Back to Journals » Drug Design, Development and Therapy » Volume 11
Morusin shows potent antitumor activity for human hepatocellular carcinoma in vitro and in vivo through apoptosis induction and angiogenesis inhibition
Authors Gao L ![]() , Wang L, Sun Z, Li H, Wang Q, Yi C, Wang X
, Wang L, Sun Z, Li H, Wang Q, Yi C, Wang X
Received 29 March 2017
Accepted for publication 9 May 2017
Published 16 June 2017 Volume 2017:11 Pages 1789—1802
DOI https://doi.org/10.2147/DDDT.S138320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ling Gao,1,* Li Wang,2,* Zhen Sun,3 Haiyan Li,3 Qiaoping Wang,3 Cheng Yi,1 Xiujie Wang3
1Department of Abdominal Cancer, 2Laboratory of Lung Cancer, Lung Cancer Center, 3Laboratory of Experimental Oncology, West China Hospital, West China Clinical Medical School, Sichuan University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Abstract: Hepatocellular carcinoma (HCC) is one of the most aggressive cancers with high mortality worldwide. Research and development of novel agents for HCC therapy is in demand, urgently. Morusin has been reported to exhibit potential cytotoxic activity in several cancer cell lines. However, whether it has potential antiangiogenic activity especially in HCC remains unclear. In the current study, we found that morusin exerted growth inhibition effects on human HCC cells (HepG2 and Hep3B) in vitro and human HCC cell (HepG2) xenografts in vivo. Moreover, apoptosis induction was observed in a dose-dependent manner after morusin treatment along with an increase in the expression of active caspase-3 and the Bax/Bcl-2 expression ratio. More importantly, morusin inhibited proliferation, migration, and tube formation of human umbilical vein endothelial cells (HUVECs) in vitro and downregulated angiogenic proteins in HCC cells and HUVECs. In vivo, tumor angiogenesis was also attenuated after morusin treatment. In addition, morusin suppressed constitutive as well as IL-6-induced STAT3 phosphorylation in HCC cells and corresponding tumor tissues. Overall, morusin has a potential anticancer effect on human HCC cells in vitro and in vivo by inducing apoptosis and inhibiting anti-angiogenesis. The corresponding mechanism might be associated with the attenuation of the IL-6/STAT3 signaling pathway. Morusin might serve as a promising novel anticancer agent in HCC therapy, and requires further study.
Keywords: morusin, human hepatocellular carcinoma, apoptosis, angiogenesis, IL-6, STAT3
Introduction
Liver cancer is the fifth most common cancer in men and the seventh most common cancer in women worldwide,1 and hepatocellular carcinoma (HCC) is one of the predominant types of liver cancer.2 Although the current curative therapeutic modalities for HCC, including surgical resection and liver transplantation, are encouraging, HCC patients have high mortality and poor prognosis. This results partly from the diagnosis in advanced stages and the consequent loss of therapeutic opportunity.3 Moreover, because of their low response rate and high toxicity, many chemotherapy agents are of limited use and provide minimal benefit to survival time in HCC patients.4,5 Therefore, there is an urgent need to explore more effective therapeutic strategies for HCC patients.
It is well known that angiogenesis occurs in solid tumors and plays an essential role in the supply of nutrient substances for the growth of cancer cells. HCC, unlike other tumor types, is a hypervascular carcinoma, and angiogenesis in HCC results from a pathologic vascularization pattern.6 VEGF, which is the most potent angiogenesis-inducing factor identified so far, is overexpressed in HCC. VEGF activates endothelial cell (EC) migration and proliferation, which results in primitive vessel formation.7 VEGFR2, a major transducer of VEGF, elicits angiogenesis.8 Therefore, antiangiogenesis by targeting VEGF/VEGFR2 is one of the promising therapeutic strategies for HCC.
STAT3, a major transducer, could play an important role in tumorigenesis and progression of HCC through the regulation of several genes involved in inflammation, apoptosis, cell cycle progression, angiogenesis, and cellular invasion.9 IL-6 is a key cytokine that can activate STAT3 signaling pathway,10 and its expression is found to be elevated in HCC.11 Therefore, the inhibition of aberrant STAT3 phosphorylation activated by IL-6 appears to be a potential therapeutic target, and the benefit has already been verified in HCC.12–14
Morusin is a prenylated flavonoid derived from the root bark of Morus alba Linn.,15 which has been reported to possess antibacterial, antioxidant, and anti-inflammatory activities.16–18 Previous studies demonstrated that morusin possesses cytotoxicity against some cancer cells, including glioblastoma, pancreatic cancer, colorectal cancer, breast cancer, cervical cancer, and other cancer cells.19–28 However, whether morusin has potential anticancer activity in human HCC in vivo and its possible mechanism of activity remain unknown. Therefore, the aim of this study was to assess the anticancer effect of morusin on human HCC cells in vitro and in vivo. Moreover, we aimed to elucidate its potential mechanism of activity.
Materials and methods
Cell lines and culture
Human normal liver cells (LO2) and human HCC cells (cell lines HepG2 and Hep3B) were obtained from Shanghai Cell Biology Institute of Chinese Academy of Sciences (Shanghai, China). Human umbilical vein ECs (HUVECs) were kindly provided by the Cancer Center and State Key Laboratory of Biotherapy of Sichuan University (purchased from ScienCell, Carlsbad, CA, USA). Cell lines were routinely cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37°C in a humidified atmosphere of 5% CO2.
Cell viability assay (MTT assay)
The cytotoxicity of morusin was investigated using the modified MTT assay.29 LO2, HepG2, and Hep3B cells and HUVECs (1×103 cells/well) were seeded in 96-well plates in triplicate with 100 μL medium per well. After overnight incubation at 37°C, cells were then treated with different concentrations of morusin (1, 2, 4, 6, and 8 μg/mL) for 24, 48, 72, 96, and 120 h. Subsequently, 20 μL of MTT (pH 4.7) was added to each well, and the cells were incubated for another 4 h. Then, 100 μL of 10% sodium dodecyl sulfate (SDS)/0.01 M HCL was added, and the cells were incubated at 37°C overnight to dissolve the formazan crystals. Absorbance was measured at 570 nm, and the effect of morusin on the viabilities of the LO2, HepG2, and Hep3B cells and HUVECs were expressed as the percentage of cell viability, using the following formula: percentage of cell viability = A570 of treated cells/A570 of control cells ×100.29 Three independent experiments were performed.
Colony formation inhibition assay
To test the effect of morusin on the colony formation of HCC cells, 300 HepG2 or Hep3B cells per well were seeded and incubated in 12-well plates overnight. Subsequently, various concentrations of morusin (0.5, 1, and 1.5 μg/mL) were added. After 12–14 days of incubation at 37°C, cells were stained with 0.5% crystal violet in absolute ethanol and the colonies (>50 cells) were counted under a microscope. Colony-forming efficiency was expressed as follows: colony-forming efficiency = (colony number of drug-treated cells/cell population) ×100.30 Three independent experiments were performed.
Morphological examination of apoptosis
4′,6-Diamidino-2-phenylindole (DAPI) (Beyotime Biotech, Shanghai, China) staining was performed to detect the apoptotic morphology–nuclear chromatin condensation.31 HepG2 or Hep3B cells (1×105 cells/well) were seeded in six-well plates, and after overnight incubation cells were treated with different concentrations of morusin (4, 6, and 8 μg/mL) for 48 h. Subsequently, the cells were harvested, fixed in 4% paraformaldehyde, treated with 0.25% Triton X-100 in Tris-buffered saline (TBS) for 15 min at room temperature, and stained with 25 μL DAPI for 30 min at room temperature. Finally, the samples of stained cells were washed with PBS and kept in the dark. They were observed under a fluorescent microscope.
Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining of apoptotic cells
Apoptotic cells were measured using an Annexin V–FITC/PI double-staining assay (Beyotime Biotech) according to the manufacturer’s protocol. Briefly, 1×105 HCC cells treated with 4, 6, and 8 μg/mL of morusin for 48 h were harvested, washed with PBS, and re-suspended in 200 μL of binding buffer. The samples were then simultaneously incubated with 10 μL of Annexin V–FITC/PI double-staining assay and 5 μL of PI for 15 min at room temperature in the dark. The apoptotic cells were analyzed using flow cytometry and Cell Quest Pro software (BD Biosciences, San Jose, CA, USA). Three independent experiments were performed.
Wound-healing assay
HUVECs (3×105 cells/well) were seeded in six-well plates and cultured in DMEM to 90% confluence. Then, the cells were scraped with a sterile tip to create a cell-free zone and washed thrice with PBS. Subsequently, the medium was replaced with DMEM containing morusin at different concentrations (1, 2, and 3 μg/mL). DMEM with 2% FBS was used for controls. The migration of cells was recorded at 0, 24, and 48 h using the Nikon Ti-U inverted microscope at 200× magnification. Cell migration was calculated as follows: migration =0 h wound width −24 or 48 h wound width. The migration index was defined as the percentage of migration considering migration in the untreated control as 100%.32 Data were analyzed using Image-Pro Plus 6.0 software.
HUVEC migration assay
The chemotactic motility of HUVECs was determined using a Transwell migration assay (Transwell; Corning Incorporated, Corning, NY, USA) with 6.5 mm-diameter polycarbonate filters (8 μm pore size). First, HUVECs were seeded into 24-well plates and pretreated with different concentrations of morusin (1, 2, and 3 μg/mL) for 48 h. Then, cells were harvested using 0.25% trypsin and re-suspended in DMEM with 2% FBS. Subsequently, 500 μL of DMEM containing 10% FBS was added into the bottom chambers. The top chambers were seeded with pretreated HUVECs (2×104 cells/well) in 200 μL of DMEM with 2% FBS. After 24 h, the non-migrated cells were removed from the top chamber using a cotton swab. The migrated cells on the bottom side of the membrane were fixed in methanol and stained with 0.1% crystal violet. Images were taken using the Nikon Ti-U inverted microscope. The cells that had invaded the lower surface of the filter were then counted in five randomly selected fields at 200× magnification, and the counts were averaged. Presented data are representative of three individual wells.33
Tube formation assay
Matrigel™ (50 μL; BD Discovery Labware, Bedford, MA, USA) was tiled on the bottom of 96-well plates at 37°C for 45 min. HUVECs, which were treated with various concentrations of morusin (1, 2, and 3 μg/mL) for 24 h and re-suspended in DMEM with 10% FBS with or without VEGF (10 ng/mL), were seeded at a density of 2×104 cells/well in 50 μL of medium. After approximately 6–10 h, the tube began to form, and the images were obtained using the Nikon Ti-U inverted fluorescence microscope. Tube formation was defined by counting the number of branch points of the formed tubes,32 and the total tube lengths in five randomly selected microscopic fields per well were quantified using Image-Pro Plus 6.0 software.33
Animal experiments
All the procedures were according to the ethical standards of the Institutional Animal Care and Use Committee of Sichuan University and were approved by this committee (Permit Number: 2016039A). Twenty-five-week-old male nude mice (BALB/C-nu/nu) that were obtained from the Animal Centre of Sichuan University were kept in specific pathogen-free conditions and were given sterile food and autoclaved water, with a 12 h light–dark cycle. Mice were acclimated to the environment for 7 days before the experiments. Then, the cultured HepG2 cells (1×106 cells) suspended in 0.1 mL PBS were injected into the right flank of these mice. After 5 days of cell inoculation, tumor-bearing mice were randomized into four groups (control group and three morusin groups, respectively; n=5 each group). Morusin at 5, 10, and 15 mg/kg in dimethylsulfoxide (DMSO) was administered as intraperitoneal injection to mice thrice weekly for 6 weeks (7 days/week). Mice in the control group were injected with the same volume of DMSO (approximately 20–25 μL/mouse). During the experiment, mice were weighed, and the tumor size was measured in two orthogonal directions using calipers, weekly, and the tumor volume (mm3) was estimated using a standard formula (length × width2 ×0.5).34 Six weeks later, the mice were sacrificed by carbon dioxide euthanasia with a gradually rising concentration of CO2 (100% CO2 at a flow rate of 20% of the chamber volume per minute [CV·min−1]). Then, the tumors were resected and weighed. The tumor inhibitory rates were calculated using the following formula: tumor inhibitory rate (%) = (mean tumor weight of the control mice - mean tumor weight of the treated mice)/mean tumor weight of the control mice ×100.
Immunohistochemistry
Paraffin-embedded tumor tissues were sliced into 4 μm sections. The sections were processed for the immunohistochemical (IHC) detection of Ki-67 (ab16667; Abcam, Cambridge, UK) or CD34 (ab81289; Abcam). The tumor tissues were incubated overnight at 4°C with anti-Ki67 antibody (1:100 dilution) or anti-CD34 antibody (1:2,500 dilution). Subsequently, the tissues were incubated with a biotinylated donkey anti-goat immunoglobulin secondary antibody (1:800 dilution; Zhongshanjinqiao, Beijing, China). Then, the slides were counterstained with hematoxylin. A goat isotype IgG (1:400 dilution; Zhongshanjinqiao) was designed to be the corresponding native control. The results of immunohistochemistry were obtained with Olympus DP72 and quantified using Image-Pro Plus 6.0 analysis software. Ten different regions for each section were chosen within random fields. The Ki-67 proliferation index was considered as the ratio of positively stained cells to the total cell count expressed as a percentage at 200× magnification. The microvessel density (MVD) was determined as previously described.35 The areas of highest neovascularization were identified and examined under 100× magnification, and one field in each of ten vascularized areas was counted at a magnification of 200×. The average of the ten areas was recorded as the MVD level of the slide. Any brown-stained EC cluster distinct from adjacent microvessels, tumor cells, or other stromal cells was considered as a single countable microvessel.35,36
Western blot analysis
The total protein from both morusin-treated HCC cells and tumor tissues was prepared as previously described.23 Equal amounts of protein from each sample were subjected to 12% SDS polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked in 5% non-fat milk at room temperature for 1 h and then incubated with the primary antibody overnight at 4°C. Primary antibodies were rabbit anti-STAT3, rabbit anti-p-STAT3 (Tyr705), rabbit anti-p-STAT3 (Ser727), rabbit anti-IL-6, rabbit anti-Bcl-2, rabbit anti-Bax, rabbit anti-active caspase-3, rabbit anti-MMP2, rabbit anti-MMP9, rabbit anti-VEGF and rabbit anti-VEGFR2, and rabbit anti-β-actin (Beijing Biosynthesis Biotechnology Co., Ltd., Beijing, China). Subsequently, the membranes were incubated with peroxidase-conjugated goat anti-rabbit IgG (H + L) secondary antibody (Zhongshan Goldenbridge Biotechnology Co., Ltd, Beijing, China) at 1:6,000 dilution. Immune complexes were detected by Chemiluminescent HRP Substrate (EMD Millipore, Billerica, MA, USA) and a Western blot analysis system (Universal Hood II; Bio-Rad Laboratories Inc., Hercules, CA, USA).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). All data were analyzed using one-way analysis of variance (ANOVA), followed by Dunnett’s test for pairwise comparison. All data were analyzed using SPSS version 22.0 (IBM Corporation, Armonk, NY, USA). A P-value of <0.05 was considered as an indication for statistical significance.
Results
Morusin inhibited the proliferation of human HCC cells and HUVECs
The cytotoxic effects of morusin on human normal liver cells (LO2), human HCC cells (HepG2 and Hep3B), and HUVECs are shown in Figure 1A–D. Morusin treatment exhibited significant inhibition in HCC cells and HUVECs in a time- and dose-dependent manner. After 72 h of exposure to morusin, the IC50 values were 12.458, 4.906, 4.196, and 4.595 μg/mL for LO2, HepG2, Hep3B cells, and HUVECs, respectively. The maximal inhibitory efficiency of the growth of human HCC cells (>80%) was approximately 8 μg/mL. The results demonstrated that morusin inhibited the proliferation of human HCC cells selectively in vitro. In addition, after 48 h of exposure to morusin, the IC20 value of HUVECs was 3.441 μg/mL, and no significant toxicity occurred until 4 μg/mL (Figure 1D). Considering the possible toxicity of morusin to HUVECs, which would affect the reliability of subsequent angiogenesis assays, we finally selected nontoxic concentrations of morusin (1, 2, and 3 μg/mL).
 | Figure 1 Morusin inhibited the proliferation of human HCC cells and HUVECs and the colony formation of HCC cells in vitro. |
Morusin inhibited colony formation of human HCC cells
After treatment with morusin (0.5, 1, and 1.5 μg/mL), the colony number of morusin-treated HCC cells decreased significantly in a dose-dependent manner. As shown in Figure 1E, the colony formation rates of HepG2 cells were 63.78%±11.53%, 50.56%±7.43% (P=0.552), 38.78%±6.93% (P=0.115), and 15.22%±4.76% (P=0.034). Similarly, the colony formation rates of Hep3B cells were 65.17%±3.69%, 63.33%±3.01% (P=0.973), 52.83%±3.40% (P=0.055), and 19.67%±4.54% (P<0.01).
Morusin induced apoptosis in human HCC cells
To determine the influence of apoptosis induction by morusin, morphological changes of human HCC cells were assessed by DAPI staining and Annexin V–FITC/PI double-staining assays. As shown in Figure 2A and B, typical apoptotic features, including nuclear condensation, nuclear fragmentation and the formation of apoptotic bodies, were observed in morusin-treated HepG2 and Hep3B cells. After treatment with 4, 6, and 8 μg/mL of morusin for 48 h, the apoptotic cells among treated HepG2 and Hep3B cells increased significantly in a dose-dependent manner (Figure 2C, P<0.01). In flow cytometry analysis, the apoptotic cells among morusin-treated HepG2 and Hep3B cells increased significantly in a dose-dependent manner (Figure 2D–F). These findings indicate that morusin induces apoptosis of HepG2 and Hep3B cells.
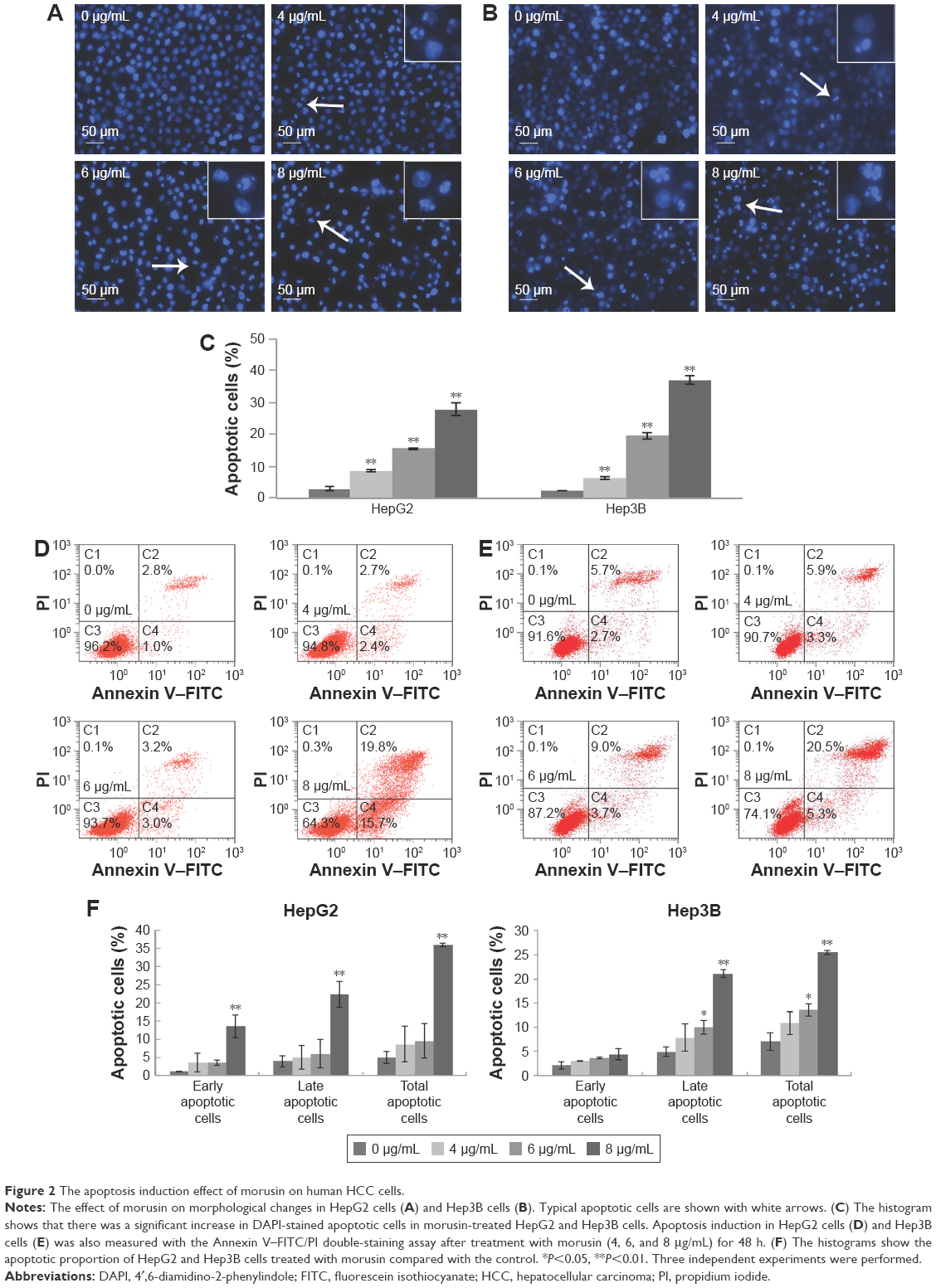 | Figure 2 The apoptosis induction effect of morusin on human HCC cells. |
Morusin inhibited HUVEC migration
EC migration is an essential step in angiogenesis, and we assessed the effect of morusin on the motility of HUVECs using wound-healing assay and Transwell assay. In the wound-healing assay, morusin significantly inhibited HUVEC migration in a dose–time-dependent manner (Figure 3A and C). In addition, in the Transwell assay, morusin exhibited potency with regard to the suppression of invasion of HUVECs (Figure 3B and D).
 | Figure 3 The migration and tube formation inhibition effects of morusin on HUVECs. |
Morusin inhibited tube formation of HUVECs
We next detected whether morusin has a potential inhibitory effect on tube formation. As shown in Figure 3E and F, morusin significantly inhibited tube formation among HUVECs in a dose-dependent manner (P<0.01), while VEGF could increase tube formation (P=0.03) when used alone and could partly reverse the inhibitory effect of morusin.
Morusin inhibited tumor growth and angiogenesis in a subcutaneous HepG2 xenograft model
To demonstrate the tumor growth inhibition of morusin in vivo, we established a nude mouse xenograft model through subcutaneous inoculation of HepG2 cells. Morusin at 5, 10, and 15 mg/kg was administered for 6 weeks. After 6 weeks, all mice were sacrificed and then the tumor masses from tumor-bearing mice were assessed (Figure 4C). As shown in Figure 4B and D, tumor volume and weight decreased in a dose-dependent manner after treatment with morusin. Tumor inhibitory rates were 34.29% (P=0.06), 35.11% (P=0.09), and 61.4% (P=0.038), respectively. More importantly, no obvious evidence of toxicity was observed in treated tumor-bearing mice by assessing the body weight of mice (Figure 4A).
 | Figure 4 The growth inhibition effect of morusin in human HCC cells xenograft model. |
Moreover, the expression levels of Ki-67 and CD34 in tumor tissues were analyzed through immunohistochemistry, and representative images are shown in Figure 4E. As expected, the number of Ki-67-labeled cells was lower in the high-dose morusin-treated group (15 mg/kg) than in the control group (P<0.01), and the Ki-67 labeling index values were 30.79%±7.38% and 70.46%±6.89%, respectively. Meanwhile, MVD, detected using anti-CD34, was also significantly lower in the high-dose group than in the control group (Figure 4F, P<0.01). These results indicate that morusin significantly inhibits tumor growth and angiogenesis in vivo.
Morusin affected apoptotic and angiogenic protein expressions in vitro and in vivo
To explore the potential molecular mechanisms underlying the growth inhibition effect of morusin, we first detected the expression levels of associative proteins in HepG2 and Hep3B cells, as well as corresponding tumor tissues after treatment with morusin. The results showed that morusin treatment resulted in the downregulation of Bcl-2 and VEGF and the upregulation of Bax and active caspase-3 (Figure 5A–E and G). Next, we analyzed the expression levels of associative proteins in HUVECs after treatment with morusin and found that the expression levels of MMP2, MMP9, and VEGFR2 in HUVECs were also downregulated in a dose-dependent manner following treatment with morusin (Figure 5F and G). Overall, these results indicate that morusin induces apoptotic death of human HCC cells in vitro and in vivo through mitochondria-based signal transduction in both HepG2 and Hep3B cells, and inhibited the migration and invasion of HUVECs and angiogenesis associated with the downregulation of VEGF, MMP2, MMP9, and VEGFR2.
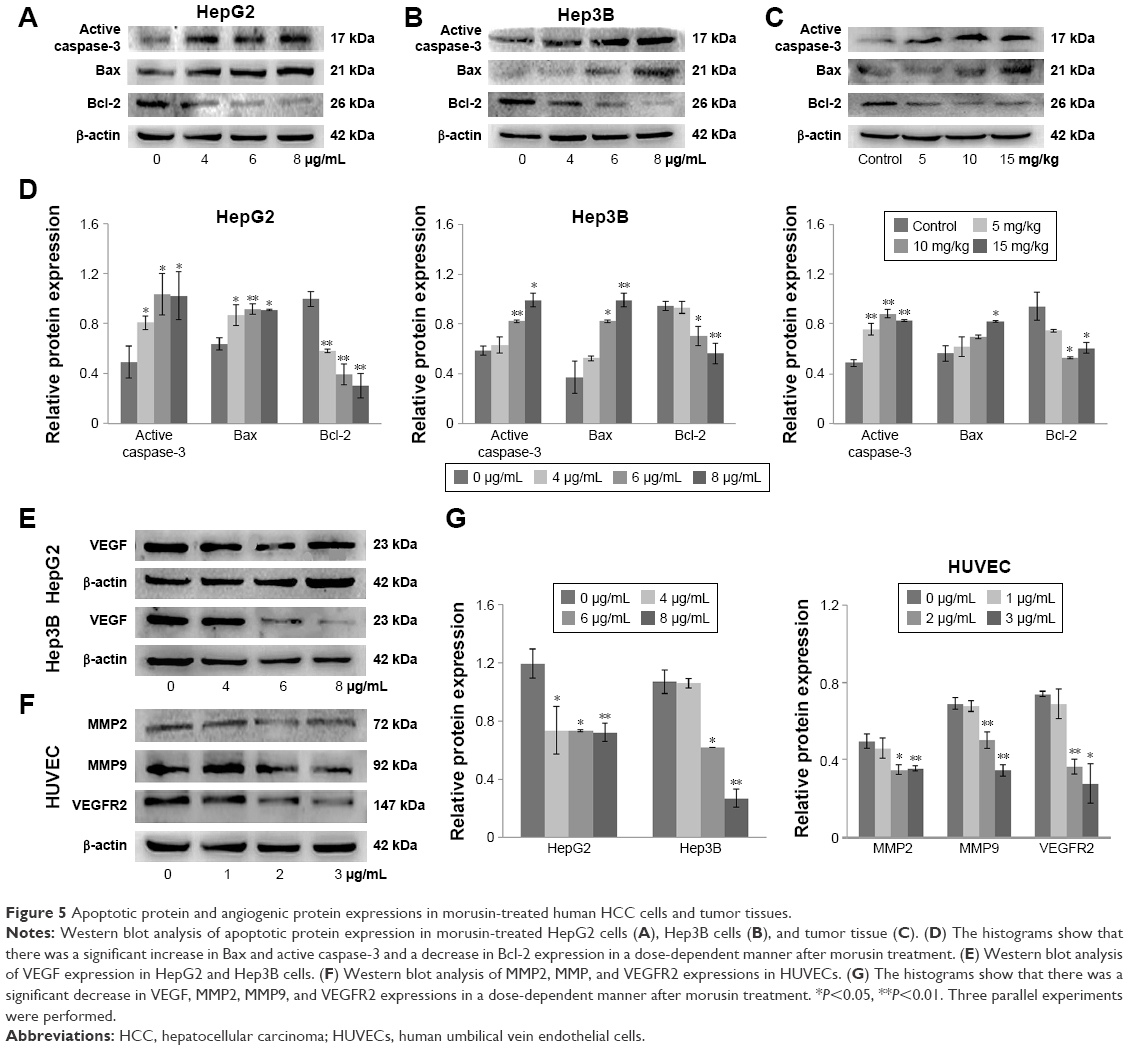 | Figure 5 Apoptotic protein and angiogenic protein expressions in morusin-treated human HCC cells and tumor tissues. |
Morusin regulated inflammatory signaling pathways by suppressing constitutive and IL-6-induced STAT3 phosphorylation
To examine whether the antitumor activity of morusin might be involved in the inhibition of the IL-6/STAT3 signaling pathway, we evaluated the effects of morusin on the expression of constitutive p-STAT3 (Tyr705) and p-STAT3 (Ser727) activation and IL-6-induced STAT3 phosphorylation in human HCC cells. As shown in Figure 6A–C, after treatment with indicated concentrations of morusin for 48 h, the levels of constitutive STAT3 phosphorylation were inhibited significantly.
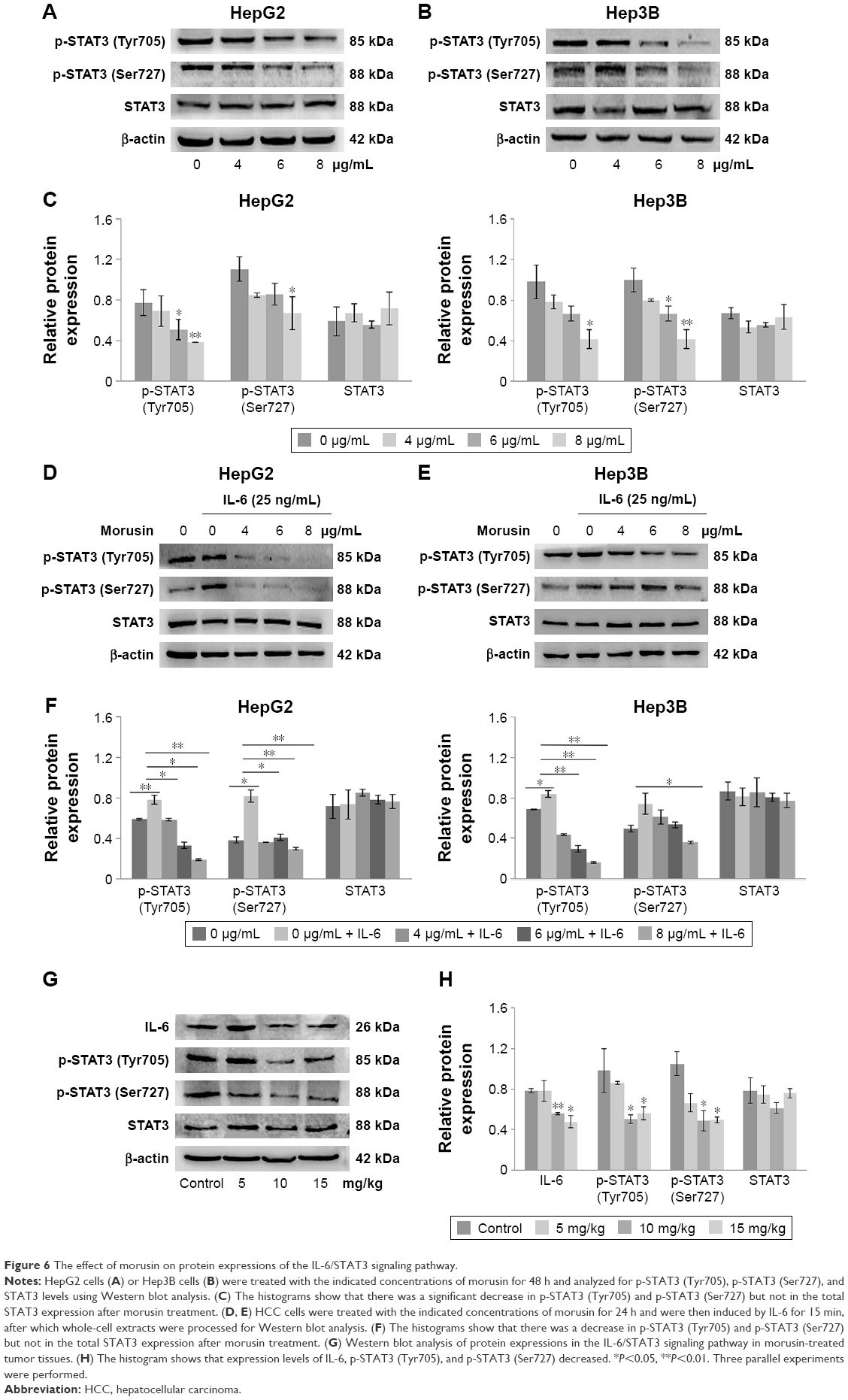 | Figure 6 The effect of morusin on protein expressions of the IL-6/STAT3 signaling pathway. |
Further, we also demonstrated that morusin could inhibit IL-6-induced STAT3 phosphorylation in HepG2 and Hep3B cells. As shown in Figure 6D–F, 25 ng/mL of IL-6 significantly activated STAT3 phosphorylation in HepG2 and Hep3B cells, and morusin significantly inhibited IL-6-induced STAT3 phosphorylation in HepG2 cells, while the inhibitory effect was weaker in Hep3B cells.
To confirm whether the antitumor efficiency of morusin on xenograft tumor growth was also mediated by blocking the IL-6/STAT3 signaling pathway, we investigated the expression levels of IL-6, total STAT3, and phosphorylated STAT3 in morusin-treated tumor tissues through Western blotting. The results showed that the expression levels of IL-6 and phosphorylated STAT3 decreased, especially in the groups treated with 10 and 15 mg/kg of morusin, while almost none was observed on the expression level of total STAT3 (Figure 6G and H). These results suggested that morusin exerted antitumor efficiency in HCC associated with the blockage of the IL-6/STAT3 signaling pathway and might be a promising antitumor agent for cancer therapy and/or prevention of HCC.
Discussion
HCC is one of the most aggressive cancers and the second leading cause of cancer-related death worldwide. Less than 20% of HCC patients are amenable to surgical resection or liver transplantation owing to the discovery or diagnosis of HCC at advanced stages. Even worse, therapeutic strategies including chemotherapy and radiation have limited impact on the survival of HCC patients because of their limitations with regard to efficacy and side effects.1,3,4 Therefore, the current study focused on identifying more effective therapeutic options for HCC.
Morusin is an important component of the traditional Chinese medicine Morus alba L. Currently, many studies, including our previous study, have focused on its anticancer activities against several kinds of cancer cells.19–28 For HCC, morusin suppressed the invasion of human HCC cells (SK-Hep1) in vitro, decreased their lung colonization in vivo,25 and inhibited human HCC cell (Bel-7402) growth in vitro via apoptosis induction.26 In addition, it exhibited an anticancer effect on murine liver cancer (H22).27 However, little attention has been paid to its growth inhibition in vivo, and its potential antiangiogenic activity remains unclear.
In the current study, we demonstrated that morusin can inhibit the growth of human HCC cells in vitro and in vivo. The results of the MTT and colony formation assays indicate selective cytotoxicity of morusin in different human HCC cells and less toxicity in normal liver cells (LO2; Figure 1). Similarly, in HepG2 mice, morusin exhibited an obviously inhibitory effect on tumor growth. Ki-67 is an important indicator that reflects the proliferative situation of cells. In this study, we observed that Ki-67-labeled cells in HepG2 xenograft tumor tissues were significantly decreased in the morusin-treated groups (Figure 4). Apoptosis induction is one of the most effective strategies to inhibit tumor cell growth. Previous studies demonstrated that flavonoids have an apoptosis induction potency on HCC cells.37–39 The increase in DAPI staining and Annexin V–FITC/PI double staining of apoptotic cells are two important hallmarks of apoptosis. As shown in Figure 2, there was a significant increase in DAPI-stained and Annexin V–FITC/PI double-stained apoptotic cells after morusin treatment, which indicated that morusin induced the apoptotic death of human HCC cells. However, the differences in early apoptotic cell proportions (Figure 2) and apoptotic protein expressions (Figure 5) between HepG2 and Hep3B cells were obvious. This may have been caused by the difference of the p53 state in the two cell lines. HepG2 expresses the wild-type p53 gene, whereas Hep3B is p53 deficient. p53 plays a key role in apoptosis through regulating a series of apoptosis-associated proteins, including PUMA and the Bcl-2 family of proteins.40 Under the condition of p53 deficiency, the cells could be influenced by other factors, such as p73 and p63, which are highly homologous to p53, for repairing injured cells or inducing apoptosis.41,42 In addition, some other protein kinases, such as MEK/ERK, JNK, and PKC, are also able to induce apoptosis via mitochondrial-associated pathways in the absence of p53.43,44 Bcl-2 family proteins, including pro-apoptotic members such as Bax and Bak, combined with antiapoptotic members such as Bcl-2 and Bcl-xL, are important regulators in the process of mitochondrial-mediated apoptosis. In addition, caspase-3, a downstream effector of apoptosis pathways, is activated to regulate the caspase-signaling cascade, and this eventually leads to apoptosis.40 In the current study, morusin upregulated the expression of active caspase-3 and the Bax/Bcl-2 ratio in both HepG2 and Hep3B cells, which indicated that p53-independent mitochondrial-mediated mechanisms of apoptosis in Hep3B cells may have caused the different apoptosis phenotypes between Hep3B and HepG2 cells. However, further research should be performed to assess these possibilities.
Abnormal angiogenesis, especially in solid tumors, plays an important role in tumor growth and metastasis.45,46 For the regulation of angiogenesis, VEGF is a key regulator of vascular permeability and promotes EC proliferation and migration.7,47,48 A number of Chinese herbal components have demonstrated anti-angiogenesis effects on targeting the VEGF-induced angiogenesis. For instance, resveratrol, a compound of Pediomelum cuspidatum, might decrease VEGF expression in HepG2 cells,49 and pedicularioside G, a phenylpropanoid glycoside isolated from Pedicularis striata, has been shown to inhibit the proliferation and migration of HUVECs.50 In the current study, we found that morusin downregulated the expressions of VEGF in human HCC cells. In addition, VEGF specifically reacted with receptor tyrosine kinases (VEGFR1, R2, and R3),51 especially VEGFR2, which is a major transducer of VEGF signals in ECs.52 The expression level of VEGFR2 decreased in HUVECs after morusin treatment, which indicated that morusin exerted antiangiogenesis, and this might be involved in targeting the VEGF/VEGFR2 signaling pathway. In addition, MMPs are zinc-dependent proteolytic enzymes and play crucial roles in the degradation of the extracellular matrix for the migration of ECs.53,54 Owing to the inhibitory effect of morusin on the migration of HUVECs, we investigated the expressions of MMP2 and MMP9. The results of Western blot analysis (Figure 5F) showed that morusin significantly decreased the expression levels of MMP2 and MMP9 in HUVECs. Moreover, the antiangiogenic effect of morusin in vivo has been confirmed. CD34 is believed to be a sensitive and specific marker for microvessels in HCC. The expression level of CD34 was downregulated in tumor tissues after morusin treatment, which indicated a decrease in MVD in morusin-treated tumor tissues (Figure 4E and F). Overall, these results suggest that morusin may have potential antiangiogenic activity in vitro and in vivo.
STAT3 has been found to be constitutively activated in a number of human cancer cell lines and primary tumors. It has been taken for granted that IL-6 was linked to constitutive or aberrant activation of STAT3 in various cancers, including HCC.55,56 STAT3 phosphorylation could regulate several genes encoding anti-apoptotic proteins such as Bcl-2 and Bcl-xL, and pro-apoptotic proteins, such as Bax and Bad.57 Moreover, the previous studies indicated that activated STAT3 is also involved in tumor angiogenesis through the regulation of angiogenesis factors (eg, VEGF and MMP9)58,59 and promotion of signal transduction of angiogenic factor receptors in ECs.9 Therefore, the IL-6/STAT3 signaling pathway has been recognized as a pleiotropic drug target. In the current study, morusin effectively induced the apoptosis of human HCC cells and inhibited angiogenesis, which may be related to the inhibition of the expression of IL-6 and phosphorylated STAT3. In addition, according to some reports, wild-type p53 protein could repress several genes including Il-6 and Stat3, while mutation of p53 resulted in the attenuation of this function.60,61 This suggests that the different results of p-STAT3 levels between the two HCC cells after morusin and IL-6 treatment may be caused by their p53 state. However, further research should be performed to assess this possibility.
Conclusion
Our findings indicate that morusin has the potential to inhibit the growth of human HCC cells in vitro and in vivo through apoptosis induction and angiogenesis inhibition. The mechanism might be involved in the suppression of the IL-6/STAT3 signaling pathway. Therefore, morusin might serve as a novel candidate for the treatment and/or prevention of human HCC cells, and it should be investigated further.
Acknowledgment
This work was partially supported by the Research Foundation of Science and Technology Bureau of Sichuan Province, China (Grant No 2014SZ0001).
Disclosure
The authors report no conflicts of interest in this work.
References
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | ||
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. | ||
Grandhi MS, Kim AK, Ronnekleiv-Kelly SM, Kamel IR, Ghasebeh MA, Pawlik TM. Hepatocellular carcinoma: from diagnosis to treatment. Surg Oncol. 2016;25(2):74–85. | ||
Connell LC, Harding JJ, Abou-Alfa GK. Advanced hepatocellular cancer: the current state of future research. Curr Treat Options Oncol. 2016;17(8):43. | ||
Simonetti RG, Liberati A, Angiolini C, Pagliaro L. Treatment of hepatocellular carcinoma: a systematic review of randomized controlled trials. Ann Oncol. 1997;8(2):117–136. | ||
Bishayee A, Darvesh AS. Angiogenesis in hepatocellular carcinoma: a potential target for chemoprevention and therapy. Curr Cancer Drug Targets. 2012;12(9):1095–1118. | ||
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–410. | ||
Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49(3):568–581. | ||
Subramaniam A, Shanmugam MK, Perumal E, et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim Biophys Acta. 2013;1835(1):46–60. | ||
May P, Schniertshauer U, Gerhartz C, Horn F, Heinrich PC. Signal transducer and activator of transcription STAT3 plays a major role in gp130-mediated acute phase protein gene activation. Acta Biochim Pol. 2003;50(3):595–601. | ||
Malaguarnera M, Di Fazio I, Laurino A, Romeo MA, Giugno I, Trovato BA. Role de l’interleukine 6 dans le carcinome hepatocellulaire. [Role of interleukin 6 in hepatocellular carcinoma]. Bull Cancer. 1996;83(5):379–384. French. | ||
Liu Y, Liu A, Li H, Li C, Lin J. Celecoxib inhibits interleukin-6/interleukin-6 receptor-induced JAK2/STAT3 phosphorylation in human hepatocellular carcinoma cells. Cancer Prev Res (Phila). 2011;4(8):1296–1305. | ||
Gu FM, Li QL, Gao Q, et al. Sorafenib inhibits growth and metastasis of hepatocellular carcinoma by blocking STAT3. World J Gastroenterol. 2011;17(34):3922–3932. | ||
Rajendran P, Ong TH, Chen L, et al. Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin Cancer Res. 2011;17(6):1425–1439. | ||
Zong YY, Ip SP, Dong TX, Che CT. [Determination of morusin in cortex mori]. Zhongguo Zhong Yao Za Zhi. 2007;32(11):1038–1040. | ||
Bellik Y, Boukraa L, Alzahrani HA, et al. Molecular mechanism underlying anti-inflammatory and anti-allergic activities of phytochemicals: an update. Molecules. 2012;18(1):322–353. | ||
Fukai T, Satoh K, Nomura T, Sakagami H. Antinephritis and radical scavenging activity of prenylflavonoids. Fitoterapia. 2003;74(7–8):720–724. | ||
Sohn HY, Son KH, Kwon CS, Kwon GS, Kang SS. Antimicrobial and cytotoxic activity of 18 prenylated flavonoids isolated from medicinal plants: Morus alba L., Morus mongolica Schneider, Broussnetia papyrifera (L.) Vent, Sophora flavescens Ait and Echinosophora koreensis Nakai. Phytomedicine. 2004;11(7–8):666–672. | ||
Guo H, Liu C, Yang L, et al. Morusin inhibits glioblastoma stem cell growth in vitro and in vivo through stemness attenuation, adipocyte transdifferentiation, and apoptosis induction. Mol Carcinog. 2016;55(1):77–89. | ||
Kim C, Kim JH, Oh EY, et al. Blockage of STAT3 signaling pathway by morusin induces apoptosis and inhibits invasion in human pancreatic tumor cells. Pancreas. 2016;45(3):409–419. | ||
Lee HJ, Lyu da H, Koo U, et al. Protection of prenylated flavonoids from Mori Cortex Radicis (Moraceae) against nitric oxide-induced cell death in neuroblastoma SH-SY5Y cells. Arch Pharm Res. 2012;35(1):163–170. | ||
Lee JC, Won SJ, Chao CL, et al. Morusin induces apoptosis and suppresses NF-kappaB activity in human colorectal cancer HT-29 cells. Biochem Biophys Res Commun. 2008;372(1):236–242. | ||
Li H, Wang Q, Dong L, et al. Morusin suppresses breast cancer cell growth in vitro and in vivo through C/EBPbeta and PPARgamma mediated lipoapoptosis. J Exp Clin Cancer Res. 2015;34:137. | ||
Lim SL, Park SY, Kang S, et al. Morusin induces cell death through inactivating STAT3 signaling in prostate cancer cells. Am J Cancer Res. 2015;5(1):289–299. | ||
Lin WL, Lai DY, Lee YJ, Chen NF, Tseng TH. Antitumor progression potential of morusin suppressing STAT3 and NFkappaB in human hepatoma SK-Hep1 cells. Toxicol Lett. 2015;232(2):490–498. | ||
Ding B, Lv Y, Zhang YQ. Anti-tumor effect of morusin from the branch bark of cultivated mulberry in Bel-7402 cells via the MAPK pathway. RSC Adv. 2016;6(21):17396–17404. | ||
Wan LZ, Ma B, Zhang YQ. Preparation of morusin from Ramulus mori and its effects on mice with transplanted H22 hepatocarcinoma. Biofactors. 2014;40(6):636–645. | ||
Wang L, Guo H, Yang L, et al. Morusin inhibits human cervical cancer stem cell growth and migration through attenuation of NF-kappaB activity and apoptosis induction. Mol Cell Biochem. 2013;379(1–2):7–18. | ||
van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–245. | ||
Wang Q, Li H, Sun Z, et al. Kukoamine A inhibits human glioblastoma cell growth and migration through apoptosis induction and epithelial-mesenchymal transition attenuation. Sci Rep. 2016;6:36543. | ||
Weerapreeyakul N, Junhom C, Barusrux S, Thitimetharoch T. Induction of apoptosis in human hepatocellular carcinoma cells by extracts of Lannea coromandelica (Houtt.) Merr. and Diospyros castanea (Craib) Fletcher. Chin Med. 2016;11:19. | ||
Wang SB, Cheng YN, Cui SX, et al. Des-gamma-carboxy prothrombin stimulates human vascular endothelial cell growth and migration. Clin Exp Metastasis. 2009;26(5):469–477. | ||
Lu H, Li X, Zhang J, Shi H, Zhu X, He X. Effects of cordycepin on HepG2 and EA.hy926 cells: potential antiproliferative, antimetastatic and anti-angiogenic effects on hepatocellular carcinoma. Oncol Lett. 2014;7(5):1556–1562. | ||
Tsai CH, Lin FM, Yang YC, et al. Herbal extract of Wedelia chinensis attenuates androgen receptor activity and orthotopic growth of prostate cancer in nude mice. Clin Cancer Res. 2009;15(17):5435–5444. | ||
Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis – correlation in invasive breast carcinoma. N Engl J Med. 1991;324(1):1–8. | ||
Zhang ZL, Liu ZS, Sun Q. Expression of angiopoietins, Tie2 and vascular endothelial growth factor in angiogenesis and progression of hepatocellular carcinoma. World J Gastroenterol. 2006;12(26):4241–4245. | ||
Jiang CP, Ding H, Shi DH, Wang YR, Li EG, Wu JH. Pro-apoptotic effects of tectorigenin on human hepatocellular carcinoma HepG2 cells. World J Gastroenterol. 2012;18(15):1753–1764. | ||
Kim BR, Jeon YK, Nam MJ. A mechanism of apigenin-induced apoptosis is potentially related to anti-angiogenesis and anti-migration in human hepatocellular carcinoma cells. Food Chem Toxicol. 2011;49(7):1626–1632. | ||
Zhang HT, Luo H, Wu J, et al. Galangin induces apoptosis of hepatocellular carcinoma cells via the mitochondrial pathway. World J Gastroenterol. 2010;16(27):3377–3384. | ||
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. | ||
Holcakova J, Ceskova P, Hrstka R, et al. The cell type-specific effect of TAp73 isoforms on the cell cycle and apoptosis. Cell Mol Biol Lett. 2008;13(3):404–420. | ||
Guan X, Zhang N, Yin Y, et al. Polymorphisms in the p63 and p73 genes are associated with ovarian cancer risk and clinicopathological variables. J Exp Clin Cancer Res. 2012;31:89. | ||
Bai L, Yoon SO, King PD, Merchant JL. ZBP-89-induced apoptosis is p53-independent and requires JNK. Cell Death Differ. 2004;11(6):663–673. | ||
Prasad S, Yadav VR, Kannappan R, Aggarwal BB. Ursolic acid, a pentacyclin triterpene, potentiates TRAIL-induced apoptosis through p53-independent up-regulation of death receptors: evidence for the role of reactive oxygen species and JNK. J Biol Chem. 2011;286(7):5546–5557. | ||
Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–936. | ||
Chiodoni C, Colombo MP, Sangaletti S. Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2010;29(2):295–307. | ||
Prager GW, Lackner EM, Krauth MT, et al. Targeting of VEGF-dependent transendothelial migration of cancer cells by bevacizumab. Mol Oncol. 2010;4(2):150–160. | ||
Gong C, Xu C, Ji L, Wang Z. A novel semi-synthetic andrographolide analogue A5 inhibits tumor angiogenesis via blocking the VEGFR2-p38/ERK1/2 signal pathway. Biosci Trends. 2013;7(5):230–236. | ||
Zhang H, Yang R. Resveratrol inhibits VEGF gene expression and proliferation of hepatocarcinoma cells. Hepatogastroenterology. 2014;61(130):410–412. | ||
Mu P, Gao X, Jia ZJ, Zheng RL. Natural antioxidant pedicularioside G inhibits angiogenesis and tumourigenesis in vitro and in vivo. Basic Clin Pharmacol Toxicol. 2008;102(1):30–34. | ||
Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18(1):4–25. | ||
Xiao D, Li M, Herman-Antosiewicz A, et al. Diallyl trisulfide inhibits angiogenic features of human umbilical vein endothelial cells by causing Akt inactivation and down-regulation of VEGF and VEGF-R2. Nutr Cancer. 2006;55(1):94–107. | ||
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. | ||
Theret N, Musso O, Turlin B, et al. Increased extracellular matrix remodeling is associated with tumor progression in human hepatocellular carcinomas. Hepatology. 2001;34(1):82–88. | ||
Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13(19):5665–5669. | ||
Liu Y, Fuchs J, Li C, Lin J. IL-6, a risk factor for hepatocellular carcinoma: FLLL32 inhibits IL-6-induced STAT3 phosphorylation in human hepatocellular cancer cells. Cell Cycle. 2010;9(17):3423–3427. | ||
Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18(2):254–267. | ||
Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–2008. | ||
Kim JE, Kim HS, Shin YJ, et al. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp Mol Med. 2008;40(5):514–522. | ||
Wang L, Rayanade RJ, Garcia D, Patel K, Pan H, Sehgal PB. Modulation of interleukin-6-induced plasma protein secretion in hepatoma cells by p53 species. J Biol Chem. 1995;270(39):23159–23165. | ||
Rayanade RJ, Patel K, Ndubuisi M, et al. Proteasome- and p53-dependent masking of signal transducer and activator of transcription (STAT) factors. J Biol Chem. 1997;272(8):4659–4662. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.