Back to Journals » Journal of Inflammation Research » Volume 18
Multi-Omics Mendelian Randomization Identifies Therapeutic Targets for Sjögren’s Disease with Clinical and Bayesian Validation
Authors He J, Li K, Guo Z, Zhou X, Tang X
Received 29 May 2025
Accepted for publication 11 November 2025
Published 27 November 2025 Volume 2025:18 Pages 16685—16698
DOI https://doi.org/10.2147/JIR.S543644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Jiale He, Kesong Li, Zilin Guo, Xinyao Zhou, Xiaopo Tang
Department of Rheumatology, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, 100053, People’s Republic of China
Correspondence: Xinyao Zhou, Email [email protected] Xiaopo Tang, Email [email protected]
Background: There are currently no effective pharmacological treatments for Sjögren’s disease (SjD). Our study aims to identify potential therapeutic targets for the condition using druggable genome-wide Mendelian randomization (MR).
Methods: Druggable genome data were obtained from the Drug-Gene Interaction Database (DGIdb) and the study by Finan et al. We then integrated these druggable genes with blood-derived cis-eQTL, cis-mQTL, and cis-pQTL datasets, each analyzed separately in two-sample MR analyses using SjD GWAS summary statistics as the outcome, applying a genome-wide MR approach focused on druggable targets. Bayesian colocalization was applied to validate shared causal genetic variants. Protein levels of prioritized genes were validated in clinical serum samples from SjD patients and controls using ELISA. Phenome-wide MR (Phe-MR) analysis was conducted across 1359 phenotypes from the UK Biobank to evaluate potential pleiotropic effects and safety profiles of the identified targets, assessed side effects and alternative indications of identified targets.
Results: Fourteen druggable genes were identified, with eight (PLAT, SIRPB1, LAIR2, NEU1, SLC22A16, RAD52, PSPH, and CDH23) demonstrating consistent causal relationships with SjD. ELISA validation supported differential protein expression for these targets. Key findings include the protective role of NEU1 and PSPH in systemic immune regulation, the pathogenic impact of PLAT, SIRPB1, BRD2, and LAIR2 on inflammation, and the potential involvement of SLC22A16 and RAD52 in metabolic stress and immune activation. Existing pharmacological compounds targeting these genes, including Aminocaproic acid, Resveratrol, Levocarnitine, Imatinib, and Zinc chloride were identified as potential therapeutic candidates. No significant adverse effects were detected through Phe-MR analysis.
Conclusion: Our research indicated PLAT, SIRPB1, LAIR2, NEU1, SLC22A16, RAD52, PSPH, and CDH23 may serve as promising targets for SjD, while the effectiveness of Aminocaproic acid, Resveratrol, Levocarnitine, Imatinib, and Zinc chloride for SjD requires further validation.
Keywords: Sjögren’s disease, Mendelian randomization, druggable genome, Bayesian colocalization, therapeutic targets, Phe-MR analysis
Introduction
Affecting an estimated 0.1–0.6% of the population worldwide, Sjögren’s Disease (SjD) is a chronic systemic autoimmune disorder characterized by lymphocytic infiltration of the salivary and lacrimal glands, leading to xerostomia and keratoconjunctivitis sicca.1 Beyond these hallmark symptoms, SjD is associated with systemic manifestations such as fatigue, arthritis, and even lymphoma.2 Despite its prevalence and debilitating nature, the molecular mechanisms underlying SjD pathogenesis remain incompletely understood, and targeted therapies are currently lacking.3,4 Current treatment strategies focus on symptom relief rather than disease modification, and recent immunosuppressive trials have failed to show consistent efficacy, likely due to limited understanding of SjD’s molecular drivers.
Recent advances in high-throughput sequencing and bioinformatics have enabled the integration of multi-omics data, including transcriptomics, epigenomics, and proteomics, to uncover disease mechanisms.5,6 Expression quantitative trait loci (eQTL), methylation QTL (mQTL), and protein QTL (pQTL) analyses have emerged as powerful tools for linking genetic variants to gene regulation and protein function.7 These approaches are particularly valuable for complex diseases such as SjD, where single-level analyses often fail to capture the intricate molecular interplay.8
The concept of a “druggable genome” refers to the subset of genes encoding proteins that can be targeted by therapeutic compounds, including small molecules and biologics.9,10 The application of druggable genome analysis in autoimmune diseases has shown promise for identifying potential therapeutic targets, leveraging existing drug databases and computational predictions, such as DrugBank and Open Targets, to prioritize candidates with clinical relevance. In this study, we focused on a curated druggable genome comprising over 6000 genes, derived from DGIdb and Finan et al, integrating this framework into multi-omics data to systematically evaluate potential targets for SjD.
While genome-wide association studies (GWAS) have identified several susceptibility loci for SjD,11 their functional relevance remains largely unexplored. Moreover, few studies have integrated multi-omics data with druggable genome frameworks to identify actionable therapeutic targets.12 Addressing this gap requires a comprehensive pipeline that combines QTL analyses, druggability assessment, causal inference, and experimental validation.
This study aims to identify druggable targets for SjD by integrating multi-omics datasets-including eQTL, mQTL, and pQTL analyses-with druggable genome data. We applied Mendelian randomization (MR) and Bayesian colocalization to infer causality and validate shared genetic signals. Additionally, we utilized Phenome-wide Mendelian Randomization (Phe-MR) to assess the side effects or alternative indications of identified drug targets and investigated actionable drugs targeting these genes using existing databases. Enzyme-linked immunosorbent assay (ELISA) was performed to validate protein-level changes in serum samples from SjD patients and controls, providing translational relevance by connecting genomic findings to clinically measurable biomarkers. By combining multi-omics data, the druggable genome, MR analysis, Bayesian colocalization, and clinical validation, this study identifies drug targets for SjD and highlights potential pathways for therapeutic intervention.
Materials and Methods
The study flow diagram is presented in Figure 1. A summary of all data sources used in this study is provided in Table 1. This study received ethical approval from the Institutional Review Board (IRB) of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (Approval No. 2022-175-KY-01). We certify that the study was performed in accordance with the 1964 Declaration of Helsinki and later amendments. All participants provided written informed consent. Comprehensive dataset details are available in the supplementary material (Supplementary Tables).
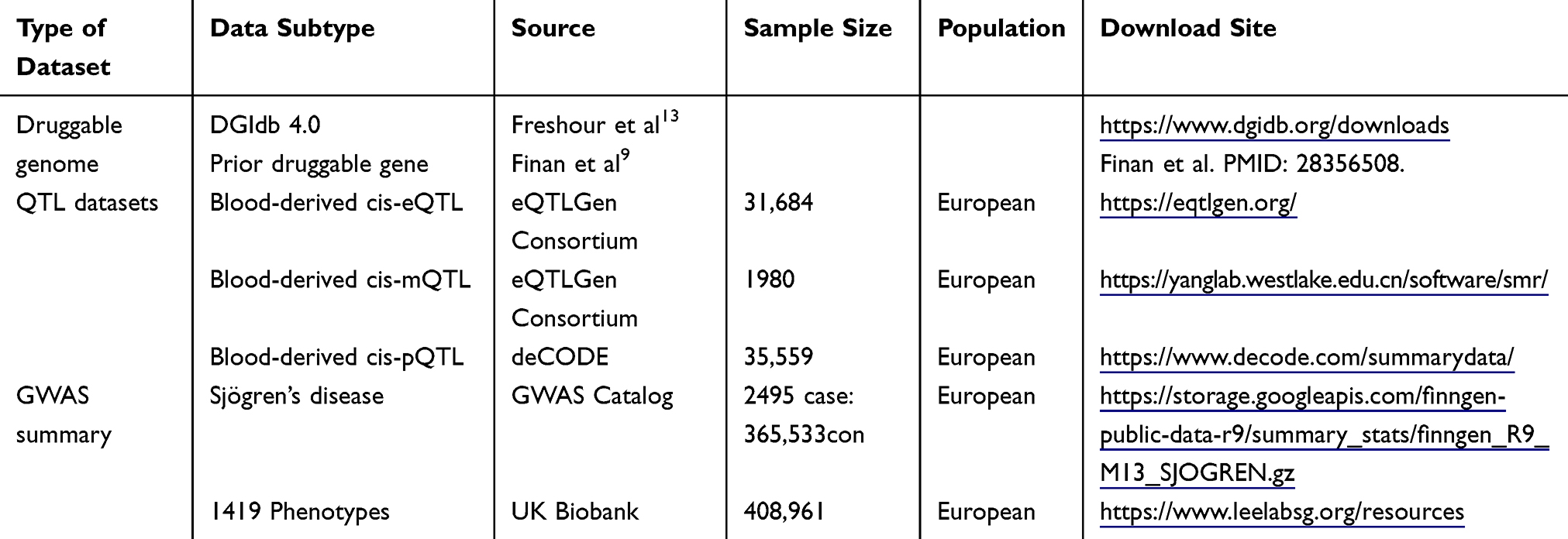 |
Table 1 Data Sources for Mendelian Randomization and Related Analyses |
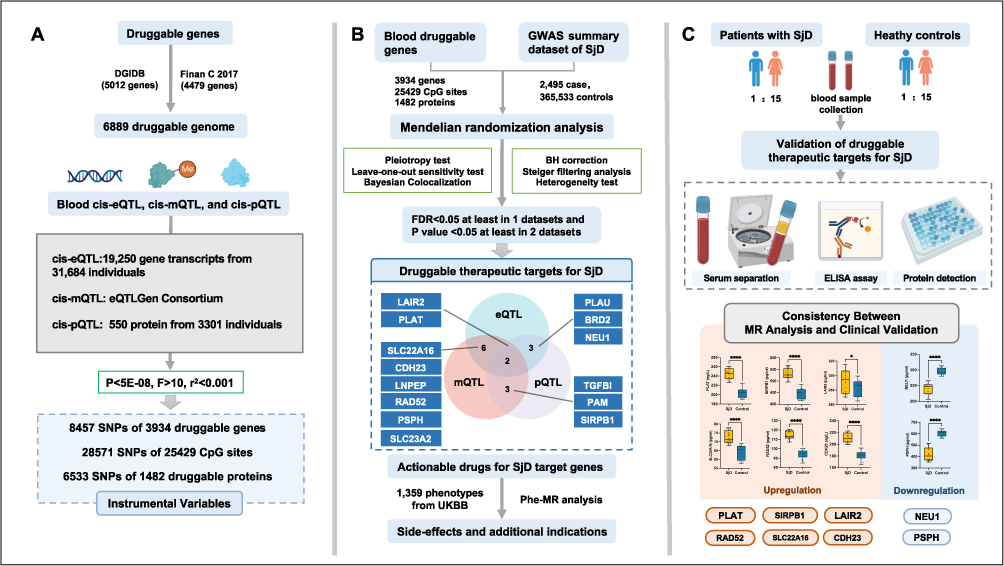 |
Figure 1 Study flow diagram. (A) A total of 6889 druggable genes were curated from the DGIdb database and previous literature. To identify potential genetic regulators, we utilized blood cis-eQTL, cis-mQTL, and cis-pQTL datasets, encompassing 19,250 gene transcripts, 25429 CpG sites, and 1482 proteins. Independent genetic variants within 1 Mb upstream or downstream of the coding region served as IVs under strict significance thresholds (P<5×10−8, F>10, r2<0.001). (B) Using MR analysis, we evaluated the causal relationship between druggable gene regulation and SjD risk. Bayesian colocalization tests confirmed shared causal variants. Genes meeting stringent criteria were classified as priority therapeutic targets, including 14 candidates. Additional Phe-MR analysis explored side effects or new indications using phenotypes from the UK Biobank GWAS database. (C) Validation was performed by quantifying serum protein levels of candidate genes in 16 SjD patients and 16 controls using ELISA. Comparative results demonstrated upregulated and downregulated genes. The consistency between MR analysis and clinical validation highlights these targets’ potential for diagnostic and therapeutic development. Abbreviations: SjD, Sjögren’s disease; eQTL, expression quantitative trait loci; mQTL, methylation quantitative trait loci; pQTL, protein quantitative trait loci; DGIdb, drug-gene interaction database; IVs, instrumental variables; Phe-MR, phenome-wide Mendelian randomization. |
Data Sources
Druggable Genome
Druggable genome data was selected from the Drug-Gene Interaction Database (DGIdb)13 and Prior druggable gene.9 DGIdb is an open web resource for drug-gene interactions and the druggable genome.13 We downloaded the drug-gene interaction data (December 2023) from DGIdb, which includes 5012 druggable genes with drug-gene interactions (Table S1). In addition, we incorporated 4479 druggable genes identified in the study by Finan et al. This study correlates complex loci associated with diseases and biomarkers identified in GWAS with druggable genes, aiming to support drug target identification and validation (Table S2).
Multi-Omics Datasets
This study utilized cis-eQTL, cis-mQTL, and cis-pQTL data from human blood, offering more accurate prediction and validation of drug targets compared to trans-regulatory elements. Blood eQTL data were obtained from the eQTLGen Consortium, comprising 19,250 transcripts across 31,684 individuals. The mQTL dataset included 28,571 SNP–CpG site pairs covering 25,429 CpG sites. The pQTL data were sourced from the deCODE database and included quantifications for 550 plasma proteins from 3301 individuals (Figure 1A).
GWAS Summary Data
Genome-wide association summary statistics for SjD were sourced from the FinnGen study via the GWAS Catalog (Figure 1B). The dataset included 2495 SjD cases and 365,533 controls, all of European ancestry (Table 1). Population stratification was accounted for using Principal Component Analysis (PCA).
Clinical Samples for ELISA
Serum samples were collected from 16 SjD patients and 16 age-sex matched controls (male to female ratio 1:15). All participants provided written informed consent prior to enrollment. The inclusion criteria for the SjD group adhered to the ACR-EULAR 2016 classification criteria,14 ensuring a consistent and reliable diagnosis, while the control group included healthy individuals without autoimmune disorders. We utilized ELISA to quantify the expression levels of proteins encoded by druggable target genes in the serum samples. Commercially available ELISA kits with established sensitivity and specificity were selected based on a thorough review of relevant literature and manufacturer recommendations. The assays were conducted according to standardized protocols, ensuring high reproducibility and minimizing technical variability (Figure 1C).
Statistical Analysis
Multi-Omics Data Integration
We selected genetic loci reaching genome-wide significance (P < 5×10−8). To ensure the strength and independence of instrumental variables (IVs), we applied additional criteria: F-statistics > 10 (to avoid weak instrument bias) and linkage disequilibrium threshold of r2 < 0.001.15 SNPs meeting these criteria were retained for downstream analysis. Volcano plots were generated to visualize the statistical significance and effect sizes of prioritized loci.
MR Analysis and Bayesian Colocalization
Selection of SNPs as instrumental variables (IVs) include those associated with significant changes in mQTLs alongside eQTLs and pQTLs. A two-sample MR design was applied, in which cis-eQTL, cis-mQTL, and cis-pQTL datasets were analyzed separately as exposures, with SjD GWAS summary data as the outcome.
Three complementary MR models were utilized to ensure robustness and minimize biases: (1) Inverse-Variance Weighted (IVW): Provided primary causal effect estimates under the assumption that all IVs were valid and uncorrelated with pleiotropic effects.15 (2) MR-Egger Regression: Adjusted for horizontal pleiotropy by estimating an intercept term that quantified average pleiotropic bias. (3) Weighted Median: Produced robust causal effect estimates even if up to 50% of the IVs were invalid.
To evaluate the validity and robustness of the causal inference, the following sensitivity analyses were performed: (1) Steiger Filtering: Ensured the correct causal direction by confirming that the genetic instruments primarily influenced the exposure rather than the outcome.16 (2) Heterogeneity Testing: Cochran’s Q statistic was used to assess consistency across IVs; significant heterogeneity indicated potential bias.17 (3) Leave-One-Out Analysis: Iteratively removed individual SNPs to determine their influence on the overall causal effect estimate. This analysis assessed whether the results were driven by any single SNP.18 All data analyses were performed using R software (version 4.1.2) and R packages (TwoSampleMR, MRPRESSO, and Rmediation).
Bayesian colocalization analysis was conducted using the coloc R package to test whether QTL and GWAS signals shared the same causal variant. A posterior probability (PPH4) > 0.8 was considered strong evidence of colocalization.19
Actionable Drug Identification and Phe-MR Analysis
We searched the DrugBank database (version 5.1.10, https://go.drugbank.com), ChEMBL database (version 33, https://www.ebi.ac.uk/chembl), and ClinicalTrials (https://www.clinicaltrials.gov) to evaluate the drug development for the identified druggable genes, including obtaining drug molecule types, indications for drug targets, and clinical development activities. The objective of our Phe-MR analysis was to determine causal relationships between the identified druggable genes and other disease traits to assess their potential associated side effects or additional indications. Phe-MR analysis was performed using 1359 phenotypes from the UK Biobank to evaluate potential side effects and additional indications (Tables 1 and S12).
Experimental Validation of Druggable Therapeutic Targets
Proteins encoded by prioritized genes were measured using ELISA in serum samples from 16 SjD patients and 16 controls matched for age and sex (Table S14). Differences in protein expression between SjD patients and controls were assessed using unpaired t-tests or Wilcoxon rank-sum tests, with multiple testing corrected by the Benjamini-Hochberg method (p<0.05). We used GraphPad Prism 10 for all graphical presentations.
Results
Druggable Genome
We identified 6889 druggable genes (Table S3) by integrating druggable genome data from DGIdb v4.2.0 (Table S1) and the Finan et al study (Table S2). These genes were then intersected with blood cis-eQTL, cis-mQTL, and cis-pQTL datasets. Genetic variants located within 1 Mb of each gene’s coding region were extracted.
After IVs selection, we obtained 8457 SNPs mapped to 3934 druggable genes from blood cis-eQTL (Table S4), 28571 SNPs mapped to 25429 CpG sites from blood cis-mQTL (Table S5), and 6533 SNPs mapped to 1482 druggable genes from blood cis-pQTL (Table S6) as IVs representing exposure for MR analysis. These SNPs were retained as IVs for MR analyses, rather than as direct disease-associated loci.
Candidate Druggable Genes for SjD
We performed MR analysis using the GWAS summary dataset for SjD as the outcome. Our results suggest that 220, 1507, and 94 druggable genes from blood eQTL, mQTL, and pQTL, respectively, have potential causal relationships with SjD (P<0.05, F>10, r2<0.001; Tables S7–S9). Volcano plots visualizing the significant loci across the eQTL, mQTL, and pQTL datasets (Figure 2A–C). These genes demonstrated statistically significant associations with SjD, making them strong candidates for further investigation.
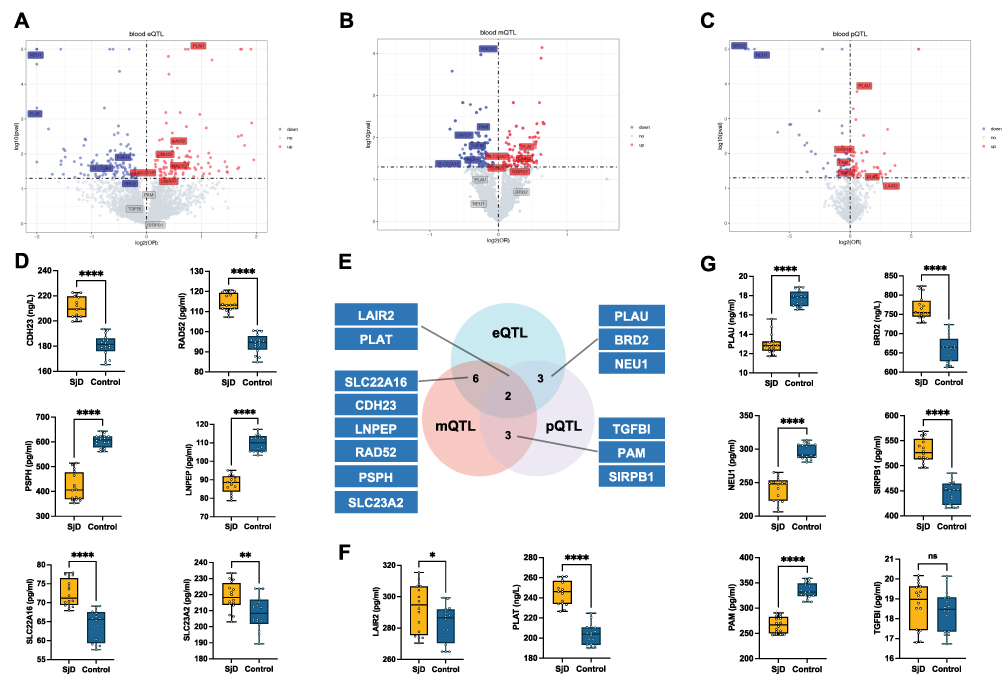 |
Figure 2 Multi-Omics Data Integration and Validation. (A–C) Volcano plots representing the association of cis-eQTL (A), cis-mQTL (B), and cis-pQTL (C) variants with SjD. Genes with significant positive associations (red) and negative associations (blue) are highlighted. Each point represents a genetic variant, and significance thresholds are indicated by dashed lines. (E) Venn diagram illustrating the overlap of significant targets identified across eQTL, mQTL, and pQTL datasets. A total of 14 druggable targets were identified, with FDR<0.05 at least in 1 datasets and P value <0.05 at least in 2 datasets (D, F and G) Box plots representing the plasma protein levels of 16 patients with SjD and 16 controls, as measured by ELISA. Serum protein levels of prioritized druggable genes (LAIR2, PLAT, PLAU, BRD2, NEU1, SIRPB1, PAM, TGFBI, SLC22A16, SLC23A2, CDH23, LNEP, PSPH, and RAD52) in SjD patients versus controls. The box plots display the median, interquartile range, and minimum-maximum values, highlighting significant differences in protein levels between the SjD and control groups. *P < 0.05, **P < 0.01, ****P < 0.0001, “ns” indicates not significant (p ≥ 0.05). |
To enhance the accuracy and robustness of our findings, we selected druggable genes present in at least two QTL datasets as potential therapeutic targets for SjD. Ultimately, we identified 14 druggable genes (LAIR2, PLAT, PLAU, BRD2, NEU1, SLC22A16, CDH23, LNPEP, RAD52, PSPH, SLC23A2, TGFBI, PAM, and SIRPB1) that show causal relationships with SjD across two or more QTL datasets (Figure 2D and E). To identify candidate drug targets sharing genetic variants with those associated with SjD, we conducted Bayesian colocalization analysis. This method was used to confirm whether the causal signals observed in the Mendelian randomization analyses originate from the same genetic variants. The results indicated that all 14 potential druggable genes passed the colocalization analysis (Table S10).
LAIR2 and PLAT were validated in all three QTL datasets (Figure 2E). LAIR2 showed significant positive correlation signals in the eQTL, mQTL, and pQTL analyses (Figures 3–5). In the eQTL dataset, transcription levels of LAIR2 exhibited a strong positive correlation with SjD risk (HR: 1.37, 95% CI: 1.093–1.716), indicating its overexpression is associated with SjD pathogenesis. A result confirmed in both the blood mQTL (HR: 1.172, 95% CI: 1.012–1.357) and pQTL datasets (HR: 6.08, 95% CI: 1.021–36.192). ELISA assays validated the elevated expression of LAIR2 at the protein level in SjD patients compared to controls (Figure 2F), further supporting these findings. However, PLAT transcription levels were negatively correlated with SjD in the blood eQTL dataset (HR: 0.25, 95% CI: 0.115–0.545), yet showed positive correlations in the blood mQTL (HR: 1.235, 95% CI: 1.036–1.471) and pQTL datasets (HR: 2.339, 95% CI: 1.077–5.081). ELISA validation experiments revealed significant differences in serum PLAT levels between SjD patients and controls (Figure 2F).
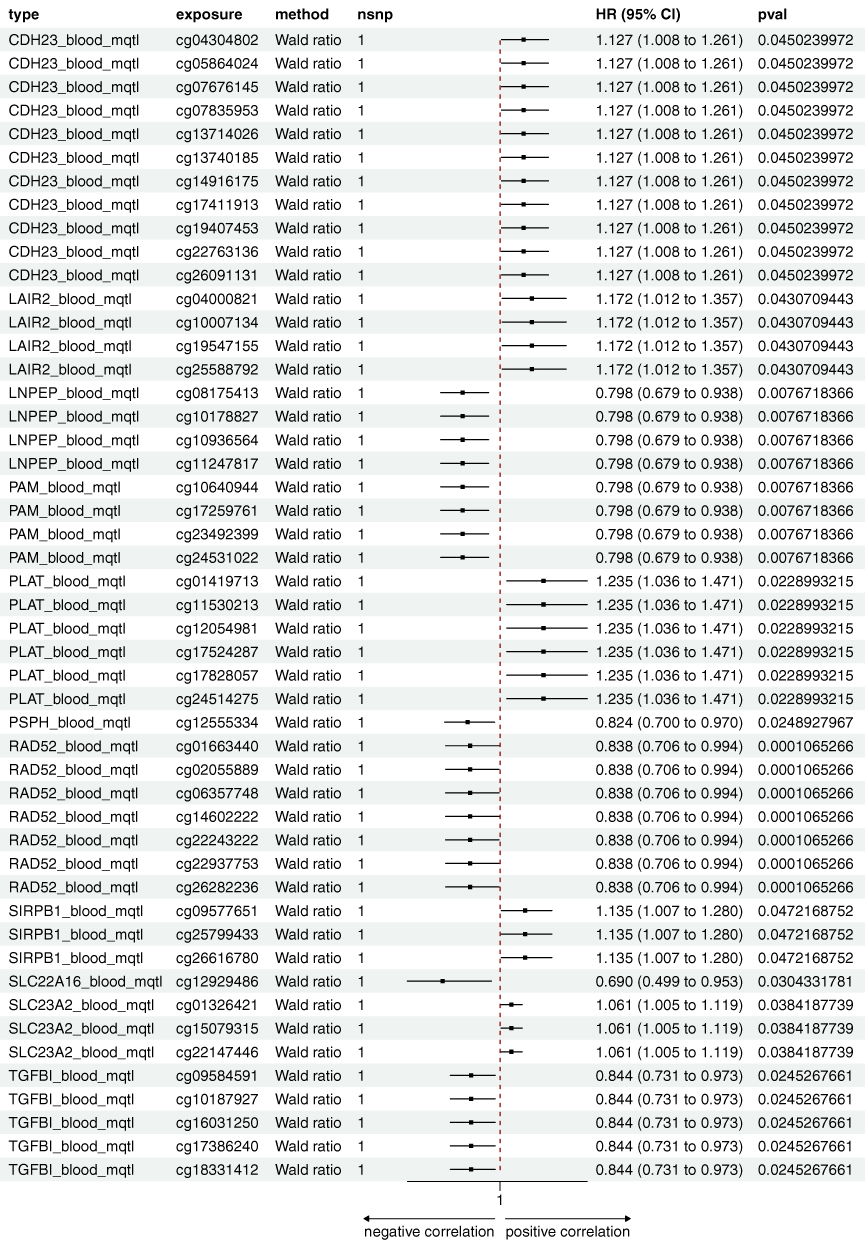 |
Figure 3 The forest plot summarizes the associations of cis-mQTL variants with SjD risk, highlighting HR, CI, and p-values for each candidate gene and variant. Abbreviations: HR, hazard ratios; CI, confidence intervals. |
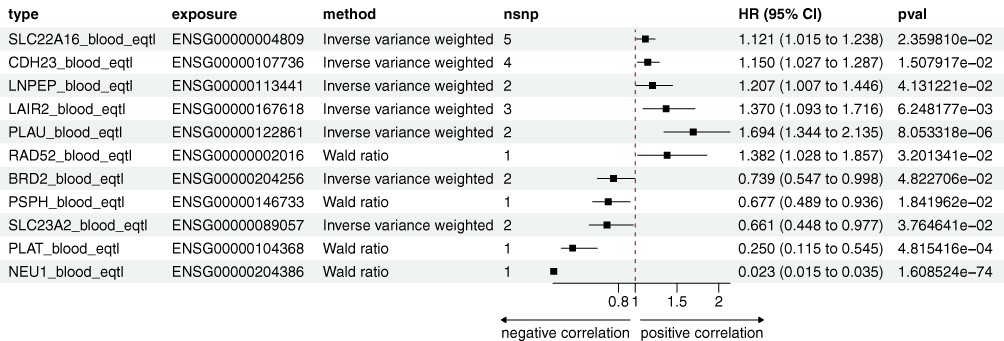 |
Figure 4 The forest plot summarizes the associations of cis-eQTL variants with SjD risk, highlighting HR, CI, and p-values for each candidate gene and variant. |
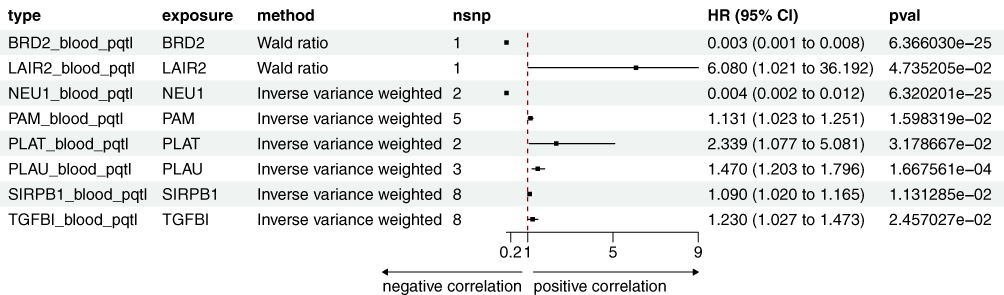 |
Figure 5 The forest plot summarizes the associations of cis-pQTL variants with SjD risk, highlighting HR, CI, and p-values for each candidate gene and variant. |
Moreover, NEU1, PLAU, and BRD2 exhibited causal relationships with SjD in both the eQTL and pQTL datasets (Figures 4 and 5). Transcription and protein levels of NEU1 negatively correlated with SjD (eQTL, HR: 0.023, 95% CI: 0.015–0.035; pQTL, HR: 0.004, 95% CI: 0.002–0.012), a finding consistent in the ELISA validation (Figure 2G), suggesting a protective role for NEU1 in SjD. PLAU were positively correlated with SjD in the blood eQTL and pQTL dataset (eQTL, HR: 1.694, 95% CI: 1.344–2.135; pQTL, HR: 1.47, 95% CI: 1.203–1.796). Conversely, the protein level of PLAU was significantly reduced in SjD patients compared to the controls (Figure 2G). A similar contradiction was also observed in BRD2. Transcription and protein levels of BRD2 negatively correlated with SjD (eQTL, HR: 0.739, 95% CI: 0.547–0.998; pQTL, HR: 0.023, 95% CI: 0.015–0.035), while compared to the control group, the protein level of BRD2 was significantly elevated in SjD patients (Figure 2G).
SIRPB1, TGFBI, and PAM were validated in the mQTL and pQTL datasets (Figures 3 and 5). SIRPB1 levels were positively correlated with SjD (mQTL, HR: 1.135, 95% CI: 1.007–1.280; pQTL, HR: 1.090, 95% CI: 1.020–1.165), suggesting an increased risk of SjD. And the serum protein level of SIRPB1 was significantly elevated in SjD patients compared to controls (Figure 2G). The methylation levels of TGFBI and PAM were negatively correlated with SjD in the mQTL datasets (TGFBI, HR: 0.844, 95% CI: 0.731–0.973; PAM, HR: 0.78, 95% CI: 0.679–0.938), while the pQTL dataset showed the opposite correlation (TGFBI, HR: 1.230, 95% CI: 1.027–1.473; PAM, HR: 1.131, 95% CI: 1.023–1.251). Moreover, ELISA validation showed no significant difference in serum TGFBI protein levels between SjD patients and the control group, while PAM levels were significantly elevated, contradicting the pQTL data (Figure 2G).
In the eQTL and mQTL datasets, SLC22A16, SLC23A2, CDH23, LNPEP, RAD52, and PSPH exhibited causal relationships with SjD (Figures 3 and 4). Transcription levels of SLC22A16, RAD52, and LNPEP were positively correlated with SjD (SLC22A16, HR: 1.121, 95% CI: 1.015–1.238; LNPEP, HR: 1.207, 95% CI: 1.007–1.446; RAD52, HR: 1.382, 95% CI: 1.028–1.857), while their methylation levels exhibited a negative correlation with SjD (SLC22A16, HR: 0.690, 95% CI: 0.499–0.953; LNPEP, HR: 0.798, 95% CI: 0.679–0.938; RAD52, HR: 0.838, 95% CI: 0.706–0.994). SLC23A2 transcription level negatively correlated with SjD in the blood eQTL dataset (HR: 0.661, 95% CI: 0.448–0.977), while its methylation level in the pQTL dataset was positively correlated (HR: 1.061, 95% CI: 1.005–1.119). These results suggest the impact of post-transcriptional methylation regulation on gene expression. The transcription and methylation levels of CDH23 were positively correlated with SjD across both blood eQTL and mQTL datasets (eQTL, HR: 1.150, 95% CI: 1.027–1.287; mQTL, HR: 1.127, 95% CI: 1.008–1.261), whereas those of PSPH were negatively correlated (eQTL, HR: 0.677, 95% CI: 0.489–0.936; mQTL, HR: 0.824, 95% CI: 0.700–0.970). In the clinical validation of SjD patients, SLC22A16, CDH23, RAD52, and PSPH were all confirmed, while the serum protein levels of SLC23A2 and LNPEP showed contradictory results.
Actionable Drugs and Phe-MR Analysis for SjD Target Genes
For this study, we define “actionable” as referring to pharmacological compounds that are FDA-approved or currently in clinical development targeting these genes. We have identified actionable drugs of 11 candidate druggable genes for SjD (Figure S1). Formulations targeting 5 druggable genes (PLAT, SIRPB1, SLC22A16, and RAD52) have been tested in clinical trials for other diseases but have not yet been used to treat SjD. We conducted a Phe-MR analysis on 1359 diseases and traits in the UK Biobank (Table S12). The IVs used for Phe-MR analysis were consistent with those previously identifying SjD-related druggable genes (79 SNPs for the 14 druggable genes, see Table S11). Causal effects in the Phe-MR analysis were considered statistically significant at FDR value<0.05. We have identified the potential side effects or additional indications of prior druggable genes (Table S13). For example, the Phe-MR analysis suggested that targeting NEU1 may also confer beneficial effects in systemic lupus erythematosus, while inhibition of PLAT was associated with increased risk of thromboembolic events, highlighting potential adverse effects.
Discussion
Based on multi-omics data and clinical validation, our MR and colocalization analyses identified potential causal evidence for 8 drug target genes (PLAT, SIRPB1, LAIR2, NEU1, SLC22A16, RAD52, PSPH, and CDH23) with SjD. The PHE-MR analysis did not identify potential adverse effects for these drug target genes. By assessing the clinical development potential of these drug target genes, we identified potential drugs for SjD treatment: Aminocaproic acid inhibits PLAT; Resveratrol inhibits SIRPB1; Levocarnitine inhibits SLC22A1; Imatinib targets RAD52, and Zinc chloride activate PSPH.
Our findings identify PLAT (tissue plasminogen activator, tPA) as a key contributor to SjD pathogenesis through its involvement in fibrinolysis and inflammation. Multi-omics analyses revealed that transcription levels of PLAT were negatively correlated with SjD risk in blood eQTL data, while pQTL and mQTL analyses demonstrated strong positive associations. Elevated PLAT protein levels in SjD patients, as validated by ELISA, suggest enhanced plasmin activity. This activity promotes extracellular matrix degradation, tissue remodeling, and pro-inflammatory signaling, exacerbating immune infiltration and chronic inflammation.20 Moreover, targeting PLAT offers a promising therapeutic strategy. Inhibitors such as aminocaproic acid, Human C1-esterase inhibitor and Conestat alfa could mitigate tissue damage and inflammation while preserving systemic hemostasis. These findings highlight PLAT as a potential dual-purpose target for biomarker development and therapeutic intervention. Future studies should investigate tissue-specific regulation of PLAT and its interactions with other inflammatory pathways to refine therapeutic approaches.
As a member of the immunoglobulin superfamily, SIRPB1 (Signal Regulatory Protein Beta 1) regulates immune cell activity and inflammatory pathways,21 including the modulation of IL1RA expression via SYK phosphorylation.22 Our findings revealed a positive correlation between SIRPB1 methylation, protein levels, and SjD risk. Serum SIRPB1 protein levels were markedly elevated in SjD patients compared to controls. Three SIRPB1 inhibitors—Resveratrol, Selisistat, and Cambinol—have been identified. Importantly, our Phe-MR analysis revealed no significant adverse effects associated with SIRPB1 inhibition. Additionally, recent evidence indicates that mitochondrial double-stranded RNAs (mt-dsRNAs) is upregulated in SjD,23 while resveratrol has been shown to alleviate the mt-dsRNA-mediated immunogenic stress response24 and improves salivary dysfunction in a non-obese diabetic (NOD) mouse model of SjD.25 SIRPB1 represents a promising therapeutic target for SjD, and its inhibitor, resveratrol, holds potential for SjD treatment; however, further studies are needed to validate its efficacy.
LAIR2 (Leukocyte-associated immunoglobulin-like receptor 2) is a soluble collagen receptor that antagonizes LAIR1-mediated signaling, thereby activating pro-inflammatory pathways and enhancing immune activation.26 Elevated LAIR2 likely contributes to chronic inflammation and tissue damage by disrupting collagen-mediated immunosuppressive signals. Studies have reported elevated levels of LAIR2 in the synovial fluid of rheumatoid arthritis (RA) patients26 and in the serum of patients with autoimmune thyroid diseases.27 Additionally, high LAIR2 expression has been observed in Treg cells within head and neck squamous cell carcinoma (HNSCC) tissues.28 Our MR analyses reveal that elevated transcription, methylation, and protein expression of LAIR2 are causally associated with an increased risk of SjD. These findings are further corroborated by the observed elevation of serum LAIR2 levels in SjD patients compared to controls. Importantly, our Phe-MR analysis did not identify any significant adverse effects associated with LAIR2 inhibition, highlighting its potential safety profile as a therapeutic target. However, no direct therapeutic agents targeting LAIR2 are currently available, and its promise as a diagnostic biomarker and therapeutic target for SjD warrants further investigation.
NEU1 (neuraminidase 1) encodes a lysosomal enzyme critical for glycoprotein modification and cellular signaling regulation.29 It plays a pivotal role in inflammatory and fibrotic processes, making it a potential therapeutic target for immune dysregulation.30 MR study results show that NEU1 gene transcription and protein levels are downregulated in SjD, contributing to an increased disease risk. Serum validation further confirmed reduced NEU1 protein levels in SjD patients. The loss or dysfunction of NEU1 may lead to an imbalance in M1/M2 macrophage polarization,31 enhancing pro-inflammatory pathways, and exacerbating extracellular matrix (ECM) remodeling.32 However, inhibitors of NEU1, such as Oseltamivir, Acetylsalicylic acid, and Celecoxib, may inadvertently elevate SjD risk by further reducing NEU1 activity. Future studies should focus on developing NEU1 activators and exploring its potential as both a therapeutic target and a biomarker for disease activity in SjD.
SLC22A16 (Solute Carrier Family 22 Member 16) encodes a critical transporter involved in the uptake of organic cations and carnitine, playing essential roles in metabolism and immune regulation.33,34 Our findings reveal a positive correlation between SLC22A16 expression and SjD risk, suggesting its potential involvement in exacerbating metabolic burden and inflammatory responses in SjD. Hypomethylation of the SLC22A16 locus further supports its overexpression, highlighting the importance of epigenetic regulation in SjD pathogenesis. From the drug screening analysis, two substrates of SLC22A16—levocarnitine and doxorubicin—were identified. Levocarnitine, as a carnitine supplement, may mitigate metabolic imbalance and inflammation,35 offering a potential therapeutic strategy for SjD. Conversely, caution is warranted with doxorubicin, a chemotherapeutic agent, as its interaction with SLC22A16 may exacerbate metabolic stress in SjD patients.
Our study revealed a positive correlation between RAD52 expression and SjD risk, with elevated serum protein levels in SjD patients. Additionally, hypomethylation at the RAD52 locus suggests epigenetic regulation contributes to its overexpression in SjD. RAD52 (RAD52 Homolog, DNA Repair Protein) encodes a key protein involved in DNA double-strand break repair through homologous recombination.36 Overexpression of RAD52 may promote aberrant DNA repair, leading to persistent genomic instability.37 This could trigger immune activation by exposing nuclear antigens, such as damaged DNA, which are recognized by the immune system as danger signals.38 Three RAD52-targeting drugs—Imatinib, Dasatinib, and Nilotinib—were identified. These inhibitors, known for targeting DNA repair pathways, may reduce RAD52 overactivity, minimizing autoantigen release and immune overactivation.39 Dasatinib, with its additional immunomodulatory properties, presents a promising dual-action therapeutic approach for SjD. However, balancing efficacy and safety in inhibiting RAD52 requires further investigation to prevent adverse effects on normal DNA repair mechanisms.
PSPH (Phosphoserine Phosphatase) is a crucial enzyme in the serine biosynthesis pathway, catalyzing the conversion of phosphoserine to serine.40 Our findings revealed a negative correlation between PSPH expression, methylation and SjD risk, supported by reduced serum PSPH protein levels in SjD patients. Dysregulation of PSPH may impair glutathione synthesis, exacerbating oxidative stress and chronic inflammation.41 PSPH-binding compounds like zinc chloride and zinc sulfate, known to modulate oxidative stress and immune responses, present promising avenues for intervention. Future studies should validate PSPH’s role as a biomarker and its therapeutic targeting in SjD.
CDH23 (Cadherin Related 23), a calcium-dependent adhesion protein, plays a key role in maintaining epithelial integrity and mediating intercellular signaling.42 Our study identified a positive correlation between CDH23 expression, methylation, and SjD risk, along with elevated serum CDH23 levels in SjD patients. There are currently no direct therapies targeting CDH23. However, CDH23 holds potential as a diagnostic biomarker or therapeutic target for SjD, meriting further research to clarify its mechanisms and translational significance.
PLAU (urokinase-type plasminogen activator, uPA) and PLAT are central regulators of the fibrinolytic system, facilitating plasmin generation to maintain homeostasis and modulate immune responses.43 By activating plasmin, PLAU regulates immune cell migration and activity, with elevated levels linked to tissue damage and fibrosis.44,45 Our study found that both PLAU transcription and protein levels are elevated in SjD, increasing the disease risk. However, serum PLAU levels were paradoxically lower in SjD patients than in controls, potentially reflecting systemic compensatory mechanisms or localized overexpression in inflamed tissues. Amiloride, a uPA receptor inhibitor, has demonstrated efficacy in reducing PLAU activity in other inflammatory diseases,46 but its therapeutic potential in SjD remains unexplored. Further studies are needed to clarify PLAU’s tissue-specific regulation and evaluate targeted therapies.
BRD2 (Bromodomain-Containing 2) is an epigenetic regulator that modulates transcription through interactions with acetylated histones, influencing immune differentiation and cytokine production.47,48 In our study, MR analysis demonstrated a negative correlation between BRD2 expression and protein levels with SjD risk, suggesting a systemic protective role in immune regulation. Elevated BRD2 expression may suppress pro-inflammatory transcription, mitigating immune overactivation.49 However, ELISA validation revealed increased serum BRD2 protein levels in SjD patients, indicating potential tissue-specific dysregulation or compensatory mechanisms. These contrasting findings highlight BRD2’s dual role in systemic protection and localized inflammation. The identification of BRD2-targeting compounds, such as Isoniazid and Cisplatin, offers actionable therapeutic strategies. As inhibitors, these agents may suppress BRD2’s transcriptional activity, potentially reducing pro-inflammatory gene expression in SjD. While these drugs are not yet used in autoimmune diseases, their repurposing warrants investigation, especially in combination with targeted delivery strategies to minimize systemic effects. Future research should clarify BRD2’s tissue-specific functions and evaluate its therapeutic potential in SjD.
SLC23A2 (Solute Carrier Family 23 Member 2), a sodium-dependent vitamin C transporter, plays a key role in regulating oxidative stress and immune homeostasis.50 In our study, SLC23A2 expression was inversely correlated with SjD risk, suggesting its protective role in reducing oxidative stress and tissue damage. Conversely, increased methylation levels of the SLC23A2 locus may suppress its expression, potentially exacerbating oxidative damage and inflammation in SjD. Ascorbic acid, commonly known as vitamin C, is the primary substrate of SLC23A2 and possesses significant antioxidant and immunoregulatory properties. Increasing vitamin C intake may restore antioxidant defenses, reduce oxidative stress, and improve immune balance, offering a promising approach for alleviating SjD symptoms.51,52 However, elevated levels of SLC23A2 were found in the serum of SjD patients, contradicting the MR results. Therefore, any suggestion of vitamin C as a therapeutic strategy for SjD should be viewed with caution and warrants further validation in functional and clinical studies before consideration as a viable treatment option.
Although our results identified a causal relationship between TGFBI, PAM, and SjD in mQTL and pQTL datasets, this was not supported by ELISA analysis of serum samples from SjD patients. TGFBI (Transforming Growth Factor Beta-Induced) is associated with corneal dystrophies,53 and its gene expression is significantly downregulated in SjD patients,54 a result that contradicts our findings. PAM (Peptidylglycine Alpha-Amidating Monooxygenase) is a key enzyme primarily involved in neuropeptide modification and the regulation of immune cell signaling, influencing neuro-immune interactions.55 Further studies are required to validate TGFBI and PAM as therapeutic targets for SjD.
This study has certain limitations. Firstly, although it utilizes GWAS data from FinnGen and UK Biobank, it may not fully capture the genetic and epigenetic diversity of the general population. This restricts the generalizability of the findings to all SjD patients, particularly those from non-European backgrounds. Secondly, the cross-sectional nature of the genetic and epigenetic datasets may not adequately reflect the dynamic changes in gene expression and methylation status that occur over the course of chronic SjD progression. Thirdly, while protein-level validation was performed in clinical serum samples, the relatively small sample size (n=16 per group) limits statistical power, and discrepancies observed between MR predictions and ELISA results for certain genes may reflect post-transcriptional regulation, tissue-specific versus systemic expression differences, or technical variability. Moreover, although the study included clinical validation, no longitudinal or functional follow-up experiments were performed. Future studies should therefore incorporate larger and more diverse cohorts, longitudinal profiling, and mechanistic experiments in cellular and animal models to elucidate the direct biological consequences of the identified genetic and epigenetic modifications, thereby enhancing the robustness and translational relevance of the findings.
Conclusion
In summary, our study identified 14 druggable genes with causal evidence for SjD through genome-wide MR and Bayesian colocalization analyses. Among these, eight prioritized genes (PLAT, SIRPB1, LAIR2, NEU1, SLC22A16, RAD52, PSPH, and CDH23) demonstrated consistent support across multi-omics datasets and clinical validation, making them the most promising therapeutic candidates. Several of these genes are linked to existing pharmacological compounds, such as Aminocaproic acid, Resveratrol, Levocarnitine, Imatinib, and Zinc chloride, which may offer opportunities for drug repurposing. Nevertheless, discrepancies observed for certain genes highlight the need for cautious interpretation and underscore the importance of further mechanistic investigation. While MR and integrative omics provide valuable causal insights, the therapeutic relevance of these targets must be validated through studies in disease-relevant tissues, functional assays in cellular and animal models, and rigorously designed clinical trials. Collectively, our findings lay the groundwork for precision medicine strategies in SjD and open new directions for the development of targeted therapies.
Data Sharing Statement
SjD GWAS summary were obtained from GWAS Catalog: https://storage.googleapis.com/finngen-public-data-r9/summary_stats/finngen_R9_M13_SJOGREN.gz. Druggable genome were obtained from DGIdb 4.0: https://www.dgidb.org/downloads, and Finan C, et al 2017: Finan C, et al. PMID: 28356508.
Ethics Approval and Consent to Participate
This study received ethical approval from the Institutional Review Board (IRB) of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (Approval No. 2022-175-KY-01). All participants provided written informed consent.
Acknowledgments
The authors would like to thank the UK Biobank for providing publicly accessible GWAS summary statistics for this analysis. Special thanks to China Academy of Chinese Medical Sciences for their support. We have published a poster abstract that was presented at EULAR 2025: European Congress of Rheumatology (https://www.sciencedirect.com/science/article/abs/pii/S0003496725037288).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received funding from the High Level Chinese Medical Hospital Promotion Project (No. HLCMHPP2023002), the National Natural Science Foundation of China (82374285), the Scientific and technological innovation project of China Academy of Chinese Medical Sciences (no. CI2021A01502, no. CI2021A01510).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Beydon M, McCoy S, Nguyen Y, Sumida T, Mariette X, Seror R. Epidemiology of Sjögren syndrome. Nat Rev Rheumatol. 2024;20(3):158–169. doi:10.1038/s41584-023-01057-6
2. Mariette X, Criswell LA. Primary Sjögren’s syndrome. N Engl J Med. 2018;378(10):931–939. doi:10.1056/NEJMcp1702514
3. Seror R, Nocturne G, Mariette X. Current and future therapies for primary Sjögren syndrome. Nat Rev Rheumatol. 2021;17(8):475–486. doi:10.1038/s41584-021-00634-x
4. Baldini C, Fulvio G, La Rocca G, Ferro F. Update on the pathophysiology and treatment of primary Sjögren syndrome. Nat Rev Rheumatol. 2024;20(8):473–491. doi:10.1038/s41584-024-01135-3
5. Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18(1):83. doi:10.1186/s13059-017-1215-1
6. Schadt EE, Björkegren JLM. NEW: network-enabled wisdom in biology, medicine, and health care. Sci Transl Med. 2012;4(115):115rv1. doi:10.1126/scitranslmed.3002132
7. Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16(4):197–212. doi:10.1038/nrg3891
8. Fairfax BP, Humburg P, Makino S, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343(6175):1246949. doi:10.1126/science.1246949
9. Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383):eaag1166. doi:10.1126/scitranslmed.aag1166
10. Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16(1):19–34. doi:10.1038/nrd.2016.230
11. Thorlacius GE, Björk A, Wahren-Herlenius M. Genetics and epigenetics of primary Sjögren syndrome: implications for future therapies. Nat Rev Rheumatol. 2023;19(5):288–306. doi:10.1038/s41584-023-00932-6
12. Bai Y, Wang J, Feng X, et al. Identification of drug targets for Sjögren’s syndrome: multi-omics Mendelian randomization and colocalization analyses. Front Immunol. 2024;15:1419363. doi:10.3389/fimmu.2024.1419363
13. Freshour SL, Kiwala S, Cotto KC, et al. Integration of the drug-gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021;49(D1):D1144–D1151. doi:10.1093/nar/gkaa1084
14. Shiboski CH, Shiboski SC, Seror R, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. 2017;76(1):9–16. doi:10.1136/annrheumdis-2016-210571
15. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
16. Schadt EE, Lamb J, Yang X, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37(7):710–717. doi:10.1038/ng1589
17. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
18. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
19. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi:10.1371/journal.pgen.1004383
20. Gliozzi M, Greenwell-Wild T, Jin W, et al. A link between interferon and augmented plasmin generation in exocrine gland damage in Sjögren’s syndrome. J Autoimmun. 2013;40:122–133. doi:10.1016/j.jaut.2012.09.003
21. Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. 2006;6(6):457–464. doi:10.1038/nri1859
22. Geng R, Zhao Y, Xu W, et al. SIRPB1 regulates inflammatory factor expression in the glioma microenvironment via SYK: functional and bioinformatics insights. J Transl Med. 2024;22(1):338. doi:10.1186/s12967-024-05149-z
23. Yoon J, Lee M, Ali AA, et al. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjӧgren’s syndrome. Mol Ther Nucleic Acids. 2022;30:257–269. doi:10.1016/j.omtn.2022.09.020
24. Yoon J, Ku D, Lee M, Lee N, Im SG, Kim Y. Resveratrol attenuates the mitochondrial RNA-mediated cellular response to immunogenic stress. Int J Mol Sci. 2023;24(8):7403. doi:10.3390/ijms24087403
25. Inoue H, Kishimoto A, Ushikoshi-Nakayama R, et al. Resveratrol improves salivary dysfunction in a non-obese diabetic (NOD) mouse model of Sjögren’s syndrome. J Clin Biochem Nutr. 2016;59(2):107–112. doi:10.3164/jcbn.16-31
26. Lebbink RJ, van den Berg MCW, de Ruiter T, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol. 2008;180(3):1662–1669. doi:10.4049/jimmunol.180.3.1662
27. Simone R, Pesce G, Antola P, Merlo DF, Bagnasco M, Saverino D. Serum LAIR-2 is increased in autoimmune thyroid diseases. PLoS One. 2013;8(5):e63282. doi:10.1371/journal.pone.0063282
28. Choi JH, Lee BS, Jang JY, et al. Single-cell transcriptome profiling of the stepwise progression of head and neck cancer. Nat Commun. 2023;14(1):1055. doi:10.1038/s41467-023-36691-x
29. Mei S, Li D, Wang A, et al. The role of sialidase Neu1 in respiratory diseases. Respir Res. 2024;25(1):134. doi:10.1186/s12931-024-02763-9
30. Gorelik A, Illes K, Mazhab-Jafari MT, Nagar B. Structure of the immunoregulatory sialidase NEU1. Sci Adv. 2023;9(20):eadf8169. doi:10.1126/sciadv.adf8169
31. Escalona E, Olate-Briones A, Albornoz-Muñoz S, et al. Neu1 deficiency and fibrotic lymph node microenvironment lead to imbalance in M1/M2 macrophage polarization. Front Immunol. 2024;15:1462853. doi:10.3389/fimmu.2024.1462853
32. Lillehoj EP, Luzina IG, Atamas SP. Mammalian neuraminidases in immune-mediated diseases: mucins and beyond. Front Immunol. 2022;13:883079. doi:10.3389/fimmu.2022.883079
33. Nigam SK. The SLC22 transporter family: a paradigm for the impact of drug transporters on metabolic pathways, signaling, and disease. Annu Rev Pharmacol Toxicol. 2018;58:663–687. doi:10.1146/annurev-pharmtox-010617-052713
34. Engelhart DC, Granados JC, Shi D, et al. Systems biology analysis reveals eight SLC22 transporter subgroups, including OATs, OCTs, and OCTNs. IJMS. 2020;21(5):1791. doi:10.3390/ijms21051791
35. Juraszek B, Nałęcz KA. SLC22A5 (OCTN2) Carnitine transporter—indispensable for cell metabolism, a Jekyll and Hyde of human cancer. Molecules. 2019;25(1):14. doi:10.3390/molecules25010014
36. Yasuhara T, Kato R, Hagiwara Y, et al. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell. 2018;175(2):558–570.e11. doi:10.1016/j.cell.2018.08.056
37. Souliotis VL, Vlachogiannis NI, Pappa M, Argyriou A, Ntouros PA, Sfikakis PP. DNA damage response and oxidative stress in systemic autoimmunity. IJMS. 2019;21(1):55. doi:10.3390/ijms21010055
38. Xu Y, Zhou H, Post G, Zan H, Casali P. Rad52 mediates class-switch DNA recombination to IgD. Nat Commun. 2022;13(1):980. doi:10.1038/s41467-022-28576-2
39. Li P, Xu Y, Zhang Q, et al. Evaluating the role of RAD52 and its interactors as novel potential molecular targets for hepatocellular carcinoma. Cancer Cell Int. 2019;19:279. doi:10.1186/s12935-019-0996-6
40. Liao L, Ge M, Zhan Q, et al. PSPH mediates the metastasis and proliferation of non-small cell lung cancer through MAPK signaling pathways. Int J Biol Sci. 2019;15(1):183–194. doi:10.7150/ijbs.29203
41. Sun L, Song L, Wan Q, et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015;25(4):429–444. doi:10.1038/cr.2015.33
42. Jaiganesh A, Narui Y, Araya-Secchi R, Sotomayor M. Beyond cell-cell adhesion: sensational cadherins for hearing and balance. Cold Spring Harb Perspect Biol. 2018;10(9):a029280. doi:10.1101/cshperspect.a029280
43. Napolitano F, Montuori N. Role of plasminogen activation system in platelet pathophysiology: emerging concepts for translational applications. Int J Mol Sci. 2022;23(11):6065. doi:10.3390/ijms23116065
44. Kanno Y. The uPA/uPAR system orchestrates the inflammatory response, vascular homeostasis, and immune system in fibrosis progression. Int J Mol Sci. 2023;24(2):1796. doi:10.3390/ijms24021796
45. Zou ML, Teng YY, Chen ZH, et al. The uPA system differentially alters fibroblast fate and profibrotic ability in skin fibrosis. Front Immunol. 2022;13:845956. doi:10.3389/fimmu.2022.845956
46. Lin H, Xu L, Yu S, Hong W, Huang M, Xu P. Therapeutics targeting the fibrinolytic system. Exp Mol Med. 2020;52(3):367–379. doi:10.1038/s12276-020-0397-x
47. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–356. doi:10.1038/nrd4286
48. Wang N, Wu R, Tang D, Kang R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021;6(1):23. doi:10.1038/s41392-020-00384-4
49. Cribbs AP, Filippakopoulos P, Philpott M, et al. Dissecting the role of BET bromodomain proteins BRD2 and BRD4 in human NK cell function. Front Immunol. 2021;12:626255. doi:10.3389/fimmu.2021.626255
50. Toyoda Y, Miyata H, Shigesawa R, Matsuo H, Suzuki H, Takada T. SVCT2/SLC23A2 is a sodium-dependent urate transporter: functional properties and practical application. J Biol Chem. 2023;299(8):104976. doi:10.1016/j.jbc.2023.104976
51. Machowicz A, Hall I, de Pablo P, et al. Mediterranean diet and risk of Sjögren’s syndrome. Clin Exp Rheumatol. 2020;38 Suppl 126(4):216–221.
52. Feng R, Xiao X, Wang Y, et al. Metabolic impact of low dose IL-2 therapy for primary Sjögren’s Syndrome in a double-blind, randomized clinical trial. Clin Rheumatol. 2024;43(12):3789–3798. doi:10.1007/s10067-024-07165-2
53. Han KE, Choi S, Kim T, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog Retin Eye Res. 2016;50:67–88. doi:10.1016/j.preteyeres.2015.11.002
54. de Paiva CS, Trujillo-Vargas CM, Schaefer L, Yu Z, Britton RA, Pflugfelder SC. Differentially expressed gene pathways in the conjunctiva of Sjögren syndrome keratoconjunctivitis sicca. Front Immunol. 2021;12:702755. doi:10.3389/fimmu.2021.702755
55. Merkler DJ, Hawley AJ, Eipper BA, Mains RE. Peptidylglycine α-amidating monooxygenase as a therapeutic target or biomarker for human diseases. Br J Pharmacol. 2022;179(13):3306–3324. doi:10.1111/bph.15815
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.