Back to Journals » Biologics: Targets and Therapy » Volume 13
Monoclonal antibody therapy of solid tumors: clinical limitations and novel strategies to enhance treatment efficacy
Received 24 February 2019
Accepted for publication 10 April 2019
Published 1 May 2019 Volume 2019:13 Pages 33—51
DOI https://doi.org/10.2147/BTT.S166310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Doris Benbrook
Esteban Cruz, Veysel Kayser
School of Pharmacy, The University of Sydney, Sydney, NSW, Australia
Abstract: Monoclonal antibodies (mAbs) have become a cornerstone in the therapeutic guidelines of a wide range of solid tumors. The targeted nature of these biotherapeutics has improved treatment outcomes by offering enhanced specificity to reduce severe side effects experienced with conventional chemotherapy. Notwithstanding, poor tumor tissue penetration and the heterogeneous distribution achieved therein are prominent drawbacks that hamper the clinical efficacy of therapeutic antibodies. Failure to deliver efficacious doses throughout the tumor can lead to treatment failure and the development of acquired resistance mechanisms. Comprehending the morphological and physiological characteristics of solid tumors and their microenvironment that affect tumor penetration and distribution is a key requirement to improve clinical outcomes and realize the full potential of monoclonal antibodies in oncology. This review summarizes the essential architectural characteristics of solid tumors that obstruct macromolecule penetration into the targeted tissue following systemic delivery. It further describes mechanisms of resistance elucidated for blockbuster antibodies for which extensive clinical data exists, as a way to illustrate various modes in which cancer cells can overcome the anticancer activity of therapeutic antibodies. Thereafter, it describes novel strategies designed to improve clinical outcomes of mAbs by increasing potency and/or improving tumor delivery; focusing on the recent clinical success and growing clinical pipeline of antibody-drug conjugates, immune checkpoint inhibitors and nanoparticle-based delivery systems.
Keywords: antibody therapy, treatment resistance, antibody–drug conjugates, immune checkpoint inhibitors, nanoparticle delivery vehicles
Introduction
Therapeutic monoclonal antibodies (mAbs) successfully entered the clinic over 25 years ago and have become one of the central components of the healthcare system.1,2 Their arrival brought about a therapeutic revolution due to their capacity to target specific molecular components, with a large number of mAbs already approved in oncology, autoimmune disorders, chronic diseases and many more conditions. Currently, over 80 antibody therapeutics have received regulatory approval in Europe and/or the United States and just in 2017 sales of therapeutic antibodies exceeded 100$ billion worldwide.3
In oncology, therapeutic antibodies offer the possibility to treat tumors in a targeted fashion and reduce the severe side effects of conventional chemotherapy. Recent developments in cancer biology have aided the discovery of molecular biomarkers in a wide range of solid malignancies that can be used as targets with beneficial therapeutic outcomes. At present, over 15 distinct monoclonal antibodies are indicated for the treatment of solid tumors.4 Notwithstanding, in spite of their remarkable clinical success some patients do not benefit from the treatment due to intrinsic resistance mechanisms or the emergence of acquired resistance following treatment initialization .5,6
In solid tumors, the development of acquired resistance mechanisms is thought to emerge primarily from continuous genetic alterations that modify the cellular phenotype and undermine the initial therapeutic efficacy. This capacity of cancer cells to overcome the anticancer effect of the antibody is facilitated by the exposure to subtherapeutic concentrations of the drug.7,8 The tumor microenvironment poses physical barriers, most notably a markedly increased hydrostatic pressure, that hinder penetration of macromolecules into the tumor following systemic administration.9,10 This reduces the overall amount of antibody molecules that reach the target tissue and exposes areas of the tumor that are difficult to penetrate to marginal doses of the antibody, leading to acquired resistance and treatment failure.8 In fact, therapeutic mAbs in oncology are more commonly administered as combination therapy in conjunction with chemotherapeutics due to relatively limited efficacy as single agents.11
Identifying and understanding primary and acquired resistance mechanisms and overcoming the barriers that impair efficient delivery of the drug into the tissue is critical to enhance therapeutic outcomes. Most of the understanding regarding primary and acquired resistance comes from the evaluation of clinical data available for early-approved blockbuster antibodies, such as trastuzumab and cetuximab. This review gives an overview of the key factors affecting tumor distribution upon systemic delivery and describes relevant mechanisms of resistance identified in trastuzumab (anti-HER2) and cetuximab (anti-EGFR) therapy. Additionally, it describes recent developments in the implementation of novel antibody-based therapeutics, such as antibody–drug conjugates (ADCs), immune checkpoint inhibitors (ICI), and antibody-targeted nanoparticles (NPs) that have the potential to improve therapeutic outcomes of solid tumors.
Limitations that impact clinical efficacy
Poor penetration and heterogeneous distribution in solid tumors
Therapeutic IgG antibodies must overcome pronounced physical and physiological obstacles in order to penetrate and distribute uniformly throughout the tumor. In solid malignancies, impaired lymphatic drainage due to the sparse presence of lymphatic vessels leads to the accumulation of macromolecules in the interstitial tissue and a consequent increase in hydrostatic pressure.9,12–14 Hence, the altered pressure differential from vascular vessels to the interstitial compartment limits convection and extravasation of macromolecules from the vascular lumen into the tumor (Figure 1).15 Moreover, antibody distribution following extravasation is further impeded by cellular internalization and subsequent endocytic clearance at the tumor edge (an effect coined the “binding-site barrier”), leading to poor penetration and regions of marginal antibody concentrations.10,16,17 The binding-site barrier suggests that higher affinity and higher antigen expression, especially at the tumor edge, can retard mAb tumor percolation and impair homogeneous distribution; although this barrier can be overcome by increasing the administered dose.
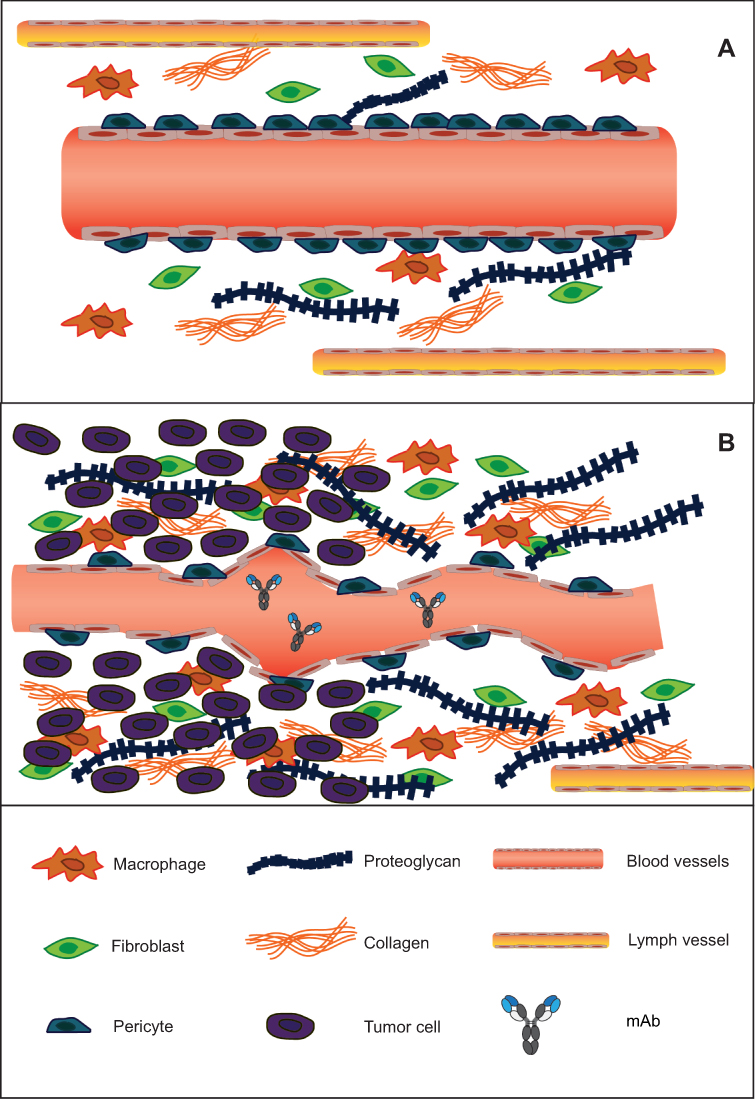 | Figure 1 Structural features of the tumor microenvironment that increase interstitial pressure and hinder mAb extravasation and distribution. (A) Blood vessels that irrigate healthy normal tissue possess a continuous inner lining of endothelial cells, enveloped by perivascular cells called pericytes that grant integrity to the vascular tube. The extracellular matrix (ECM) contains a lax network of collagen and proteoglycan fibers, and the presence of macrophages and fibroblasts is scarce. Lymph vessels efficiently remove and prevent the accumulation of macromolecules and interstitial fluid. (B) Increased demand of oxygen and nutrients in tumor tissues causes blood vessels to form defectively and irregularly shaped. The lack of pericytes makes the vascular tube unstable and leaky. The abundant presence of fibroblasts and infiltrating macrophages promote the formation of a dense ECM, with a condensed network of collagen and proteoglycan fibers. The paucity of lymph vessels leads to the accumulation of macromolecules and an increase in interstitial fluid pressure (IFP). The fibrotic nature of the ECM and the altered pressure differential between the vascular lumen and the tumor hinder antibody convection into the targeted tissue. |
A vast body of research studying some of the blockbuster therapeutic mAbs has highlighted the significance of increasing tissue penetration to improve the outcome of antibody therapy.15,18 A study on cetuximab and trastuzumab in mouse xenografts confirmed that tumor distribution can be improved with an increase in dose; however, hypoxic areas remained difficult to reach even at higher doses. Moreover, xenografts expressing intermediate levels of ErbB1 (cognate antigen for cetuximab) displayed more homogeneous distribution of cetuximab compared to xenografts with higher ErbB1 expression.19
An alternative approach consists in improving diffusion by employing smaller antibody fragments, such as Fab fragments (~50 kDa), single-chain variable fragments (scFv ~30 kDa) and single-domain antibodies (sdAb 12–15 kDa). Yet, while these formats indeed possess higher diffusion rates, the tumor distribution achieved in physiological settings is poor because the clearance rates for smaller fragments is markedly higher relative to full-size antibody molecules.15,20 IgG immunoglobulins undergo salvage recycling through interaction of the Fc region with the neonatal Fc receptor (FcRn), leading to prolonged half-lives of >20 days for most therapeutic mAbs.21 Conversely, antibody fragments lacking an Fc region display half-lives of hours, or even minutes for formats below the glomerular filtration cutoff (30–50 kDa). The high elimination rates upon systemic delivery prevent most antibody fragments from saturating the tumor and achieving uniform distributions.22,23 Increasing tumor tissue penetration thus poses significant challenges given the intricate pharmacokinetic properties of IgGs.
Resistance to monoclonal antibody therapy
Understanding the resistance mechanisms that affect monoclonal antibody therapy in cancer has proven to be a strenuous task, insofar as the antitumor activity of mAbs stems from a multiplicity of molecular mechanisms – eg, signaling pathway disruption, antibody-dependent cellular cytotoxicity (ADCC), antibody dependent cellular phagocytosis (ADCP) and complement dependent cytotoxicity (CDC). To this day, the clinical contribution of the various modes of action involved in the anticancer activity of most mAbs remains controversial.24–26 On that account, intrinsic phenotypic variations in tumor cells or tumor-related cells affecting any of the involved modes of action can compromise treatment efficacy. Moreover, adaptive phenotypic modifications can arise following repeated exposure to sub-optimal doses of the biotherapeutic resulting in acquired resistance.27,28
Most of the current understanding of the contributing factors in the development of intrinsic or acquired resistance and their clinical significance comes from preclinical and clinical trials of benchmark therapeutic antibodies. Notwithstanding that the modes of action of different mAbs are not identical, the vast clinical data available for these benchmark antibodies are pivotal to comprehend host response and optimize monoclonal antibody therapy. The next sections briefly discuss resistance mechanisms identified in clinical settings for trastuzumab in HER2 positive breast cancer, and for cetuximab in colorectal cancer as archetypes of solid tumor treatment.
Resistance to trastuzumab (anti-HER2 therapy)
Trastuzumab was the first therapeutic monoclonal antibody to be approved for a solid carcinoma (FDA approval in the year 1998).29 Trastuzumab targets the extracellular domain (ECD) of the human epidermal growth factor receptor 2 (HER2/Neu or ErbB2) that is overexpressed in a broad range of malignancies. HER2 overexpression is detected in 15–20% of breast cancers, and this subset is associated with poor prognosis and higher rates of recurrence.30,31
HER2 exists primarily as a monomeric receptor that can form heterodimers with other members of the ErbB family of receptors (HER1, HER3 and HER4) upon ligand-mediated activation of the latter. Heterodimerization activates the MAPK and PI3K/AKT/mTor intracellular pathways, inducing cell proliferation and inhibition of apoptosis, respectively.32,33 Direct binding of trastuzumab with HER2 can hinder heterodimerization and promote proteolysis of the receptor through receptor-mediated endocytosis. This interaction inhibits downstream signaling and causes cell cycle arrest by accumulation of the cyclin-dependent kinase inhibitor p27.34 Additionally, trastuzumab can mediate ADCC35,36 and ADCP.26,36,37 Induction of CDC has also been documented in in vitro experiments, but it is thought to contribute only minimally to the anticancer effect in patients.36,38
Intrinsic alterations of the HER2 receptor involving regions associated with the binding epitope of trastuzumab have been linked to intrinsic (or primary) resistance mechanisms. For instance, alternate transcription initiation sites can result in the expression of a truncated variant of the receptor (p95-HER2) that lacks the cognate epitope for trastuzumab.39 Insertions and point mutations in the tyrosine kinase domain of HER2 have been identified in various cancers, some of them associated with resistance to trastuzumab and lapatinib, however evidence of such mutations in HER2 overexpressing breast cancers has not been reported to date.40,41 A further alteration resulting in impaired target binding comes from the overexpression of mucin-4, which has been shown to induce association with HER2 causing steric hindrance to abrogate trastuzumab binding to HER2.42
Additional intrinsic and acquired resistance mechanisms predominantly involve alterations in the P13K/Akt/mTOR axis, activation of other ErbB receptors (especially EGFR and HER3) by increased ligand production, and circumvention of HER2 binding by activation of the PI3K cascade through alternative pathways. Mutations in PIK3CA and function impairment of PTEN (both downstream of HER2 signaling) have been implicated in bypassing HER2 blockade.43,44 Overexpression of the insulin-like growth factor (IGF-IR) has been documented as an adaptive response to trastuzumab by some tumors, resulting in resistance to the antibody. IGF-1R can form heterodimers and heterotrimers with HER2 and HER3 in breast cancer cells resistant to trastuzumab.45,46 Similarly, increased levels of EpoR, EpHA2 and RTK MET can activate P13K/Akt/mTOR by interacting with other members of the ErbB family or through activation of intracellular kinases.46–48
Resistance to cetuximab (anti-EGFR therapy)
The epidermal growth factor receptor (EGFR; HER1; ErbB1) forms part of the ErbB family of receptors. EGFR is pivotal in modulating proliferative mechanisms and has been implicated in a broad range of cancers.49–51 Cetuximab (chimeric IgG1) was the first anti-EGFR mAb to receive regulatory approval in 2004.52 Since then, two more anti-EGFR mAbs (panitumumab and necitumumab) and six anti-EGFR small molecule inhibitors (gefitinib, erlotinib, lapatinib, neratinib, vandetanib and osimertinib) have obtained regulatory approval for various cancers.4
The anticancer activity of cetuximab partially resembles that of trastuzumab in that it targets another member of the ErbB family of receptors with intrinsic protein tyrosine kinase activity. Accordingly, dimerization of EGFR can activate the PI3K/AKT/mTOR, RAS/RAF/MAPK and JAK/STAT signaling pathways to promote cell growth and proliferation.53,54 In contrast to HER2, EGFR can undergo a conformational transition triggered by binding of specific ligands, predominantly EGF and TGFα, that promotes the formation of homodimers and heterodimers with other members of the HER family.55 Cetuximab can block ligand activation of EGFR by binding directly to the ECD III of the receptor and inducing receptor internalization and proteolysis.56 A further contributing mechanism of action involves suppression of VEGF (a pro-angiogenic factor) production resulting in impaired angiogenesis.57 Moreover, ADCC and CDC are also believed to contribute to cetuximab efficacy in EGFR over-expressing cancers.58,59
There is vast documentation of primary and acquired resistance to anti-EGFR therapy in patients with colorectal and head and neck cancer. Indeed, roughly 80% of metastatic colorectal cancer patients do not display susceptibility to EGFR blockade.60 This low response rate has been linked to a broad spectrum of alterations in several of the components of the downstream signaling pathways. Specifically, mutations in the PIK3CA,61 NRAS, BRAF and KRAS62 genes that confer constitutive activation of the EGFR are among the best studied contributing factors in intrinsic and acquired resistance. Further alterations such as low EGFR copy numbers or low expression of specific EGFR-ligands (eg, EREG and AREG) have been implicated in resistance to EGFR therapy.63,64
EGFR downregulation and structural modifications in the binding region can also compromise treatment efficacy.65 The role of mutations in the ECD of EGFR in cetuximab resistance remains unclear. Recent publications have identified several point mutations that abrogate cetuximab binding to the receptor.66 Still, RAS mutations are found more frequently in refractory patients than ECD mutations and have been associated with worst clinical outcomes.67
Novel approaches to enhance efficacy
Increasing the therapeutic index with antibody-drug conjugates
ADCs were conceived as an approach to enhance the therapeutic window of its primary components, namely the targeted antibody and a cytotoxin or an immunotoxin covalently attached to the antibody. Endowing the drug with specificity toward a molecular target – by virtue of the attachment of an antibody – allows for the utilization of highly potent cytotoxic compounds, that otherwise display intolerable systemic toxicity.
ADCs increase the intrinsic potency of the targeted treatment – relative to the antibody agent, therefore lower doses are required to reach the tumor to effectively destroy the targeted cells. Moreover, depending on the chemical nature of the drug and its release in the tumor (either intracellular or extracellular), some payloads can subsequently diffuse and kill surrounding cells (“bystander killing”).68,69 Consequently, these features could ameliorate the drawbacks of the heterogeneous tumor distributions of therapeutic antibodies and decrease the risk of developing resistance.
Despite the potential of the concept, the clinical implementation of ADCs has met with significant challenges, mostly regarding off-target toxicity. To date, only four ADCs (Mylotarg, Adcetris, Kadcyla and Besponsa) have received regulatory approval. Gemtuzumab ozogamicin (Mylotarg) (anti-CD33) was the first to enter the market in 2000 under an accelerated approval process.70 It was originally approved as stand-alone treatment for refractory CD33-positive acute myeloid leukemia, but it was voluntarily withdrawn in 2010 after failure to display benefits relative to standard therapies in a phase III comparative controlled clinical trial (NCT00085709 or SWOG-0106).71 Moreover, Mylotarg caused a significantly higher rate of fatal induction toxicity in this confirmatory trial. Gentuzumab ozogamicin had previously raised hepatotoxicity concerns due to high incidence (~20%) of Grade 3 or 4 liver transaminitis and hyperbilirubinemia, and reports of hepatic veno-occlusive disease.72 Mylotarg received FDA approval once again in 2017 following a careful review of the dosing regimen, whereby fractionated lower-dose regimens demonstrated a decrease in early mortality without compromise in complete remission rate.73,74 Brentuximab vedotin (Adcetris) (anti-CD30 for Hodgkin lymphoma and anaplastic large cell lymphoma) and ado-trastuzumab emtansine (Kadcyla) (anti-HER2 for HER2-positive metastatic breast cancer) gained approvals in 201175 and 2013,76 respectively. More recently, the FDA granted approval to inotuzumab ozogamicin (Besponsa) (anti-CD22) for treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL) in 2017.77
The clinical development of ADCs has been hampered predominantly by systemic toxicity due to off-target release of the payload. Most adverse effects reported in clinical reports are ascribed to the potent cytotoxicity of the payload, underlining the importance of improving ADC design to enhance therapeutic index.78,79 On that account, the linker chemistry plays a crucial role in determining plasma stability to prevent premature release.
Linker chemistry
Earlier ADC formats carried mostly chemically labile linkers, such as pH-labile moieties intended to be released within the cell. These linkers should be stable at the neutral pH of the blood (pH 7.3–7.5) and undergo hydrolysis once they are internalized within the cell by receptor-mediated endocytosis, where the more acidic environment of the endosome (pH 5.0–6.5) or the lysosome (pH 4.5–5.0) trigger the release of the payload.80–82 Both Mylotarg and Besponsa employ a pH-labile hydrazone linker. Other early constructs bore reducible disulfide linkers that enable payload delivery in the intracellular reducing environment. The higher concentrations of glutathione in the intracellular compartment induce disulfide bond reduction and cytotoxin release.83
Since then, plasma stability has been improved by the implementation of alternative release strategies. Most commonly, the linker is designed to possess a dipeptide sequence that is recognized and cleaved by lysosomal proteases following receptor-mediated endocytosis. Most ADCs currently in development employ this approach.82,84 Specifically, the dipeptide valine-citrulline group – recognized and cleaved by cathepsin B (lysosomal protease) – is the most widely implemented technology in the current clinical pipeline.84 A further approach consists in utilizing non-cleavable linkers, whereby release of the drug requires cellular uptake and proteolysis (Figure 2).
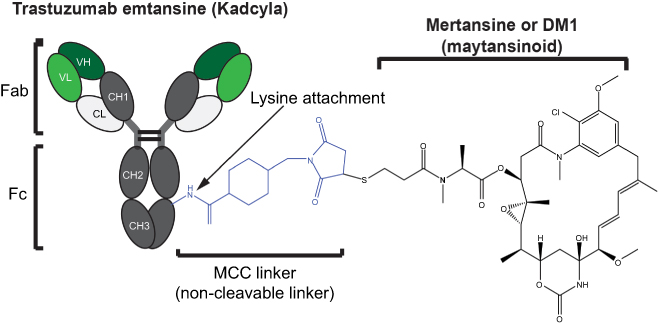 | Figure 2 Structural components of an antibody–drug conjugate. Trastuzumab emtansine is a commercially approved anti-Her2 antibody with a potent maytansinoid payload attached to lysines in the mAb polypeptide chain through a non-cleavable linker. |
Conjugation methods
Most of the ADC formats that have entered clinical trials employ stochastic conjugation methods to lysine residues in the antibody, or to free SH groups in cysteines obtained by partial reduction of the interchain disulfide bonds. These techniques, although widely used, suffer from several disadvantages. In IgG molecules, lysine side chains are abundant and lysine conjugation yields consequently highly heterogeneous drug attachments, some of them occurring on residues where attachment can be detrimental to the physicochemical stability of the antibody.85 Additionally, highly heterogeneous drug-to-antibody-ratios (DAR) are obtained, where the ADCs with high DARs (>8) show more narrow therapeutic indices.86 Conjugation to free SH groups offers greater homogeneity as the maximum amount of available SH groups after partial reduction of the interchain disulfide bonds is limited to 8. Nonetheless, the disruption of these bonds can result in alterations in the quaternary structure of the IgG molecule.87 The impact of these conjugation techniques on the physicochemical stability of ADCs is thoroughly described in Ref. 88.
Novel developments in linker technologies intend to enhance the homogeneity of ADCs by providing site-specific attachment of the drug-linker to the antibody, thereby controlling the number of drugs affixed as well as preventing attachment to regions in the antibody that may impair binding to the cognate epitope or to Fc receptors on immune effector cells. The THIOMAB platform, developed by Genentech, was the first site-specific technique to be implemented, and it consists of the insertion of engineered unpaired cysteines on protein surface.89 Site-specific methods also include recombinant techniques to introduce unnatural amino acids – eg, p-acetylphelylalanine, N6-((2-azidoethoxy)carbonyl)-L-lysine, selenocysteine – in the primary sequence of the antibody that can be readily modified.89–91 Furthermore, other formats have employed short peptide tags or specific attachment to the glycan moiety in the CH2 domain.92,93 Several preclinical studies have reported superiority in efficacy and safety of site-specific homogeneous ADCs compared to conventional lysine or cysteine-conjugation chemistry.94,95 Site-specific conjugates currently account for approximately 15% of ADC formats in development.96
Cytotoxic payloads
A further increasing trend in ADC optimization focuses on the development and employment of more potent payloads. In particular, DNA alkylators – predominantly calicheamycins, pyrrolobenzodiazepines and duocarmycins – have seen a significant increase in popularity in the development of novel ADC platforms.97 This strategy gained relevance after several ADCs failed to demonstrate adequate efficacy in clinical trials early in the decade. In 2013, 80% of the clinical pipeline was made up of conjugates bearing antimitotic agents, namely auristatins or maytansinoids (mostly DM1, DM4, MMAE and MMAF). Since then, this fraction has dropped by >15% owing to the introduction of novel formats carrying DNA alkylating agents and other novel cytotoxic compounds, eg trastuzumab deruxtecan,98 trastuzumab duocarmazine,99 vadastuximab talirine.100,101
Further optimization of ADC design is sure to bring about major improvements to the field of antibody therapeutics and precision medicine. The field has grown dramatically in recent years and will likely continue to experience major developments in the near future as novel technologies and strategies are implemented in preclinical and clinical development. The ADC field will also benefit from advancements in the identification of novel target antigens.
Engaging the immune system
Immune checkpoint blockade
One of the most important recent developments in antibody therapy in oncology has been the introduction of ICI in the clinic. ICI therapy consists in the utilization of monoclonal antibodies to disrupt key signaling pathways involved in the suppression of immune effector cells.102 Releasing the brakes of the immune system in this way can trigger potent and durable antitumor responses. One of the most advantageous features of ICI therapy is the capability of eliciting antitumor responses in a wide range of malignancies, since the treatment engages the immune machinery as opposed to traditional targeted therapy that is specific to antigens expressed in cancer cells. A further key feature of immune checkpoint blockade is the observed long-term durability of the anticancer response.103
Two crucial inhibitory pathways have been exploited in the development of these therapeutics, namely the cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and the programmed cell death (PD-1) receptor or its ligand PD-L1. The first FDA approval was granted in 2011 to ipilimumab (anti-CTLA-4) for late-stage melanoma, following the review of a phase III randomized, controlled trial that included 676 melanoma patients (stage III or IV) and demonstrated an increase in overall survival rate. This was the first drug to achieve a significant improvement in overall survival in advanced melanoma, and it marked a key development in the field of cancer immunotherapy.104 Following the first approval of ipilimumab the field has experienced a remarkable expansion. Anti-PD-1 antibodies pembrolizumab and nivolumab received regulatory approval in 2014. More recently, the anti-PD-L1 atezolizumab entered the clinic in 2016 and anti-PD-L1 mAbs avelumab and durvalumab in 2017 (Table 1).
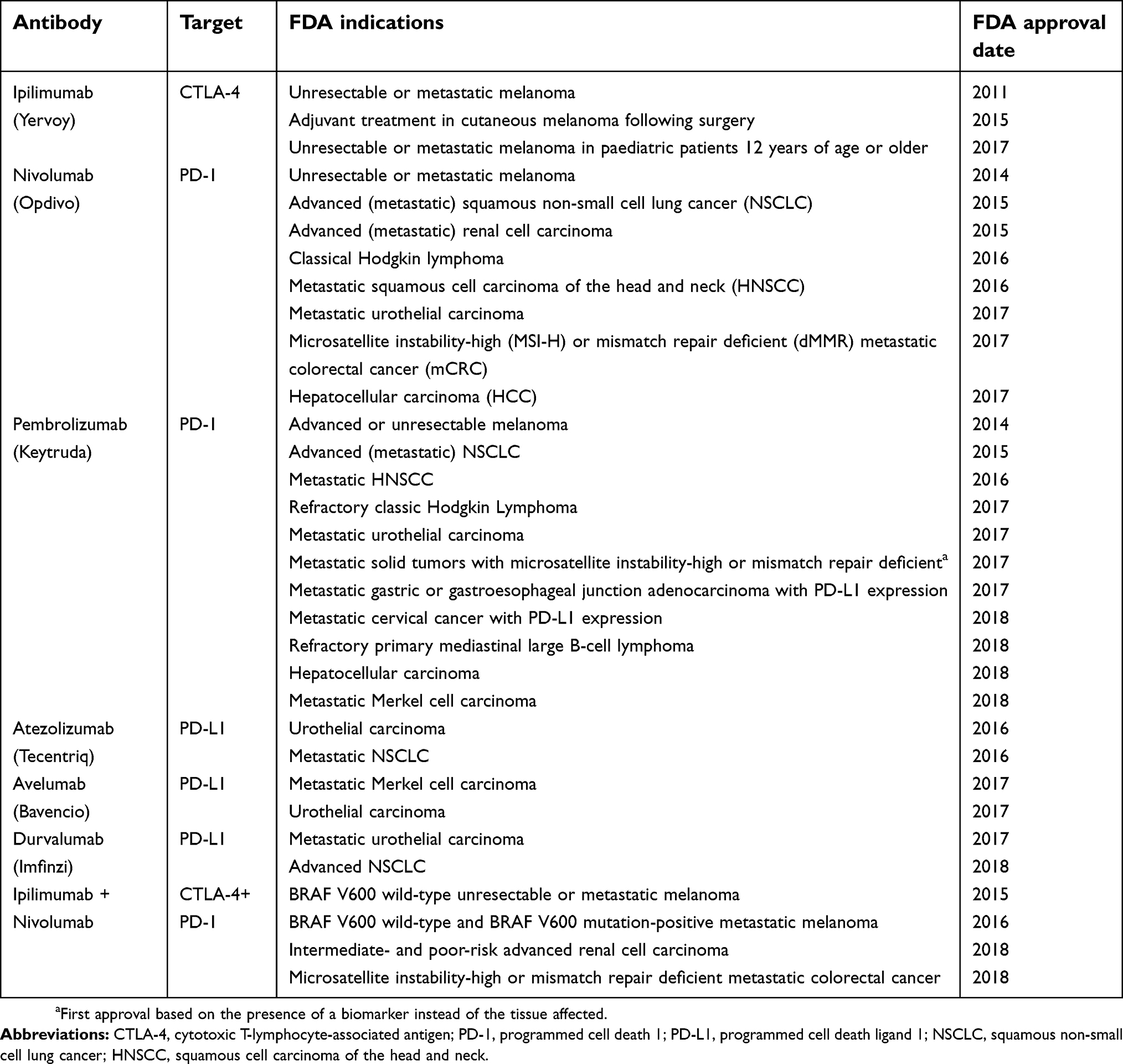 | Table 1 Approved immune checkpoint inhibitors and FDA indications |
The CTLA-4 and PD-1 immunosuppressive checkpoints are key regulatory mechanisms in immune response modulation and self-tolerance. In cancer, the presentation of neoantigens by antigen presenting cells (mainly dendritic cells) in the lymph nodes induces an initial activation of naïve T cells that leads to expansion and proliferation of cytotoxic and helper T cells specific to tumor antigens. These activated T cells can subsequently infiltrate the tumor and mount a local immune response against cancer cells. The initial activation that takes place in the lymph nodes requires two co-stimulatory events: (1) T cell receptor (TCR) activation through interaction with an major histocompatibility complex (MHC)-peptide complex on the APC and (2) co-stimulation through T cell CD28 and APC B7 ligand (CD80 or CD86) interaction.105 Upon T cell activation, CTLA-4 (CD152), which is localized in intracellular vesicles in naïve T cells, is upregulated and translocates to the cellular membrane.106–108 CTLA-4 is a homolog of CD28 with higher affinity towards CD80 (or B7-1) and CD86 (B7-2), therefore its exposure on the cell surface can lead to disruption of CD28-CD80 stimulation and T cell suppression through CD80-CTLA-4 signaling (Figure 3).102,109 CTLA-4 works as a signal damper of T cell activation and compromises the potency of the immune antitumor response. Recent data indicate that the therapeutic efficacy of anti-CTLA-4 antibodies in oncology could also stem from a selective depletion of intratumoral regulatory T cells (Treg) through ADCC or ADCP, mediated by antibody binding to overexpressed CTLA-4 in these Tregs.110–112 Comprehensive reviews of the mechanism of action of anti-CTLA-4 therapy can be found in.113,114
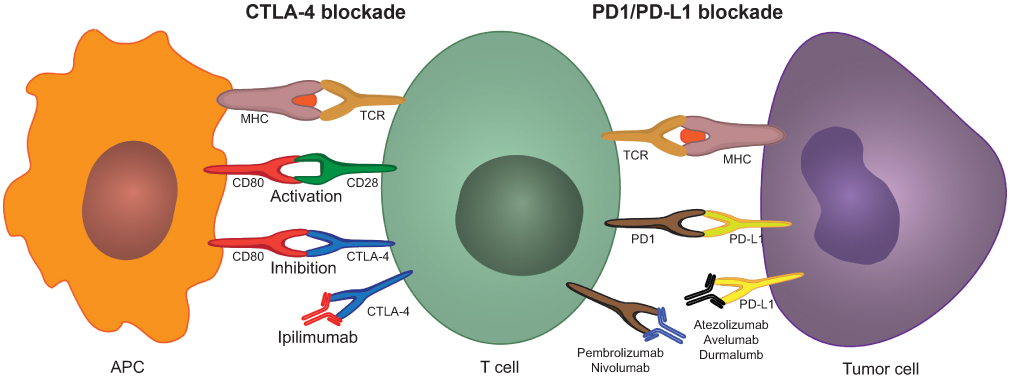 | Figure 3 CTLA-4 and PD1/PD-L1 blockade using immune checkpoint inhibitors. Dendritic cells process and present tumor neoantigens through the MHC to the TCR on T-cells in the draining lymph nodes. T-cell activation further requires a co-stimulatory signal by CD80-CD28 binding. Upon T-cell activation, CTLA-4 can be upregulated in T-cells. CTLA-4 has a higher affinity towards CD80 than CD28; therefore, the overexpression of CTLA-4 interferes with the co-stimulatory CD80-CD28 signal preventing T-cell activation. Ipilimumab prevents this mechanism by binding to CTLA-4 thus blocking its interaction with CD80. Once activated T-cells migrate to the tumor to mount an immune anti-tumor response, tumor cells and macrophages can upregulate PD-L1 and suppress the immune response by interacting with the upregulated PD-1 on T-cells. Anti-PD1 and anti-PD-L1 antibodies inhibit this adaptive immune resistance mechanism.Abbreviations: CTLA-4, cytotoxic T-lymphocyte-associated antigen; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; MHC, major histocompatibility complex; TCR, T cell receptor. |
The PD-1/PD-L1 pathway plays a crucial role in adaptive immune responses. PD-1 is expressed by activated T cells, B cells, macrophages, natural killer (NK) cells and several APCs.115 PD-1 expression on naïve T cells is induced upon TCR stimulation or TGF-β and cytokine (eg, IL-2, IL-7, IL-15, IL-21) autocrine/paracrine signaling. When activated tumor-specific T cells infiltrate the tumor, TCRs are triggered by recognition of the MHC-cognate antigen complex, resulting in the release of interferon-γ (IFN-γ) and other inflammatory cytokines. Secretion of IFN-γ can induce the expression of PD-L1, and PD-L2 to a lesser extent (PD-1 ligands), on the cell surface of tumor cells and tumor macrophages.116 PD-1 binding to PD-L1 suppresses the T cell response of previously activated T cells at the tumor-invasive margin, leading to adaptive immune resistance (Figure 3).117,118 The proposed mechanism of action of PD-1/PD-L1 inhibitors thus consists of suppression of the PD-1 regulatory signal exerted on activated tumor-infiltrating T cells.119 Nonetheless, further mechanisms of action have been suggested and are reviewed elsewhere.119–121
Targeting PD-1 or PD-L1 has been presumed to be a more tumor-specific approach than CTLA-4 blockade, given the involvement of the former in restoring T cell function at the effector stage which requires previous tumor-specific T-cell activation. This is supported by clinical data showing improved outcomes and a lower rate of grade 3–4 adverse events with anti-PD-1 therapy compared to ipilimumab (anti-CTLA-4).122 The open-label, randomized, phase III clinical trial KEYNOTE-006 provided a head-to-head comparison of advanced melanoma treatment with ipilimumab or two different dose regimens of pembrolizumab (anti-PD-1). Pembrolizumab treatment achieved a more than twofold increase in 24-month progression-free survival rates compared to ipilimumab while the 24-month overall survival rate was 55% (pembrolizumab) to 43% (ipilimumab).122,123 Moreover, pembrolizumab has shown clinical efficacy in advanced melanomas refractory to ipilimumab by increasing progression-free survival.124 A clinical trial comparing nivolumab (anti-PD-1) to ipilimumab in advanced melanoma also reported substantial improvements in overall survival and progression-free survival rates with PD-1 therapy.125
Moving forward with ICI
Despite the remarkable clinical outcomes of ICI therapy, immune checkpoint blockade is still a relatively new concept and is undergoing extensive efforts for optimization. Key limitations being addressed include low objective response rates and primary and acquired resistance to treatment. Low objective response rates are presumably associated with primary resistance mechanisms. Achieving higher response rates will likely come from a better understanding of tumor biology and the elucidation of biomarkers that can identify patients that are more likely to respond to specific immunotherapeutics.
Moreover, since CTLA-4 and PD-1 are non-redundant inhibitory mechanisms, combination therapy targeting both pathways can significantly increase objective response rates. This was shown in a phase II trial where nivolumab plus ipilimumab therapy displayed a 61% objective response rate compared to 11% with ipilimumab mono-therapy.126 Dual immune checkpoint inhibition has shown great promise in increasing therapeutic efficacy and nivolumab plus ipilimumab combination has already gained approval for metastatic melanoma, renal cell carcinoma and microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer (Table 1). Notwithstanding, combined therapy also seems to increase the frequency of immune-related toxicities.127 Thorough reviews on strategies and novel concepts for combination therapy can be found in.128,129 Additionally, alternative inhibitory pathways of the antitumor immune response are also being targeted for clinical development; for example, blockade of LAG-3, TIM-3, TIGIT, VISTA, and others have started early clinical trials; and are reviewed elsewhere.130
Bispecific antibodies (BsAbs)
A conceptually different strategy to engage the immune system in tumor cell depletion consists in the use of bispecific antibodies (BsAbs), wherein one arm of the BsAb targets a tumor cell antigen while the other arm recruits and activates T cells, or other immune effector cells. Additionally, various BsAb formats have been designed for therapeutic approaches that do not involve direct immunomodulation; eg, cross-linking or inhibition of two different receptors.131,132 A plethora of bispecific antibody formats have started clinical development, and have been reviewed by others.133–135 Nonetheless, only two bispecific formats have obtained approval by established regulatory agencies for cancer therapy. The first case – catumaxomab – comprises a hybrid rat-mouse full-size mAb with specificity toward tumor-expressed EpCAM and to the CD3 T cell co-receptor. Catumaxomab was approved by the EMA in 2009 for treatment of malignant ascites in EpCAM positive carcinomas.136 Conversely, the other marketed BsAb – blinatumomab – comprises two scFv proteins connected by a peptide linker; a BsAb format called Bispecific T cell Engagers (BiTE). Blinatumomab binds to CD19 expressed on malignant B lymphocytes, while also engaging the CD3 co-receptor to recruit T cells. Blinatumomab was approved by the FDA in 2014 under the accelerated approval program, for use in precursor B-cell ALL.122
Another flourishing strategy in cancer immunotherapy with bispecifics involves the recruitment and activation of NK cells. Analogous to BiTEs, Bispecific Killer cell Engagers (BiKEs) possess two scFv fragments; one directed towards a tumor antigen and another scFv that engages FcγRIIIa (CD16) on NK cells. Moreover, trispecific formats (TriKEs) have been created by incorporating an additional scFv fragment targeting another tumor antigen;137 or alternatively containing IL-15 to induce NK cell expansion.138 Several BiKEs and TriKEs are undergoing preclinical development.139,140 Other strategies to target NK cells for tumor eradication have been reviewed in.141
Nanoparticle delivery vehicles to improve tumor delivery
In cancer therapy, NP delivery systems offer the possibility to modify the pharmacokinetic profile of small molecule cytotoxins and increase tumor targeting as a means to improve therapeutic indices and safety profiles. NP delivery systems are typically in the 10–100 nm range, making them susceptible to accumulation in tumor tissues as a consequence of the EPR effect. The EPR refers to the enhanced accumulation of nanostructures in tumor tissue following extravasation through the endothelium that irrigates the neoplasm. The vasculature in these sites is formed rapidly due to an increased demand of oxygen and nutrients and secretion of vascular effectors, leaving large fenestrations or endothelial gaps that allow diffusion of NPs that are otherwise too large to penetrate through healthy capillaries.142–144 Moreover, accumulation of NPs is further enhanced by a decrease in lymphatic drainage.142,145 Preferential accumulation due to the EPR effect is termed passive targeting and is an inherent property of nano-sized materials. Importantly, the contribution of the EPR effect in preclinical and clinical settings has been debated and it is known to depend on myriad factors relating to tumor characteristics, including localization, stage, vascular density, fibrotic tumor microenvironment, lymphatic drainage and vascular architecture.143,146,147 Still, the EPR remains a fundamental principle behind the design and development of NP delivery strategies for solid tumors. Several NPs have also been developed as imaging agents; however, this section discusses only those formats intended for therapeutic purposes in oncology.
Since the first reports of the EPR effect in 1986,142 interest in the development of NP-delivery platforms has increased substantially and has led to the approval of several NP formulations. At present, liposomal delivery systems comprise the vast majority of NP-based therapeutics approved for clinical use in oncology and those undergoing clinical development.148 Doxil (doxorubicin encapsulated in PEGylated liposomes) was the first nano-carrier to be licensed in the US in 1995 for treatment of AIDS-related Kaposi’s sarcoma. The first approval of Doxil served as a benchmark for the validation of NP systems in oncology, and the formulation is currently also FDA approved in ovarian cancer and multiple myeloma.149 Importantly, the approval granted for the aforementioned indications was based on superior safety profiles compared to established therapy, and it also demonstrated superior efficacy in Kaposi’s sarcoma.150–152 Thereafter, 10 other nanotherapeutics have entered the clinic (Table 2). Except for Abraxane (albumin-bound paclitaxel) and NanoTherm (iron oxide NPs), all other approved nanomedicines consist of liposomal chemotherapeutics.148,153
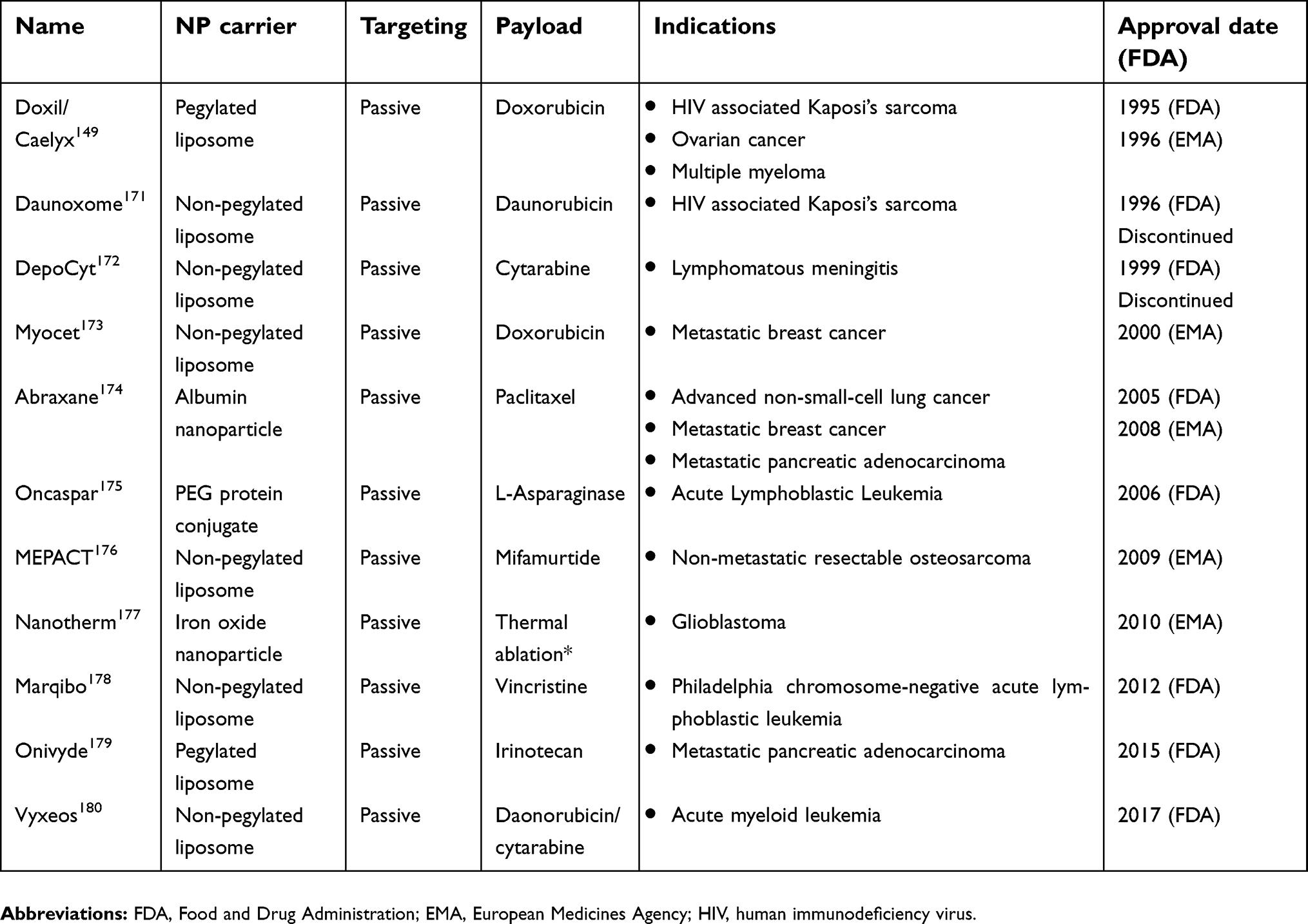 | 3.6. Approved nanoparticles in oncology |
Subsequent advancements in NP synthesis and engineering have allowed for the development of multifunctional NP delivery platforms with expanded therapeutic capabilities. For instance, a highly attractive characteristic of NPs is the possibility to functionalize their surface with multiple bioactive substances that can aid in tumor localization, treatment and diagnosis. A representative case is CYT-6091, a construct composed of PEGylated gold NPs carrying tumor necrosis factor alpha (TNFα) on its surface that has shown promising results in a phase I clinical trial, wherein the maximum tolerated dose of nano-formulated TNFα exceeded that of native TNFα by threefold due to enhanced localization in tumors.154 Furthermore, NPs can be functionalized with biomolecules that target the tumor stroma to induce changes in the extracellular matrix (ECM) and facilitate uptake. This strategy is conceptually appealing, and it is thought to hold great promise, yet it requires further understanding of the cross-talk between the multiple paracrine interactions that take place during the formation of the ECM.155,156 Additionally, the physicochemical properties of the NP format can be tailored to enable controlled release of a drug cargo upon exposure to tumor-specific or external stimuli. Examples of these NP vehicles include pH-responsive polymeric micelles, temperature responsive polyN-isopropylacrylamide NPs, light responsive mesoporous silica NPs and redox-responsive copolymer-based micelles.157–160
Active-targeting to increase specificity
Conceptually, the targeting capacity of NPs can be further enhanced through the attachment of target-specific biomolecules, such as mAbs, antibody fragments, aptamers, affimers and peptides. In this modality, NPs can initially accumulate in tumor tissue due to passive targeting and subsequently engage in high-affinity interactions with tumoral targets (Figure 4).161–163 Most commonly, upregulated cell surface receptors (eg, HER2, EGFR, transferrin receptor, folate receptor) are targeted, wherein the multivalent presentation of targeting agent on the NP surface can cause receptor cross-linking and induce receptor-mediated endocytosis – an advantageous feature for intracellular drug delivery.164 Monoclonal antibodies play a pivotal role in this strategy due to their exquisite specificity; as well as to the existence of well-established techniques – primarily phage display – that allow for high-throughput development of mAbs to specific antigens. Additionally, the prevailing clinical success of monoclonal antibodies is favorable for regulatory approval.
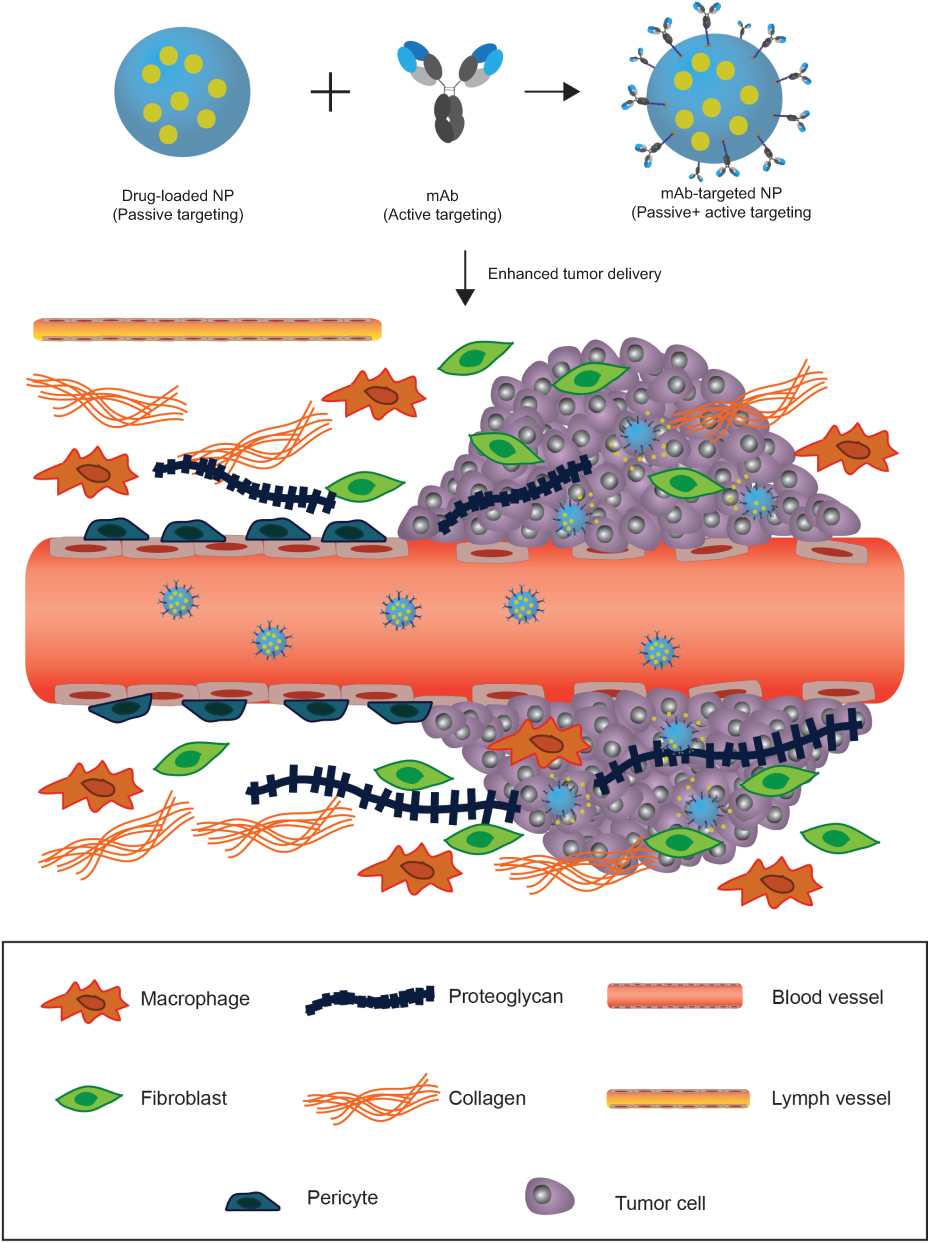 | Figure 4 Harnessing the EPR effect to improve tumor delivery using nanoparticle carriers. Blood vessels that irrigate the tumor tissue are defective. The lack of pericytes and altered structural features make the vessels less stable and leaky. Larger fenestrations between the endothelial cells allow nanoparticles to extravasate into the tumor. The fibrotic extracellular matrix lacks proper lymphatic drainage, therefore, nanoparticles can accumulate in the tissue following extravasation (passive targeting). Nanoparticles (NP) can be functionalized with monoclonal antibodies, or other active targeting agents, to promote specific internalization and drug delivery into targeted cells (cancer cells or other cells in the tumor microenvironment) once they accumulate in the tumor through passive targeting. |
At present, a plethora of active-targeted NPs have undergone preclinical development, however only a few have initiated clinical trials. A notable example is BIND-014, a docetaxel-containing polymeric NP targeting the prostate-specific membrane antigen that has recently completed phase II clinical trials for various cancers, where it has demonstrated clinical efficacy and acceptable safety profiles.165 Recently, paclitaxel solid lipid NPs conjugated to various antibodies as targeting agents have demonstrated remarkable pharmacokinetic properties and efficacy in mouse models, and have started clinical development.166
In its inception, active targeting was intended to aid tumor localization and retention in conjunction with passive targeting. Notwithstanding, experimental data have demonstrated that while engagement and internalization within cancer cells are significantly increased, tumor accumulation is only marginally improved.20,163,167 An extensive analysis of in vivo data published from 2005 to 2015 showed that passive targeting results in 0.6% (median) of the injected dose accumulating in tumor tissue, compared to 0.9% with active-targeted NPs.146 It is noteworthy to underscore that these data were obtained with several different NP formats administered in a wide variety of solid tumors. Still, it suggests that accumulation via passive targeting is essential for enhanced delivery of payloads through active-targeting. Consequently, successful clinical implementation of both passive and active-targeted NPs will require a better understanding of the physiological factors that determine the extent of EPR accumulation in order to identify patients that can benefit from this approach.168 Alternatively, therapeutic strategies to increase EPR-related accumulation can be implemented, such as administration of angiotensin-II receptor blockers to increase vessel perfusion, or sonoporation to promote vascular permeability.169,170
Conclusion
Improving tumor penetration and distribution upon systemic delivery are crucial requirements in mAb therapy to improve clinical outcomes and prevent the emergence of acquired resistance mechanisms. Preventing treatment failure due to intrinsic resistance will require a better understanding of cancer biology and the identification of novel biomarkers for a better selection of therapeutic agents and treatment regimens. The clinical pipeline of alternative mAb-based approaches to enhance clinical efficacy has experienced a marked expansion in the last decade. The formats discussed in this review – ADC, immune-checkpoint inhibitors and NP-delivery systems – are among key strategies with demonstrated clinical benefits in the treatment of solid tumors. Of note is the accelerated growth of the ICI class having obtained regulatory approval for 6 distinct antibodies since the year 2011 (first approval) and a remarkable broadening of clinical indications. Despite their clinical success, these therapeutics are based on relatively new technologies that are still undergoing extensive efforts to optimize therapeutic potential. Numerous ICI antibodies targeting alternative targets for immune inhibition (eg, LAG-3, TIM-3, TIGIT, VISTA, B7-H3) are in phase I/II clinical trials. Safety concerns inherent to the high potency and structural versatility of ADCs and NPs have been prominent barriers in their implementation, but clinical validation of novel designs could bring about major breakthroughs in these fields in the coming years.
Acknowledgments
EC acknowledges the Ministry of Science, Technology and Telecommunications of the Republic of Costa Rica for postgraduate scholarship.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 2017;122:2–19. doi:10.1016/j.addr.2016.11.004
2. Chung J. Special issue on therapeutic antibodies and biopharmaceuticals. Exp Mol Med. 2017;49:e304. doi:10.1038/emm.2017.46
3. Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2018;37(1):9–16.
4.
5. De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. doi:10.1016/S1470-2045(10)70130-3
6. Jin MH, Nam AR, Park JE, Bang JH, Bang YJ, Oh DY. Resistance mechanism against trastuzumab in HER2-positive cancer cells and its negation by src inhibition. Mol Cancer Ther. 2017;16(6):1145–1154. doi:10.1158/1535-7163.MCT-16-0669
7. Baker JHE, Kyle AH, Reinsberg SA, et al. Heterogeneous distribution of trastuzumab in HER2-positive xenografts and metastases: role of the tumor microenvironment. Clin Exp Metastasis. 2018;35(7):691–705. doi:10.1007/s10585-018-9929-3
8. Pallasch CP, Leskov I, Braun CJ, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell. 2014;156(3):590–602. doi:10.1016/j.cell.2013.12.041
9. Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 1989;37(1):77–104.
10. Juweid M, Neumann R, Paik C, et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52(19):5144–5153.
11. Bhutani D, Vaishampayan UN. Monoclonal antibodies in oncology therapeutics: present and future indications. Expert Opin Biol Ther. 2013;13(2):269–282. doi:10.1517/14712598.2012.758705
12. Netti PA, Baxter LT, Boucher Y, Skalak R, Jain RK. Time-dependent behavior of interstitial fluid pressure in solid tumors: implications for drug delivery. Cancer Res. 1995;55(22):5451–5458.
13. Young JS, Llumsden CE, Stalker AL. The significance of the “tissue pressure” of normal testicular and of neoplastic (Brown-Pearce carcinoma) tissue in the rabbit. J Pathol Bacteriol. 1950;62(3):313–333.
14. Less JR, Posner MC, Boucher Y, Borochovitz D, Wolmark N, Jain RK. Interstitial hypertension in human breast and colorectal tumors. Cancer Res. 1992;52(22):6371–6374.
15. Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60(12):1421–1434. doi:10.1016/j.addr.2008.04.012
16. Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31(7):1191–1198.
17. Saga T, Neumann RD, Heya T, et al. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. Proc Natl Acad Sci U S A. 1995;92(19):8999–9003.
18. Thurber GM, Zajic SC, Wittrup KD. Theoretic criteria for antibody penetration into solid tumors and micrometastases. J Nucl Med. 2007;48(6):995–999. doi:10.2967/jnumed.106.037069
19. Lee CM, Tannock IF. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer. 2010;10:255. doi:10.1186/1471-2407-10-663
20. Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther. 2009;8(10):2861–2871. doi:10.1158/1535-7163.MCT-09-0195
21. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. doi:10.2165/11531280-000000000-00000
22. Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29(2):57–61. doi:10.1016/j.tips.2007.11.004
23. Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol. 1999;17:780. doi:10.1038/11717
24. Weiner GJ. Monoclonal antibody mechanisms of action in cancer. Immunol Res. 2007;39(1–3):271–278.
25. Gennari R, Menard S, Fagnoni F, et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res. 2004;10(17):5650. doi:10.1158/1078-0432.CCR-04-1002
26. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi:10.1038/74704
27. Reslan L, Dalle S, Dumontet C. Understanding and circumventing resistance to anticancer monoclonal antibodies. mAbs. 2009;1(3):222–229.
28. Viloria-Petit A, Crombet T, Jothy S, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61(13):5090–5101.
29. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi:10.1056/NEJM200103153441101
30. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182.
31. Tovey SM, Brown S, Doughty JC, Mallon EA, Cooke TG, Edwards J. Poor survival outcomes in HER2-positive breast cancer patients with low-grade, node-negative tumours. Br J Cancer. 2009;100(5):680–683. doi:10.1038/sj.bjc.6604940
32. Mitri Z, Constantine T, O‘Regan R. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012;2012:743193. doi:10.1155/2012/743193
33. Yamamoto T, Ikawa S, Akiyama T, et al. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature. 1986;319(6050):230–234. doi:10.1038/319230a0
34. Le XF, Claret FX, Lammayot A, et al. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem. 2003;278(26):23441–23450. doi:10.1074/jbc.M300848200
35. Barok M, Isola J, Palyi-Krekk Z, et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol Cancer Ther. 2007;6(7):2065–2072. doi:10.1158/1535-7163.MCT-06-0766
36. Petricevic B, Laengle J, Singer J, et al. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Transl Med. 2013;11:307. doi:10.1186/1479-5876-11-307
37. Shi Y, Fan X, Deng H, et al. Trastuzumab triggers phagocytic killing of high HER2 cancer cells in vitro and in vivo by interaction with Fcgamma receptors on macrophages. J Immunol. 2015;194(9):4379–4386. doi:10.4049/jimmunol.1402891
38. Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol. 2013;7(3):580–594. doi:10.1016/j.molonc.2013.02.011
39. Scaltriti M, Rojo F, Ocana A, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–638. doi:10.1093/jnci/djk134
40. Ben–Baruch NE, Bose R, Kavuri SM, Ma CX, Ellis MJ. HER2 mutated breast cancer responds to treatment with single agent neratinib, a second generation HER2/EGFR tyrosine kinase inhibitor. J Natl Compr Canc Netw. 2015;13(9):1061–1064.
41. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17(1):1–16.
42. Nagy P, Friedlander E, Tanner M, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65(2):473–482.
43. Black JD, Lopez S, Cocco E, et al. PIK3CA oncogenic mutations represent a major mechanism of resistance to trastuzumab in HER2/neu overexpressing uterine serous carcinomas. Br J Cancer. 2015;113(7):1020–1026. doi:10.1038/bjc.2015.306
44. Kim C, Lee C-K, Chon HJ, et al. PTEN loss and level of HER2 amplification is associated with trastuzumab resistance and prognosis in HER2-positive gastric cancer. Oncotarget. 2017;8(69):113494–113501. doi:10.18632/oncotarget.23054
45. Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128. doi:10.1158/0008-5472.CAN-04-3841
46. Zhuang G, Brantley-Sieders DM, Vaught D, et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010;70(1):299–308. doi:10.1158/0008-5472.CAN-09-1845
47. Liang K, Esteva FJ, Albarracin C, et al. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell. 2010;18(5):423–435. doi:10.1016/j.ccr.2010.10.025
48. Shattuck DL, Miller JK, Carraway KL
49. Foekens JA, Klijn JGM, Schmitz PIM, Berns PMJJ. The clinical significance of epidermal growth factor receptor (EGF-R) in human breast cancer: a review on 5232 patients. Endocr Rev. 1992;13(1):3–17. doi:10.1210/edrv-13-1-3
50. Lu SH, Hsieh LL, Luo FC, Weinstein IB. Amplification of the EGF receptor and c-myc genes in human esophageal cancers. Int J Cancer. 1988;42(4):502–505.
51. Ochiai A, Takanashi A, Takekura N, et al. Effect of human epidermal growth factor on cell growth and its receptor in human gastric carcinoma cell lines. Jpn J Clin Oncol. 1988;18(1):15–25.
52. Kirkpatrick P, Graham J, Muhsin M. Cetuximab. Nat Rev Drug Discov. 2004;3:549. doi:10.1038/nrd1445
53. Andl CD, Mizushima T, Oyama K, Bowser M, Nakagawa H, Rustgi AK. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am J Physiol. 2004;287(6):G1227–G1237. doi:10.1152/ajpgi.00253.2004
54. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. 2017;9(5):52. doi:10.3390/cancers9050052
55. Walker F, Rothacker J, Henderson C, et al. Ligand binding induces a conformational change in epidermal growth factor receptor dimers. Growth Factors. 2012;30(6):394–409. doi:10.3109/08977194.2012.739619
56. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. doi:10.1016/j.ccr.2005.03.003
57. Luwor RB, Lu Y, Li X, Mendelsohn J, Fan Z. The antiepidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia-inducible factor-1 alpha, leading to transcriptional inhibition of vascular endothelial growth factor expression. Oncogene. 2005;24(27):4433–4441. doi:10.1038/sj.onc.1208625
58. Chen S, Li X, Chen R, Yin M, Zheng Q. Cetuximab intensifies the ADCC activity of adoptive NK cells in a nude mouse colorectal cancer xenograft model. Oncol Lett. 2016;12(3):1868–1876. doi:10.3892/ol.2016.4835
59. Milano GA. ADCC and treatment outcome in colorectal cancer patients receiving cetuximab-based therapy. J Clin Oncol. 2016;34(15_suppl):e15085–e15085. doi:10.1200/JCO.2016.34.15_suppl.e15085
60. Zhao B, Wang L, Qiu H, et al. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget. 2016;8(3):3980–4000.
61. Xu JM, Wang Y, Wang YL, et al. PIK3CA mutations contribute to acquired cetuximab resistance in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23(16):4602–4616. doi:10.1158/1078-0432.CCR-16-2738
62. Hsu HC, Thiam TK, Lu YJ, et al. Mutations of KRAS/NRAS/BRAF predict cetuximab resistance in metastatic colorectal cancer patients. Oncotarget. 2016;7(16):22257–22270. doi:10.18632/oncotarget.8076
63. Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6(5):279–286. doi:10.1016/S1470-2045(05)70102-9
64. Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–3237. doi:10.1200/JCO.2006.10.5437
65. Lu Y, Li X, Liang K, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67(17):8240–8247. doi:10.1158/0008-5472.CAN-07-0589
66. Braig F, Marz M, Schieferdecker A, et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget. 2015;6(14):12035–12047. doi:10.18632/oncotarget.3574
67. Van Emburgh BO, Arena S, Siravegna G, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun. 2016;7:13665. doi:10.1038/ncomms13665
68. Li F, Emmerton KK, Jonas M, et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 2016;76(9):2710–2719. doi:10.1158/0008-5472.CAN-15-1795
69. Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16(3):888–897. doi:10.1158/1078-0432.CCR-09-2069
70. Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–1496.
71. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854. doi:10.1182/blood-2013-01-466706
72. Giles FJ, Kantarjian HM, Kornblau SM, et al. Mylotarg™ (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer. 2001;92(2):406–413.
73. Norsworthy KJ, Ko CW, Lee JE, et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist. 2018. doi:10.1634/theoncologist.2017-0604
74. Jen EY, Ko CW, Lee JE, et al. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed cd33-positive acute myeloid leukemia. Clin Cancer Res. 2018;24(14):3242–3246. doi:10.1158/1078-0432.CCR-17-3179
75. Deng C, Pan B, Connor OA. Brentuximab Vedotin. Clin Cancer Res. 2013;19(1):22. doi:10.1158/1078-0432.CCR-12-0290
76. Approves FDA. Kadcyla for breast cancer. Cancer Discov. 2013;3(4):366.
77. Yurkiewicz IR, Muffly L, Liedtke M. Inotuzumab ozogamicin: a CD22 mAb-drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des Devel Ther. 2018;12:2293–2300. doi:10.2147/DDDT.S150317
78. Saber H, Leighton JK. An FDA oncology analysis of antibody-drug conjugates. Regul Toxicol Pharmacol. 2015;71(3):444–452. doi:10.1016/j.yrtph.2015.01.014
79. Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. 2018;36(1):121–135. doi:10.1007/s10637-017-0520-6
80. Hu Y-B, Dammer EB, Ren R-J, Wang G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener. 2015;4:18. doi:10.1186/s40035-015-0041-1
81. Greenfield RS, Kaneko T, Daues A, et al. Evaluation in vitro of adriamycin immunoconjugates synthesized using an acid-sensitive hydrazone linker. Cancer Res. 1990;50(20):6600.
82. Hamann PR, Hinman LM, Hollander I, et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody−calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13(1):47–58.
83. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280. doi:10.1158/0008-5472.CAN-08-1776
84. Wang Y, Fan S, Zhong W, Zhou X, Li S. Development and properties of valine-alanine based antibody-drug conjugates with monomethyl auristatin e as the potent payload. Int J Mol Sci. 2017;18(9):1860.
85. Wakankar AA, Feeney MB, Rivera J, et al. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: changes due to modification and conjugation processes. Bioconjug Chem. 2010;21(9):1588–1595. doi:10.1021/bc900434c
86. Sun X, Ponte JF, Yoder NC, et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjug Chem. 2017;28(5):1371–1381. doi:10.1021/acs.bioconjchem.7b00062
87. Seegan GW, Smith CA, Schumaker VN. Changes in quaternary structure of IgG upon reduction of the interheavy-chain disulfide bond. Proc Natl Acad Sci U S A. 1979;76(2):907–911.
88. Ross PL, Wolfe JL. Physical and chemical stability of antibody drug conjugates: current status. J Pharm Sci. 2016;105(2):391–397. doi:10.1016/j.xphs.2015.11.037
89. Axup JY, Bajjuri KM, Ritland M, et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc National Acad Sci. 2012;109(40):16101. doi:10.1073/pnas.1211023109
90. VanBrunt MP, Shanebeck K, Caldwell Z, et al. Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody-drug conjugates using click cycloaddition chemistry. Bioconjug Chem. 2015;26(11):2249–2260. doi:10.1021/acs.bioconjchem.5b00359
91. Hofer T, Thomas JD, Burke TR
92. Strop P, Liu SH, Dorywalska M, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20(2):161–167. doi:10.1016/j.chembiol.2013.01.010
93. Li X, Fang T, Boons G-J. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew Chem Int Ed Engl. 2014;53(28):7179–7182. doi:10.1002/anie.201402606
94. Behrens CR, Ha EH, Chinn LL, et al. Antibody-drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharm. 2015;12(11):3986–3998. doi:10.1021/acs.molpharmaceut.5b00432
95. Bryant P, Pabst M, Badescu G, et al. In vitro and in vivo evaluation of cysteine rebridged trastuzumab-mmae antibody drug conjugates with defined drug-to-antibody ratios. Mol Pharm. 2015;12(6):1872–1879. doi:10.1021/acs.molpharmaceut.5b00116
96. Lambert JM, Berkenblit A. Antibody–drug conjugates for cancer treatment. Annu Rev Med. 2018;69(1):191–207. doi:10.1146/annurev-med-061516-121357
97. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–337. doi:10.1038/nrd.2016.268
98. Iwata H, Tamura K, Doi T, et al. Trastuzumab deruxtecan (DS-8201a) in subjects with HER2-expressing solid tumors: long-term results of a large phase 1 study with multiple expansion cohorts. J Clin Oncol. 2018;36(15_suppl):2501. doi:10.1200/JCO.2018.36.15_suppl.2501
99. Saura C, Thistlethwaite F, Banerji U, et al. A phase I expansion cohorts study of SYD985 in heavily pretreated patients with HER2-positive or HER2-low metastatic breast cancer. J Clin Oncol. 2018;36(15_suppl):1014. doi:10.1200/JCO.2018.36.15_suppl.1014
100. Stein EM, Walter RB, Erba HP, et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood. 2018;131(4):387–396. doi:10.1182/blood-2017-06-789800
101. Drake PM, Rabuka D. Recent developments in ADC technology: preclinical studies signal future clinical trends. BioDrugs. 2017;31(6):521–531. doi:10.1007/s40259-017-0254-1
102. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736.
103. Pons-Tostivint E, Latouche A, Vaflard P, et al. Comparative analysis of durable responses on immune checkpoint inhibitors versus other systemic therapies: a pooled analysis of phase III trials. JCO Precis Oncol. 2019;(3):1–10. doi:10.1200/PO.18.00114
104. Hodi FS, O‘Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
105. Sharpe AH. Mechanisms of costimulation. Immunol Rev. 2009;229(1):5–11. doi:10.1111/j.1600-065X.2009.00784.x
106. Perkins D, Wang Z, Donovan C, et al. Regulation of CTLA-4 expression during T cell activation. J Immunol. 1996;156(11):4154–4159.
107. Alegre ML, Noel PJ, Eisfelder BJ, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157(11):4762–4770.
108. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16(1):23–35.
109. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1(9):793–801.
110. Du X, Tang F, Liu M, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018;28(4):416–432. doi:10.1038/s41422-018-0011-0
111. Bulliard Y, Jolicoeur R, Windman M, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210(9):1685–1693. doi:10.1084/jem.20130573
112. Ingram JR, Blomberg OS, Rashidian M, et al. Anti–CTLA-4 therapy requires an Fc domain for efficacy. Proc National Acad Sci. 2018;115(15):3912. doi:10.1073/pnas.1801524115
113. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018. doi:10.1158/2159-8290.CD-18-0367
114. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8(86). doi:10.3389/fonc.2018.00086
115. Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: friend or foe for immunotherapy? Oncoimmunology. 2017;7(1):e1364828–e1364828. doi:10.1080/2162402X.2017.1364828
116. Kil SH, Estephan R, Sanchez J, et al. PD-L1 is regulated by interferon gamma and interleukin 6 through STAT1 and STAT3 signaling in cutaneous T-cell lymphoma. Blood. 2017;130(Suppl 1):1458.
117. Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883–895. doi:10.1084/jem.20051776
118. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034.
119. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 – potential mechanisms of action. Nat Rev Immunol. 2014;15:45. doi:10.1038/nri3790
120. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv324. doi:10.1126/scitranslmed.aaf0746
121. LaFleur MW, Muroyama Y, Drake CG, Sharpe AH. Inhibitors of the PD-1 pathway in tumor therapy. J Immunol. 2018;200(2):375–383. doi:10.4049/jimmunol.1701044
122. Przepiorka D, Ko C-W, Deisseroth A, et al. FDA approval: blinatumomab. Clin Cancer Res. 2015;21(18):4035. doi:10.1158/1078-0432.CCR-15-0612
123. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390(10105):1853–1862. doi:10.1016/S0140-6736(17)31601-X
124. Schadendorf D, Dummer R, Hauschild A, et al. Health-related quality of life in the randomised KEYNOTE-002 study of pembrolizumab versus chemotherapy in patients with ipilimumab-refractory melanoma. Eur J Cancer. 2016;67:46–54. doi:10.1016/j.ejca.2016.07.018
125. Wan MT, Ming ME. Nivolumab versus ipilimumab in the treatment of advanced melanoma: a critical appraisal: ORIGINAL ARTICLE: wolchok JD, Chiarion-Sileni V, Gonzalez R et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017; 377:1345-56. Br J Dermatol. 2018;179(2):296–300. doi:10.1111/bjd.16785
126. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi:10.1056/NEJMoa1504030
127. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi:10.1056/NEJMoa1709684
128. Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–598. doi:10.1016/j.ccell.2018.03.005
129. Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–433. doi:10.1016/j.immuni.2018.03.007
130. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11(1):39. doi:10.1186/s13045-018-0582-8
131. Huang S, Li C, Armstrong EA, et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73(2):824–833. doi:10.1158/0008-5472.CAN-12-1611
132. Patnaik A, Gordon M, Tsai F, et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother Pharmacol. 2018;82(3):407–418. doi:10.1007/s00280-018-3623-7
133. Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67(2,Part A):95–106. doi:10.1016/j.molimm.2015.01.003
134. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20(7):838–847. doi:10.1016/j.drudis.2015.02.008
135. Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307
136. Heiss MM, Murawa P, Koralewski P, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer. 2010;127(9):2209–2221. doi:10.1002/ijc.25423
137. Gleason MK, Verneris MR, Todhunter DA, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther. 2012;11(12):2674–2684. doi:10.1158/1535-7163.MCT-12-0692
138. Arvindam US, van Hauten P, Hallstrom C, et al. CD16-IL15-CLEC12A Trispecific Killer Engager (TriKE) drives NK cell expansion, activation, and antigen specific killing of cancer stem cells in acute myeloid leukemia. Blood. 2018;132(Suppl 1):1454.
139. Wiernik A, Foley B, Zhang B, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. 2013;19(14):3844–3855. doi:10.1158/1078-0432.CCR-13-0505
140. Don Yun H, Felices M, Vallera DA, et al. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv. 2018;2(13):1580. doi:10.1182/bloodadvances.2017010645
141. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025. doi:10.1038/ni.3518
142. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392.
143. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. doi:10.1016/j.addr.2015.01.002
144. Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. doi:10.1016/j.jconrel.2010.08.027
145. Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25–37. doi:10.1007/978-1-60761-609-2_3
146. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014. doi:10.1038/natrevmats.2016.14
147. Nichols JW, Bae YH. EPR: evidence and fallacy. J Control Release. 2014;190:451–464. doi:10.1016/j.jconrel.2014.03.057
148. Tran S, DeGiovanni P-J, Piel B, Rai P. Cancer nanomedicine: a review of recent success in drug delivery. Clin Transl Med. 2017;6(1):44. doi:10.1186/s40169-017-0175-0
149. Barenholz Y. Doxil(R)–the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi:10.1016/j.jconrel.2012.03.020
150. Northfelt DW, Dezube BJ, Thommes JA, et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi‘s sarcoma: results of a randomized phase III clinical trial. J Clin Oncol. 1998;16(7):2445–2451. doi:10.1200/JCO.1998.16.7.2445
151. Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19(14):3312–3322. doi:10.1200/JCO.2001.19.14.3312
152. Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol. 2007;25(25):3892–3901. doi:10.1200/JCO.2006.10.5460
153. Ventola CL. Progress in nanomedicine: approved and investigational nanodrugs. P T. 2017;42(12):742–755.
154. Libutti SK, Paciotti GF, Byrnes AA, et al. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res. 2010;16(24):6139–6149. doi:10.1158/1078-0432.CCR-10-0978
155. Yang S, Gao H. Nanoparticles for modulating tumor microenvironment to improve drug delivery and tumor therapy. Pharmacol Res. 2017;126:97–108. doi:10.1016/j.phrs.2017.05.004
156. Zhang B, Hu Y, Pang Z. Modulating the tumor microenvironment to enhance tumor nanomedicine delivery. Front Pharmacol. 2017;8(952). doi:10.3389/fphar.2017.00952
157. Liao Z-S, Huang S-Y, Huang -J-J, et al. Self-assembled pH-responsive polymeric micelles for highly efficient, noncytotoxic delivery of doxorubicin chemotherapy to inhibit macrophage activation: in vitro investigation. Biomacromolecules. 2018;19(7):2772–2781. doi:10.1021/acs.biomac.8b00380
158. Naha PC, Bhattacharya K, Tenuta T, et al. Intracellular localisation, geno- and cytotoxic response of polyN-isopropylacrylamide (PNIPAM) nanoparticles to human keratinocyte (HaCaT) and colon cells (SW 480). Toxicol Lett. 2010;198(2):134–143. doi:10.1016/j.toxlet.2010.06.011
159. Chai S, Guo Y, Zhang Z, Chai Z, Ma Y, Qi L. Cyclodextrin-gated mesoporous silica nanoparticles as drug carriers for red light-induced drug release. Nanotechnology. 2017;28(14):145101. doi:10.1088/1361-6528/aa5e74
160. Ma YC, Wang JX, Tao W, et al. Redox-responsive polyphosphoester-based micellar nanomedicines for overriding chemoresistance in breast cancer cells. ACS Appl Mater Interfaces. 2015;7(47):26315–26325. doi:10.1021/acsami.5b09195
161. Friedman AD, Claypool SE, Liu R. The smart targeting of nanoparticles. Curr Pharm Des. 2013;19(35):6315–6329.
162. Bazak R, Houri M, El Achy S, Kamel S, Refaat T. Cancer active targeting by nanoparticles: a comprehensive review of literature. J Cancer Res Clin Oncol. 2015;141(5):769–784. doi:10.1007/s00432-014-1767-3
163. Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. doi:10.1158/0008-5472.CAN-05-4199
164. Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145. doi:10.1038/nnano.2008.30
165. Autio KA, Dreicer R, Anderson J, et al. Safety and efficacy of BIND-014, a docetaxel nanoparticle targeting prostate-specific membrane antigen for patients with metastatic castration-resistant prostate cancer: a phase 2 clinical trial. JAMA Oncol. 2018;4(10):1344–1351. doi:10.1001/jamaoncol.2018.2168
166. Kim J-H, Kim Y, Bae KH, Park TG, Lee JH, Park K. Tumor-targeted delivery of paclitaxel using low density lipoprotein-mimetic solid lipid nanoparticles. Mol Pharm. 2015;12(4):1230–1241. doi:10.1021/mp500737y
167. Sykes EA, Chen J, Zheng G, Chan WCW. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8(6):5696–5706. doi:10.1021/nn500299p
168. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y
169. Theek B, Baues M, Ojha T, et al. Sonoporation enhances liposome accumulation and penetration in tumors with low EPR. J Control Release. 2016;231:77–85. doi:10.1016/j.jconrel.2016.02.021
170. Chauhan VP, Martin JD, Liu H, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi:10.1038/ncomms3516
171. Forssen EA. The design and development of DaunoXome® for solid tumor targeting in vivo. Adv Drug Deliv Rev. 1997;24(2):133–150. doi:10.1016/S0169-409X(96)00453-X
172. Glantz MJ, Jaeckle KA, Chamberlain MC, et al. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin Cancer Res. 1999;5(11):3394–3402.
173. Batist G, Barton J, Chaikin P, Swenson C, Welles L. Myocet (liposome-encapsulated doxorubicin citrate): a new approach in breast cancer therapy. Expert Opin Pharmacother. 2002;3(12):1739–1751. doi:10.1517/14656566.3.12.1739
174. Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99–105.
175. Dinndorf PA, Gootenberg J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (ALL). Oncologist. 2007;12(8):991–998. doi:10.1634/theoncologist.12-8-991
176. Frampton JE. Mifamurtide. Pediatr Drugs. 2010;12(3):141–153. doi:10.2165/11204910-000000000-00000
177. Maier-Hauff K, Ulrich F, Nestler D, et al. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J Neurooncol. 2011;103(2):317–324. doi:10.1007/s11060-010-0389-0
178. Silverman JA, Deitcher SR. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–564. doi:10.1007/s00280-012-2042-4
179. Lamb YN, Scott LJ. Liposomal irinotecan: a review in metastatic pancreatic adenocarcinoma. Drugs. 2017;77(7):785–792. doi:10.1007/s40265-017-0741-1
180. Krauss AC, Gao X, Li L, et al. FDA approval summary: (daunorubicin and cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin Cancer Res. 2018. doi:10.1158/1078-0432.CCR-18-2990
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.