Back to Journals » Journal of Inflammation Research » Volume 18
MPO-ANCA-Associated Hypertrophic Pachymeningitis Mimicking IgG4-Related Disease: A Case Report and Literature Review
Received 7 February 2025
Accepted for publication 13 May 2025
Published 24 May 2025 Volume 2025:18 Pages 6673—6680
DOI https://doi.org/10.2147/JIR.S521138
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yuxue Chen,1 Lu Liu,2 Cuihong Xie3
1Department of Rheumatology and Immunology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 2Department of Pharmacy, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 3Department of Emergency and Critical Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
Correspondence: Cuihong Xie, Department of Emergency and Critical Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095th Jiefang Avenue, Wuhan, 430030, People’s Republic of China, Tel +86-13628699465, Fax +86-27-83665264, Email [email protected]
Abstract: Hypertrophic pachymeningitis (HP) is a rare and chronic clinical disease characterized by thickening of the dura mater, leading to persistent headache, cranial neuropathy, seizures, and other neurological symptoms. Immune-mediated causes, particularly antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis and IgG4-related disease (IgG4-RD), are among the most common etiologies. We report a case of a 54-year-old female with recurrent headache, blepharoptosis, hearing loss, and markedly elevated inflammatory markers. Blood tests, and serum levels of IgG4 were within normal ranges. Contrast enhanced cranial MRI revealed thickening and enhancement of bilateral cerebral hemispheres and tentorial dural maters. Additional findings included mild left lacrimal gland enlargement, bilateral middle ear mastoiditis, and tympanic tegmen destruction. Abdominal high-resolution computed tomography (CT) showed enlarged retroperitoneal lymph nodes. Histopathology demonstrated dense lymphoplasmacytic and neutrophilic infiltration with 80 IgG4-positive plasma cells per high-power field and an IgG4+/IgG+ cell ratio of 20%. An initial diagnosis of possible IgG4-RD was made. However, the patient’s symptoms responded poorly to prednisolone (20 mg/day), and fever ensued. Pseudomonas aeruginosa, nocardia malleis, and leptocyclus virus were found in the cerebrospinal fluid measured by NGS. Subsequent laboratory testing showed positive p-ANCA and anti-myeloperoxidase antibodies (anti-MPO), with a negative anti-nuclear antibodies panel, leading to a revised diagnosis of MPO-ANCA-associated HP. Treatment was escalated to intravenous methylprednisolone (40 mg/day), cyclophosphamide, and anti-infectious agents, leading to improved symptoms and decreased inflammatory markers. However, there was a recurrence during the taper of prednisolone. The addition of rituximab achieved complete remission. MPO-ANCA-associated HP is a rare inflammatory disorder that brings diagnostic challenges and requires comprehensive differential diagnosis. In relapsed or refractory cases, rituximab may be a valuable therapeutical option.
Keywords: IgG4-related disease, hypertrophic pachymeningitis, ANCA-associated vasculitis, rituximab
Introduction
Hypertrophic pachymeningitis (HP) is a rare chronic inflammatory disorder characterized by focal or diffuse thickening of the dura mater. Clinically, HP presents with a range of neurological symptoms, including chronic headache, cranial nerve palsies, ataxia, seizures, and other signs of intracranial hypertension. The disorder can significantly impact a patient’s quality of life, and its varied presentation makes early diagnosis challenging. Its etiology is diverse, with varying global incidence patterns. It is hypothesized that HP may result from intracranial infections (such as tuberculosis, syphilis, fungal infections, or sinusitis complication), trauma, malignancies (eg lymphoma, metastatic tumors, multiple myeloma, histiocytosis), and systemic autoimmune diseases. Among the autoimmune causes, systemic lupus erythematosus, sarcoidosis, rheumatoid arthritis, Sjogren’s syndrome, antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), and immunoglobulin G4-related disease (IgG4-RD), are commonly implicated. AAV, which accounts for approximately 30–50% of HP cases, and IgG4-RD, which accounts for about 8.8%, are two of the most frequently recognized inflammatory causes of HP.1–4 Additionally, 25%-44% of cases are classified as idiopathic HP, where the underlying cause remains unidentified.5 This highlights the multifactorial nature of HP, with diverse etiologies ranging from autoimmune and infectious origins to cases with no apparent cause. The global incidence of HP is likely influenced by factors such as healthcare access, diagnostic advancements, and the varying prevalence of underlying diseases across regions. More epidemiological studies are needed to clarify disease distribution and to optimize diagnostic and therapeutic strategies.
The estimated prevalence of immune-mediated HP in Japan is 0.949 per 100,000 individuals,6 with ANCA positivity observed in 30%–50% of cases.1,2 AAV is characterized by predominant inflammation of small vessel walls, leading to tissue and endothelial damage. IgG4-RD is recognized as a systemic inflammatory infiltrative disorder that can affect any organ system and often presents with features mimicing other autoimmune disease. Both of them are among the most common causes of HP.7 HP secondary to AAV is more commonly observed in patients with granulomatosis with polyangiitis (GPA).8 However, the term ANCA-associated HP has been broadly applied to those patients with HP who test positive for ANCA, irrespective of whether they meet the full classification criteria for AAV.9 Thus, early differential diagnosis between IgG4-RD and AAV can be challenging for clinicians, particularly since two conditions share overlapping serological features as well as clinical manifestations. This overlap makes differential diagnosis particularly challenging in cases where both AAV and IgG4-RD are suspected. Here, we present a clinical case of a 54-year-old female whose initial presentation strongly suggested possible IgG4-RD. However, after further investigation, including serum markers and imaging studies, the final diagnosis was made as MPO-ANCA-associated hypertrophic pachymeningitis, highlighting the diagnostic complexity and need for careful clinical and laboratory evaluation.
Case Presentation
A 54-year-old Chinese woman was admitted to the Department of Neurology with a one-year history of otitis media with effusion. The patient reported recurrent headaches and progressively worsening hearing loss. Her medical history revealed that she had undergone bilateral middle ear catheterization 20 days prior to admission. Six months before admission, the patient began experiencing recurrent, stabbing and burning pain in the right temporal region, with an intensity of 8 out of 10 on the numerical pain scale. Three months later, these symptoms worsened, and she developed additional symptoms, including nausea, vomiting, weight loss, bilateral hearing loss, and onset of left eyelid blepharoptosis. The episodes of these symptoms lasted from 10 minutes to several hours and did not respond to self-care or oral analgesics.
Clinical examination revealed bilateral hearing loss (complete on the left), left-sided blepharoptosis, vestibular dysfunction, and left facial (cranial nerve VII) and auditory (cranial nerve VIII) nerve paralysis, with associated hypesthesia. Laboratory tests showed a markedly elevated erythrocyte sedimentation rate (ESR) of 116 mm/H and hypersensitive C-reactive protein (hsCRP) of 104.5 mg/L, indicating chronic inflammatory. Blood cell count was 11.72×109/L, and hemoglobin concentration was 95 g/L. Test for anti-nuclear antibodies, liver and kidney function, urine routine, coagulation function, antiphospholipid antibody, serum immunoglobulin, and serum complements showed no significant abnormalities. Cerebral spinal fluid (CSF) analysis showed increased protein (0.84 g/L), albumin (0.38 g/L), glucose (4.52 mmol/L), along with a slightly decreased chlorine level (117.0 mmol/L). CSF immunoglobulin G was elevated at 243 mg/dL, with increased immunoglobulin A at 26.3 mg/L. Cultures for mycobacterium tuberculosis, bacterial, and fungal were negative. Contrast-enhanced cranial MRI revealed thickening and enhancement of bilateral cerebral hemispheres and tentorial dural maters. Additionally, mild enlargement of the left lacrimal gland, bilateral middle ear mastoiditis, and destruction of the tympanic tegmen were observed. Thrombophlebitis and phlebitis were identified in the left sigmoid sinus, superior and inferior petrosal sinuses, and cavernous sinus, leading to thickening and edema of the left optic nerve (Figures 1A–C and 2A). Abdominal high-resolution computed tomography (CT) showed enlarged retroperitoneal lymph nodes. Electroencephalogram and transcranial Doppler ultrasound were unremarkable.
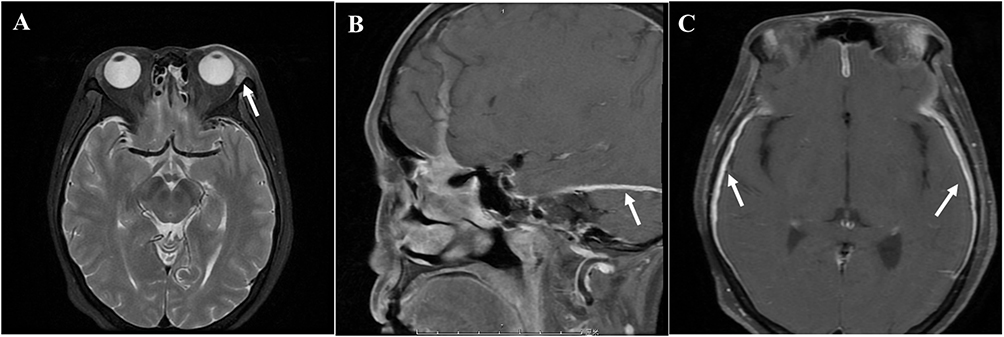 |
Figure 1 Cranial enhanced magnetic resonance imaging of the patient. (A) The left lacrimal gland is slightly larger than that of the opposite side (arrow pointing). (B and C) MRI revealed a thickening and enhancement of bilateral cerebral hemispheres and tentorial dural maters (arrow pointing). |
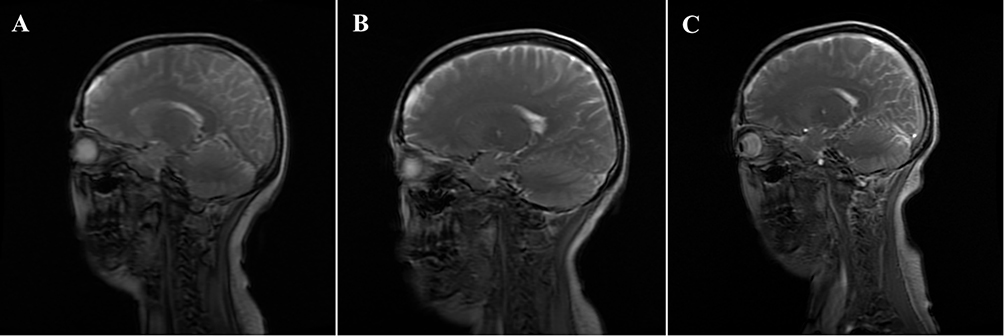 |
Figure 2 Comparison of enhanced magnetic resonance imaging before and after treatment. (A) Before treatment, bilateral cerebral hemispheres and tentorial dural thickening and enhancement; (B) After 2 month of treatment, the bilateral frontoparietotemporal and tentorium of the cerebellum were slightly thickened, which was better than before; (C) After 2 years of treatment, the right tentorium of the cerebellum was slightly thickened. |
To further investigate the cause of hearing loss and bone destruction, the patient underwent left ear tympanoplasty. A small amount of purulent secretion were found in the mastoid process, tympanic sinus, and tympanic cavity. Enlargement of the geniculate ganglion of the facial nerve was also noted. Histopathological examination revealed a dense lymphoplasmacytic and neutrophilic inflammatory infiltrate, including 80 IgG4-positive plasma cells per high-power field, with an IgG4+/IgG+ plasma cell ratio of 20%. However, there was no evidence of storiform fibrosis, vascular occlusion, or obliterative phlebitis (Figure 3). However, serum level of IgG4 and IgE were within normal ranges. Based on the histopathological and serological findings, an initial diagnosis of possible IgG4-related disease involving the lacrimal gland, middle ear/mastoid, dura mater, and retroperitoneal lymph nodes was made.
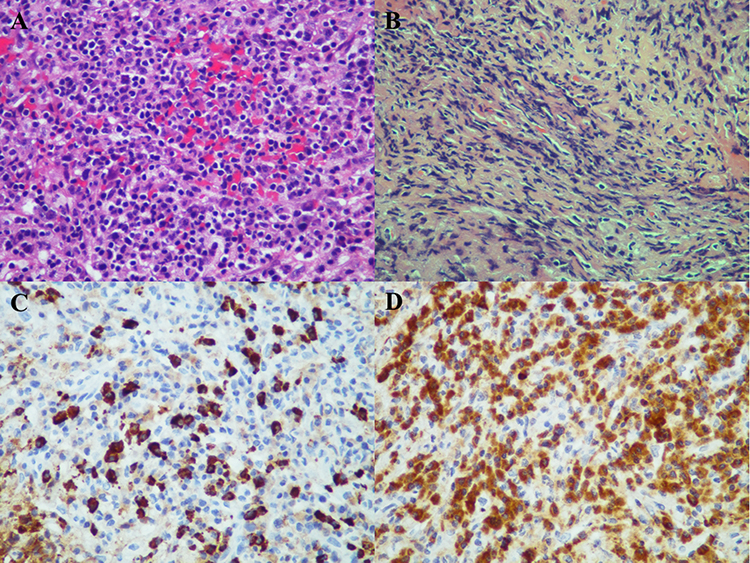 |
Figure 3 Histopathological examination of middle ear granulation. (A) large number of granulation tissue hyperplasia, fibrous tissue with acute and chronic inflammatory cell infiltration (A and B), such as plasma cells, foam-like histiocytes and neutrophils. Immunohistochemistry showed a large number of IgG4-positive plasma cells infiltrated (> 80 /HPF), and the IgG4+/IgG+ plasma cell ratio was about 20% (C and D). |
The patient was initially treated with prednisolone at a dose of 20 mg/day (0.6 mg/kg/d). However, the effectiveness was limited, with a slight improvement in hearing loss and headaches (from 8/10 to 7/10 intensity on the numerical pain scale). Antibiotic (moxifloxacin) was also used for this patient. Additionally, the patient’s condition worsened with the onset of a fever, reaching a peak temperature at 38.8 degrees. Following transfer to the Department of Rheumatology, a repeated lumbar puncture was conducted. CSF was analyzed using next-generation sequencing (NGS) for further evaluation, which detected pseudomonas aeruginosa (sequence number 161, relative abundance 0.8%), nocardia malleis (sequence number 3, relative abundance 0.1%), and leptocyclus virus (sequence number 3, relative abundance 83.7%). Lung CT revealed scattered nodules and nodular shadows in both lungs (Figure 4A). Furthermore, Serologic testing showed positive p-ANCA, accompanied with elevated anti-myeloperoxidase antibodies (anti-MPO) at 41.89 RU/mL. Based on the constellation of polyneuropathy (cranial nerves II, VII, and VIII), granulomatous inflammation, nasosinusitis, thrombophlebitis, middle ear mastoiditis, pulmonary nodules, dural thickening, and MPO-ANCA positivity, a final diagnosis of MPO-ANCA-associated HP was established.
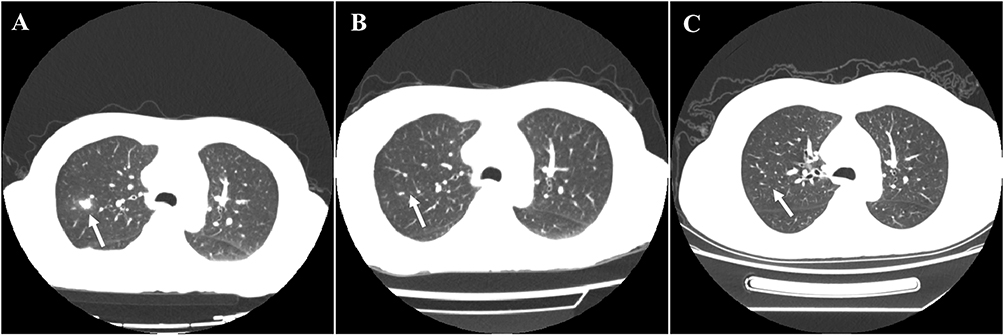 |
Figure 4 Comparison of lung CT before and after treatment. (A) Before treatment, multiple nodules and film shadows were scattered in both lungs (arrow pointing); (B) After 2-month treatment, less nodules and film shadows in both lungs and the absorption improved significantly (arrow pointing); (C) lung CT after 2-year treatment (arrow pointing). |
Oral prednisolone was withdrawn, and intravenous methylprednisolone was initiated at 40 mg/day, followed by intravenous cyclophosphamide (CYC, 500 mg/m2 body surface area) due to the severity and complexity of the clinical manifestation. Sulfameprozole and other antibiotics (cefoperazone sodium and Sulbactam sodium) were applied to manage the presumed intracranial infection. Following this regimen, the patient showed marked clinical improvement, with headaches intensity reduced to 2/10 and partial improvement of hearing loss. The patient was subsequently discharged from hospitalization with biweekly intravenous CYC.
Unfortunately, the patient suffered a recurrence of severe headache (pain score 6/10) and worsening hearing loss when glucocorticoid dose was tapered to oral prednisolone 20mg/day. Despite continuing pulse CYC therapy and increasing her prednisolone dosage to 40 mg/day on her own, there was no significant improvement. Each CYC cycle provided only brief, transient relief of headache symptoms, particularly when combined with hydromorphone for analgesia. Due to refractory symptoms, immunotherapy was intensified with rituximab at a dosage of 500 mg every 2 weeks for twice. Two months later, the patient reported complete remission of headaches and significant improvement in hearing loss. Analgesic medication was discontinued, and glucocorticoid tapering was successfully achieved. Follow-up brain enhanced MRI (Figure 2B) and lung CT (Figure 4B) showed significant reduction in meningeal thickening and pulmonary nodules. Anti-MPO antibody levels decreased from 41.89 RU/mL to 29.31 RU/mL. A maintenance regimen of rituximab at 100mg every six months was initiated. At the two-year follow-up, the patient remained in complete remission from headaches while maintaining a reduced prednisone of 2.5mg/day. Hearing loss remained stable, with mild improvement on audiometry. Imaging showed no significant changes in the previously thickened dura or pulmonary nodules (Figures 2C and 4C). The latest laboratory results revealed negative MPO-ANCA and normalized ESR and hsCRP. The treatment regimen was well tolerated, and no side effects were reported.
Discussion
Hypertrophic pachymeningitis (HP) was first identified by Charcot and Joffrov in 1869 as a rare neurological disorder characterized by local or diffuse thickening of the dura mater. Clinical manifestations typically include headache, cranial nerve deficits, seizures, and ataxia. HP can be categorized into idiopathic and secondary etiologies. Possible causes of secondary HP include autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis, Sjogren’s syndrome, sarcoidosis, IgG4-related diseases (IgG4-RD), and ANCA associated vasculitis (AAV), in addition to infectious diseases or neoplastic conditions. When no identifiable cause is found, it is classified as idiopathic HP. According to a Japanese epidemiological survey, the incidence of HP was 0.949 per 100,000 population, with a mean age of onset of 58 years.4 AAV is the most common secondary etiology, accounting for 30–50% of HP cases, with MPO-ANCA-positive observed in up to 27.7%.2,6
This case presents a rare instance of MPO-ANCA-associated HP that initially mimicked IgG4-related disease (IgG4-RD). IgG4-RD is a chronic inflammatory fibrosing disease characterized by histopathological features, including storiform fibrosis, obliterative phlebitis, and dense infiltration of IgG4-positive plasma cells. Differentiating ANCA-related vasculitis and IgG4-RD is crucial due to the overlapping clinical features but distinct pathogenesis, treatment approaches, and prognosis. Both conditions might present with similar symptoms, while have different underlying mechanisms and treatments. Studies have shown a degree of overlap between IgG4-RD and AAV, where patients with AAV may exhibit elevated serum IgG4 levels, while some patients with IgG4-RD may test positive for ANCA.10,11 AAV typically manifests with invasive sinonasal disease, pulmonary cavitary nodules, and crescentic glomerulonephritis, whereas IgG4-RD tends to involve non-invasive sinus lesions, pulmonary nodules, ground glass, interstitial lung lesions, interstitial nephritis, or membranous nephropathy. Histopathological findings in AVV often include necrotizing vasculitis, granuloma, and neutrophil infiltration. Compared with AAV, patients with IgG4-RD generally show less fevers and have lower C-reactive protein levels. Distinguishing these conditions is full of challenges. A comprehensive evaluation that integrates both clinical and pathological analysis is necessary.
The diagnostic for AAV relies on detecting ANCAs (PR3 or MPO antibodies) and histopathology showing necrotizing vasculitis. The diagnostic for IgG4-RD via biopsy demonstrating IgG4+ plasma cell infiltration, storiform fibrosis, and obliterative phlebitis. Elevated serum IgG4 levels are also a diagnostic criterion, but can also be seen in other diseases. Patients with AAV may be rapidly progressive, especially with kidneys or lungs are involved, leading to organ failure if not treated timely. IgG4-RD tends to be more indolent with growing masses, often cause chronic damage due to fibrosis. So differentiating them early is crucial to apply the correct treatment and prevent irreversible damage.
Despite abundant IgG4-positive plasma cells in the pathological tissue of this patient (80 per high-power field), the IgG4+/IgG+ ratio was only 20%, which falls below the commonly accepted diagnostic threshold for IgG4-RD. Moreover, histology revealed prominent neutrophilic infiltration and granulation tissue hyperplasia, without storiform fibrosis or obliterative phlebitis. Meantime, the serum IgG4 level of the patient was within the normal range. According to the 2019 ACR/EULAR classification and diagnosis criteria for IgG4-RD,12 the presence of primary granulomatous inflammation is considered as a pathological exclusion criterion. Furthermore, the presence of fever, bone destruction in the tympanic tegmental region, and imaging findings of bilateral cerebral and tentorial dural thickening were atypical for IgG4-RD. Taken together with pANCA and MPO positivity, as well as enhanced head magnetic resonance imaging (MRI) findings of bilateral cerebral hemispheres and tentorial dural thickening and strengthening, a final diagnosis of MPO-ANCA-associated HP was determined.
Interestingly, this patient was also found to have cerebral nocardiosis, supported by NGS detection of Nocardia mallei in the cerebrospinal fluid and clinical improvement following targeted antibiotic therapy. Nocardia is an exogenous pathogen typically acquired via the respiratory tract or cutaneous entry points, particularly in immunocompromised individuals. This infection frequently targets lungs and causes suppurative inflammation and lung necrosis, which has similar symptoms to tuberculosis. Nocardia can spread through the bloodstream, causing meningitis and brain abscess in approximately one-third of infected patients. Brazilian Nocardia has strong pathogenicity and can cause outbreaks in healthy individuals or animals. Nocardia mallei, known for being the highest virulence among the species, can invade the brain, lungs, and skin. The diagnosis mainly relies on culture or histopathological identification, although yield is low due to its fastidious nature. However, it is difficult to find Nocardia in blood culture due to its fastidious characters, which has led to historically low diagnostic rates. In recent years, advances of NGS technology have greatly improved detection rates. However, the tough cell wall of Nocardia complicates DNA extraction, and low sequence abundance does not exclude clinical significance.13 In this case, the positive NGS results and clinical course supported a diagnosis of Nocardia infection, likely secondary to systemic spread. The patient responded well to sulfamethoxazole-trimethoprim (SMZ/TMP), which remains the first-line therapy for Nocardia despite emerging resistance trends.
At present, the knowledge of how MPO-ANCA-positive associates with HP still remains insufficient. One of the possible pathogenesis hypothesis suggests that MPO-ANCA binds to antigen or Fc receptors on neutrophils and monocytes, leading to their activation and a cascade of inflammatory responses.14 Clinically, MPO-ANCA-associated HP predominantly occurs in elderly women, with subacute onset and involvement of the dural and upper respiratory tract, presenting with headache, chronic sinusitis, otitis media, or mastoiditis. Headache is the most common and may be the only symptom of the patient, which is possibly caused by inflammation of the dural membrane or cranial hypertension. Another classic symptom is cranial nerve palsy. All cranial nerves can be involved, particularly cranial nerves II or VIII. Others manifestations include seizures, ataxia, venous sinus thrombosis, and so on. Currently, dural biopsy remains as the gold standard for diagnosis, typically revealing dural thickening with fibrosis, necrotizing granulomatous inflammation, and vasculitis. Immunohistochemical findings indicate the infiltration of inflammatory cells such as CD4+/CD8+ T cells, CD20+ B cells, neutrophils, eosinophils, plasma cells, monocytes, and macrophages.1 However, due to the invasive nature of the procedure and the high risk in elderly or systemically ill patients, biopsies are often declined. Enhanced MRI is the most specific diagnostic method for HP, showing band-like enhancement corresponding to thickened dura. Laboratory tests often show MPO-ANCA positivity, sometimes accompanied by antinuclear antibodies or rheumatoid factor. Elevated erythrocyte sedimentation rate and C-reactive protein, as well as protein elevation in CSF, are common. Meantime, the inflammatory indicators, MPO-ANCA level, or MRI dural thickening of most patients are significantly correlated with the degree of disease activity, suggesting that the above indicators can be used for monitoring disease progress and treatment responses.6,15
Till now, there is no standardized therapy protocol for MPO-ANCA-associated HP, and current diagnosis standards and managements are based on AAV guidelines. Glucocorticoids remain the cornerstone of treatment, often combined with immunosuppressants, such as cyclophosphamide (CYC), methotrexate (MTX), mycophenolate morphenate (MMF), and azathioprine (AZA). For relapsed, refractory, or organ/life-threatening patients, rituximab (RTX) is increasingly recognized as effective in relapsing, refractory, or organ/life-threatening cases.15,16 Organ/life-threatening manifestations include glomerulonephritis, pulmonary hemorrhage, central nervous system vasculitis, meningeal involvement, multiple mononeuritis, heart involvement, mesenteric ischemia, and limb/finger ischemia. In this case, the patient’s symptoms initially responded to glucocorticoids and CYC but relapsed during steroid tapering. Rituximab therapy was then introduced for the meningeal involvement,17 leading to complete remission and normalization of inflammatory markers. Based on findings from the MAINRITSAN 2 trial, which showed no significant difference in relapse rates between fixed-schedule and tailored RTX regimens, a maintenance dose of 100 mg every six months was chosen.18–20 This regimen was well tolerated without adverse effects.
In conclusion, HP is a crucial neurological manifestation of AAV that presents considerable diagnostic challenges. Given the clinical overlap with IgG4-RD, particularly in cases with overlapping serological or histopathological features, differential diagnosis is essential. In the new classification criteria for IgG4-related diseases developed by ACR/EULAR in 2019,12 ANCA positivity (specifically targeting PR3 and MPO) was specifically pointed out as the exclusion criterion, underscoring the importance of comprehensive clinical, serologic, imaging, and histologic evaluation. Currently, there is little understanding of MPO-ANCA-related HP, and the diagnosis rate is relatively low. The reasons are possibly as follows: 1) The occurrence of MPO-ANCA-related HP is rare and with an insidious onset, which makes clinical diagnosis difficult and leads to misdiagnosis or missed diagnosis frequently. Meanwhile, there is a lack of clear clinical diagnosis and treatment guidelines. 2) The main clinical manifestations of MPO-ANCA-related HP are headache and multiple cranial nerve injuries, so most patients seek treatment in neurology or otolaryngology. 3) The diagnosis of MPO-ANCA-related HP relies on dural biopsy, which has a low acceptance rate among patients. 4) The status of MPO-ANCA-related HP in AAV is still controversial. Therefore, multidisciplinary collaboration is essential to enhance diagnostic accuracy and optimize treatment outcomes for this complex and under-recognized condition.
Conclusions
AAV may occasionally present with clinical, serological, and even histopathological features similar to those of IgG4-RD. Both conditions are recognized as common causes of HP. In order to differentiate between these diseases, clinicians should undertake comprehensive analyses of the pathological findings and consider the full clinical scenarios. At present, there are no standardized treatment protocols for MPO-ANCA-associated HP. Glucocorticoids combined with immunosuppressive agents are typically the first-line treatment. For refractory cases, rituximab has shown promise as an effective therapeutic option.
Data Sharing Statement
The data used this article are available from Cuihong Xie and Yuxue Chen, without undue reservation.
Ethics Approval and Consent to Participate
Ethical approval is not necessary because this is a case report based on the clinical data during the patient’s hospitalization other than research or experiment. The patient has given her consent and authorized her clinical data in our report and signed the Patient Consent Form.
Consent for Publication
The patient has given her consent and authorized her clinical data in our report to be published.
Acknowledgments
The authors would like to thank this patient participated in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Acquisition of data: Yuxue Chen; Article writing: Yuxue Chen; Article revision: Lu Liu; Critically reviewed the article: Cuihong Xie.
Funding
This work was supported by Scientific and Technological Innovation Projects of Hubei Province (2021CFB058, 2023AFB876, 2024AFB604), and Science Foundation of Tongji Hospital (2018A09).
Disclosure
The authors have no conflicts of interest for this study.
References
1. Yokoseki A, Saji E, Arakawa M, et al. Hypertrophic pachymeningitis: significance of myeloperoxidase anti-neutrophil cytoplasmic antibody. Brain. 2014;137(2):520–536. doi:10.1093/brain/awt314
2. Ikeda J, Shimojima Y, Usami Y, Ueno KI, Kishida D, Sekijima Y. Cerebrospinal fluid biomarkers implicated in the pathogenesis of anti-neutrophil cytoplasmic antibody-related hypertrophic pachymeningitis. Clin Rheumatol. 2020;39(6):1803–1811. doi:10.1007/s10067-020-04971-2
3. Abrantes FF, Moraes MPM, Rezende Filho FM, Pedroso JL, Barsottini OGP. A clinical approach to hypertrophic pachymeningitis. Arq Neuropsiquiatr. 2020;78(12):797–804. doi:10.1590/0004-282x20200073
4. Yonekawa T, Murai H, Utsuki S, et al. A nationwide survey of hypertrophic pachymeningitis in Japan. J Neurol Neurosurg Psych. 2014;85(7):732–739. doi:10.1136/jnnp-2013-306410
5. Lu C, Lv PP, Zhu XY, Han YM. Cogan’s Syndrome Combined with Hypertrophic Pachymeningitis: a Case Report. J Inflamm Res. 2024;17:1839–1843. doi:10.2147/JIR.S453071
6. Shimojima Y, Sekijima Y. Hypertrophic pachymeningitis in ANCA-associated vasculitis: clinical and immunopathological features and insights. Autoimmun Rev. 2023;22(6):103338. doi:10.1016/j.autrev.2023.103338
7. Gautier F, Neumann L, Adle-Biassete H, et al. Pachymeningitis associated with IgG4-related disease and ANCA positivity: case report and review of the literature. Autoimmun Rev. 2023;22(4):103285. doi:10.1016/j.autrev.2023.103285
8. Seror R, Mahr A, Ramanoelina J, Pagnoux C, Cohen P, Guillevin L. Central nervous system involvement in Wegener granulomatosis. Medicine. 2006;85(1):54–65. doi:10.1097/01.md.0000200166.90373.41
9. Hahn LD, Fulbright R, Baehring JM. Hypertrophic pachymeningitis. J Neurol Sci. 2016;367:278–283. doi:10.1016/j.jns.2016.06.024
10. Alamino PR, Martínez C, Espinoza LR. IgG4-associated vasculitis. Curr Rheumatol Rep. 2013;15(8):348. doi:10.1007/s11926-013-0348-9
11. Xia C, Li P. IgG4-related hypertrophic pachymeningitis with ANCA-positivity: a case series report and literature review. Front Neurol. 2022;13:986694. doi:10.3389/fneur.2022.986694
12. Wallace ZS, Naden RP, Chari S, et al. The 2019 American college of rheumatology/European league against rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020;72(1):7–19. doi:10.1002/art.41120
13. LaHue SC, Guterman EL, Mikhail M, Li Y, Cha S, Richie MB. Clinical and radiographic characteristics of nocardia vs non-nocardia brain abscesses. Neurol Clin Pract. 2023;13(2):e200134. doi:10.1212/CPJ.0000000000200134
14. Shimojima Y, Kishida D, Hineno A, Yazaki M, Sekijima Y, Ikeda SI. Hypertrophic pachymeningitis is a characteristic manifestation of granulomatosis with polyangiitis: a retrospective study of anti-neutrophil cytoplasmic antibody-associated vasculitis. Int J Rheum Dis. 2017;20(4):489–496. doi:10.1111/1756-185X.13046
15. Chung SA, Langford CA, Maz M, et al. 2021 American college of rheumatology/vasculitis foundation guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol Hoboken NJ. 2021;73(8):1366–1383. doi:10.1002/art.41773
16. Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis. 2024;83(1):30–47. doi:10.1136/ard-2022-223764
17. Jang Y, Lee S, Jung K, Chu K, Lee SK. Rituximab treatment for idiopathic hypertrophic pachymeningitis. J Clin Neurosci. 2017;13(2):155–161.
18. Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771–1780. doi:10.1056/NEJMoa1404231
19. Smith RM, Jones RB, Specks U, et al. Rituximab versus azathioprine for maintenance of remission for patients with ANCA-associated vasculitis and relapsing disease: an international randomised controlled trial. Ann Rheum Dis. 2023;82(7):937–944. doi:10.1136/ard-2022-223559
20. Wallace ZS, Miloslavsky EM. Management of ANCA associated vasculitis. BMJ. 2020;8(368):m421. doi:10.1136/bmj.m421
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.