, Hu J, Sun Y. Mesenchymal stem cells: a potential targeted-delivery vehicle for anti-cancer drug, loaded nanoparticles. Nanomedicine. 2013;9(2):174–184. | ||
Xu Y, Hu N, Jiang W, Yuan HF, Zheng DH. Curcumin-carrying nanoparticles prevent ischemia-reperfusion injury in human renal cells. Oncotarget. 2016;7(52):87390–87401. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.