Back to Journals » Journal of Pain Research » Volume 15
Moving Toward a Multimodal Analgesic Regimen for Acute Sickle Cell Pain with Non-Opioid Analgesic Adjuncts: A Narrative Review
Received 11 October 2021
Accepted for publication 4 February 2022
Published 31 March 2022 Volume 2022:15 Pages 879—894
DOI https://doi.org/10.2147/JPR.S343069
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
Martha O Kenney,1 Wally R Smith2
1Division of Pediatric Anesthesiology, Department of Anesthesiology, The University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC, USA; 2Division of General Medicine, Department of Internal Medicine, Virginia Commonwealth University School of Medicine, Richmond, VA, USA
Correspondence: Martha O Kenney, Division of Pediatric Anesthesiology, Department of Anesthesiology, The University of North Carolina at Chapel Hill School of Medicine, N2198 UNC Hospitals, Campus Box 7010, Chapel Hill, NC, 27599, USA, Tel +1 919-966-5136, Email [email protected]
Purpose of Review: Sickle cell disease (SCD) is an inherited hemoglobinopathy with potential life-threatening complications that affect millions of people worldwide. Severe and disabling acute pain, referred to as a vaso-occlusive crisis (VOC), is a fundamental symptom of the disease and the primary driver for acute care visits and hospitalizations. Despite the publication of guidelines for VOC management over the past decade, management of VOCs remains unsatisfactory for patients and providers.
Recent Findings: Acute SCD pain includes pain secondary to VOCs and other forms of acute pain. Distinguishing VOC from non-VOC pain may be challenging for both patients and clinicians. Further, although opioids have been the gold-standard for VOC pain management for decades, the current highest standard of care for all acute pain is a multimodal approach that is less dependent on opioids, and, instead incorporates analgesics and adjuvants from different mechanistic pathways. In this narrative review, we focus on a multimodal pharmacologic approach for acute SCD pain management and explore the evidence for existing non-opioid pharmacological adjuncts. Moreover, we present an explanatory model of pain, which is not only novel in its application to SCD pain but also captures the multidimensional nature of the SCD pain experience and supports the need for such a multimodal approach. This model also highlights opportunities for new investigative and therapeutic targets – both pharmacological and non-pharmacological.
Summary: Multimodal pain regimens that are less dependent on opioids are urgently needed to improve acute pain outcomes for individuals with SCD. The proposed explanatory model for SCD pain offers novel opportunities to improve acute pain management for SCD patients.
Keywords: acute pain, sickle cell disease, non-opioid analgesia, multimodal analgesia, neuromatrix pain theory
Introduction
Sickle cell disease (SCD) is an inherited hemoglobinopathy that affects over 6 million individuals of African and Mediterranean descent worldwide.1 Recurrent and severe pain, known as a vaso-occlusive crisis (VOC), is the primary clinical manifestation of SCD. SCD pain can be ever-present, unpredictable, severe2–5 and can significantly impair a patient’s health-related quality of life (HRQOL).6
Pathophysiology of Sickle Cell Pain
Sickle cell pain begins during childhood in response to vaso-occlusive ischemia.7 Ischemia is sensed by fast-firing nociceptive neurons8 via elevations of substance P9 and other neurotransmitters. However, pain is maintained by slower, repetitively firing neurons in response to a complicated array of inflammation and other mechanistic pathways.7 Studies in Berkeley transgenic mouse model of SCD (HbSS-BERK) suggest that ischemia-induced mast cell activation results in the release of inflammatory cytokines and neuropeptides. This then leads to nociceptor activation and neuropeptide release from peripheral nerve terminals, and, therefore, sustains pain once it is triggered.10,11 In turn, nociceptor hypersensitivity to mechanical, heat and cold stimuli may be further exacerbated by hypoxia/reoxygenation during repeated VOCs.12 Later, SCD pain derives from neuropathic mechanism in as many as 37% of the patients aged ≥14 years.13,14 Central sensitization (CS) – nociceptive hyperexcitability known to intensify and maintain clinical pain – likely arises from years of repetitive vaso-occlusive, inflammatory, or other stimulation from SCD and then leads to nociceptor neuronal injury.15
Definition of Acute Sickle Cell Pain
While classic VOCs have long been a focus of research and treatment, more recently an expert panel defined three overarching classifications of sickle cell pain: acute, chronic, and acute-on-chronic. Acute SCD pain encompasses not only VOCs but also other forms of acute pain. The core diagnostic criteria for acute SCD pain are defined as
- “Diagnosis of SCD as confirmed by laboratory testing.
- Reports of increased pain that lasts ≥2hours and started in the past 10 days.
- Must display ≥1 of the following signs:
- Palpation of region of reported pain elicits pain or tenderness
- Movement of region of pain elicits focal pain
- Decreased range of motion or weakness in region of reported pain
- Pain is not entirely explained by specific physical examination findings (leg ulcers, priapism …).”16
Chronic SCD pain is defined differently and based primarily on pain being present during a majority of days during a 3- to 6-month interval.17 The prevalence of chronic SCD pain in a four-city cohort of patients presenting with acute SCD pain to either emergency departments (ED) or infusions centers was 68%.18 Thus, chronic SCD pain often colors the presentation of acute SCD pain. Specifically, VOCs (as part of acute-on-chronic pain) are more frequent among SCD patients with chronic pain.16,17
Admittedly, applying these new definitions to interpret SCD pain raises a misclassification problem. SCD patients may still refer to all SCD pain, acute and chronic, as “crises.” It may also be unclear whether, causally, acute SCD pain is a VOC, is an acute exacerbation of otherwise chronic pain or is due to opioid withdrawal or opioid-induced hyperalgesia. The focus of this review will be on acute SCD pain secondary to a VOC.
The majority of SCD pain – whether acute, chronic, or acute-on-chronic – is self-managed at home.2,4,19,20 However, healthcare utilization for SCD pain presenting as a VOC is highly prevalent among the subset of SCD patients aged 18 to 34 years.21 In fact, VOCs account for as much as 85% of hospitalizations and readmissions for SCD. In 2016 alone, hospitalizations for SCD accounted for $811.4 million in cost, with a length of stay averaging 5 days.21–23
The State of Sickle Cell Pain Management
Management of acute SCD pain remains unsatisfactory.24,25 First, there is ignorance of and barriers to implementation of guidelines. Two published acute SCD pain management guidelines, the National, Heart, Lung and Blood Institute’s (NHLBI) recommendations26 for treatment of VOCs and the American Society of Hematology’s (ASH) guidelines for acute SCD pain management27 call for swift administration of analgesics upon arrival to hospitals. However, a recent publication suggested swift analgesic administration was far from universal.28 Second, while not universally guideline-recommended, most experts agree that individualized pain treatment plans are the highest standard of SCD care.29,30 Third, general pain guidelines published by the International Association for the Study of Pain (IASP)31 and others strongly advocate for a multimodal and multidisciplinary approach to SCD pain treatment.32 However, most SCD centers and providers have trouble amassing resources to provide such multidisciplinary treatment.33
We propose, within the framework of multidisciplinary treatment, that strong incorporation of multimodal analgesia is needed to treat acute SCD pain. Multimodal analgesia is defined as including two or more drugs that provide analgesia via different mechanisms.34,35 Multimodal analgesia is a growing standard in pain management for chronic painful disease.31,36,37 Failure to use multimodal analgesia is likely another contributing factor to patient and provider dissatisfaction with SCD pain treatment. Despite the complex pathophysiology of SCD pain described above, patients and providers have relied for decades on treatment of acute nociceptive pain, using primarily opioid therapy, as the oversimplified approach to treating acute SCD pain,38 ignoring other pain pathways and analgesic mechanisms.
Opioids’ Path to Becoming the “Gold Standard” Analgesic for Acute Sickle Cell Pain
Opioids became the “gold standard” for analgesic or palliative therapy for acute SCD pain based on evidence extrapolated from cancer research in the 1980s. One particularly influential extrapolation was published in 1992, entitled “Treating sickle cell pain like cancer pain.”39 The extrapolation was based on some evidence. Severe acute SCD pain treated with continuous, long-acting opioids, in addition to intermittent, short-acting opioids, reduced return rates to an urban emergency department for acute SCD pain. The article promoted this apparent efficacy as similar to the efficacy that had been previously demonstrated in treatment of cancer pain.39 Despite the initially negative reaction that this publication created,40,41 pain guidelines published around that time urged the treatment of acute SCD pain as an emergency and urged aggressive treatments, primarily opioids.42,43 Opioid manufacturers seized on this opportunity to suggest their products were safe, effective, and even evidence-based for the management of not only VOCs and chronic SCD pain but also many other acute pain states.44 While this article demonstrated reduced return rates for pain care after initiation of long-acting opioids, it was unfortunately mistitled and misleading.
Unlike cancer pain, the clinical efficacy of opioids for SCD pain had not at the time of this article, and still has not, been determined through large randomized controlled trials (RCTs). Rather, clinical experience and small, low powered studies dominate the literature searches conducted to date.26 Even more pointedly, there have been no RCTs demonstrating which of several commonly used opioids in SCD is most beneficial (eg, morphine versus hydromorphone).45 Even weaker is the evidence for the benefit of opioids in chronic or acute-on-chronic SCD pain18,46 that was likely depicted in the above 1992 study. The few available studies suggest that the magnitude of opioids’ benefit for this pain, like that of all chronic noncancer pain, is low.47
In contrast to the analgesic benefits, opioids are clearly known to be associated with several harms. Opioid-related adverse effects (ORAEs) include nausea, vomiting, constipation, respiratory depression, sedation, dependence and withdrawal.48 And while the prevalence of opioid use disorder among sickle cell patients is similar to that of the general public,49 patients with SCD are at risk for prescription opioid related mortality.46,50 Due to the recurrent nature of VOCs, SCD patients are also frequently exposed to opioids, both in the hospital and at home.4,51 Opioid tolerance – where increased doses of opioids are needed for pain relief – is frequently observed among SCD patients, although the exact prevalence is unknown.52 In addition, chronic opioid use can lead to the paradoxical phenomenon of opioid-induced hyperalgesia (OIH), where escalating doses of opioids lead to increased perception of pain.53
Multimodal Analgesic Therapy: An Untested Strategy in SCD Management
Pain experts are now advocating for a shift away from a “one size fits all” pain management approach to a targeted approach, based on the underlying pain mechanism(s) of a disease process.54 The moderate evidence for the clinical efficacy of opioids for relief of acute SCD pain, as well as the concern for ORAEs, raise an urgent need for clinical trials of multimodal approaches to the management of acute SCD pain. Such trials should determine whether these approaches are efficacious in SCD, as is being done for other acute-on-chronic painful diseases such as rheumatic diseases (eg. rheumatoid arthritis55,56 and ankylosing spondylarthritis57) and inflammatory bowel disease.58,59
Meanwhile, as we wait for future trials on the efficacy of multimodal analgesia for SCD pain, we advocate that the balance of benefits and harms demands empiric use of multimodal therapies now for acute SCD pain. Based on evidence in other disease states, we advocate for a multimodal or multi-mechanistic-based therapeutic approach with empiric use but also careful monitoring for effective analgesia despite decreased treatment with opioids. The potential benefits include decreased ORAEs and harm reduction from potential opioid misuse and diversion.
In this review, we support this advocacy by providing a summary of the evidence of the benefits of non-opioid pharmacological analgesics for management of acute SCD pain. Like the pool of opioid studies, the pool of studies of non-opioid pharmacological interventions for acute SCD pain is small, and the evidence is primarily found in retrospective studies and small prospective trials (Table 1). Therefore, the review will draw attention to analgesics that although may require further clinical exploration to demonstrate their efficacy for acute SCD pain, can currently be used as adjuncts to opioids. The review will also draw attention to non-opioid analgesics with evidence of efficacy in other painful conditions. Lastly, our review will propose a novel explanatory model for SCD pain, adapted from the work of pain pioneer Richard Melzack, to support multimodal analgesia as well as multidisciplinary treatment of all types of SCD pain. A comprehensive understanding of SCD pain is not only critical to development of new interventions but also important in the assessment of the clinical efficacy of existing interventions, such as the ones highlighted in this review.
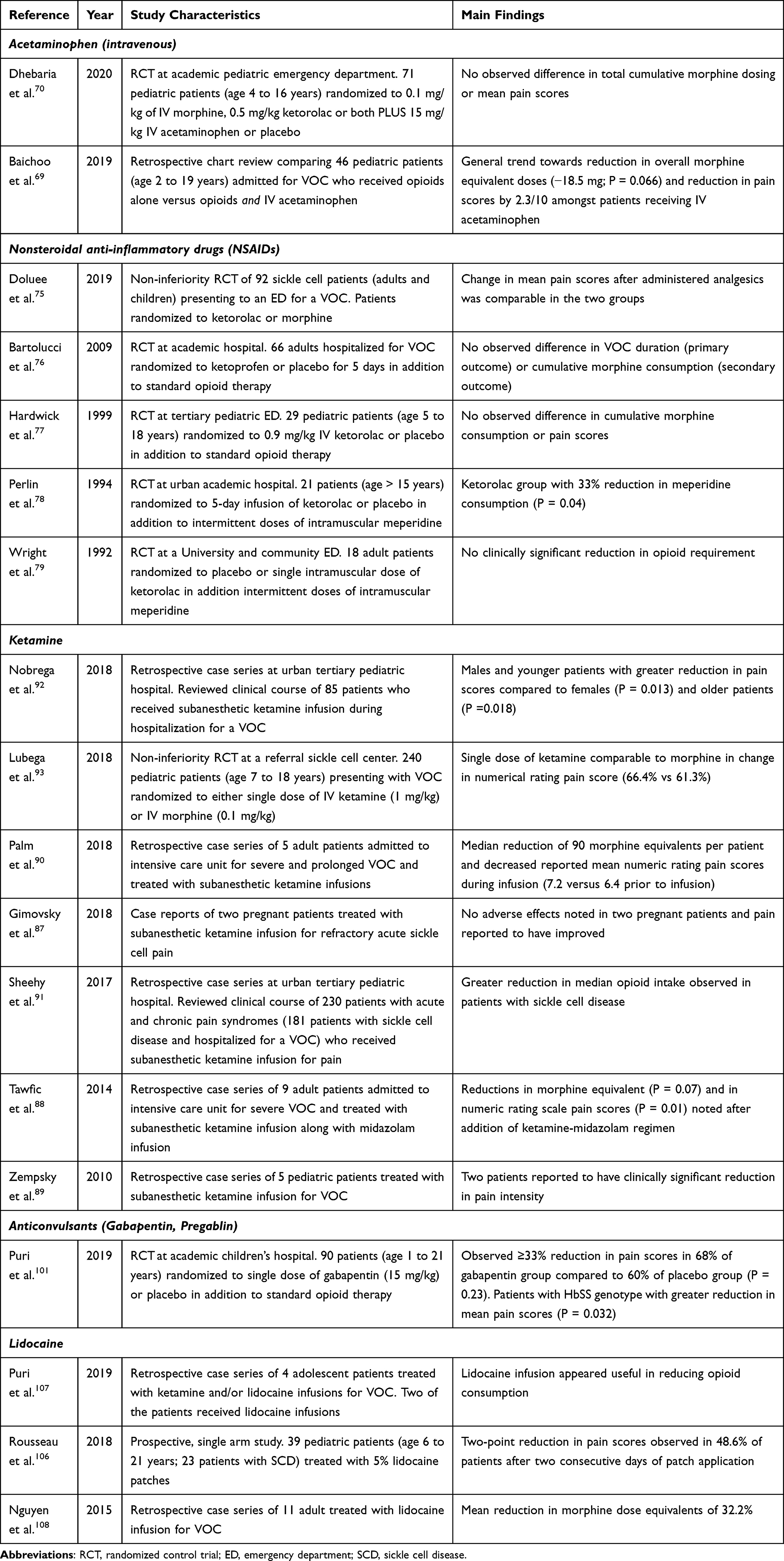 |
Table 1 Characteristics of Studies of Identified in Literature Review of Non-Opioid Analgesic Adjuncts for Management of Sickle Cell Vaso-Occlusive Crisis (VOC) |
Methods
We conducted a narrative review of peer-reviewed literature on the use of non-opioid analgesics for treatment of acute SCD pain. We limited our search to non-opioid analgesics that have established use in other painful conditions and are accessible and used with some frequency within SCD clinical practice.27,60,61 Although non-pharmacological interventions are an important component of multimodal pain management, such interventions were not included in our literature search for the following reasons: 1) these interventions are usually inaccessible in the acute clinical setting for management of acute SCD pain, 2) sickle cell providers lack familiarity with these interventions, and 3) the evidence for these interventions for acute SCD pain is sparse.27,60,62,63 In addition, because this review is focused on acute SCD pain, non-opioid adjuncts, such as antidepressants, which may be used in the outpatient setting for chronic SCD pain61 were not included in our literature search. Based on these considerations, our list of non-opioid analgesic adjuncts was narrowed to intravenous acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), ketamine, anticonvulsants (gabapentin and pregabalin) and lidocaine.
To identify literature relevant to this list of non-opioid pharmacological interventions, we performed a literature search from January 2021 to March 2021. Searches were performed in Embase and PubMed for English language articles using terms relating to sickle cell, sickle anemia, or vaso-occlusive in conjunction with a list of terms for each drug. Additional searches for evidence were performed in UpToDate, Micromedex, Google Scholar, and Google. No date limits were applied for the search.
The titles and abstracts were reviewed, and articles were selected for inclusion based on the following criteria: 1) published in English, 2) peer-reviewed article or abstract, and 3) included any of the following study design: case series, retrospective cohort study, prospective cohort study or RCT. Articles where any of the drugs of interest were administered to SCD patients only in the outpatient setting were excluded.
The primary author searched the relevant literature in consultation with the senior author. Relevant items were extracted and included in the review. The search identified eight RCTs, one single arm prospective study, and nine case series or retrospective cohort studies, which are subsequently presented in this review.
Non-Opioid Pharmacological Interventions
As we consider the list of non-opioid analgesic adjuncts (Table 1), it is important to consider where during an acute sickle cell pain crisis each adjunct would be most effective. In 1995, SCD pain pioneer, Samir Ballas64 proposed four distinct phases of a severe SCD pain crisis – the prodromal, initial, established, and resolving phases.64,65 Ideally, a non-opioid analgesic adjunct, particularly one with opioid-sparing properties, should be initiated during the prodromal phase of an acute pain crisis – that is, prior to the initiation of severe pain. However, the prodromal phase is challenging to identify in patients because of a lack of laboratory abnormalities and its occurrence prior to a patient’s encounter with the healthcare system.64 Therefore, we considered the three latter phases for initiation of the proposed list of non-opioid adjuncts (Figure 1).
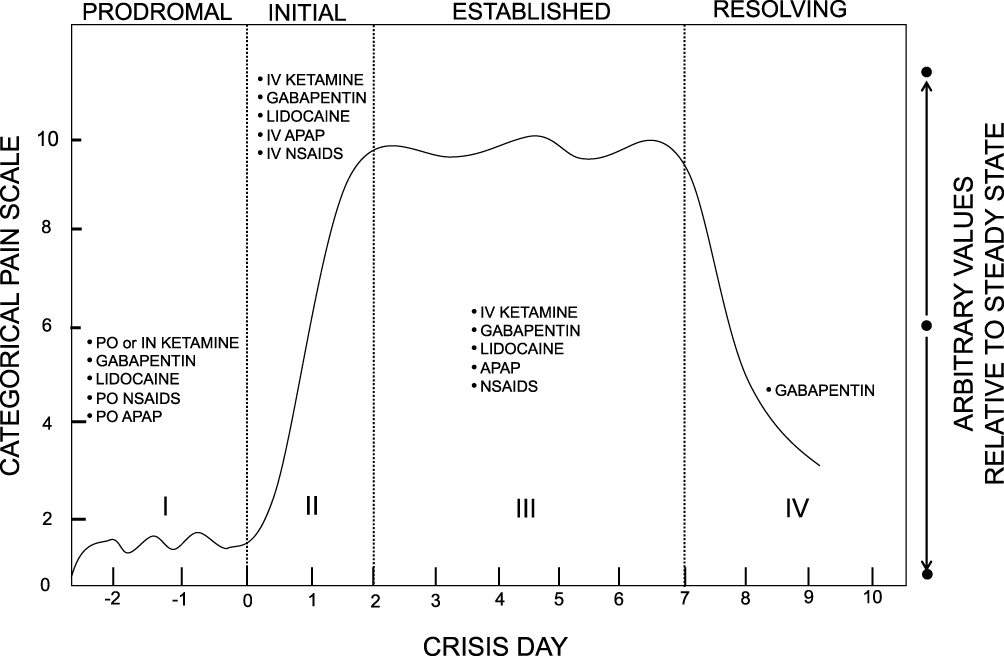 |
Figure 1 Non-opioid analgesics within the four distinct phases of a severe SCD pain crisis or VOC. I = Prodromal phase or pre-crisis phase. Patients may experience numbness or paresthesia and/or premonition of an on-coming crisis. II = Initial phase, characterized by gradual increase in severity of pain. III = Established phase, persistent, severe pain averaging 4 to 5 days. IV = Resolving phase, pain severity gradually decreases. Adapted and reproduced with permission from Taylor and Francis, Ltd. Originally published in Ballas Sk. The Sickle Cell Painful Crisis in Adults: Phases and Objective Signs. Hemoglobin. 1995;19(6):323–333 (Taylor & Francis Ltd, http://www.tandfonline.com).64 Abbreviations: PO, by mouth; IN, intranasal; APAP, acetaminophen; NSAIDs, non-steroidal anti-inflammatory drugs. |
Intravenous Acetaminophen
Acetaminophen (APAP) is the most widely available and used analgesic for mild-to-moderate pain worldwide. Even though it has been used for many years, acetaminophen’s mechanism of action has not been clearly elucidated. It is believed to exert its analgesic effect centrally via cyclooxygenase (COX) inhibition with no significant peripheral anti-inflammatory effects.66 Oral and suppository formulations were previously the only available forms. However, since it became commercially available, intravenous (IV) APAP has grown in popularity for acute pain analgesia.67 Intravenous APAP has been demonstrated to have an early onset of clinical analgesia within 5 minutes of administration. It is usually administered as a 15-minute infusion to minimize injection site pain. This formulation is also believed to reach a higher peak plasma concentration compared to its oral equivalent.68
There are few studies examining acetaminophen’s clinical benefits for acute SCD pain. Baichoo et al69 reviewed the charts of 46 children with SCD who received opioid or opioid and IV APAP for treatment of VOC. The authors reported a mean reduction in pain scores of 2.4 in their analysis of administered IV APAP doses without a concomitant analgesic (total of 49 doses). In an another study, Dhebaria et al70 conducted an RCT with pediatric patients admitted with a VOC to an academic urban ED. Seventy-one patients were randomized to receive either 0.1 mg/kg of IV morphine, 0.5 mg/kg ketorolac, or both along with 15 m/kg of IV APAP or placebo. The authors noted no change in morphine consumption in the APAP group nor clinical or statistically significant differences in admission or 72-hr ED return visit. This study was limited by the small number of study subjects and short duration of the interventions. Moreover, once an SCD patient has entered the initial phase of a crisis, a single dose of APAP may not alter their course. At the time of our literature search, there were no published RCTs examining the clinical efficacy of IV APAP as an adjunct for VOC treatment in adults.
Until recently, the price tag of $33/g of IV APAP67 limited its use. With the new availability of a generic and less cost-prohibitive formulation, further exploration of its clinical efficacy as adjunctive analgesia for acute SCD pain through large, prospective, randomized studies is needed.
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
Inflammation plays a vital role in many painful disease states, such as SCD.10 NSAIDs are used for both their anti-inflammatory and analgesic properties through its inhibition of cyclooxygenase (COX) enzymes and, thus, its suppression of prostaglandin production.71 Non-aspirin NSAIDS – ibuprofen and ketorolac – are the most commonly encountered NSAIDs in the treatment regimen for acute SCD pain.72 Many of the potential beneficial effects of NSAIDs are extrapolated from its use for postoperative and cancer pain. NSAIDs act on peripheral and central pain-mediated sites not involved in the opioid-mu receptor system pathways. When administered in conjunction with opioids such as morphine, NSAIDs are believed to have opioid sparing effects, therefore, reducing incidence of nausea/vomiting and sedation in patients with acute, post-operative pain.73,74
However, the studies assessing the clinical efficacy of NSAIDs in SCD patients are limited. Five randomized control studies (range of 18–92 patients) studying the efficacy of NSAIDs for acute SCD pain have been published from 1992 to 2019.75–79 Bartolucci et al76 randomized 66 patients with severe VOC to ketoprofen, a non-selective NSAID, or placebo in conjunction with intravenous morphine for total of 5 days. They found no significant difference between the two groups with regards to their primary outcome of VOC duration (p=0.61). In another study, Doluee et al75 randomized 92 patients presenting to a university-affiliated emergency department in Iran to receive intravenous ketorolac or morphine. They reported the two analgesics to be comparable in terms of mean pain scores. Neither study provides conclusive evidence regarding the analgesic effectiveness of NSAIDs for acute sickle cell pain. Moreover, they were limited by the small sample size and number of observed VOCs.
Without further prospective studies, the use of NSAIDs in SCD is based on expert opinion and clinical experience. The potential gastrointestinal and renal adverse effects of NSAIDs preclude the use of NSAIDS chronically or for prolonged duration. Understandably, given that renal disease is a common comorbidity and a complication of SCD, clinicians are reluctant to use NSAIDs regularly for SCD pain.72 NSAIDs, however, can be an important component of a multimodal analgesia to address mild-to-moderate acute SCD pain. Short-term use (less than 5 days) in SCD patients without renal insufficiency (creatine <1.0) can be considered on an individualized basis, taking into consideration the patient’s other predisposing factors.80 Brief use of NSAIDs should be considered from a mechanistic perspective as a component of the multimodal analgesic regimen even as preclinical studies in transgenic mice with SCD continue to look at the potential of other COX-2 inhibitors in the treatment of SCD pain without the undesired renal adverse effects.81,82
Ketamine
Used for over 50 years as an anesthetic, ketamine is a phencyclidine analogue with potent analgesic properties. It is cheap and widely accessible even in low resource settings and available in multiple formulations, including intravenous, oral, intramuscular, intrarectal or intranasal routes. Ketamine is a competitive antagonist of the N-methyl-D-aspartate (NMDA) receptor, which is a glutamate receptor and ion channel found in nerve cells.83 Preclinical and clinical studies have highlighted that NMDA receptors are involved in central sensitization.84 In fact, activation of NMDA receptors is believed to play a key role in inducing and maintaining central sensitization. Moreover, significant cross-talk exists between mu-opioid receptors and NMDA receptors. Mechanistic studies provide strong evidence that antagonism of NMDA receptors plays a preventive role in the hyperalgesia pathway that is triggered by interaction of opioids with their receptors.84,85
Thus, ketamine is a unique non-opioid adjunct with both analgesic properties and potential opioid sparing effects. In addition, ketamine interacts with several other binding sites, including mu-opioid receptors, cholinergic and muscarinic receptors, although the clinical contribution of these interactions has not been clearly elucidated.83
Ketamine use, at subanesthetic concentrations, has grown significantly over the past decade. Refractory depression and chronic pain are now common conditions where subanesthetic ketamine is employed. Although supported by less compelling evidence, subanesthetic ketamine as a sole analgesic or adjunct to opioid therapy for acute pain has also risen in popularity. Recently published consensus guidelines from a pain and anesthesiology expert panel on the use of subanesthetic ketamine for acute pain management included acute pain secondary to VOCs as an indication for subanesthetic ketamine as an analgesic adjunct, although the supporting evidence was of low certainty.86 Moreover, even more importantly, guidelines published for management of sickle cell pain by the ASH expert panel included subanesthetic ketamine infusion for refractory sickle cell pain.27
Much of the early evidence for subanesthetic ketamine infusions as an adjunctive analgesic for a VOC is from small case series involving two to nine patients.87–90 Two more recent published studies from a tertiary pediatric hospital have the largest cohort of patients receiving subanesthetic ketamine infusion in the non-perioperative setting. One reviewed the hospital course of 230 patients, 181 of whom had SCD, who received subanesthetic ketamine infusions and found that patients with pain secondary to cancer or inflammation appeared to have the greatest reduction in pain scores.91 Another study from the same center evaluated the course of 85 SCD patients admitted for VOC who received sub-anesthetic ketamine infusions ranging from 0.05 to 0.4 mg/kg/h. The study noted an associated decrease in pain intensity and opioid consumption. In addition, longer infusion duration, younger age and male sex appeared to be independent predictors of greater pain reduction (p=0.018).92 All of these studies used ketamine infusions as an adjunctive analgesia – meaning in addition to standard opioid therapy.87–92
At the time of our literature search, there were no published prospective studies on the use of subanesthetic ketamine infusions for VOC management. However, there is one prospective, noninferiority trial that randomized 240 pediatric patients who presented to a large teaching hospital in Uganda to receive either a single dose of 1 mg/kg of IV ketamine or 0.1 mg/kg of IV morphine. This study found no statistical difference in pain scores between the two randomized groups. The ketamine group was associated with a faster onset of analgesia, but greater likelihood of side effects – nystagmus and dysphoria being the most common.93 It is important to note that a ketamine dose of 1 mg/kg is associated with greater side effects.94 In addition, evidence from the perioperative setting suggests that there is no significant analgesic benefit from a single dose of ketamine.95
Currently, the data for ketamine as an analgesic for acute SCD pain are lacking and inconclusive. However, if one considers what is known about the mechanism of SCD pain thus far and the mechanistic target of ketamine, ketamine may have potential benefits for acute SCD pain for the following reasons. In general, central sensitization does not play a role in acute pain.85 However, acute SCD pain is unique from other acute pain entities (eg, acute perioperative pain) in that it occurs as a symptomatic manifestation of a chronic painful disease – that is sickle cell disease. Moreover, in SCD, central sensitization along with inhibition of descending inhibitory pathways is postulated to play a key role in the development of chronic pain, which is highly prevalent among SCD patients.15 In summary, when an SCD patient presents in the acute setting for pain, it is possible they are experiencing acute on chronic pain.18 For this patient, ketamine, from a mechanistic perspective, could have analgesic and opioid-sparing benefits.
Ketamine, as a subanesthetic infusion adjunct for a VOC, is deserving of further clinical exploration, particularly as up-front adjunct to standard therapy, during the initial phase of the pain crisis (Figure 1), rather than as a rescue when standard opioid therapy has failed because of its mechanistic targets. As we wait for clinical efficacy data on ketamine for acute SCD pain, it is important that dosing is appropriate to minimize dysphoric side effects. In general, when used at subanesthetic concentrations, ketamine boluses should be 0.35 mg/kg or less, and infusions should not exceed 1.2 mg/kg/h in non-intensive care settings.86
Anticonvulsants (Gabapentin and Pregabalin)
Over the past decade, a deeper understanding of VOCs and the neurobiology of SCD has revealed that acute sickle cell pain has nociceptive, inflammatory, and neuropathic components.15,96 Neuropathic pain is generally defined as pain secondary to injury/lesion or disease of the somatosensory system. Hyperalgesia – increased sensitivity to a painful stimulus – and allodynia – pain that is provoked by a stimulus that is normally non-painful – are defining descriptors of neuropathic pain. The mechanism for sickle cell-related neuropathic pain is believed to be vaso-occlusion of the vessels supplying blood to nerves, then leading to peripheral nerve injury.65
In general, neuropathic pain is not responsive to analgesics such as opioids, NSAIDs or acetaminophen. Gabapentin and pregabalin – analogues of the neurotransmitter, gamma-aminobutyric acid (GABA) – are the two most widely prescribed analgesics for neuropathic pain and are considered first-line treatment for neuropathic pain. The specific mechanism of gabapentin (and pregabalin) is unknown, but it is believed to bind to α2δ-1 subunit of voltage gated calcium channels, leading to decreased central sensitization.97,98 Adverse effects include sedation, dizziness, and peripheral edema. Dosing should be adjusted in patients with renal disease due to the drugs’ renal excretion.98
Preclinical studies in SCD mice found that a single dose of gabapentin alleviated acute sickle cell pain induced by hypoxia/reoxygenation tissue damage.99 Human studies, however, provide limited evidence about the anticonvulsant’s effectiveness in SCD. To date, there have been two published RCTs100,101 examining the role of either gabapentin or pregabalin for treatment of acute sickle cell pain101 or chronic sickle cell pain.100 Puri et al101 randomized 90 pediatric sickle cell patients with an acute VOC to single dose of gabapentin (15 mg/kg) or placebo plus standard opioid therapy. The study reported a ≥33% decrease in pain scores in 68% of the gabapentin group compared to 60% of the placebo group (p = 0.23). Interestingly, the study also found that patients with the more severe SCD genotype (HbSS) had a significantly greater reduction in mean absolute pain scores (p = 0.032). It is important to highlight that the Puri et al study did not assess for the presence of neuropathic pain among study participants, and thus the differences in the efficacy of gabapentin among SCD patients with neuropathic pain compared to those without neuropathic pain could not be determined.
In designing future efficacy studies, it is important to take into consideration that analgesic effects of gabapentin are not immediate and may not be observed after a single dose.102 For studying these anticonvulsants’ clinical efficacy in acute sickle cell pain, a study design that initiates gabapentin or pregabalin at the onset of the VOC and continues for at least 2–3 weeks, that is through the entire duration of the pain crisis (Figure 1), would be optimized to detect any analgesic benefit of the drugs over placebo.
Lidocaine
Lidocaine is a local anesthetic that is known to exert its mechanistic effects through the blockade of voltage-gated sodium channels. It was first established as an antiarrhythmic drug. In the 1960s, the analgesic properties of systemic lidocaine were published in primarily anesthesia journals. Both systemic and transdermal lidocaine are second-line treatment for neuropathic pain.103 There is growing basic science evidence that lidocaine has several molecular targets beyond sodium channels, including NMDA receptors and G-protein coupled receptors, which are contributors to its analgesic and anti-hyperalgesic effects.103–105
The literature regarding the use of lidocaine in sickle cell patients is sparse. A prospective, single arm, multicenter trial of 5% lidocaine topical patches in 39 pediatric patients (23 with SCD) in France is the only published prospective study examining the use of lidocaine in SCD. Patients included in the study were characterized as experiencing either neuropathic pain or boney, localized, superficial pain secondary to a VOC. At least a two-point reduction in pain scores was observed in 48.6% of the patients after 12-hour application of the patch for two consecutive days. Clinical efficacy, however, could not be assessed in this study, given the small number of patients and lack of randomization to placebo.106 Moreover, a subgroup analysis of the pain outcomes of SCD patients was not reported.
Systemic lidocaine administered as infusions is even more rare among sickle cell patients. Lidocaine’s narrow therapeutic index raises some challenges related to patient safety and medication administration error, especially if administered in settings where clinical staff lacks familiarity with the drug. Nevertheless, there are two published case series involving a combined total of 13 sickle cell patients who received systemic lidocaine infusions for refractory pain secondary to VOC.107,108
A New Explanatory Model for Sickle Cell Pain
Multimodal analgesia for the treatment of SCD pain presupposes a multi-mechanistic model of SCD pain causality and relief. In infants, SCD pain initially occurs in response to vaso-occlusive ischemia or tissue injury.7 However, there is evidence this initial casual model becomes far more complex with time and repeated pain experiences. Further, SCD patients may report severe acute pain without abnormalities in routine laboratory tests often used as a marker of tissue vaso-occlusion or ischemia.109,110 Moreover, patients can experience pain out of proportion to their laboratory abnormalities, which is not only frustrating to both physicians and patients but also provokes mistrust in patients’ self-reported pain scores.111 A new explanatory model of sickle cell pain should fully capture the multidimensional nature of pain experienced by patients. It should suggest new SCD pain targets as well as new measures of analgesic efficacy for SCD.
In 1989, Richard Melzack, a pain psychologist, proposed a new explanatory model for pain – the neuromatrix theory of pain.112,113 Several decades prior, Melzack, along with his colleague, Patrick Wall, developed the gate theory, which emphasized the role of the central nervous system in pain perception.114 In this new explanatory model, Melzack expanded upon the gate theory to suggest that multiple parts of the central nervous system (CNS) – the “body-self neuromatrix” – work together to generate pain, and that pain can be produced independently of peripheral sensory input.115 Melzack also suggested that the pattern of neural networks that make up the neuromatrix is genetically predetermined but later influenced by environmental factors.
Melzack’s neuromatrix theory shifts away from the long-held and accepted Cartesian pain model where pain is a unique biomarker of tissue damage or inflammation that is detected by the peripheral nervous system. In this older model, the CNS only plays a passive role as a receiver of pain signals.116 However, Melzack’s analysis of phantom limb pain, where periphery sensory input is absent but the experience of pain is quite real, led him to propose that pain can be felt in the absence of input from the body to the CNS.112,117 He suggested that the components of the CNS that make up the “body-self neuromatrix” receive three types of inputs – the sensory-discriminative, cognitive-evaluative, and motivational-affective– to generate pain perception. The sensory-discriminative input consists of cutaneous, musculoskeletal, visceral, visual, and other sensory inputs, which define the location and quality of the pain perception. Cognitive-evaluative input provides interpretation or meaning for the experience. This includes factors such as memories of past experiences, cultural learning, etc. The motivational-affective domain adds an emotion or “feeling” to the experience. These three domains interact to generate pain perception as one of three potential outputs. Pain can accompany two other outputs: voluntary and involuntary motor actions or stress. These two outputs can also occur without pain perception.112,113,115
The evidence supporting Melzack’s neuromatrix model is strong,118–120 and includes functional imaging studies.121 This multidimensional conception of pain perception can also serve as a new explanatory model for SCD pain (Figure 2). Within SCD pain, the sensory input from vaso-occlusive ischemia has been extensively studied. However, there is growing evidence that this is not the sole factor for generating SCD pain. The clinical understanding of SCD pain has evolved to acknowledge the potential roles that other factors, such as anxiety, depression,20,122 and catastrophizing,123 have in the perception of SCD pain. Adapting Melzack’s model, these factors contribute to the cognitive-evaluative domain of the neuromatrix. The limbic system and its associated regulation of autonomic and endocrine responses provide an emotional input in the model. The literature is scarce with regards to this area within SCD pain.124,125 Endocrine and autonomic nervous system alterations in SCD pain require further exploration. Last, the role of motor/physical function126,127 and stress (or biological markers of stress)128–130 in SCD continues to be studied, although the literature in this area is also not robust.
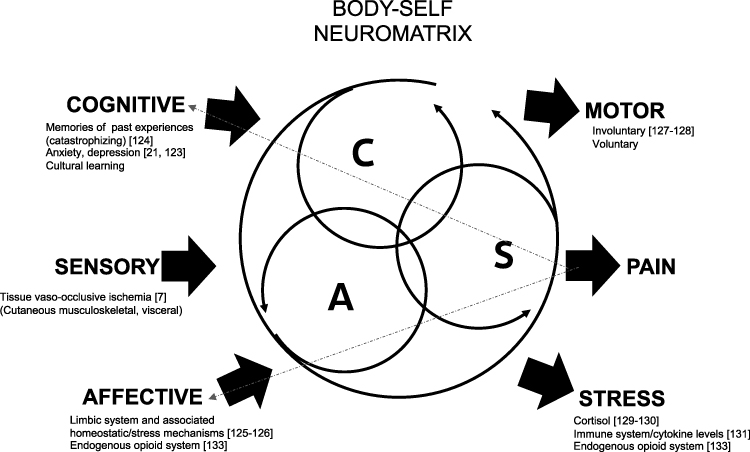 |
Figure 2 Three patterns of input, cognitive (C), sensory (S), and affective (A), interact to generate three potential outputs, pain perception, motor and stress. Cognitive-evaluative domain provides interpretation of the sensory experience. Memories of past experiences, underlying anxiety, depression and cultural/familial learning can influence this interpretation. Limbic system along with resultant endocrine and autonomic regulations contributes an emotion/affective to the experience. The output of pain perception can feed back into the cognitive and affective inputs. Adapted and modified with permission from Melzack R. Pain and the neuromatrix in the brain. J Dent Educ. 2001;65(12):1378–1382. © American Dental Education Association.112 Abbreviation: SCD, sickle cell disease. |
We advocate an application of Melzack’s neuromatrix model to SCD pain. This has several implications for future analgesic trials, particularly non-opioid analgesics. First, comprehensive SCD pain phenotyping requires an assessment of all three output domains. This necessitates patient reported outcomes (PROs) that have been validated in the SCD population to comprehensively measure outcomes in each of these domains. For example, PROs that assess catastrophizing, somatization, sleep, depression, and anxiety could provide a multidimensional assessment of the pain experiences of individuals with SCD.131 Objective or biological markers are also needed. Focusing solely on pain perception as an outcome may lead to the exclusion of an effective adjunct, which may not change pain scores, but it may improve physical function, and decrease markers of stress – and, thus, improve patient reported quality of life. Second, this new explanatory model for SCD pain (Figure 2) offers additional interventions. For instance, antidepressants may warrant further exploration in addressing the cognitive-evaluative input to pain perception. The model also offers support for the incorporation of non-pharmacological interventions into the therapeutic regimen for SCD pain. The efficacy of non-pharmacological interventions for management of SCD pain is unknown because of the lack of robust and high-quality studies.62 However, a multimodal approach that incorporates non-opioid analgesics and non-pharmacological interventions is needed to treat SCD pain.
Lastly, there is some evidence that the endogenous opioid system plays a role in the emotional and stress responses.132 It is unclear the degree to which exposure to high-dose exogenous opioids, such as in the case of SCD, modifies some factors in this explanatory model, and this may be the area that requires further investigation.
Conclusions and Future Directions
An adaptation of Richard Melzack’s multidimensional pain model –the neuromatrix theory– as a framework for conceptualizing sickle cell pain indicates that management of acute sickle cell pain should be multimodal and individualized. However, significant gaps remain in analgesic management of acute SCD pain. Standard analgesic management for VOCs has been based on little evidence, and expert consensus has evolved mostly from expert-panel-guided opinions and reviews of the scant literature evidence. This is especially true for opioids, long considered the mainstay of treatment for acute SCD pain. Many analgesic drugs, such as intravenous lidocaine, ketamine, pregabalin and gabapentin, are relatively untested in SCD. They rely on non-opioid mechanisms. However, at the moment, they are used inconsistently. Given the complex pathophysiology of SCD pain, it is imperative to test whether their widespread use is warranted, by conducting well-designed multi-center RCTs. It is encouraging that, since the completion of our literature search, several studies assessing the efficacy of these adjuncts for sickle cell pain have been published.133–136 These analgesics have the potential to expand treatment for VOCs to a true multimodal approach, and as a result, attenuate some of the possible adverse effects of the opioid analgesics. Our explanatory pain model for SCD captures the multidimensional nature of the pain experience and suggests new SCD pain targets as well as new measures of efficacy.
Acknowledgments
We are grateful for Elizabeth Moreton (Librarian, UNC-Chapel Hill) who conducted the literature search for this review.
Funding
MOK was supported in part by funding from the National Heart, Lung, and Blood Institute’s Programs to Increase Diversity Among Individuals engaged in Health-Related Research (PRIDE): R25HL106365.
Disclosure
Dr Wally R Smith reports personal fees as a consultant from Emmaus Pharmaceutical, Pfizer; consultant, speakers’ bureau for Global Blood Therapeutics; grants from Health Resources and Services Administration, National Heart Lung and Blood Institute, NIH, Patient-Centered Outcomes Research Institute; personal fees, as a consultant and/or DSMB member for Novartis Pharmaceuticals, Novo-Nordisk; grants as an investigator, pharmaceutical trial from Imara, Forma, and Agios, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi:10.1038/nrdp.2018.10
2. Dampier C, Ely E, Brodecki D, O’Neal P. Home management of pain in sickle cell disease: a daily diary study in children and adolescents. J Pediatr Hematol Oncol. 2002;24(8):643–647. doi:10.1097/00043426-200211000-00008
3. Dampier C, Ely B, Brodecki D, O’Neal P. Characteristics of pain managed at home in children and adolescents with sickle cell disease by using diary self-reports. J Pain. 2002;3(6):461–470. doi:10.1054/jpai.2002.128064
4. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148(2):94–101. doi:10.7326/0003-4819-148-2-200801150-00004
5. Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325(1):11–16. doi:10.1056/nejm199107043250103
6. Dampier C, LeBeau P, Rhee S, et al. Health-related quality of life in adults with sickle cell disease (SCD): a report from the comprehensive sickle cell centers clinical trial consortium. Am J Hematol. 2011;86(2):203–205. doi:10.1002/ajh.21905
7. Ballas SK. Pathophysiology and principles of management of the many faces of the acute vaso‐occlusive crisis in patients with sickle cell disease. Eur J Haematol. 2015;95(2):113–123. doi:10.1111/ejh.12460
8. Fishman SM, Rathmell JP. Bonica’s Management of Pain. Lippincott Williams & Wilkins; 2010.
9. Brandow AM, Wandersee NJ, Dasgupta M, et al. Substance P is increased in patients with sickle cell disease and associated with haemolysis and hydroxycarbamide use. Br J Haematol. 2016;175(2):237–245. doi:10.1111/bjh.14300
10. Kohli DR, Li Y, Khasabov SG, et al. Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids. Blood. 2010;116(3):456–465. doi:10.1182/blood-2010-01-260372
11. Hillery CA, Kerstein PC, Vilceanu D, et al. Transient receptor potential vanilloid 1 mediates pain in mice with severe sickle cell disease. Blood. 2011;118(12):3376–3383. doi:10.1182/blood-2010-12-327429
12. Cain DM, Vang D, Simone DA, Hebbel RP, Gupta K. Mouse models for studying pain in sickle disease: effects of strain, age, and acuteness. Br J Haematol. 2012;156(4):535–544. doi:10.1111/j.1365-2141.2011.08977.x
13. Antunes FD, Propheta VGS, Vasconcelos HA, Cipolotti R. Neuropathic pain in patients with sickle cell disease: a cross-sectional study assessing teens and young adults. Ann Hematol. 2017;96(7):1121–1125. doi:10.1007/s00277-017-2984-z
14. Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer. 2014;61(3):512–517. doi:10.1002/pbc.24838
15. Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: a systematic review of the literature. Pediatr Blood Cancer. 2015;62(9):1501–1511. doi:10.1002/pbc.25574
16. Field JJ, Ballas SK, Campbell CM, et al. AAAPT diagnostic criteria for acute sickle cell disease pain. J Pain. 2019;20(7):746–759. doi:10.1016/j.jpain.2018.12.003
17. Dampier C, Palermo TM, Darbari DS, Hassell K, Smith W, Zempsky W. AAPT diagnostic criteria for chronic sickle cell disease pain. J Pain. 2017;18(5):490–498. doi:10.1016/j.jpain.2016.12.016
18. Lanzkron S, Little J, Field J, et al. Increased acute care utilization in a prospective cohort of adults with sickle cell disease. Blood Adv. 2018;2(18):2412–2417. doi:10.1182/bloodadvances.2018018382
19. Smith WR, Scherer M. Sickle-cell pain: advances in epidemiology and etiology. Hematology Am Soc Hematol Educ Program. 2010;2010:409–415. doi:10.1182/asheducation-2010.1.409
20. Osunkwo I, Andemariam B, Minniti CP, et al. Impact of sickle cell disease on patients’ daily lives, symptoms reported, and disease management strategies: results from the international Sickle Cell World Assessment Survey (SWAY). Am J Hematol. 2021;96(4):404–417. doi:10.1002/ajh.26063
21. Fingar KR, Owens PL, Reid LD, Mistry KB, Barrett ML. Characteristics of inpatient hospital stays involving sickle cell disease, 2000–2016: statistical brief #251. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); 2006.
22. Shah N, Bhor M, Xie L, Paulose J, Yuce H. Sickle cell disease complications: prevalence and resource utilization. PLoS One. 2019;14(7):e0214355. doi:10.1371/journal.pone.0214355
23. Yusuf HR, Atrash HK, Grosse SD, Parker CS, Grant AM. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999–2007. Am J Prev Med. 2010;38(4Suppl):S536–S541. doi:10.1016/j.amepre.2010.01.001
24. Evensen CT, Treadwell MJ, Keller S, et al. Quality of care in sickle cell disease: cross-sectional study and development of a measure for adults reporting on ambulatory and emergency department care. Medicine. 2016;95(35):e4528. doi:10.1097/md.0000000000004528
25. Kanter J, Gibson R, Lawrence RH, et al. Perceptions of US adolescents and adults with sickle cell disease on their quality of care. JAMA Netw Open. 2020;3(5):e206016. doi:10.1001/jamanetworkopen.2020.6016
26. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. doi:10.1001/jama.2014.10517
27. Brandow AM, Carroll CP, Creary S, et al. American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv. 2020;4(12):2656–2701. doi:10.1182/bloodadvances.2020001851
28. Linton EA, Goodin DA, Hankins JS, et al. A survey-based needs assessment of barriers to optimal sickle cell disease care in the emergency department. Ann Emerg Med. 2020;76(3s):S64–s72. doi:10.1016/j.annemergmed.2020.08.013
29. Kanter J, Smith WR, Desai PC, et al. Building access to care in adult sickle cell disease: defining models of care, essential components, and economic aspects. Blood Adv. 2020;4(16):3804–3813. doi:10.1182/bloodadvances.2020001743
30. Tanabe P, Silva S, Bosworth HB, et al. A randomized controlled trial comparing two vaso-occlusive episode (VOE) protocols in sickle cell disease (SCD). Am J Hematol. 2018;93(2):159–168. doi:10.1002/ajh.24948
31. International Association for the Study of Pain. Pain treatment services. Available from: https://www.iasp-pain.org/Education/Content.aspx?ItemNumber=1381.
32. Flor H, Fydrich T, Turk DC. Efficacy of multidisciplinary pain treatment centers: a meta-analytic review. Pain. 1992;49(2):221–230. doi:10.1016/0304-3959(92)90145-2
33. National Academies of Sciences E. Medicine, health. In: Martinez RM, Osei-Anto HA, McCormick M, editors. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. National Academies Press (US) Copyright 2020 by the National Academy of Sciences. All rights reserved; 2020.
34. Rosero EB, Joshi GP. Preemptive, preventive, multimodal analgesia: what do they really mean? Plast Reconstr Surg. 2014;134(4Suppl 2):85s–93s. doi:10.1097/prs.0000000000000671
35. Raffa RB, Pergolizzi JV
36. Argoff CE, Albrecht P, Irving G, Rice F. Multimodal analgesia for chronic pain: rationale and future directions. Pain Med. 2009;10(Suppl 2):S53–66. doi:10.1111/j.1526-4637.2009.00669.x
37. Walsh DA, McWilliams DF. Mechanisms, impact and management of pain in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10(10):581–592. doi:10.1038/nrrheum.2014.64
38. Miller ST, Kim HY, Weiner D, et al. Inpatient management of sickle cell pain: a ‘snapshot’ of current practice. Am J Hematol. 2012;87(3):333–336. doi:10.1002/ajh.22265
39. Brookoff D, Polomano R. Treating sickle cell pain like cancer pain. Ann Intern Med. 1992;116(5):364–368. doi:10.7326/0003-4819-116-5-364
40. Portenoy RK. Treating sickle cell pain like cancer pain. Ann Intern Med. 1992;117(3):264–265.
41. Ballas SK, Rubin RN, Gabuzda TC. Treating sickle cell pain like cancer pain. Ann Intern Med. 1992;117(3):
42. Agency for Health Care Policy and Research. Acute pain management in infants, children, and adolescents: operative and medical procedures. Clin Pract Guidel Quick Ref Guide Clin. 1992;1b:1–22.
43. Agency for Health Care Policy and Research, US Department of Health and Human Services. Clinicians’ quick reference guide to acute pain management in infants, children, and adolescents: operative and medical procedures. Pain Management Guideline Panel. J Pain Symptom Manage. 1992;7(4):229–242. doi:10.1016/0885-3924(92)90079-w
44. Kolodny A, Courtwright DT, Hwang CS, et al. The prescription opioid and heroin crisis: a public health approach to an epidemic of addiction. Annu Rev Public Health. 2015;36:559–574. doi:10.1146/annurev-publhealth-031914-122957
45. Jacobson SJ, Kopecky EA, Joshi P, Babul N. Randomised trial of oral morphine for painful episodes of sickle-cell disease in children. Lancet. 1997;350(9088):1358–1361. doi:10.1016/s0140-6736(97)08462-6
46. Finan PH, Carroll CP, Moscou-Jackson G, et al. Daily opioid use fluctuates as a function of pain, catastrophizing, and affect in patients with sickle cell disease: an electronic daily diary analysis. J Pain. 2018;19(1):46–56. doi:10.1016/j.jpain.2017.08.010
47. Busse JW, Wang L, Kamaleldin M, et al. Opioids for chronic noncancer pain: a systematic review and meta-analysis. JAMA. 2018;320(23):2448–2460. doi:10.1001/jama.2018.18472
48. Machelska H, Celik M. Advances in achieving opioid analgesia without side effects. Front Pharmacol. 2018;9:1388. doi:10.3389/fphar.2018.01388
49. Ruta NS, Ballas SK. The opioid drug epidemic and sickle cell disease: guilt by association. Pain Med. 2016;17(10):1793–1798. doi:10.1093/pm/pnw074
50. Smith WR, McClish DK, Roberts JD, et al. Prescription opioid misuse index in sickle cell patients: a brief questionnaire to assess at-risk for opioid abuse. J Opioid Manag. 2019;15(4):323–331. doi:10.5055/jom.2019.0517
51. Smith WR, McClish DK, Dahman BA, et al. Daily home opioid use in adults with sickle cell disease: the PiSCES project. J Opioid Manag. 2015;11(3):243–253. doi:10.5055/jom.2015.0273
52. Ballas SK. How I treat acute and persistent sickle cell pain. Mediterr J Hematol Infect Dis. 2020;12(1):e2020064. doi:10.4084/mjhid.2020.064
53. Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145–161. doi:10.36076/ppj.2011/14/145
54. Rekatsina M, Paladini A, Piroli A, Zis P, Pergolizzi JV, Varrassi G. Pathophysiologic approach to pain therapy for complex pain entities: a narrative review. Pain Ther. 2020;9(1):7–21. doi:10.1007/s40122-019-00147-2
55. Chandanwale AS, Sundar S, Latchoumibady K, et al. Efficacy and safety profile of combination of tramadol-diclofenac versus tramadol-paracetamol in patients with acute musculoskeletal conditions, postoperative pain, and acute flare of osteoarthritis and rheumatoid arthritis: a Phase III, 5-day open-label study. J Pain Res. 2014;7:455–463. doi:10.2147/jpr.S67817
56. Fitzcharles MA, Shir Y. Management of chronic pain in the rheumatic diseases with insights for the clinician. Ther Adv Musculoskelet Dis. 2011;3(4):179–190. doi:10.1177/1759720x11408999
57. Regel A, Sepriano A, Baraliakos X, et al. Efficacy and safety of non-pharmacological and non-biological pharmacological treatment: a systematic literature review informing the 2016 update of the ASAS/EULAR recommendations for the management of axial spondyloarthritis. RMD Open. 2017;3(1):e000397. doi:10.1136/rmdopen-2016-000397
58. Dalal RS, Palchaudhuri S, Snider CK, et al. A multimodal intervention using nonopioid analgesics is associated with reduced intravenous opioid exposure among hospitalized patients with inflammatory bowel diseases. Am J Gastroenterol. 2020;115(9):1474–1485. doi:10.14309/ajg.0000000000000806
59. Dalal RS, Palchaudhuri S, Snider CK, Lewis JD, Mehta SJ, Lichtenstein GR. Exposure to intravenous opioids is associated with future exposure to opioids in hospitalized patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2020;18(10):2269–2278.e3. doi:10.1016/j.cgh.2019.12.024
60. Cooper TE, Hambleton IR, Ballas SK, Johnston BA, Wiffen PJ. Pharmacological interventions for painful sickle cell vaso-occlusive crises in adults. Cochrane Database Syst Rev. 2019;2019(11):Cd012187. doi:10.1002/14651858.CD012187.pub2
61. Ballas SK. Pain management of sickle cell disease. Hematol Oncol Clin North Am. 2005;19(5):785–802, v. doi:10.1016/j.hoc.2005.07.008
62. Lakkakula B, Sahoo R, Verma H, Lakkakula S. Pain management issues as part of the comprehensive care of patients with sickle cell disease. Pain Manag Nurs. 2018;19(6):558–572. doi:10.1016/j.pmn.2018.06.004
63. Moody K, Abrahams B, Baker R, et al. A randomized trial of yoga for children hospitalized with sickle cell vaso-occlusive crisis. J Pain Symptom Manage. 2017;53(6):1026–1034. doi:10.1016/j.jpainsymman.2016.12.351
64. Ballas SK. The sickle cell painful crisis in adults: phases and objective signs. Hemoglobin. 1995;19(6):323–333. doi:10.3109/03630269509005824
65. Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647–3656. doi:10.1182/blood-2012-04-383430
66. Jahr JS, Lee VK. Intravenous Acetaminophen. Anesthesiol Clin. 2010;28(4):619–645. doi:10.1016/j.anclin.2010.08.006
67. Nguyen LP, Nguyen L, Austin JP. A quality improvement initiative to decrease inappropriate intravenous acetaminophen use at an academic medical center. Hosp Pharm. 2020;55(4):253–260. doi:10.1177/0018578719841054
68. Moller PL, Sindet-Pedersen S, Petersen CT, Juhl GI, Dillenschneider A, Skoglund LA. Onset of Acetaminophen analgesia: comparison of oral and intravenous routes after third molar surgery. Br J Anaesth. 2005;94(5):642–648. doi:10.1093/bja/aei109
69. Baichoo P, Asuncion A, El-Chaar G. Intravenous acetaminophen for the management of pain during vaso-occlusive crises in pediatric patients. P T. 2019;44(1):5–8.
70. Dhebaria T, Sivitz A, Tejani C. Does intravenous acetaminophen reduce opioid requirement in pediatric emergency department patients with acute sickle cell crises? Acad Emerg Med. 2020;28:639–646. doi:10.1111/acem.14149
71. Brune K, Hinz B. The discovery and development of antiinflammatory drugs. Arthritis Rheum. 2004;50(8):2391–2399. doi:10.1002/art.20424
72. Han J, Saraf SL, Lash JP, Gordeuk VR. Use of anti-inflammatory analgesics in sickle-cell disease. J Clin Pharm Ther. 2017;42(5):656–660. doi:10.1111/jcpt.12592
73. Elia N, Lysakowski C, Tramèr MR. Does multimodal analgesia with Acetaminophen, nonsteroidal antiinflammatory drugs, or selective cyclooxygenase-2 inhibitors and patient-controlled analgesia morphine offer advantages over morphine alone? Meta-analyses of randomized trials. Anesthesiology. 2005;103(6):1296–1304. doi:10.1097/00000542-200512000-00025
74. Straube S, Derry S, McQuay HJ, Moore RA. Effect of preoperative Cox-II-selective NSAIDs (coxibs) on postoperative outcomes: a systematic review of randomized studies. Acta Anaesthesiol Scand. 2005;49(5):601–613. doi:10.1111/j.1399-6576.2005.00666.x
75. Doluee MT, Kakhki BR, Mir HH, Fateminayyeri M, Madanitorbati F, Hosseini S. Pain relief in the sickle-cell crisis: intravenous morphine versus ketorolac; a double-blind, randomized clinical trial. Article. Iran Red Crescent Med J. 2019;21(4). doi:10.5812/ircmj.83614
76. Bartolucci P, El Murr T, Roudot-Thoraval F, et al. A randomized, controlled clinical trial of ketoprofen for sickle-cell disease vaso-occlusive crises in adults. Blood. 2009;114(18):3742–3747. doi:10.1182/blood-2009-06-227330
77. Hardwick WE
78. Perlin E, Finke H, Castro O, et al. Enhancement of pain control with ketorolac tromethamine in patients with sickle cell vaso-occlusive crisis. Am J Hematol. 1994;46(1):43–47. doi:10.1002/ajh.2830460108
79. Wright SW, Norris RL, Mitchell TR. Ketorolac for sickle cell vaso-occlusive crisis pain in the emergency department: lack of a narcotic-sparing effect. Ann Emerg Med. 1992;21(8):925–928. doi:10.1016/s0196-0644(05)82929-4
80. Baker M, Perazella MA. NSAIDs in CKD: are they safe? Am J Kidney Dis. 2020;76(4):546–557. doi:10.1053/j.ajkd.2020.03.023
81. Sadler KE, Stucky CL. Blocking COX-2 for sickle cell pain relief. Blood. 2019;133(18):1924–1925. doi:10.1182/blood-2019-03-900944
82. Singh J, Sharma D, Bansal R. Pyridazinone: an attractive lead for anti-inflammatory and analgesic drug discovery. Future Med Chem. 2017;9(1):95–127. doi:10.4155/fmc-2016-0194
83. Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci Ther. 2013;19(6):370–380. doi:10.1111/cns.12099
84. Raja SN, Sivanesan E, Guan Y. Central sensitization, N-methyl-d-aspartate receptors, and human experimental pain models. Anesthesiology. 2019;131(2):233–235. doi:10.1097/aln.0000000000002808
85. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926. doi:10.1016/j.jpain.2009.06.012
86. Schwenk ES, Viscusi ER, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for acute pain management from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):456–466. doi:10.1097/AAP.0000000000000806
87. Gimovsky AC, Fritton K, Viscusi E, Roman A. Evaluating the use of ketamine for pain control with sickle cell crisis in pregnancy: a report of 2 cases. A a Pract. 2018;10(1):20–22. doi:10.1213/xaa.0000000000000624
88. Tawfic QA, Faris AS, Kausalya R. The role of a low-dose ketamine-midazolam regimen in the management of severe painful crisis in patients with sickle cell disease. J Pain Symptom Manage. 2014;47(2):334–340. doi:10.1016/j.jpainsymman.2013.03.012
89. Zempsky WT, Loiselle KA, Corsi JM, Hagstrom JN. Use of low-dose ketamine infusion for pediatric patients with sickle cell disease-related pain: a case series. Clin J Pain. 2010;26(2):163–167. doi:10.1097/AJP.0b013e3181b511ab
90. Palm N, Floroff C, Hassig TB, Boylan A, Kanter J. Low-dose ketamine infusion for adjunct management during vaso-occlusive episodes in adults with sickle cell disease: a case series. J Pain Palliat Care Pharmacother. 2018;32(1):20–26. doi:10.1080/15360288.2018.1468383
91. Sheehy KA, Lippold C, Rice AL, Nobrega R, Finkel JC, Quezado ZM. Subanesthetic ketamine for pain management in hospitalized children, adolescents, and young adults: a single-center cohort study. J Pain Res. 2017;10:787–795. doi:10.2147/jpr.S131156
92. Nobrega R, Sheehy KA, Lippold C, Rice AL, Finkel JC, Quezado ZMN. Patient characteristics affect the response to ketamine and opioids during the treatment of vaso-occlusive episode-related pain in sickle cell disease. Pediatr Res. 2018;83(2):445–454. doi:10.1038/pr.2017.197
93. Lubega FA, DeSilva MS, Munube D, et al. Low dose ketamine versus morphine for acute severe vaso occlusive pain in children: a randomized controlled trial. Scand J Pain. 2018;18(1):19–27. doi:10.1515/sjpain-2017-0140
94. Gorlin AW, Rosenfeld DM, Ramakrishna H. Intravenous sub-anesthetic ketamine for perioperative analgesia. J Anaesthesiol Clin Pharmacol. 2016;32(2):160–167. doi:10.4103/0970-9185.182085
95. Himmelseher S, Durieux ME. Ketamine for perioperative pain management. Anesthesiology. 2005;102(1):211–220. doi:10.1097/00000542-200501000-00030
96. Puri L, Nottage KA, Hankins JS, Anghelescu DL. State of the art management of acute vaso-occlusive pain in sickle cell disease. Pediatr Drugs. 2018;20(1):29–42. doi:10.1007/s40272-017-0263-z
97. Luo ZD, Chaplan SR, Higuera ES, et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21(6):1868–1875. doi:10.1523/JNEUROSCI.21-06-01868.2001
98. Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. Nat Rev Dis Primers. 2017;3:17002. doi:10.1038/nrdp.2017.2
99. Sadler KE, Langer SN, Menzel AD, et al. Gabapentin alleviates chronic spontaneous pain and acute hypoxia-related pain in a mouse model of sickle cell disease. Br J Haematol. 2019;187(2):246–260. doi:10.1111/bjh.16067
100. Schlaeger JM, Molokie RE, Yao Y, et al. Management of sickle cell pain using pregabalin: a pilot study. Pain Manag Nurs. 2017;18(6):391–400. doi:10.1016/j.pmn.2017.07.003
101. Puri L, Anghelescu D, Hankins J, et al. Gabapentin for pain in sickle cell disease: results of a randomized Phase II clinical trial. Conference Abstract. Pediatr Blood Cancer. 2019;66:S7. doi:10.1002/pbc.27713
102. Chincholkar M. Gabapentinoids: pharmacokinetics, pharmacodynamics and considerations for clinical practice. Br J Pain. 2020;14(2):104–114. doi:10.1177/2049463720912496
103. Hermanns H, Hollmann MW, Stevens MF, et al. Molecular mechanisms of action of systemic lidocaine in acute and chronic pain: a narrative review. Br J Anaesth. 2019;123(3):335–349. doi:10.1016/j.bja.2019.06.014
104. Boas RA, Covino BG, Shahnarian A. Analgesic responses to i.v. lignocaine. Br J Anaesth. 1982;54(5):501–505. doi:10.1093/bja/54.5.501
105. Bartlett EE, Hutserani O. Xylocaine for the relief of postoperative pain. Anesth Analg. 1961;40:296–304. doi:10.1213/00000539-196105000-00011
106. Rousseau V, Morelle M, Arriuberge C, et al. Efficacy and tolerance of lidocaine 5% patches in neuropathic pain and pain related to vaso-occlusive sickle cell crises in children: a Prospective Multicenter Clinical Study. Pain Pract. 2018;18(6):788–797. doi:10.1111/papr.12674
107. Puri L, Morgan KJ, Anghelescu DL. Ketamine and lidocaine infusions decrease opioid consumption during vaso-occlusive crisis in adolescents with sickle cell disease. Curr Opin Support Palliat Care. 2019;13(4):402–407. doi:10.1097/SPC.0000000000000437
108. Nguyen NL, Kome AM, Lowe DK, Coyne P, Hawks KG. Intravenous lidocaine as an adjuvant for pain associated with sickle cell disease. J Pain Palliat Care Pharmacother. 2015;29(4):359–364. doi:10.3109/15360288.2015.1082009
109. Lopez BL, Griswold SK, Navek A, Urbanski L. The complete blood count and reticulocyte count–are they necessary in the evaluation of acute vasoocclusive sickle-cell crisis? Acad Emerg Med. 1996;3(8):751–757. doi:10.1111/j.1553-2712.1996.tb03510.x
110. Chapman JI, El-Shammaa EN, Bonsu BK. The utility of screening laboratory studies in pediatric patients with sickle cell pain episodes. Am J Emerg Med. 2004;22(4):258–263. doi:10.1016/j.ajem.2004.04.014
111. Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. 2020;105(3):237–246. doi:10.1111/ejh.13430
112. Melzack R. Pain and the neuromatrix in the brain. J Dent Educ. 2001;65(12):1378–1382. doi:10.1002/j.0022-0337.2001.65.12.tb03497.x
113. Melzack R. Evolution of the neuromatrix theory of pain. The Prithvi Raj Lecture: presented at the third World Congress of World Institute of Pain, Barcelona 2004. Pain Pract. 2005;5(2):85–94. doi:10.1111/j.1533-2500.2005.05203.x
114. Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150(3699):971–979. doi:10.1126/science.150.3699.971
115. Melzack R. From the gate to the neuromatrix. Pain. 1999;Suppl 6:S121–s126. doi:10.1016/s0304-3959(99)00145-1
116. Trachsel LA, Cascella M. Pain theory. In: StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC; 2021.
117. Melzack R. Phantom limbs and the concept of a neuromatrix. Trends Neurosci. 1990;13(3):88–92. doi:10.1016/0166-2236(90)90179-e
118. Arntz A, Claassens L. The meaning of pain influences its experienced intensity. Pain. 2004;109(1–2):20–25. doi:10.1016/j.pain.2003.12.030
119. Moseley GL, Arntz A. The context of a noxious stimulus affects the pain it evokes. Pain. 2007;133(1–3):64–71. doi:10.1016/j.pain.2007.03.002
120. Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288(5472):1769–1772. doi:10.1126/science.288.5472.1769
121. Derbyshire SW. Exploring the pain “neuromatrix. Curr Rev Pain. 2000;4(6):467–477. doi:10.1007/s11916-000-0071-x
122. Levenson JL, McClish DK, Dahman BA, et al. Depression and anxiety in adults with sickle cell disease: the PiSCES project. Psychosom Med. 2008;70(2):192–196. doi:10.1097/PSY.0b013e31815ff5c5
123. Hollins M, Stonerock G, Kisaalita N, Jones S, Orringer E, Gil K. Hypervigilance and catastrophizing, but not temporal summation, are elevated in sickle cell disease. J Pain. 2010;11(4):S20. doi:10.1016/j.jpain.2010.01.086
124. el-Hazmi MA, Bahakim HM, al-Fawaz I. Endocrine functions in sickle cell anaemia patients. J Trop Pediatr. 1992;38(6):307–313. doi:10.1093/tropej/38.6.307
125. Veluswamy S, Shah P, Denton CC, Chalacheva P, Khoo MCK, Coates TD. Vaso-occlusion in sickle cell disease: is autonomic dysregulation of the microvasculature the trigger? J Clin Med. 2019;8(10):1690. doi:10.3390/jcm8101690
126. Hyacinth O, Machado Silva-Rodrigues F, Darezzo Rodrigues Nunes M, et al. Pain experience, physical function, pain coping, and catastrophizing in children with sickle cell disease who had normal and abnormal sensory patterns. J Pain Symptom Manage. 2020;60(6):1079–1091. doi:10.1016/j.jpainsymman.2020.07.006
127. Zempsky W, Corsi J, Loiseiie K, Hagstrom J, Palermo T, Caseiia J. Functional assessment for children hospitalized for sickle cell pain: a new paradigm. J Pain. 2010;11(4):S2. doi:10.1016/j.jpain.2010.01.012
128. Jhun EH, Sadhu N, Yao Y, et al. Glucocorticoid receptor single nucleotide polymorphisms are associated with acute crisis pain in sickle cell disease. Pharmacogenomics. 2018;19(13):1003–1011. doi:10.2217/pgs-2018-0064
129. Akinlade KS, Atere AD, Olaniyi JA, Rahamon SK, Adewale CO. Serum copeptin and cortisol do not accurately predict sickle cell anaemia vaso-occlusive crisis as C-reactive protein. PLoS One. 2013;8(11):e77913. doi:10.1371/journal.pone.0077913
130. Silva-Junior AL, Garcia NP, Cardoso EC, et al. Immunological hallmarks of inflammatory status in vaso-occlusive crisis of sickle cell anemia patients. Front Immunol. 2021;12:559925. doi:10.3389/fimmu.2021.559925
131. Knisely MR, Pugh N, Kroner B, et al. Patient-reported outcomes in sickle cell disease and association with clinical and psychosocial factors: report from the sickle cell disease implementation consortium. Am J Hematol. 2020. doi:10.1002/ajh.25880
132. Bodnar RJ. Endogenous opiates and behavior: 2014. Peptides. 2016;75:18–70. doi:10.1016/j.peptides.2015.10.009
133. Alshahrani MS, AlSulaibikh AH, ElTahan MR, et al. Ketamine administration for acute painful sickle cell crisis: a randomized controlled trial. Acad Emerg Med. 2021. doi:10.1111/acem.14382
134. Anghelescu DL, Morgan KJ, Frett MJ, et al. Lidocaine infusions and reduced opioid consumption-Retrospective experience in pediatric hematology and oncology patients with refractory pain. Pediatr Blood Cancer. 2021;68(11):e29215. doi:10.1002/pbc.29215
135. Hall EA, Sauer HE, Davis MS, Anghelescu DL. Lidocaine infusions for pain management in pediatrics. Paediatr Drugs. 2021;23(4):349–359. doi:10.1007/s40272-021-00454-2
136. Puri L, Nottage K, Hankins J, et al. Gabapentin for acute pain in sickle cell disease: a randomized double-blinded placebo-controlled phase II clinical trial. EJHaem. 2021;2(3):327–334. doi:10.1002/jha2.188
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.