Back to Journals » Journal of Inflammation Research » Volume 18
Multi-Niche Microbial Profiling in Papillary Thyroid Carcinoma and Thyroid Nodules: Linking Oral, Gut, and Tissue Microbiota
Authors Chang W, Kang L, Lan T, Zhou C, Pang B ![]() , Jiang X, Zhao Z
, Jiang X, Zhao Z
Received 22 July 2025
Accepted for publication 10 October 2025
Published 17 October 2025 Volume 2025:18 Pages 14391—14404
DOI https://doi.org/10.2147/JIR.S552936
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Junhao Wang
Wang Chang,1,2,* Longfei Kang,1,3,* Tianhao Lan,1 Chuanmin Zhou,1,3 Bo Pang,1,3 Xia Jiang,1,3 Zengren Zhao1,3
1Department of Gastrointestinal Disease Diagnosis and Treatment Center, The First Hospital of Hebei Medical University, Shijiazhuang, Hebei, People’s Republic of China; 2Department of Head and Neck Surgery, Tangshan Gongren Hospital, Tangshan, Hebei, People’s Republic of China; 3Department of General Surgery, Hebei Key Laboratory of Colorectal Cancer Precision Diagnosis and Treatment, The First Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zengren Zhao, Department of Gastrointestinal Disease Diagnosis and Treatment Center, The First Hospital of Hebei Medical University, Shijiazhuang, Hebei, People’s Republic of China, Email [email protected] Bo Pang, Department of Gastrointestinal Disease Diagnosis and Treatment Center, The First Hospital of Hebei Medical University, Shijiazhuang, Hebei, People’s Republic of China, Email [email protected]
Purpose: Microbial communities have emerged as crucial regulators in the initiation and progression of thyroid cancer. However, most studies focus on single microbial sources, and the interplay between microbes across different ecological niches and their impact on thyroid carcinogenesis are largely unknown.
Patients and Methods: In this study, we collected tissue, oral, and fecal samples from 32 patients with benign thyroid nodules (BTN) and 32 patients with papillary thyroid carcinoma (PTC). The oral and fecal samples were subjected to 16S rRNA sequencing, while the tissue samples were analyzed using 5R 16S sequencing to comprehensively characterize the microbial communities.
Results: Clustering analysis using the Dirichlet Multinomial Mixture model with Laplace approximation identified two distinct oral microbial community types (O_1 and O_2) and three fecal types (F_1, F_2, and F_3). Microbial diversity patterns in thyroid tissues mirrored those observed in oral and fecal samples, suggesting potential microbial translocation or systemic interactions. Inflammatory markers were significantly elevated in PTC patients relative to BTN controls. Notably, the genus Veillonella, a potential anti-tumor biomarker, was significantly reduced in PTC samples across niches.
Conclusion: This study highlights the pivotal role of oral and intestinal microbiota in PTC development, emphasizing the interplay between microbial composition, inflammatory processes, and immune regulation in tumor progression. The discovery of Veillonella as a potential anti-tumor microbe, along with evidence of microbial translocation, opens new possibilities for targeted therapeutic strategies.
Keywords: papillary thyroid carcinoma, multi-niche microbiota, 16S rRNA sequencing, inflammatory factors, Veillonella
Introduction
Dysfunction of the thyroid gland, the largest endocrine gland in the human body, not only leads to hyperthyroidism or hypothyroidism but is also closely associated with various autoimmune diseases (such as Hashimoto’s thyroiditis and Graves’ disease) and thyroid cancer.1–4 The pathogenesis of thyroid cancer is highly complex, involving genetics, radiation, thyroid hormone (TH) imbalance, and trace element dysregulation.5,6 In particular, TH dysfunction may indirectly affect the function of other organs.7 The incidence of thyroid cancer, predominantly papillary thyroid carcinoma (PTC), has dramatically increased, making it one of the fastest-growing cancer types worldwide.8 According to the World Cancer Research Fund, approximately 821,214 new global thyroid cancer cases were diagnosed in 2022, accounting for 9.1% of all new cancer diagnoses that year. The average age at diagnosis is 45 years, with women at higher risk of differentiated thyroid cancer than men.9 The incidence of thyroid cancer in women is 10.2 per 100,000, three times that in men, and has continued to rise in many countries since the 1980s.10 The subtle and nonspecific clinical presentation of PTC often hinders its early diagnosis, leading to a delay in treatment initiation and potentially affecting patient outcomes.11 Although fine-needle aspiration biopsy (FNAB) is the primary method for distinguishing between benign and malignant nodules in clinical practice, up to 30% of thyroid FNAB results remain indecisive.12 Consequently, there is an urgent need for the discovery and validation of innovative biomarkers that can aid in the early and accurate diagnosis of PTC.
The human microbiome, the collection of microorganisms that inhabit various body sites, has emerged as a significant factor in health and disease, including cancer.13 The microbiota is known to influence host immunity, metabolism, and even genetic expression, suggesting its potential role in cancer development.14 Recent studies indicate an association between alterations in gut, oral, or intratumoral microbiota and the pathogenesis of various malignancies.15,16 The gut microbiome has been implicated in the development of colorectal cancer, with certain microbial signatures proposed as early diagnostic indicators.17,18 Oral microbiota has been linked to oral squamous cell carcinoma and head and neck squamous cell cancer.19,20 Guo et al analyzed the microbiota across three subtypes of pancreatic cancer. They found that the basal-like subtype not only contains a distinct microbial community but also demonstrates significantly higher abundances of Acinetobacter, Pseudomonas, and Sphingomonas.21 There is a growing body of evidence suggesting that the microbiota may play a role in thyroid cancer development. Yuan et al demonstrated that Pseudomonas was the dominant genus in PTC, followed by Rhodococcus, Ralstonia, Acinetobacter, and Sphingomonas.22 Patients with differentiated thyroid carcinoma (DTC) exhibit greater gut microbial species richness and diversity than healthy individuals. LEfSe analysis further identified that f_Oscillospiraceae, g_Subdoligranulum, and p_Actinobacteriota are significantly enriched in patients with DTC.23 Beyond the gut, the oral microbiota composition is also altered in patients with thyroid cancer. Compared to healthy controls, their oral microbiome exhibits a distinct compositional shift.24 However, most studies have focused on a single niche within the body, and a comprehensive analysis of microbiota from different sources remains limited, especially for PTC and benign thyroid nodules (BTN).
In the present study, we investigate whether unique microbial community signatures exist prior to the clinical diagnosis of PTC. To achieve this objective, we employed 16S rRNA gene sequencing analysis to comparatively assess the microbiota in tissue, fecal, and oropharyngeal swab samples from patients with PTC and those with BTN. We focused on the enrichment of PTC-associated species within thyroid tumor tissues, and conducted an in-depth exploration of their potential associations with the oral and gut microbiomes. The findings of this study are significant because they highlight potential biomarkers for the early detection of thyroid cancer and contribute to unraveling the role of microbial communities in the pathogenesis of thyroid carcinoma.
Materials and Methods
Participants
We recruited 64 patients at the Tangshan Gongren Hospital, of whom 32 had PTC and 32 had BTN. All participants provided written informed consent, and the Tangshan Gongren Hospital’s Ethical Committee approved the study. We conducted the study in accordance with the appropriate ethical guidelines. Clinical data, including age, sex, body mass index, and relevant medical history, were collected using structured questionnaires.
Inclusion and Exclusion Criteria
Participants were enrolled according to the following inclusion and exclusion criteria. Inclusion criteria: 1) age ≥18; 2) Patients who underwent surgery at our hospital and met the criteria of the latest “Guidelines for the Diagnosis and Treatment of Thyroid Nodules and Differentiated Thyroid Cancer (2023, Second Edition); and 3) Patients who had not been administered antibiotics within the prior three months.
Exclusion criteria: 1) Pregnant and lactating women; 2) Patients with multiple primary malignancies, previous chemotherapy for metastatic disease, or unknown radiotherapy history, multicentricity status, or extrathyroidal extension status; and 3) Patients with oral diseases, including Sjögren’s syndrome, Mikulicz’s syndrome, atrophic glossitis, oral leukoplakia, or oral fungal infections.
Sample Collection
Oropharyngeal swabs and matching fecal samples were collected for 16S rRNA gene sequencing. Participants were instructed to rinse their mouths before collecting oropharyngeal swab samples during non-meal times, and they used sterile cotton swabs to obtain lingual surface samples. Fecal samples were collected in the morning after an overnight fast of more than eight hours. All samples were collected in sterile tubes and stored at −80°C before analysis.
Thyroid nodule tissues and PTC tissues were collected for an in-depth analysis using 5R 16S rRNA gene sequencing. All samples were obtained during surgery and immediately frozen in liquid nitrogen before being stored at −80°C. During the sample collection process, we strictly adhered to aseptic operation procedures. We set up a blank control group to ensure the accuracy of the experimental results and minimize the risk of cross-contamination.
Peripheral venous blood was collected for the analysis of thyroid function and hematological parameters. Participants were instructed to fast and refrain from water intake after 8:00 PM on the day prior to blood collection. Peripheral venous blood samples were drawn the following morning and subsequently subjected to laboratory analysis. Serum samples were collected for the assessment of thyroid function. We collected 5 mL of blood in serum-separating tubes. The blood was spun at 2500 × g for 10 minutes. Samples were then frozen at −80°C for enzyme-linked immunosorbent assay.
DNA Extraction
DNA was extracted from oral and fecal samples using the Hi-Swab DNA Kit (DP362, TIANGEN, Inc., China) and the TIANamp Stool DNA Kit (DP328, TIANGEN, Inc., China), respectively, according to the manufacturer’s instructions. DNA was extracted from frozen tissue samples using the cetyltrimethylammonium bromide method. All negative controls were processed using the same protocols as the tissue samples. The total DNA was eluted in 50 μL of elution buffer and stored at −80°C prior to analysis. The quality and concentration of the DNA were assessed using a 2% agarose gel before amplification.
16S rRNA and 5R 16S rRNA Gene Sequencing
For oral and gut DNA, the 16S rRNA gene V3–V4 region was amplified with conserved primers. The polymerase chain reaction (PCR) program commenced with initial denaturation at 98°C for 30 seconds, followed by 35 cycles of denaturation at 98°C for 10 seconds, annealing at 54°C for 30 seconds, and extension at 72°C for 45 seconds; and a final extension at 72°C for 10 minutes was then performed. For the intratumor microbiota, variable regions V2, V3, V5, V6, and V8 of the 16S rRNA gene were amplified in a multiplex reaction with specific primers. In the first round of PCR, amplification was performed using region-specific forward and reverse primers. The amplification protocol was as follows: an initial denaturation step at 98°C for 30 seconds, followed by 30 cycles of denaturation at 98°C for 10 seconds, annealing at 62°C for 15 seconds, and extension at 72°C for 35 seconds, concluding with a final extension at 72°C for 10 minutes. The product of the first PCR round was diluted 10-fold and utilized as the template for the second PCR round, employing region-specific forward primers and a common specific reverse primer. The specific amplification protocol for the second round was as follows: 98°C for 30 seconds; 35 cycles of 98°C for 10 seconds, 64°C for 15 seconds, and 72°C for 25 seconds; and 72°C for 5 minutes. The PCR products were purified by AMPure XT beads (Beckman Coulter Genomics, Danvers, MA, USA) and quantified using Qubit (Invitrogen, USA). Subsequently, we constructed the amplicon library for sequencing. We assessed its size and quantity using the Agilent 2100 Bioanalyzer (Agilent, USA) and the Library Quantification Kit for Illumina (Kapa Biosciences, Woburn, MA, USA). Sequencing was conducted on the Illumina NovaSeq platform provided by Biotree Biotechnology Co., Ltd (Shanghai, China). All the primer sequences are presented in Supplementary Table 1.
Statistical Analysis
The data analysis for both 16S and 5R 16S sequencing began with quality control to ensure the production of high-quality reads. For 16S, paired-end reads were assembled according to distinct barcodes, with barcodes and primer sequences removed beforehand, followed by merging using fast length adjustment of short reads. Quality filtering of the raw reads was performed with fqtrim (v0.94) under defined conditions to yield high-quality clean tags. Chimeric sequences were eliminated using the Vsearch software (v2.3.4), and amplicon sequence variants (ASVs) were generated by denoising the trimmed reads via Divisive Amplicon Denoising Algorithm 2 (DADA2). These ASVs were then taxonomically classified using the naive Bayes consensus taxonomy classifier in Quantitative Insights Into Microbial Ecology 2 (QIIME2) alongside the SILVA 16S rRNA database (v. 138). Sequence alignment was accomplished using Blast, while the SILVA database provided annotations for the feature sequences corresponding to each representative sequence. The QIIME2 process was employed to analyze both alpha diversity and beta diversity. For 5R 16S sequencing, we utilized the Short Multiple Regions Framework (SMURF) to integrate and analyze sequences from five amplified regions for bacterial taxonomic identification. Briefly, the algorithm applies an expectation-maximization framework to integrate short reads derived from multiple hypervariable regions, reconstructing the most probable full-length 16S rRNA sequence for each ASV. The reconstructed sequences were subsequently used for taxonomic assignments and calculation of relative abundance, yielding an initial community profile. To mitigate the influence of low-abundance noise, all samples were rarefied to the same sequencing depth, and samples with less than 1000 high-quality reads (including negative controls) were excluded. ASVs with relative abundance consistently below 1 × 10–4 across the entire data set were likewise discarded. We performed a presence-absence analysis of negative controls to identify potential contaminants: genera detected in ≥ 50% of the negative-control samples were classified as putative contaminants and removed from all experimental samples. All downstream analyses of diversity, as well as differential abundance, were conducted on the resulting decontaminated data set.
Results
Oral and Fecal Microbiome Dynamics
A total of 4,738,596 high-quality reads, with Q30 values greater than 92.56% and an average GC content of 52.29%, were obtained from the 16S rRNA gene sequences of oral specimens (Supplementary Table 2). These reads were processed into 5600 ASVs using DADA2, of which 1596 were shared between the two groups, 2123 were unique to PTC, and 1881 were unique to BTN (Supplementary Figure 1A), representing 35 phyla and 653 genera. Similarly, from stool samples, 4,234,287 high-quality reads, with Q30 values greater than 92.58% and an average GC content of 53.76%, were obtained (Supplementary Table 2). These reads were processed into 7750 ASVs, with 2451 shared between the groups, 2425 unique to PTC, and 2874 unique to BTN (Supplementary Figure 1B), representing 36 phyla and 709 genera.
We used the Dirichlet Multinomial Mixture (DMM) model with the lowest Laplace approximation for clustering analysis, identifying two distinct oral microbial community types (O_1 and O_2) and three fecal microbial community types (F_1, F_2, and F_3) (Figure 1A). Enrichment analysis revealed that fecal samples from patients with PTC (F_ZL) were significantly enriched in the F_2 cluster, while oral samples from BTN patients (O_LX) showed significant enrichment in the O_1 cluster. In contrast, oral samples from PTC patients (O_ZL) were significantly enriched in the O_2 cluster (Figure 1B). Nonmetric multidimensional scaling (NMDS) based on Bray–Curtis distance further confirmed significant differences between oral and fecal microbial communities, highlighting the distinct differences in microbial composition between the groups (Figure 1C). Alpha diversity in the F_1 and F_2 clusters was significantly lower than in the F_3 cluster, while the O_1 cluster showed a significantly lower alpha diversity than the O_2 cluster (Figure 1D and Supplementary Table 3). In contrast, the F_2 cluster exhibited the highest evenness, which was significantly higher than that of the F_1 and F_3 clusters, and the evenness in the O_1 cluster was significantly higher than that in the O_2 cluster (Figure 1E and Supplementary Table 3). No significant differences in richness and evenness were observed between the fecal and oral samples from patients with benign lesions and patients with cancers within each cluster, indicating the homogeneity of samples within each cluster (Supplementary Figure 2A and B). The beta diversity of the oral and gut microbiota was significantly different, with notable variations between the clusters within each microbiota (Figure 1F and Supplementary Table 4). The above results indicate that the occurrence of thyroid cancer alters the composition of gut and oral microbial communities.
 |
Figure 1 DMM clustering of 16S rRNA gene sequencing data for oral and gut microbiota. (A) Heatmap showing the relative abundance of the 30 most dominant bacterial genera per DMM cluster. (B) The number of patients with benign nodules or PTC corresponding to each cluster. (C) Nonmetric multidimensional scaling (NMDS) visualization of DMM clusters using Bray–Curtis distance of oral and gut bacterial genera. (D) The α diversity (richness) per DMM cluster. (E) The α diversity per DMM cluster. (F) The β diversity per DMM cluster. * p < 0.05, *** p < 0.001, **** p < 0.0001; ns, not significant. |
Microbiome Dynamics in BTN and PTC Tissues
For tissue samples, 11,939,357 high-quality reads, with Q30 values exceeding 94.52% and an average GC content of 56.11%, were obtained from the 5R 16S rRNA gene sequences (Supplementary Table 5). The reads represent 14 phyla and 302 genera. The tissue microbiota of the corresponding patients was analyzed and compared based on the clustering patterns of gut and oral microbiota. Species richness was assessed using the Menhinick and Margalef indices, two key parameters for evaluating alpha diversity, both of which normalize the observed number of ASVs against the total sequence count using different formulas. The results demonstrated that the microbiota alpha diversity of tissue was consistent with gut microbiota alpha diversity, showing no significant difference between the T_F_1 and T_F_2 clusters, both of which had lower alpha diversity than the T_F_3 cluster (Figure 2A). The alpha diversity of tissue in the T_O_1 cluster was significantly lower than that in the T_O_2 cluster when based on oral microbiota clustering (Figure 2B). No significant differences in alpha diversity were observed between patients with BTN and those with PTC within the corresponding tissue subclusters, indicating homogeneity within each tissue sample cluster (Supplementary Figure 3A–D). For beta diversity, tissue microbiota exhibited significant differences among clusters based on gut microbiota clustering (Figure 2C), whereas no significant differences were observed when clustered by oral microbiota (Figure 2D). To further explore the relationship between tumor microbiota (F_ tumor), gut microbiota (F_ gut), and oral microbiota (F_ oral), nonlinear logistic regression models were constructed. The results visually demonstrate that F_ tumor aligns more closely with F_ gut than F_ oral (Figure 2E). The results of the quantitative analysis further supported this observation, with the gut Menhinick index exhibiting a significant positive association with the intratumoral microbiota of the tissue (Supplementary Table 6). In contrast, the oral Menhinick index had no significant relationship with it. These results indicate that the occurrence of thyroid cancer also alters the composition of tumor microbial communities.
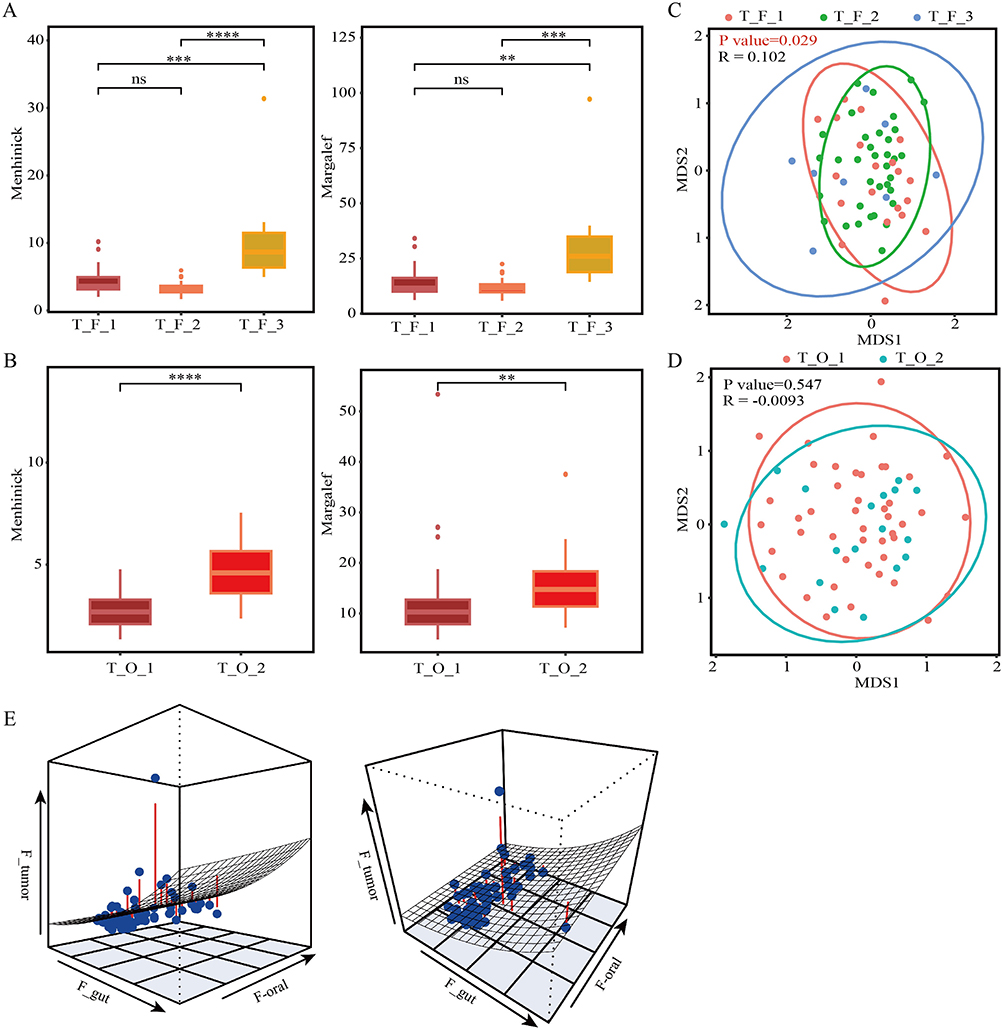 |
Figure 2 The alpha diversity and beta diversity of tissue microbiota. (A) The α diversity (richness) of tissue DMM clusters corresponding to the clustering of gut microbiota. (B) The α diversity of tissue DMM clusters corresponding to the clustering of oral microbiota. (C) The β diversity of tissue DMM clusters corresponding to the clustering of gut microbiota. (D) The β diversity of tissue DMM clusters corresponding to the clustering of oral microbiota. (E) Three-dimensional graph of the nonlinear regression model for the community structures of oral, intestinal, and tumor microbiota. **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant. |
The Correlation Between Clinical Features and Inflammatory Factors with PTC
We further analyzed the correlation between the microbiota of tissue samples and clinical indicators or inflammatory factors. In the tissue samples corresponding to oral clusters, monocytes and thymidine kinase 1 (TK1) levels were significantly higher in the T_O_2 cluster than in the T_O_1 cluster (Figure 3A). In the T_O_1 subcluster, tumor samples exhibited significantly higher levels ofTK1, midkine (MK), interleukin 6 (IL-6), monocytes, neutrophils, and white blood cells than in the benign nodule, while CXCR4, free triiodothyronine (FT3), interferon γ (IFN-γ), interleukin 17 (IL-17), and tumor necrosis factor alpha (TNF-α) levels were significantly lower in the cancer samples. In contrast, cancer samples from the T_O_2 subcluster showed significantly higher levels of interleukin 1β (IL-1β) and IL-6 compared to benign nodule samples (Figure 3B). In the tissue samples corresponding to fecal clusters, monocytes were significantly lower in the T_F_2 cluster than in the T_F_1 cluster, while leukocytes were significantly higher in the T_F_2 cluster (Figure 3C). In the T_F _1 subcluster, cancer samples showed significantly lower levels of CXCR4, FT3, IFN-γ, IL-17, and TNF-α, while IL-1β, IL-6, and MK levels were significantly higher in these samples than in benign nodule samples. Cancer samples in the T_F_2 subcluster exhibited significantly lower levels of IL-17 and TNF-α than benign nodule samples, while IL-1β, MK, monocytes, neutrophils, and TK1 levels were significantly higher. Cancer samples in the T_F_3 subcluster had significantly lower levels of CXCR4 and IL-17 than benign nodule samples (Figure 3D). These results suggested that the microbial communities may influence the nature (benign or malignant) of thyroid nodules and their biological behavior by remotely regulating the immune balance and inflammatory levels within thyroid tissue.
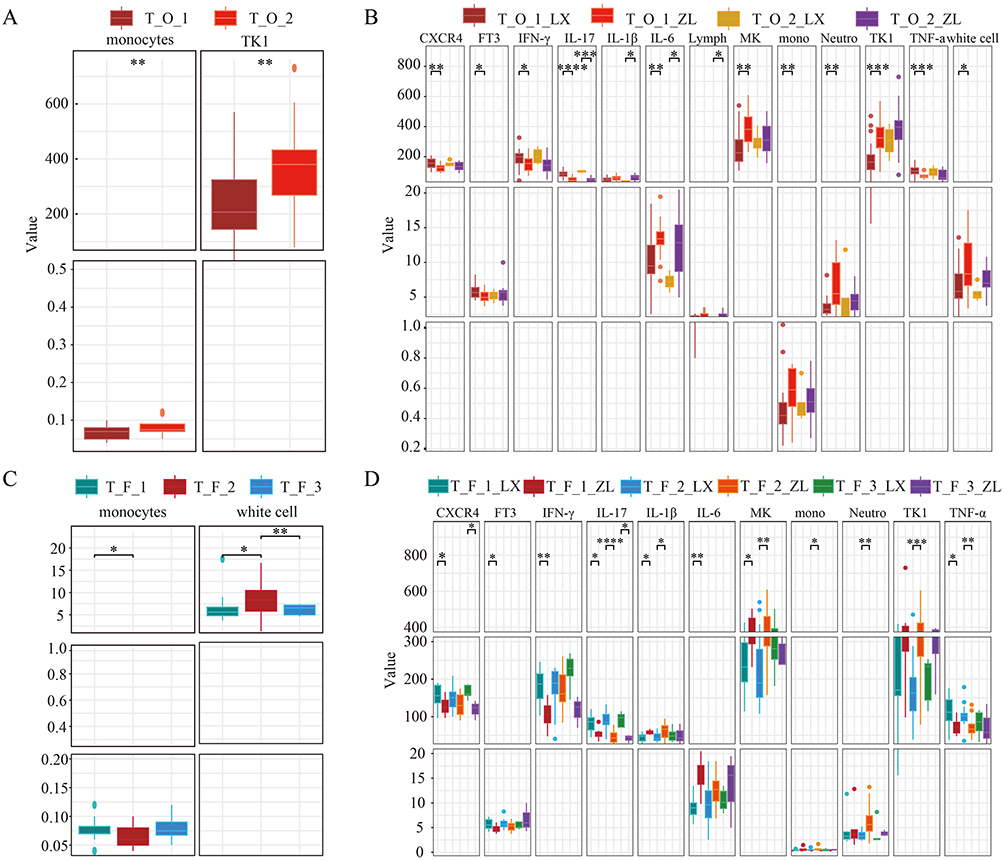 |
Figure 3 Association of inflammatory factors and clinical indicators with tissue microbiota clusters and subclusters. (A) Relationship of inflammatory factor levels and clinical indicators with tissue microbiota clusters corresponding to oral microbiota clusters. (B) Relationship of inflammatory factor levels and clinical indicators with tissue microbial subclusters corresponding to oral microbial clusters. (C) Relationship of inflammatory factor levels and clinical indicators with tissue microbiota clusters corresponding to gut microbiota clusters. (D) Relationship of inflammatory factor levels and clinical indicators with tissue microbial subclusters corresponding to gut microbial clusters. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Indicator Analysis of Microbiota in Oral, Fecal, and Tumor Samples
To confirm the relationship between variations in microbial composition in oral and gut microbiomes and the thyroid gland, community type-specific indicator taxa were identified based on the top 30 microbial genera. Among the three clusters of gut microbiota, Lactobacillus and Ligilactobacillus were identified as shared genera across all three clusters. Muribaculaceae_unclassified and Phascolarctobacterium were common to both F1 and F2. Ruminococcus_gravis was shared between F1 and F3. F1 was characterized by the exclusive presence of Escherichia-Shigella and Klebsiella; F2 uniquely harbored Akkermansia, Clostridiales_UCG-014_unclassified, Bacteroides, Firmicutes_unclassified, Faecalibacterium, stomatobaculum, and Ruminococcus _torques; and the presence of Bifidobacterium distinguished F3 as its unique genus. For oral microbiota, Alloprevotella, Veillonella, and Leptotrichia were exclusively associated with the O_1 cluster, while Gemella and Streptococcus were uniquely identified in the O_2 cluster (Figure 4A). Bifidobacterium is a typical probiotic that is beneficial for gut health and immune regulation.25–27 Escherichia-Shigella and Klebsiella bacteria include opportunistic pathogens, which could cause infections when immunity is compromised.28,29 Further analysis of the gut and oral microbiota in patients with BTN and PTC across different subclusters revealed distinct microbial signatures. Specifically, Faecalibacterium was significantly enriched in the F_3_LX subcluster, while Alloprevotella and Prevotella_7 were markedly enriched within the O_2_LX cluster in the oral samples of patients with benign nodules (Figure 4B). Studies have demonstrated that these three bacterial genera can produce short-chain fatty acids (SCFAs), which are known to exert anti-tumor effects.30–32 Next, we performed a comparative analysis of tissue samples corresponding to each cluster. Notably, Veillonella was significantly enriched in both the T_ F_3 cluster and T_F_3_LX subcluster (Figure 4C). A similar enrichment was observed in cluster T_O, where Veillonella showed marked accumulation in T_O_3_LX (Figure 4D). The presence of Veillonella in the tissue suggests communication between the microbiome through translocation.
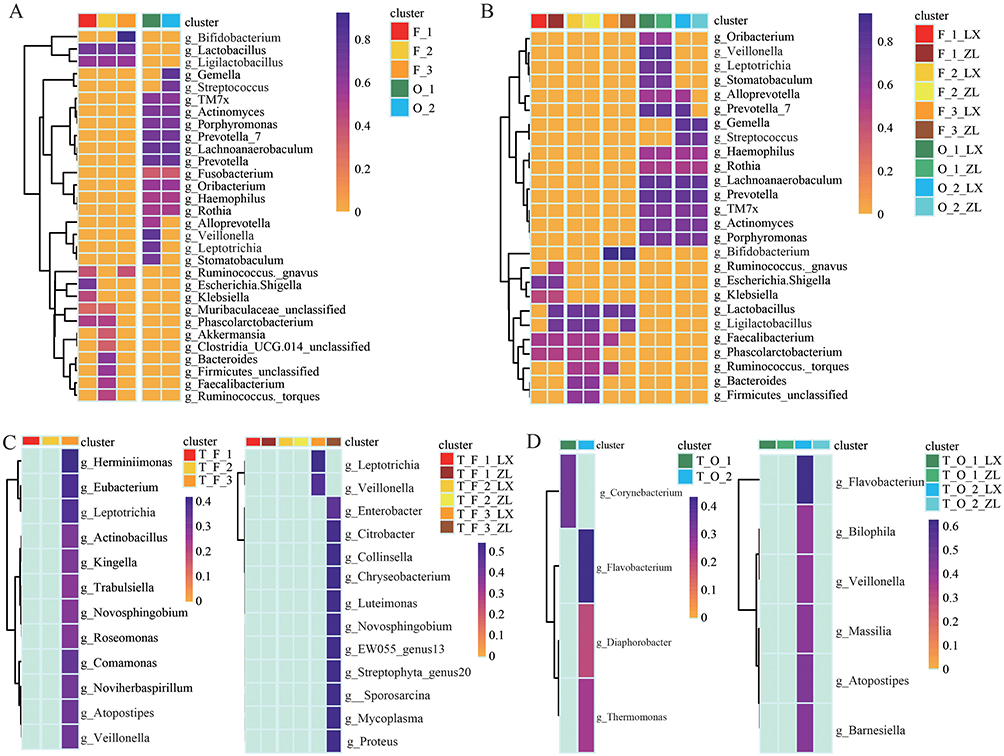 |
Figure 4 Microbial indicators across different microbial clusters and subclusters. (A) Microbial indicators of the top 30 genera in gut and oral microbial clusters. (B) Microbial indicators of the top 30 genera in gut and oral microbial subclusters. (C) Microbial indicators of tissue microbial community clusters and subclusters corresponding to gut clustering. (D) Microbial indicators of tissue microbial community clusters and subclusters corresponding to oral clustering. |
Bacteria-Bacteria Co-Occurrence Networks
To further validate the relationship between microbiota and PTC, we conducted a co-occurrence network analysis to explore the relationships between microbiota from different anatomical sites (oral, gut, and thyroid tissue) and their associations with inflammatory and immune markers. Microbiota within the same ecological niche exhibited strong co-occurrence relationships, indicating functional interactions and potential cross-talk between microbial communities. We observed significant microbial exchange between the oral, gut, and thyroid tissue, suggesting a potential role of microbial translocation in the pathogenesis of PTC (Figure 5). Specifically, within the F_3 and O_2 clusters, Veillonella demonstrated a positive correlation with monocytes, a key immune cell type involved in inflammatory responses (Figure 5). This observation may reflect a dynamic immune-microbial interaction following thyroid lesions. After the onset of a thyroid pathology, there is a marked increase in monocyte levels, likely as part of the body’s innate immune response to tissue damage and inflammation. Concurrently, Veillonella appears to migrate toward the affected thyroid tissue. This migration could represent a compensatory anti-inflammatory mechanism, where Veillonella contributes to modulating the local immune environment.
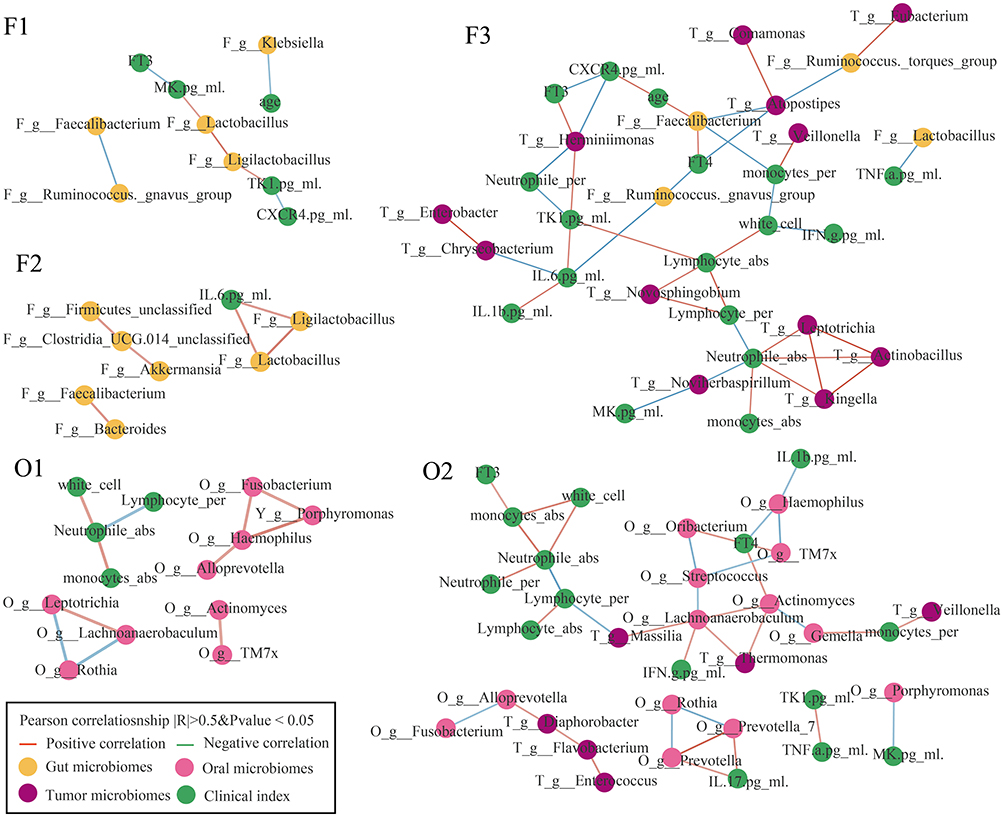 |
Figure 5 Co-occurrence network of gut, oral, and tissue microbiota, inflammatory factors, and clinical indicators. The Pearson correlation coefficient was used to compute the correlation coefficient based on their relative abundances and levels. Co-occurring pairs that met the criteria of an FDR-adjusted P-value of less than 0.05 and a correlation coefficient of greater than 0.7 were selected for display and visualized using Cytoscape version 3.8.0. |
Discussion
The present study provides novel insights into the role of oral and gut microbiota in the development and progression of PTC. By analyzing microbial communities in oral, fecal, and thyroid tissue, we identified significant differences in microbial composition and diversity between patients with BTN and those with PTC. Our findings revealed distinct microbial community types in oral and gut microbiota. There were three microbial clusters in fecal samples, while two clusters were enriched in oral samples. Furthermore, nonlinear regression models revealing that changes in the composition and function of gut microbiota. Notably, these changes exhibit a more significant association with the development of thyroid tumors. Our study revealed significant correlations between microbial communities and inflammatory markers, highlighting the interplay between microbiota, inflammation, and immune responses in PTC. Notably, Veillonella was significantly enriched in both oral and gut microbiota of patients with BTN and was negatively correlated with PTC development, suggesting its potential anti-tumor role.
The oral and gut microbiota have emerged as critical players in cancer development.33 Although the thyroid is anatomically closer to the oral cavity, our data suggest that gut microbiota may exert a more profound influence on PTC. Gut microbiota likely plays an important role in the pathogenesis of thyroid tumors by modulating the host’s systemic immune responses.34,35 Metabolites derived from gut microbiota, such as SCFAs, can influence distant organs, including the thyroid, via the circulatory system.36 Gut microbiota is involved in the activation and inactivation of thyroid hormones, and its dysregulation may directly disrupt thyroid function, thereby promoting tumorigenesis.37 In contrast, while oral microbiota may impact the thyroid through local inflammation or bacterial translocation, its systemic effects appear to be relatively weak. The oral microbiota typically exerts its effects by influencing the gut microbiota, a relationship known as the oral - gut axis. P. gingivalis is capable of inducing gut microbiota dysbiosis, which subsequently modulates insulin resistance and systemic inflammation.38 This observation suggests that the systemic effects exerted by P. gingivalis are indirect and rely on the amplifying effect of the gut microbiota.
Dysbiosis in microbial communities can influence tumorigenesis through inflammation and immune responses.39,40 The F_1_ZL and O_2_ZL subclusters exhibited elevated levels of IL-1β and IL-6, which are key mediators of chronic inflammation and tumor progression.41 In thyroid cancer, elevated IL-6 levels have been linked to tumor aggressiveness and a poor prognosis.42 Monocytes may serve as a critical link connecting microbial dysbiosis, chronic inflammation, and tumor progression. In patients with cancer, elevated levels of inflammatory cytokines and monocytes are interrelated, synergistically promoting tumor progression. Inflammatory cytokines activate monocytes, driving their differentiation into tumor-associated macrophages or myeloid-derived suppressor cells, which further secrete pro-inflammatory factors, establishing a positive feedback loop that exacerbates chronic inflammation and immune suppression.43 Microbial dysbiosis contributes to this process by activating Toll-like receptor signaling pathways and releasing metabolites, thereby promoting cytokine production and monocyte recruitment, ultimately fostering a pro-tumor microenvironment.44 In thyroid cancer, elevated IL-6 and TNF-α levels are associated with monocyte activation, supporting angiogenesis, immune evasion, and metastasis.
In our study, the enrichment of Escherichia-Shigella and Klebsiella in the F_1 cluster, both of which are opportunistic pathogens, may contribute to a pro-tumorigenic environment by promoting chronic inflammation and immune suppression.45 Hu et al reported that the abundance of Escherichia-Shigella is positively and significantly correlated with both C-reactive protein levels and peripheral white blood cell counts, suggesting that this bacterial taxon is a potential driver of host inflammatory responses.27 Specifically, Shigella dysenteriae infection enhances the activity of GSK-3β kinase, which subsequently phosphorylates β-catenin, leading to its degradation via ubiquitination. This process also upregulates IL-8 expression through the activation of NF-κB, ultimately resulting in inflammation.46 Geller et al reported that the intratumoral presence of Klebsiella in melanoma, where Klebsiella and other commensal bacteria activate Toll-like receptor 4 (TLR4) signaling to upregulate interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), thereby impairing CD8+ T-cell infiltration and effector function and culminating in resistance to immune-checkpoint blockade.47 Conversely, the presence of Faecalibacterium and Bifidobacterium, known for their anti-inflammatory and immunomodulatory properties, may protect against tumor development.48,49 Faecalibacterium exerts systemic anti-inflammatory effects by secreting butyrate, which interrupts the IL-6-driven STAT3 signaling axis and down-regulates the transcription of the pro-inflammatory cytokine IL-17, thereby attenuating chronic intestinal inflammation typified by inflammatory bowel disease.50 In experimental models of rheumatoid arthritis, enrichment of F. prausnitzii reduces the number of IL-17-producing lymphocytes, elevates luminal butyrate concentrations, and remodels the gut microbial ecosystem, collectively ameliorating systemic inflammatory manifestations.51
The presence of Veillonella in the thyroid gland suggests microbial translocation from the oral cavity and gut to the tumor microenvironment. Microbiota translocation has been increasingly recognized as a potential mechanism linking microbiota to cancer development.52 As members of the gut and oral microbiota, Veillonella utilize lactate and metabolize it into SCFAs, such as acetate and propionate.53 Previous studies have indicated that Veillonella may enhance athletic endurance by utilizing host-derived lactate and producing propionate.54 In contrast, numerous additional studies have demonstrated that this genus is closely associated with the onset and progression of diverse human diseases. Lang et al reported elevated levels of Veillonella in patients with alcoholic hepatitis.55 In vitro data further showed that Veillonella-derived lipopolysaccharide induces the secretion of IL-6, IL-1β, IL-10, and TNF-α from human peripheral blood mononuclear cells.56 In contrast, the abundance of Veillonella was significantly lower in 35 individuals with an autism spectrum disorder group compared to the typical development group.57 Notably, coculture of Streptococcus and Veillonella reduced the production of inflammatory cytokines, highlighting a potential synergistic anti-inflammatory effect between these microbial species.58 In line with these findings, we found a lower abundance of Veillonella in PTC in our study. Our co-occurrence network analysis further supports the role of Veillonella in immune modulation, as it demonstrated a positive correlation with monocytes, a key immune cell type involved in inflammatory responses. This suggests that Veillonella may play a compensatory role in mitigating inflammation and tumor progression following thyroid lesions.
Limitation
Although this study has obtained preliminary findings in the field of the association between PTC and microbiota, it still has several limitations. First, this study only confirms the association between microbiota and PTC, but cannot establish a causal relationship between the two. Future longitudinal studies are needed to clarify the temporal sequence and causal directionality. Second, this study is still in the exploratory stage; the limited sample size of the single-center cohort may result in some potential associations not being fully identified, and the study conclusions require further verification by large-scale, multi-center cohort studies. Finally, this study failed to collect key data from participants, such as dietary patterns, usage of medications other than antibiotics, and periodontal health status. These unincorporated variables may act as potential confounders affecting the association between PTC and microbiota, and thus need to be supplemented in subsequent studies to reduce bias.
Conclusion
In summary, our study highlights the significant role of oral and gut microbiota in PTC pathogenesis, emphasizing the importance of microbial community structure, inflammation, and immune responses in tumor development. The identification of Veillonella in tumors, along with the observed microbial translocation, representing a potential pathway that may influence the tumor microenvironment. Together, the identification of Veillonella and the observed microbial translocation provide promising avenues for potential therapeutic interventions in PTC and lay a solid foundation for translating these observations into clinical applications.
Data Sharing Statement
The full dataset supporting the findings of this study, including raw sequencing data and processed bioinformatics tables such as the ASV abundance table and taxonomic composition summaries at the phylum and genus levels, can be obtained from the corresponding author (Zengren Zhao) upon reasonable request.
Ethics Statement
This study was approved by the Ethics Committee of Tangshan Gongren Hospital (Approval Number: 2024-KJ-01). All participants were informed and signed the written informed consent. This research was conducted in accordance with guidelines outlined in the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (U23A20432, 82203623 and 82302523); the Natural Science Foundation of Hebei (H2025206752, H2023206290 and H2024206117); Hebei Provincial Department of Human Resources and Social Security (C20230353, A20240020 and C2024016); Hebei Municipal Government Funded the Training of Clinical Medical Talents (ZF2025063, ZF2025042 and ZF2025038); PI Team Supporting Fund of the First Hospital of Hebei Medical University (03000191); National Key R&D Program of China (2024YFA1307100, 2024YFA1307102 and 2024YFA1307103); Key Research and Development Project of Universities in Hebei Province (241460083A).
Disclosure
The authors declare that they have no competing interest.
References
1. Zhi J, Yi J, Tian M, et al. Immune gene signature delineates a subclass of thyroid cancer with unfavorable clinical outcomes. Aging. 2020;12(7):5733–5750. doi:10.18632/aging.102963
2. Park M, Kim JY, Kang JM, et al. PRL-1 overexpressed placenta-derived mesenchymal stem cells suppress adipogenesis in Graves’ ophthalmopathy through SREBP2/HMGCR pathway. Stem Cell Res Ther. 2021;12(1):304. doi:10.1186/s13287-021-02337-2
3. Magnusson L, Barcenilla H, Pihl M, et al. Mass cytometry studies of patients with autoimmune endocrine diseases reveal distinct disease-specific alterations in immune cell subsets. Front Immunol. 2020;11:288. doi:10.3389/fimmu.2020.00288
4. Klecha AJ, Barreiro Arcos ML, Frick L, Genaro AM, Cremaschi G. Immune-endocrine interactions in autoimmune thyroid diseases. Neuroimmunomodulation. 2008;15(1):68–75. doi:10.1159/000135626
5. Mirkatouli NB, Hirota S, Yoshinaga S. Thyroid cancer risk after radiation exposure in adults-systematic review and meta-analysis. J Radiat Res. 2023;64(6):893–903. doi:10.1093/jrr/rrad073
6. Bryliński Ł, Kostelecka K, Woliński F, et al. Effects of trace elements on endocrine function and pathogenesis of thyroid diseases-a literature review. Nutrients. 2025;17(3). doi:10.3390/nu17030398
7. Chen C, Xie Z, Shen Y, Xia SF. The roles of thyroid and thyroid hormone in pancreas: physiology and pathology. Int J Endocrinol. 2018;2018:2861034. doi:10.1155/2018/2861034
8. Peitao Z, Haixia G, Shukai Y, et al. Targeting myeloid derived suppressor cells reverts immune suppression and sensitizes BRAF-mutant papillary thyroid cancer to MAPK inhibitors. Nat Commun. 2022. doi:10.1038/s41467-022-29000-5
9. Xue T, Chang L, Jingjing L, et al. Analysis of the relevance of the ultrasonographic features of papillary thyroid carcinoma and cervical lymph node metastasis on conventional and contrast-enhanced ultrasonography. Front Oncol. 2021;11. doi:10.3389/fonc.2021.794399
10. Chen YG, Liu HX, Hong Y, et al. PCNP is a novel regulator of proliferation, migration, and invasion in human thyroid cancer. Int J Biol Sci. 2022;18(9):3605–3620. doi:10.7150/ijbs.70394
11. Chang Z, Ji G, Huang R, et al. PIWI-interacting RNAs piR-13643 and piR-21238 are promising diagnostic biomarkers of papillary thyroid carcinoma. Aging. 2020;12(10):9292–9310. doi:10.18632/aging.103206
12. Mahmoudian-Sani MR, Amrollahi-Sharifabadi M, Taheri A, Hosseini SM, Tahmasebi K, Mobini GR. Diagnostic value of the candidate microRNAs in thyroid fine-needle aspiration biopsy (FNAB) samples. Horm Mol Biol Clin Investig. 2020;41(2). doi:10.1515/hmbci-2019-0027
13. Szamosi JC, Forbes JD, Copeland JK, et al. Assessment of inter-laboratory variation in the characterization and analysis of the mucosal microbiota in crohn’s disease and ulcerative colitis. Front Microbiol. 2020;11:2028. doi:10.3389/fmicb.2020.02028
14. Nikolaieva N, Sevcikova A, Omelka R, Martiniakova M, Mego M, Ciernikova S. Gut microbiota-MicroRNA interactions in intestinal homeostasis and cancer development. Microorganisms. 2022;11(1):107. doi:10.3390/microorganisms11010107
15. Rao BC, Lou JM, Wang WJ, et al. Human microbiome is a diagnostic biomarker in hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2020;19(2):109–115. doi:10.1016/j.hbpd.2020.01.003
16. Cai J, Zhang W, Zhu S, et al. Gut and Intratumoral microbiota: key to lung cancer development and immunotherapy. Int Immunopharmacol. 2025;156:114677. doi:10.1016/j.intimp.2025.114677
17. Nakatsu G, Li X, Zhou H, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727. doi:10.1038/ncomms9727
18. Coker OO, Liu C, Wu WKK, et al. Altered gut metabolites and microbiota interactions are implicated in colorectal carcinogenesis and can be non-invasive diagnostic biomarkers. Microbiome. 2022;10(1):35. doi:10.1186/s40168-021-01208-5
19. Lee WH, Chen HM, Yang SF, et al. Bacterial alterations in salivary microbiota and their association in oral cancer. Sci Rep. 2017;7(1):16540. doi:10.1038/s41598-017-16418-x
20. Kwak S, Wang C, Usyk M, et al. Oral microbiome and subsequent risk of head and neck squamous cell cancer. JAMA Oncol. 2024;10(11):1537–1547. doi:10.1001/jamaoncol.2024.4006
21. Guo W, Zhang Y, Guo S, et al. Tumor microbiome contributes to an aggressive phenotype in the basal-like subtype of pancreatic cancer. Commun Biol. 2021;4(1):1019. doi:10.1038/s42003-021-02557-5
22. Yuan L, Yang P, Wei G, et al. Tumor microbiome diversity influences papillary thyroid cancer invasion. Commun Biol. 2022;5(1):864. doi:10.1038/s42003-022-03814-x
23. Jiang X, Liu Q, Xu D, Pang H, Shi Y. Integrated microbiome and metabolome analysis reveals a novel interplay between gut microbiota and metabolites in differentiated thyroid carcinoma. BMC Microbiol. 2025;25(1):346. doi:10.1186/s12866-025-03877-w
24. Jiao J, Zheng Y, Zhang Q, Xia D, Zhang L, Ma N. Saliva microbiome changes in thyroid cancer and thyroid nodules patients. Front Cell Infect Microbiol. 2022;12:989188. doi:10.3389/fcimb.2022.989188
25. Tan J, Dong L, Jiang Z, et al. Probiotics ameliorate IgA nephropathy by improving gut dysbiosis and blunting NLRP3 signaling. J Transl Med. 2022;20(1):382. doi:10.1186/s12967-022-03585-3
26. Portincasa P, Celano G, Serale N, et al. Clinical and metabolomic effects of lactiplantibacillus plantarum and pediococcus acidilactici in fructose intolerant patients. Nutrients. 2022;14(12):2488. doi:10.3390/nu14122488
27. Zhang J, Li Q, Zhang X, et al. Bisdemethoxycurcumin alleviates dextran sodium sulfate-induced colitis via inhibiting NLRP3 inflammasome activation and modulating the gut microbiota in mice. Antioxidants. 2022;11(10):1994. doi:10.3390/antiox11101994
28. Hu J, Cheng S, Yao J, et al. Correlation between altered gut microbiota and elevated inflammation markers in patients with Crohn’s disease. Front Immunol. 2022;13:947313. doi:10.3389/fimmu.2022.947313
29. Wang X, Xiao Y, Xu X, et al. Characteristics of fecal microbiota and machine learning strategy for fecal invasive biomarkers in pediatric inflammatory bowel disease. Front Cell Infect Microbiol. 2021;11:711884. doi:10.3389/fcimb.2021.711884
30. Tomofuji Y, Kishikawa T, Maeda Y, et al. Whole gut virome analysis of 476 Japanese revealed a link between phage and autoimmune disease. Ann Rheum Dis. 2022;81(2):278–288. doi:10.1136/annrheumdis-2021-221267
31. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. doi:10.1111/1462-2920.13589
32. Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi:10.1126/science.1208344
33. Oliva M, Schneeberger PHH, Rey V, et al. Transitions in oral and gut microbiome of HPV+ oropharyngeal squamous cell carcinoma following definitive chemoradiotherapy (ROMA LA-OPSCC study). Br J Cancer. 2021;124(9):1543–1551. doi:10.1038/s41416-020-01253-1
34. Liang Q, Zhao Q, Hao X, et al. The effect of flammulina velutipes polysaccharide on immunization analyzed by intestinal flora and proteomics. Front Nutr. 2022;9:841230. doi:10.3389/fnut.2022.841230
35. Liang H, Dai Z, Liu N, et al. Dietary L-tryptophan modulates the structural and functional composition of the intestinal microbiome in weaned piglets. Front Microbiol. 2018;9:1736. doi:10.3389/fmicb.2018.01736
36. Sun L, Pang Y, Wang X, et al. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm Sin B. 2019;9(4):702–710. doi:10.1016/j.apsb.2019.02.004
37. Yu X, Jiang W, Kosik RO, et al. Gut microbiota changes and its potential relations with thyroid carcinoma. J Adv Res. 2022;35:61–70. doi:10.1016/j.jare.2021.04.001
38. Tan X, Wang Y, Gong T. The interplay between oral microbiota, gut microbiota and systematic diseases. J Oral Microbiol. 2023;15(1):2213112. doi:10.1080/20002297.2023.2213112
39. Liu M, Liu Q, Fan S, et al. LncRNA LTSCCAT promotes tongue squamous cell carcinoma metastasis via targeting the miR-103a-2-5p/SMYD3/TWIST1 axis. Cell Death Dis. 2021;12(2):144. doi:10.1038/s41419-021-03415-2
40. Garrett WS. The gut microbiota and colon cancer. Science. 2019;364(6446):1133–1135. doi:10.1038/s41419-021-03415-2
41. Wang M, Chen S, He X, Yuan Y, Wei X. Targeting inflammation as cancer therapy. J Hematol Oncol. 2024;17(1):13. doi:10.1186/s13045-024-01528-7
42. Zhang L, Xu S, Cheng X, et al. Inflammatory tumor microenvironment of thyroid cancer promotes cellular dedifferentiation and silencing of iodide-handling genes expression. Pathol Res Pract. 2023;246:154495. doi:10.1016/j.prp.2023.154495
43. Friedman-DeLuca M, Karagiannis GS, Condeelis JS, Oktay MH, Entenberg D. Macrophages in tumor cell migration and metastasis. Front Immunol. 2024;15:1494462. doi:10.1016/j.prp.2023.154495
44. Garrett WS. Immune recognition of microbial metabolites. Nat Rev Immunol. 2020;20(2):91–92. doi:10.1038/s41577-019-0252-2
45. Liang Z, Hao Y, Yang L, et al. The potential of Klebsiella and Escherichia-Shigella and amino acids metabolism to monitor patients with postmenopausal osteoporosis in northwest China. BMC Microbiol. 2023;23(1):199. doi:10.1186/s12866-023-02927-5
46. Gopal A, Chidambaram IS, Devaraj N, Devaraj H. Shigella dysenteriae infection activates proinflammatory response through β-catenin/NF-κB signaling pathway. PLoS One. 2017;12(4):e0174943. doi:10.1371/journal.pone.0174943
47. Zhu G, Su H, Johnson CH, Khan SA, Kluger H, Lu L. Intratumour microbiome associated with the infiltration of cytotoxic CD8+ T cells and patient survival in cutaneous melanoma. Eur J Cancer. 2021;151:25–34. doi:10.1016/j.ejca.2021.03.053
48. Coutzac C, Jouniaux JM, Paci A, et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat Commun. 2020;11(1):2168. doi:10.1038/s41467-020-16079-x
49. Liu Q, Li F, Zhuang Y, et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019;11:1. doi:10.1186/s13099-018-0281-6
50. Zhou L, Zhang M, Wang Y, et al. Faecalibacterium prausnitzii produces butyrate to maintain Th17/Treg balance and to ameliorate colorectal colitis by inhibiting histone deacetylase 1. Inflamm Bowel Dis. 2018;24(9):1926–1940. doi:10.1093/ibd/izy182
51. Moon J, Lee AR, Kim H, et al. Faecalibacterium prausnitzii alleviates inflammatory arthritis and regulates IL-17 production, short chain fatty acids, and the intestinal microbial flora in experimental mouse model for rheumatoid arthritis. Arthritis Res Ther. 2023;25(1):130. doi:10.1186/s13075-023-03118-3
52. Messaritakis I, Vogiatzoglou K, Tsantaki K, et al. The prognostic value of the detection of microbial translocation in the blood of colorectal cancer patients. Cancers. 2020;12(4):1058. doi:10.3390/cancers12041058
53. Zhang X, Chen S, Zhang M, et al. Effects of fermented milk containing lacticaseibacillus paracasei strain shirota on constipation in patients with depression: a randomized, double-blind, placebo-controlled trial. Nutrients. 2021;13(7). doi:10.3390/nu13072238
54. Lundberg JO, Moretti C, Benjamin N, Weitzberg E. Symbiotic bacteria enhance exercise performance. Br J Sports Med. 2021;55(5):243. doi:10.1136/bjsports-2020-102094
55. Lang S, Fairfied B, Gao B, et al. Changes in the fecal bacterial microbiota associated with disease severity in alcoholic hepatitis patients. Gut Microbes. 2020;12(1):1785251. doi:10.1080/19490976.2020.1785251
56. Matera G, Muto V, Vinci M, et al. Receptor recognition of and immune intracellular pathways for Veillonella parvula lipopolysaccharide. Clin Vaccine Immunol. 2009;16(12):1804–1809. doi:10.1128/cvi.00310-09
57. Cui J, Cui H, Yang M, et al. Tongue coating microbiome as a potential biomarker for gastritis including precancerous cascade. Protein Cell. 2019;10(7):496–509. doi:10.1007/s13238-018-0596-6
58. Li M, Li K, Tang S, et al. Restoration of the gut microbiota is associated with a decreased risk of hepatic encephalopathy after TIPS. JHEP Rep. 2022;4(5):100448. doi:10.1007/s13238-018-0596-6
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.