Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Molecular Functions of Ceruloplasmin in Metabolic Disease Pathology
Authors Liu Z, Wang M, Zhang C, Zhou S, Ji G ![]()
Received 28 October 2021
Accepted for publication 4 February 2022
Published 3 March 2022 Volume 2022:15 Pages 695—711
DOI https://doi.org/10.2147/DMSO.S346648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Zhidong Liu,1 Miao Wang,1 Chunbo Zhang,2 Shigao Zhou,1 Guang Ji3
1Department of Internal Medicine of Traditional Chinese Medicine, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, 200032, People’s Republic of China; 2School of Pharmacy, Nanchang University, Nanchang, Jiangxi, 330031, People’s Republic of China; 3Institute of Digestive Diseases, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, 200032, People’s Republic of China
Correspondence: Guang Ji, Institute of Digestive Diseases, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, 725 South Wanping Road, Shanghai, 200032, People’s Republic of China, Tel +86 18156416071, Fax +86 21-64385700, Email [email protected]
Abstract: Ceruloplasmin (CP) is a multicopper oxidase and antioxidant that is mainly produced in the liver. CP not only plays a crucial role in the metabolic balance of copper and iron through its oxidase function but also exhibits antioxidant activity. In addition, CP is an acute-phase protein. In addition to being associated with aceruloplasminemia and neurodegenerative diseases such as Wilson’s disease, Alzheimer’s disease, and Parkinson’s disease, CP also plays an important role in metabolic diseases, which are caused by metabolic disorders and vigorous metabolism, mainly including diabetes, obesity, hyperlipidemia, etc. Based on the physiological functions of CP, we provide an overview of the association of type 2 diabetes, obesity, hyperlipidemia, coronary heart disease, CP oxidative stress, inflammation, and metabolism of copper and iron. Studies have shown that metabolic diseases are closely related to systemic inflammation, oxidative stress, and disorders of copper and iron metabolism. Therefore, we conclude that CP, which can reduce the formation of free radicals in tissues, can be induced during inflammation and infection, and can correct the metabolic disorder of copper and iron, has protective and diagnostic effects on metabolic diseases.
Keywords: ceruloplasmin, physiological function, iron, oxidative stress, inflammatory state, metabolic disease
Introduction
Ceruloplasmin (CP), also known as copper oxidase, is a blue-looking copper (Cu) glycoprotein that was first purified from human serum a2-globulin in 1948 by Holmberg and Laurell. CP exists in two molecular isoforms: secreted CP (sCP) and a membrane glycosylphosphatidylinositol (GPI)-anchored form of CP (GPI-CP); sCP is mainly produced by the liver,1,2 while GPI-CP has been found in glial and sustentacular cells.3 CP has multiple physiological functions (Figure 1). It carries 40–70% of Cu in plasma and plays important roles in Cu transport, iron (Fe) regulation, free radical scavenging, and antioxidant processes. It also catalyzes the oxidation of a variety of substrates, such as Cu, Fe, and other organic substrates. It is closely related to Wilson's disease, aceruloplasminemia neurodegenerative disorders and other diseases.4–6
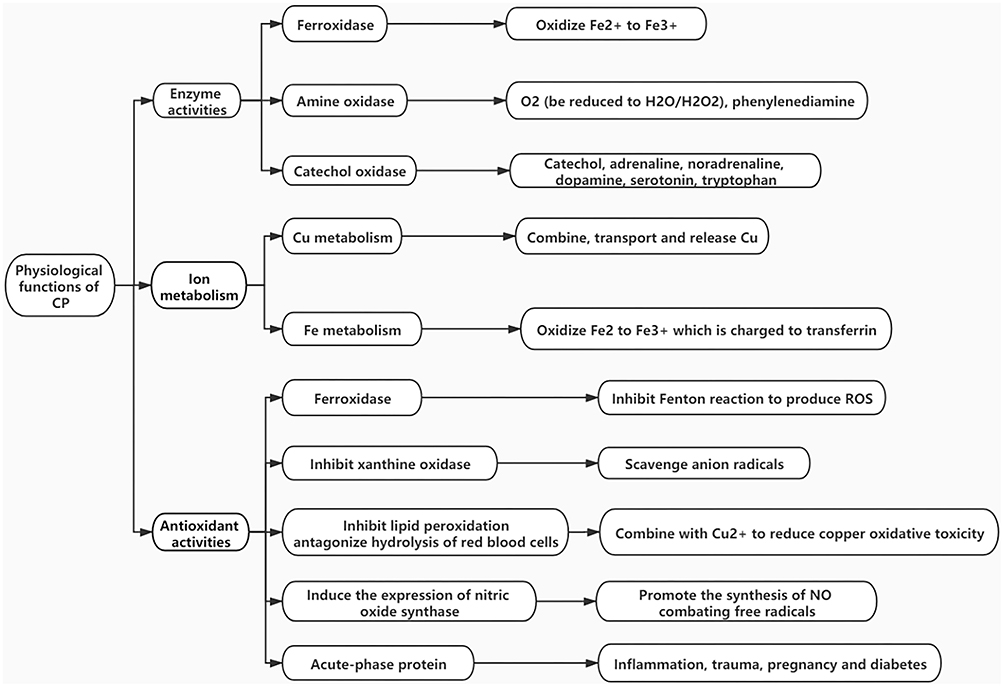 |
Figure 1 CP physiological functions and mechanisms. |
Growing evidence shows that the abnormal metabolism of Cu and Fe, as well as the abnormal expression of CP, has been observed in metabolic diseases such as diabetes and obesity,7,8 indicating that CP may have diagnostic and therapeutic potential in metabolic diseases. Herein, we summarize the latest studies on CP and discuss its role in metabolic diseases.
CP Structure and Distribution
The human CP gene, located on chromosome 8, is 65 kb in length and contains 20 exons.9 Human CP protein is a single polypeptide chain composed of 1046 amino acids and 4 glucosamine oligosaccharides, with a relative molecular weight of approximately 132 kDa.10 The beta strand and beta turn account for approximately 50% of the CP peptide chain with almost no α-helix structure. A single polypeptide chain can be hydrolyzed by protease into 3 groups of isomorphic units. The relative molecular weights are 67 kDa (480 amino acid residues), 50 kDa (405 amino acid residues), and 19 kDa (159 amino acid residues). In the complete polypeptide chain, the three units are connected by single amino acid residues, arginine R and lysine K.11
The 3D structure of CP is shown in Figure 2. CP has six compact domains that can bind to six Cu atoms, and three of those six Cu atoms exist in the second, fourth, and sixth domains as mononuclear forms, which are three “type I Cu (T1Cu)”. The other three Cu atoms also form a trinuclear Cu cluster at the interface of the first and sixth structural domains, which are one “type II Cu (T2Cu)” and two “type III Cu (T3Cu)”. Trinuclear Cu clusters not only play an important role in the catalytic activity of CP but also contribute to the stability of the CP structure.12–14 The CP that is combined with six Cu is very unstable and loses at least one T1Cu in less than a day at 37 °C, while the trinuclear Cu cluster remains intact. In addition to Cu binding sites, CP also has metal ion binding sites such as sodium, Fe, calcium, and so on.
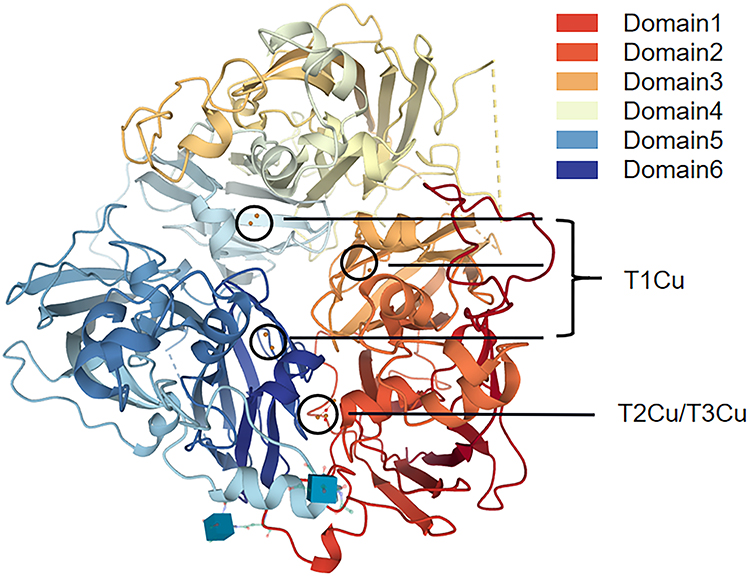 |
Figure 2 CP three-dimensional structure. T1Cu: type I Cu, T2Cu: type II Cu, T3Cu: type III Cu. |
CP in vertebrates is mainly synthesized by the liver; fat, brain, placenta, yolk sac, breast, kidney, and Sertoli cells can also synthesize CP independently.8,10 First, pro-CP is synthesized in the endoplasmic reticulum (ER) of hepatocytes and then combines with Cu in the Golgi apparatus to form total CP. CP is transported from the liver through general circulation and ingested by other tissues and organs, or excreted directly through the bile into the stools.15–17 CP is also produced in the process of macrophage and monocyte inflammation in the blood.18,19 Arner et al20 found that cultured adipose tissue could secrete CP and that the level of CP was higher in the adipose tissue of obese individuals. It has been suggested that CP could be used as a new adipose factor. The normal circulating level of plasma CP in adults is approximately 300 mg/L, and apo-CP accounts for approximately 10% of the total; apo-CP is unstable and has no enzyme catalytic activity, and it is decomposed and metabolized rapidly after half-life.4 CP in plasma is mainly secreted and synthesized by the liver and cannot pass through the blood–brain barrier. CP synthesized in the brain is mainly in the form of glycosylphosphatidylinositols and bound to the membrane of astrocytes.21,22 A study found that CP in the brain is associated with neurodegeneration, such as Parkinson’s disease and Alzheimer’s disease.23,24 In the serum of AD patients, although the concentration of CP was not different from healthy control, the structure of CP was fragmented, resulting in altered activity of CP. Lower CP activity was similarly found in the CSF of AD patients.25 This alteration may be caused by oxidative damage from incorrect or overloaded Cu into the protein, or by up-regulation of oxidoreductactive enzymes leading to increased oxidative stress, or downregulation of enzymes that regulate oxidative stress.26–28 GPI-CP is expressed not only in the brain but also in the spleen, kidney, heart, liver, and testes in relatively small amounts.29
The activity and levels of CP depend on several major factors, including Cu deficiency, inflammatory cytokines, and estrogen or progesterone. Although studies on radioactive Cu have shown that Cu does not affect the rate of synthesis or secretion rate of CP, CP is highly sensitive to Cu deficiency. Under normal physiological conditions, the increase in Cu reserves in the liver can cause a persistent increase in CP concentration, and the decrease in CP concentration will be significant when Cu reserves are deficient.30,31 In the acute phase, as an inflammatory factor, the levels of CP increase due to the response to infection and inflammation.32 The role of CP in body immunity may be related to the elimination of free radicals, the oxidation and apoptosis of neutrophil granulocytes, and the inflammatory process.2,33,34 Studies have found that estrogen can increase the synthesis of CP; with elevated estrogen levels during pregnancy, the concentration of CP can increase 3- to 4-fold.35 On the other hand, Guller et al suggested that the high expression of CP in preeclampsia is related to its role in alleviating reperfusion injury, with Fe oxidase activity.36 Dey et al37 found that CP may predict the development of preeclampsia. Although much research is still needed to explore the exact role of CP in pregnancy, it may provide a new research direction for the diagnosis and treatment of gynecological diseases.
Physiological Function of CP
Enzyme Activity
CP is a member of the multi-Cu oxidase family and one of the few important enzymes in this family that can bind to molecular oxygen and reduce it to water. Substrate electrons may be received at its single Cu ion center and transferred to the multi-Cu ion center for molecular oxygen binding and reduction to water.38 In this process, the Cu atoms of CP undergo the linear arrangement of a functional unit of redox centers, and the T2/3 site in the functional unit can take up a single electron from the substrate, transport it to the tricyclic group, and use the obtained electron to reduce the molecular oxygen into water39 as shown in Figure 3. During the transformation from the T2/3 site to the oxygen bond site, the electrons in CP can consume and oxidize various substrates without releasing reactive oxygen species (ROS). Metal ions, such as Cu and Fe, can be used as substrates. CP can oxidize Fe2+ and Cu1+ to Fe3+ and Cu2+, respectively,40 so they can be transported and metabolized in the body. In addition, CP has the effect of amine oxidase on other organic substrates, such as phenylenediamine.41 The amine oxidase action of CP can oxidize molecular oxygen to water or hydrogen peroxide. When the pH value of the reaction system is 5.2, its activity is the best, and the normal physiological concentration of chloride ions plays a strong role in promoting amine oxidase.42 CP has the effect of oxidase in catechol and its analogs, such as dopamine, epinephrine, norepinephrine, 5-hydroxytryptamine, and tryptophan.40,43
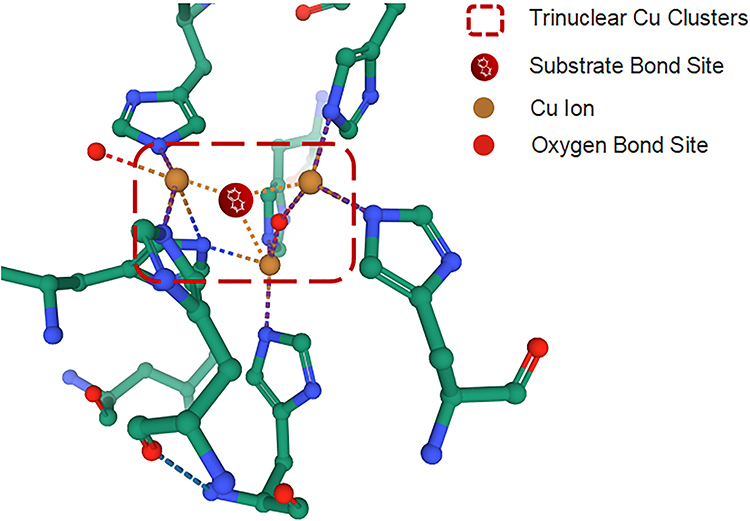 |
Figure 3 The Cu atom site of CP in the functional unit can take up a single electron from the substrate, transport it to the tricyclic group, and use the obtained electron to reduce the molecule. |
Cu and Fe Stability
Cu and Fe are essential metals that exist in an oxidized state and have high redox activity as enzyme auxiliary factors; further, Cu and Fe interact with each other in metabolism.44,45 The deficiency or excess of both elements can lead to the impairment of cell function, which will eventually lead to cell death.46,47 CP is involved in the process of Cu and Fe transport, and it is capable of oxidizing Fe2+ to Fe3+, facilitating the incorporation of the latter into transferrin (TF), as shown in Figure 4.
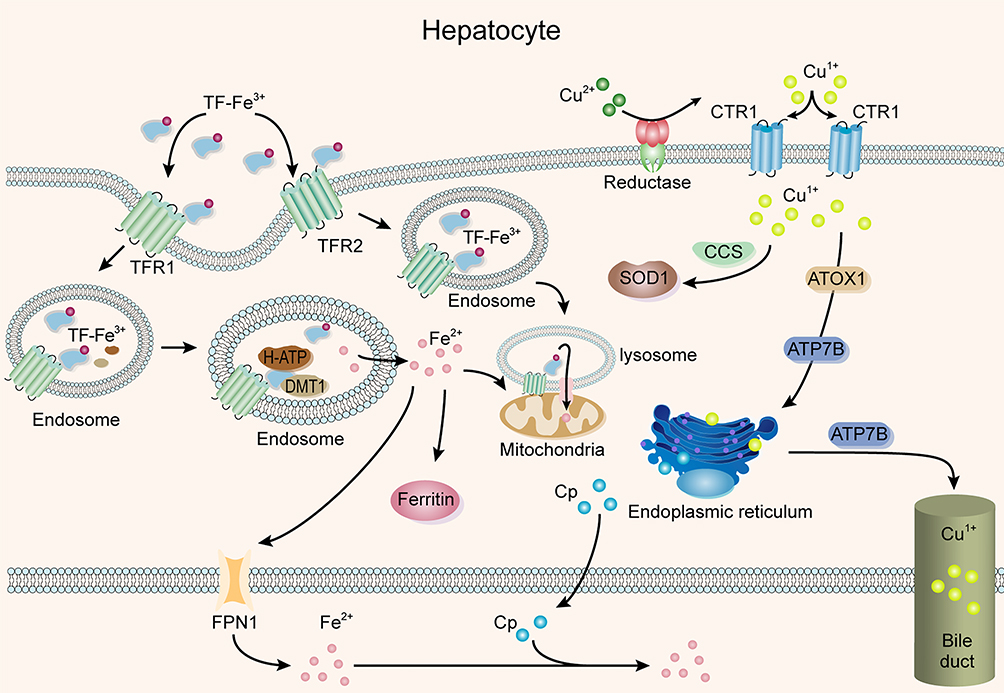 |
Figure 4 CP participates in the transport of Cu and Fe in the liver. |
CP plays an important role in Cu metabolism. Cu in the diet is mainly absorbed into the blood through ATP7A (a P-type ATP enzyme) in the small intestine, binds to albumin or α-2 macroglobulin, and is delivered to hepatocytes by Cu transporter 1 (CTR1).48–50 After entering hepatocytes, CTR1 donates Cu to Copper chaperone for superoxide dismutase; COX17 transfers Cu to mitochondria to synthesize cytochrome oxidase; antioxidant protein 1 (Atox1), as a Cu molecular chaperone, directs Cu to ATP7B (Wilson’s disease protein) in the trans-Golgi network (TGN) and then incorporates Cu into CP.51,52 In addition, ATP7B conveys excessive Cu tubule membranes and mediates the excretion of Cu into bile.53,54 CP binding with Cu is the main carrier of Cu in serum. When CP reaches the surface of target cells, it interacts with corresponding receptors to release Cu, which is absorbed and utilized by target cells. Through the binding and release of Cu by CP, the distribution of Cu in the body is realized.44 CP without Cu binding is an allosteric protein, which leads to changes in the sedimentation rate and electrophoresis mobility when it binds to Cu, but the secondary structure remains unchanged. This allosteric activity not only releases it from the ER of hepatocytes but also protects it from the acidic environment caused by bile. In the subsequent process, the structure can still be combined with Cu, indicating that the structure of Cu-free CP will also affect the metabolism of Cu.55
Cu non-bound to ceruloplasmin (nCp-Cu), also known as “free” Cu, can bind to albumin (or human serum albumin), alpha-2-macroglobulin (also reported as trancopprin), and squamous cell carcinoma. These bindings form a Cu exchangeable pool. Cu homeostasis is well regulated in the body, and the increase in exchangeable nCp-Cu is a symptom of this homeostatic disruption, and if not structurally bound to the enzyme or coordinated by proteins, Cu generates free radicals through the Harper–Weiss or Fenton reactions. Abnormal nCp-Cu levels have been recently reported in Parkinson’s disease and diabetes, as well as in abnormalities in the acute inflammatory response and stroke injury.56
Fe is very important to a variety of functions in the body, including DNA synthesis, gene expression, and the synthesis of hemoglobin and various enzymes. An increasing number of studies have found that Fe metabolism disorder leads to insulin resistance and obesity.57,58 The Fe in the diet is dominated by Fe3+, which is reduced to Fe2+ by duodenal cytochrome B (DcytB) on the top membrane of intestinal epithelial cells and then transported by divalent metal transporter-1 (DMT1) through intestinal epithelial cells and by TF to the liver through portal vein circulation.59,60 After entering the liver, Fe is used to synthesize Fe-containing proteins, and the remainder is oxidized.61 As ferrous oxidase, CP plays an important role in the regulation of Fe balance in vivo. The transmembrane transport of Fe in vivo requires a specific protein carrier, ferroportin (Fpn). The CP-ferroportin system is the main output pathway of intracellular Fe.62 Although many proteins are involved in the absorption of Fe, the only confirmed output system of Fe in the cell is the CP-Fpn system.63 CP in plasma oxidizes Fe2+ to Fe3+, which could bind to Fpn and then transport it to target cells. After endocytosis, Fe is metabolized or stored.62 The liver is the first organ to receive Fe from the intestine, and it is also an important target organ for Fe toxicity. Fe staining in patients with liver cancer shows that excessive Fe is deposited in nontumor tissues,64 and the expression of CP in nontumor tissues is significantly higher than that in tumor tissues. Fe deposition occurs in 60–95% of epithelial parenchyma cells of the liver. Histology shows that Fe deposition decreases from the portal vein to the lobular center, and the expression of CP also decreases gradually. This indicates that the high expression of CP in Fe deposition is related to the involvement of CP in iron oxidation.64 In addition, Cu levels in individuals with CP-deficient genes and aceruloplasminemia are normal or decreased, but Fe metabolism is seriously dysfunctional, and the use of Fe-chelating agents could play a therapeutic role.65 These results show the indispensable role of CP in the process of Fe transport.
Antioxidant Activities
The antioxidant mechanism of plasma CP is the activity of ferrous oxidase, cuprous oxidase, and glutathione peroxidase and its ability to scavenge ROS. CP is an important antioxidant that can convert divalent Fe into less toxic trivalent Fe without releasing ROS. At the same time, CP can further utilize the antioxidant effect, reduce metal toxicity in vivo, and avoid tissue damage and dysfunction of the body.66,67 As early as 1982, Goldstein et al68 found that normal concentrations of CP in serum can inhibit the reduction of n-ferritin C regulated by xanthine oxidase, which is similar to the scavenging effect of superoxide dismutase (SOD) on O2-. In general, the Fenton reaction can occur with Fe2+ in H2O2 (Fe2++H2O2→Fe3++OH−+OH+), but when CP exists, Fe2+ can be oxidized to Fe3+ through its Fe oxidase activity, so the Fenton reaction can be eliminated and the antioxidant effect can be realized.69 Compared with the scavenging effect of SOD on free radicals, the effect of CP is weak, but it is relatively constant; even in the case of denaturation of the protein, it still maintains the effect of scavenging free radicals. This phenomenon may be the result of a direct chemical reaction between CP and O2-. Both active CP and denatured CP can bind to Cu2+ in blood and tissue fluid, significantly inhibit lipid peroxide, and effectively antagonize erythrocyte hydrolysis induced by Cu2+.70 In addition, CP can promote the formation of s-nitrosoglutathione (s-GSNO). When added to cultured monocytes, CP can promote the expression of inducible nitric oxide (NO) synthase. Physiologically, NO can react with hydroxyl radicals (-OH) to form nitrous acid (HONO) and reduce the damage of hydroxyl radicals to the tissue.71 Paradis et al72 believed that this indirect effect may provide cellular protection by protecting mercaptan from irreversible oxidation.
CP Gene Variants
CP gene variants were found and discussed in several recent studies. Gene mutations in CP may disrupt the expression of CP. This protein generated by mutant gene expression may degrade immediately after release from the ER, and also may remain within the ER with abnormal structure, but lacks normal physiological functions, such as inability to bind copper atoms, lack of oxidase, etc.73–75 To date, 172 cases of CP gene variants have been reported worldwide, and 56 were considered pathogenic; most of the cases occurred in Japan, without typical clinical symptoms such as neurological involvement and hepatic iron load.76 At the same time, heterozygous patients with the same mutation may or may not be symptomatic, indicating incomplete penetrance of CP gene, and that environmental and other genetic factors may affect CP functions. Corradini et al77 found that CP gene variants may be the cause of methemoglobinemia and iron overload in patients with non-alcoholic fatty liver disease, but Pelucchi et al78 found that variants may have different effects under other clinical conditions.
CP in Metabolic Disease
Relationship with Type 2 Diabetes
Diabetes is a common metabolic disease characterized by elevated blood glucose levels. Patients often have carbohydrate, fat, and protein metabolic disorders due to insulin deficiency or (and) insulin resistance.79 Diabetes is also a global pandemic. According to the 2017 Chin3ese Guidelines for the Prevention and Treatment of Type 2 Diabetes, the prevalence of diabetes in China soared from 0.67 in 1980 to 10.4 in 2013.80 Aberrant alterations in CP are seen in patients with type 2 diabetes mellitus (T2DM), and the results are inconsistent. Chacko et al81 found that serum CP levels are elevated in patients with T2DM, and the CP levels in patients with complications are higher than those in patients without complications. At the same time, in a similar population, Sarkar et al82 showed that plasma CP and protein thiols are significantly reduced, but the level of Cu2+ is abnormally elevated. Those studies all show a link between CP and diabetes:83,84 diabetes is associated with abnormalities in inflammation, oxidation, and trace elements, and CP is involved in these abnormalities.
As early as the end of the 20th century, Pickup and Crook proposed that, although the innate immune system recovers quickly after the acute stimulus, activation of the innate immune system, stimulated by the living environment of long-term hyperglycemia, promotes insulin resistance, obesity, diabetes, and its complications instead.85 Since then, many clinical studies have shown that diabetes is often accompanied by elevated concentrations of a variety of inflammatory factors. A large number of clinical epidemiological investigations further confirmed that a variety of inflammatory factors can predict the occurrence of diabetes. Inflammation mainly acts on the structure and function of endothelial cells and islet B cells and signal transduction of insulin receptors, which affect glucose metabolism and ultimately lead to diabetes.86–88 Anti-inflammatory therapy has been shown to inhibit the development of diabetes.89–91 CP is an acute-phase reactive protein; its concentration in plasma increases two- to three-fold during infection or injury. CP plays a decisive role in the regulation of innate and specific immune responses, which are the main components of the active immune system and reflect the immune-inflammatory state of the human body. It is believed that CP is an anti-inflammatory factor that inhibits the production of harmful substances during inflammation.92 Therefore, as an inflammatory factor, CP may provide new diagnostic and therapeutic effects for diabetes, but more research is needed.
Oxidative stress is an important factor in the occurrence and development of T2DM.93 In a physiological state, the human body has antioxidant defense systems that can clear oxidation production, such as ROS and reactive nitrogen species (RNS). With these systems, oxidation production will balance production and clearance. Oxidative stress refers to a disruption in that balance that damages tissue and biological macromolecules such as proteins and nucleic acids.94 Oxidative stress can lead to damage of islet B cell function and peripheral insulin resistance, induce diabetes, and even lead to severe complications such as diabetic neuropathy,95 diabetic retinopathy,96 and diabetic cardiovascular disease.97 Experiments in vitro showed that the nonenzymatic glycation reaction in hyperglycemia fragments and deactivates CP, and the release of Cu2+ participates in the Fenton reaction to produce ROS. At the same time, ROS can deactivate CP again to form a vicious cycle.98 Shukla et al99 suggested that unbound Cu induces excessive ROS production through the Haber–Weiss reaction and Fenton reaction in the aortic tissues of diabetic rabbit models; this phenomenon was not found in nondiabetic rabbit models. Sarkar et al82 and Jeppu et al100 found that the level of serum CP is inversely proportional to fasting blood glucose in patients with T2DM. This may indicate that, in the case of hyperglycemia, increased oxidative stress leads to an increased availability of transition metals such as Cu released from storage sites, which are more likely to participate in the Fenton and Haber–Weiss reactions to generate ROS.82,100,101
In recent years, it has been found that trace elements (Cr, Zn, Fe, Se, Mg, Cu) are related to glucose metabolism. Trace elements play an important role in the synthesis, excretion, storage, activity of insulin, and energy metabolism.102 CP is mainly involved in the metabolism of Cu and Fe. Many studies suggest that people with T2DM have elevated levels of Cu and Fe.82,101,103 The redox effect of Cu and Fe is involved in the production of ROS, which is one of the causes of diabetes.93 In addition to the redox effect, Cu can also affect the action of zinc (Zn). Because Zn and Cu are a pair of antagonistic trace elements, they compete for the same carrier protein, metallothionein, during intestinal absorption. When the level of Cu increases, it will affect the absorption of Zn, which will lead to a large loss of Zn in the body; the reduction of Zn will promote the occurrence of diabetes.46,104 Moreover, Cu and some Cu enzymes are involved in the synthesis of a specific protein on the surface of pancreatic islet B cells, GIUT2, which promotes insulin production. When the body is seriously short of Cu, the synthesis of GIUT2 protein is insufficient, affecting the production of insulin. The secretion of insulin is also regulated by the central nervous system. Cu plays an important role in maintaining the functional stability of the central system, and its deficiency can affect nerve transmission and reduce insulin secretion, thus causing or aggravating diabetes.102 Lee et al and other studies also found that when Cu2+ combines with human amylin (HA), this combination can stabilize the nontoxic conformation of HA and block the polymerization and apoptosis of cells, suggesting that the complex of Cu2+ and HA may protect islet cells.105
An increasing number of studies have shown that Fe overload can increase the risk of diabetes. Fe deposition in the liver leads to oxidative stress disorder, increases apoptosis, decreases the expression of IRS2 and GIUT2 in the liver, causes insulin resistance, and eventually leads to abnormal glucose metabolism.106 Moreover, Fe is involved in the synthesis of hemoglobin in vivo, and Fe deficiency can induce anoxia of pancreatic tissue, which can affect the synthesis and release of insulin.107 There are many proteins involved in Fe transport, not only CP. When CP is dysfunctional, the body has a strong compensation effect on Fe metabolism, but CP is involved in the main process of Fe excretion.63,108 When CP is dysfunctional, it will cause Fe accumulation in tissue and lead to diabetes mellitus. To study the effect of tea polyphenols on glucose metabolism in CP gene knockout mice, it was found that CP gene knockout mice suffer from insulin resistance and abnormal glucose metabolism more easily due to Fe overload.106
CP also plays an important role in diabetic complications. It has been found that serum CP can be used as an independent predictor of type 2 diabetic nephropathy.109 This may be because the increase in oxidative stress and the oxidative modification of low-density lipoprotein are related to the progression of diabetes,110 and CP can reflect the degree of oxidation.98 It has also been found that the selective Cu2+-chelating agent trientine can significantly increase ventricular ejection fraction and decrease left ventricular mass index in patients with T2DM complicated by left ventricular hypertrophy. This shows that CP can also improve T2DM with left ventricular hypertrophy, but clinical research is still needed to verify this hypothesis.111 To summarize the articles on diabetic retinopathy, we found that CP, as a biological enzyme, plays a key role in the pathogenesis of diabetic retinopathy.112
In summary, CP has a close relation with the generation and development of diabetes and may play a protective role in abnormal glucose metabolism, providing a new research direction for the diagnosis and treatment of diabetes.
Relationship with Hyperlipidemia
Hyperlipidemia is a pathological state of lipid metabolism disorder. The clinical manifestations are elevated levels of serum total cholesterol (TC), triglyceride (TG), and low-density lipoprotein (LDL) and decreased levels of serum high-density lipoprotein (HDL). According to a study, the total prevalence of hyperlipidemia in patients over 18 years old in China is 40.40%; the prevalence of LDL-C was the highest (33.9%), followed by high TG (13.1%).113 Studies indicate that between 2010 and 2030 the number of patients with cardiovascular diseases (CVDs) in China will increase by 9.2 million.114 Hyperlipidemia is divided into primary and secondary categories. Primary hyperlipidemia has a familial tendency.115 Secondary hyperlipidemia is dyslipidemia caused by other diseases, such as diabetes and hypertension. In addition, age, weight, and lifestyle factors, such as diet, exercise, and mental stress, can also affect blood lipid levels. It is now believed that the pathological mechanism of hyperlipidemia is related to endoplasmic reticulum (ER) stress, gene polymorphism, inflammatory state, oxidative stress, intestinal flora, and trace elements.116 Studies have shown that CP is related to dyslipidemia and can participate in multiple stages of hyperlipidemia.
ER stress plays an important role in lipid metabolism and protein synthesis.117 Various physiological and pathological disturbances can affect the folding process of primary synthetic proteins in the ER cavity, causing the increase and accumulation of unfolded and misfolded proteins, which is ER stress.118 SR-BI is a major receptor for HDL, and ER stress induces downregulation of SR-BI gene expression, leading to lipid metabolism disorders.119,120 GRP78 is a molecular marker of ER stress. Zhou et al121 found that the mRNA and protein expression of GRP78 in hyperlipidemic rats was significantly decreased, and, after treatment, serum TC, TG, and LDL-C were significantly decreased, and GRP78 gene expression and protein content were significantly increased. All these studies indicated that ER stress plays an important role in the pathogenesis of hyperlipidemia.121 Studies on Cu-loaded hepatocytes cultured in vitro showed significant ER stress in hepatocytes, so the damage to Cu-loaded hepatocytes is closely related to excessive ER stress.122 Moreover, Kono et al123 observed that individuals with aceruloplasminemia have ER stress leading to cell death. Therefore, we speculated that CP could avoid ER stress caused by increased Cu levels by regulating Cu metabolism, thus reducing the occurrence of hyperlipidemia. However, the preventive effect of CP on hyperlipidemia still needs to be confirmed by relevant studies.
It is now believed that the inflammatory response is accompanied by the occurrence and development of hyperlipidemia, which can accelerate the accumulation of fat in liver cells. The accumulation of fat continues to aggravate the inflammatory response in a vicious cycle, resulting in lipid disorders. Studies have shown that the level of related inflammatory factors such as C-reactive protein in patients with hyperlipidemia is significantly increased and is positively correlated with TC, TG, and LDL-C.124 CP, as an acute reactive protein, may play a certain role in predicting the occurrence and prognosis of hyperlipidemia. The exact role of CP in inflammation needs further study.
In patients with hyperlipidemia, the level of oxidative stress in vivo increases, while the overall antioxidant capacity decreases. Therefore, it is likely that the mechanism of oxidative stress is involved in the occurrence of abnormal lipid metabolism. Hydroxyl radicals, oxidative products, can react directly with lipids, inducing lipid peroxidation and cause structural and functional damage to various biomolecular membranes,125,126 ultimately accelerating the process of atherosclerosis and increasing the risk of coronary heart disease.127 As an important antioxidant, CP has a therapeutic effect on oxidative stress in the body. Studies have found that CP can significantly eliminate hydroxyl radicals and improve the lipid peroxidation state.128 However, studies still show that oxidative stress may change CP from a protective factor to a vascular pathological factor.129 These data showed that CP, based on its structure and integrity in combination with Cu, could play an effective oxidant role in LDL rather than having an antioxidant effect. This also proves that the destruction of this combination may change the antioxidant function of CP.130 Therefore, CP may have preventive and predictive effects on hyperlipidemia, but the corresponding pathophysiological mechanism has not been studied.
Relationship with Obesity
Obesity is a chronic metabolic disease that is usually caused by the interaction of heredity, environment, and other factors, such as weight gain caused by abnormal fat distribution or excessive fat accumulation in the body. Obesity can cause a variety of complications and is closely related to the incidence of various acute and chronic diseases and symptoms, such as dyslipidemia, metabolic syndrome, T2DM, atherosclerosis, and CVD. According to a 2015 survey, obesity and overweight rates among children aged 6 to 17 in China reached 6.4% and 9.6%, respectively, which were 5.1 and 4.3 percentage points higher than percentages in 2002.131,132 The study found that CP is associated with obesity, and Tajik et al observed a decrease in plasma CP levels in obese women after losing weight through diet.133 CP can participate in the inflammatory response and oxidative stress in the occurrence and development of obesity, and it can also affect obesity by regulating intestinal flora and complications.
Current research suggests that obesity is a chronic low-grade systemic inflammation that results from the interaction between adipocytes, macrophages, and other immune cells that permeate and dilate adipose tissue. The inflammatory development of obesity leads to adipocyte hypertrophy, which is the most representative feature of adipose tissue dysfunction, and this feature increases the production of proinflammatory cytokines.134 CP, as an inflammatory factor, can be used to measure the degree of inflammation and distinguish inflammatory diseases. Kim et al,8 using the protein differential display technique, found that an increase in CP is significantly associated with obesity, indicating that CP may be used as a biomarker of obesity. Moreover, compared with fibrinogen, C-reactive protein (CRP), and IL-6, CP is a better predictor of long-term prognosis for obesity inflammation.135 However, whether CP plays an important mediating or inducing role in obesity inflammation, whether its increase can affect the status of obesity inflammation, or whether it is only a simple marker still needs research.
Many studies have shown that the level of oxidative stress in patients with obesity is increased for many reasons, in which mitochondrial function changes play a decisive role. Mitochondrial dysfunction of adipose tissue in patients with obesity is characterized by decreased mitochondrial biosynthesis and activity, excessive production of ROS, and increased autophagy.136–138 All these factors can adversely affect adipose tissue function. CP can promote metabolism by regulating the metabolism of Cu and Fe, promoting mitochondrial biosynthesis and activity, improving oxidative stress in adipose tissue, and inhibiting autophagy.139 Studies have found that intestinal flora is involved in the metabolic process of human nutrition and energy. Intestinal flora can mediate the occurrence and development of obesity not only by affecting the absorption of energy metabolism and intestinal wall permeability but also by participating in the metabolic process of the body and interacting with human tissues and organs.140 The disorder of trace elements such as Cu and Fe can also affect the composition and function of intestinal flora, including the function of lipid metabolism.141,142 However, the role of CP in intestinal flora is still incompletely understood.
The World Cancer Research Fund concluded in 2007 that obesity is associated with an increased risk of pancreatic (postmenopausal) breast, endometrial, and renal cancer.143 A study found that CP is a novel adipokine with increased expression in the adipose tissue of obese subjects and cells of obesity-related cancers.144 Whether there is a causal relationship between overexpression of CP and cancer development in patients with obesity still needs further study. When Safavi et al145 observed the relationship between serum CP level and obesity, they found that there was no correlation, but the serum CP level was positively correlated with serum triglyceride level. The relationship between CP and obesity still needs much research.
Relationship with Other Metabolic Diseases
In addition to diabetes and obesity, CP is also associated with other metabolic diseases, such as coronary heart disease (CHD). Göçmen et al146 found that CP levels increase in patients with CHD. In their study, they found that CP level is an independent risk factor for CVD.146 Mori et al147 separated the risk contributed by CP from that of inflammation (α1-antitrypsin, α1-acid glycoprotein, α2-macroglobulin, haptoglobin, fibrinogen, C4b binding protein, lipoprotein, and CRP) and suggested that CP could serve as an independent risk factor for coronary atherosclerosis and as a marker for the severity of disease.147 Many studies have found an association between CP and CHD, but have not reached a unified conclusion about the mechanisms for the role of CP in CHD. Some studies have suggested that the oxidation of LDL leads to the initiation or acceleration of the process of atherosclerosis, and CP is an effective catalyst for the oxidation of LDL. CP, by influencing NO levels, can reduce the bioavailability of NO in plasma, inhibiting its protective effect on cardiac ischemia and failure. However, there are also studies suggesting that CP is an antioxidant that plays a protective role in the development of CHD. For a better observation on achievements about the role of CP in CVD gathered from clinical studies, we report in brief the main points of relevant researches in recent years, as shown in Table 1. To explore the latest research trends, Web of Science was used to retrieve CP studies published from 2016 to 2020. The search yielded 2098 original studies and reviews, which were exported to CiteSpace for burst analysis, as shown in Figure 5.
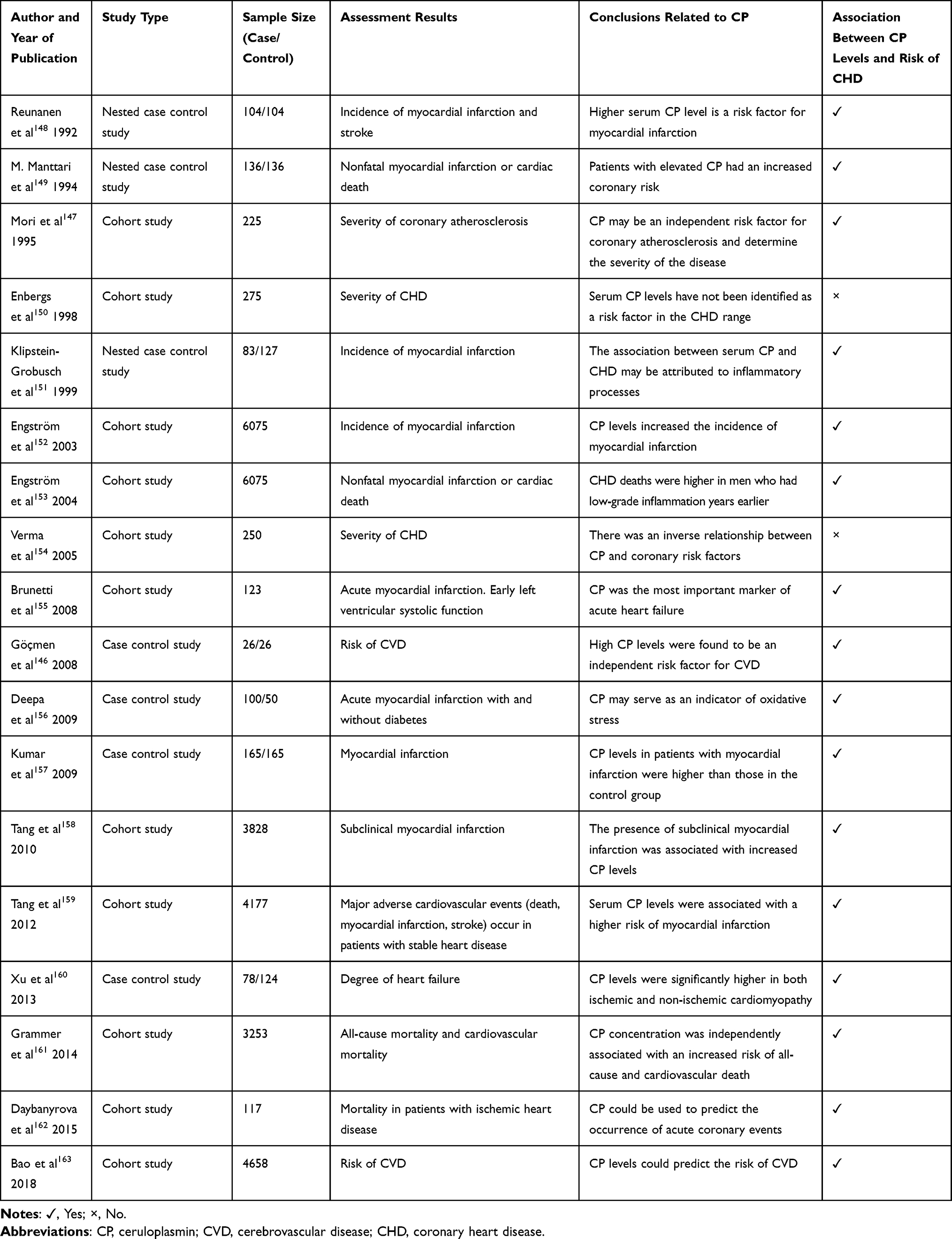 |
Table 1 Summary of Achievements About the Role of CP in CVD Gathered from Clinical Studies |
 |
Figure 5 Top 10 important CP keywords with high burst values. Bold text indicates the year of the keyword appearance, blue indicates the timeline, and red indicates the time occupied by the keyword. |
As shown, studies on CP in the past five years have focused on in vitro experiments, antioxidation, immune response, and metabolic diseases. Moreover, since 2018, research on immunity and diabetes has become a hot topic and trend for CP. At present, an association between CP and metabolic diseases such as diabetes has been found, and it is believed that CP mainly plays a role in diseases by regulating copper and iron metabolism, oxidative stress, and inflammation. Although the mechanism of CP in the metabolism of copper and iron has been thoroughly studied, questions remain. Does CP mainly work as an antioxidant or oxidant in oxidative stress? Does it play an anti-inflammatory role in the inflammatory response? Why do studies of CP, using different research methods on similar populations, find different or even opposite research results? Furthermore, the stability of CP’s physiological functions has not been determined. Therefore, future research might study the mechanism of CP in metabolic diseases, especially its involvement in oxidative stress and the immune response to uncover the specific link between CP and metabolic disease and find the precise target for its function.
Conclusion
An increasing number of studies have found correlations between CP and metabolic diseases such as diabetes and hyperlipidemia and have observed that CP can be involved in the physiological and pathological processes of these diseases. A large number of studies have shown that CP plays an important role in the balance of Cu and Fe through its oxidase activity. CP exhibits antioxidant activity and can protect tissue from oxidative damage. The study found that the level of CP increased in the inflammatory state, and attenuated the activation of neutrophils, indicating that CP can be used as a predictor and antagonist of inflammation.66 At present, it is believed that CP plays a protective role in metabolic diseases, mainly by participating in oxidative stress and the metabolism of Cu and Fe and acts as an inflammatory factor to predict those diseases. However, some studies have also found that CP plays the role of antioxidant. The causal relationship between CP and metabolic diseases in the human body is not clear. While a large number of studies have found a correlation between CP and metabolic diseases, future research should focus on solving the molecular mechanism of CP in metabolic diseases and studying its other roles.
Statement of Ethics
This article does not contain any studies with human or animals performed by any of the authors.
Acknowledgments
We would like to thank the researchers for their contributions. This paper was supported by the Clinical Research Plan of SHDC (No. SHDC2020CR3028A and No. SHDC12019X16).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Brown MA, Stenberg LM, Mauk AG. Identification of catalytically important amino acids in human ceruloplasmin by site-directed mutagenesis. FEBS Lett. 2002;520(1–3):8–12. doi:10.1016/S0014-5793(02)02652-2
2. Linder MC. Ceruloplasmin and other copper binding components of blood plasma and their functions: an update. Metallomics. 2016;8(9):887–905. doi:10.1039/C6MT00103C
3. Rouault TA, Cooperman S. Brain iron metabolism. Semin Pediatr Neurol. 2006;13(3):142–148. doi:10.1016/j.spen.2006.08.002
4. Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439–458. doi:10.1146/annurev.nutr.22.012502.114457
5. Kirsipuu T, Zadorožnaja A, Smirnova J, et al. Copper (II)-binding equilibria in human blood. Sci Rep. 2020;10(1):5686. doi:10.1038/s41598-020-62560-4
6. Chapman AL, Mocatta TJ, Shiva S, et al. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J Biol Chem. 2013;288(9):6465–6477. doi:10.1074/jbc.M112.418970
7. Golizeh M, Lee K, Ilchenko S, et al. Increased serotransferrin and ceruloplasmin turnover in diet-controlled patients with type 2 diabetes. Free Radic Biol Med. 2017;113:461–469. doi:10.1016/j.freeradbiomed.2017.10.373
8. Kim OY, Shin MJ, Moon J, Chung JH. Plasma ceruloplasmin as a biomarker for obesity: a proteomic approach. Clin Biochem. 2011;44(5–6):351–356. doi:10.1016/j.clinbiochem.2011.01.014
9. Daimon M, Yamatani K, Igarashi M, et al. Fine structure of the human ceruloplasmin gene. Biochem Biophys Res Commun. 1995;208(3):1028–1035. doi:10.1006/bbrc.1995.1437
10. Yang F, Naylor SL, Lum JB, et al. Characterization, mapping, and expression of the human ceruloplasmin gene. Proc Natl Acad Sci U S A. 1986;83(10):3257–3261. doi:10.1073/pnas.83.10.3257
11. Bento I, Peixoto C, Zaitsev VN, Lindley PF. Ceruloplasmin revisited: structural and functional roles of various metal cation-binding sites. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 2):240–248. doi:10.1107/S090744490604947X
12. Bakhautdin B, Goksoy Bakhautdin E, Fox PL. Ceruloplasmin has two nearly identical sites that bind myeloperoxidase. Biochem Biophys Res Commun. 2014;453(4):722–727. doi:10.1016/j.bbrc.2014.09.134
13. Samygina VR, Sokolov AV, Bourenkov G, et al. Ceruloplasmin: macromolecular assemblies with iron-containing acute phase proteins. PLoS One. 2013;8(7):e67145. doi:10.1371/journal.pone.0067145
14. Mukhopadhyay BP. Insights from molecular dynamics simulation of human ceruloplasmin (ferroxidase enzyme) binding with biogenic monoamines. Bioinformation. 2019;15(10):750–759. doi:10.6026/97320630015750
15. Terada K, Kawarada Y, Miura N, Yasui O, Koyama K, Sugiyama T. Copper incorporation into ceruloplasmin in rat livers. Biochim Biophys Acta. 1995;1270(1):58–62. doi:10.1016/0925-4439(94)00072-X
16. Hellman NE, Kono S, Mancini GM, Hoogeboom AJ, De Jong GJ, Gitlin JD. Mechanisms of copper incorporation into human ceruloplasmin. J Biol Chem. 2002;277(48):46632–46638. doi:10.1074/jbc.M206246200
17. Ramos D, Mar D, Ishida M, et al. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS One. 2016;11(3):e0149516. doi:10.1371/journal.pone.0149516
18. Marques L, Auriac A, Willemetz A, et al. Immune cells and hepatocytes express glycosylphosphatidylinositol-anchored ceruloplasmin at their cell surface. Blood Cells Mol Dis. 2012;48(2):110–120. doi:10.1016/j.bcmd.2011.11.005
19. Banha J, Marques L, Oliveira R, et al. Ceruloplasmin expression by human peripheral blood lymphocytes: a new link between immunity and iron metabolism. Free Radic Biol Med. 2008;44(3):483–492.
20. Arner E, Forrest AR, Ehrlund A, et al. Ceruloplasmin is a novel adipokine which is overexpressed in adipose tissue of obese subjects and in obesity-associated cancer cells. PLoS One. 2014;9(3):e80274. doi:10.1371/journal.pone.0080274
21. Patel BN, David S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem. 1997;272(32):20185–20190. doi:10.1074/jbc.272.32.20185
22. Patel BN, Dunn RJ, David S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem. 2000;275(6):4305–4310. doi:10.1074/jbc.275.6.4305
23. Ryan F, Zarruk JG, Lößlein L, David S. Ceruloplasmin plays a neuroprotective role in cerebral ischemia. Front Neurosci. 2018;12:988. doi:10.3389/fnins.2018.00988
24. Jeong SY, David S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem. 2003;278(29):27144–27148. doi:10.1074/jbc.M301988200
25. Capo CR, Arciello M, Squitti R, et al. Features of ceruloplasmin in the cerebrospinal fluid of Alzheimer’s disease patients. Biometals. 2008;21(3):367–372. doi:10.1007/s10534-007-9125-4
26. Squitti R, Quattrocchi CC, Salustri C, Rossini PM. Ceruloplasmin fragmentation is implicated in “free” copper deregulation of Alzheimer’s disease. Prion. 2008;2(1):23–27. doi:10.4161/pri.2.1.6297
27. Squitti R, Quattrocchi CC, Forno GD, et al. Ceruloplasmin (2-D PAGE) pattern and copper content in serum and brain of Alzheimer disease patients. Biomark Insights. 2007;1:205–213.
28. Siotto M, Simonelli I, Pasqualetti P, et al. Association between serum ceruloplasmin specific activity and risk of Alzheimer's disease. J Alzheimers Dis. 2016;50(4):1181–1189. doi:10.3233/JAD-150611
29. Mostad EJ, Prohaska JR. Glycosylphosphatidylinositol-linked ceruloplasmin is expressed in multiple rodent organs and is lower following dietary copper deficiency. Exp Biol Med (Maywood). 2011;236(3):298–308. doi:10.1258/ebm.2010.010256
30. Broderius MA, Prohaska JR. Differential impact of copper deficiency in rats on blood cuproproteins. Nutr Res. 2009;29(7):494–502. doi:10.1016/j.nutres.2009.06.006
31. Broderius M, Mostad E, Wendroth K, Prohaska JR. Levels of plasma ceruloplasmin protein are markedly lower following dietary copper deficiency in rodents. Comp Biochem Physiol C Toxicol Pharmacol. 2010;151(4):473–479. doi:10.1016/j.cbpc.2010.02.005
32. Vasilyev VB. Interactions of caeruloplasmin with other proteins participating in inflammation. Biochem Soc Trans. 2010;38(4):947–951. doi:10.1042/BST0380947
33. Golenkina EA, Viryasova GM, Galkina SI, Gaponova TV, Sud’ina GF, Sokolov AV. Fine regulation of neutrophil oxidative status and apoptosis by ceruloplasmin and its derivatives. Cells. 2018;7(1). doi:10.3390/cells7010008
34. Sokolov AV, Pulina MO, Ageeva KV, et al. Interaction of ceruloplasmin, lactoferrin, and myeloperoxidase. Biochemistry (Mosc). 2007;72(4):409–415. doi:10.1134/S0006297907040074
35. Ganaraja B, Pavithran P, Ghosh S. Effect of estrogen on plasma ceruloplasmin level in rats exposed to acute stress. Indian J Med Sci. 2004;58(4):150–154.
36. Guller S, Buhimschi CS, Ma YY, et al. Placental expression of ceruloplasmin in pregnancies complicated by severe preeclampsia. Lab Invest. 2008;88(10):1057–1067. doi:10.1038/labinvest.2008.74
37. Dey M, Arora D, Narayan N, Kumar R. Serum cholesterol and ceruloplasmin levels in second trimester can predict development of pre-eclampsia. N Am J Med Sci. 2013;5(1):41–46. doi:10.4103/1947-2714.106198
38. Mukhopadhyay BP. Putative role of conserved water molecules in the hydration and inter-domain recognition of mono nuclear copper centers in O2-bound human ceruloplasmin: a comparative study between X-ray and MD simulated structures. Bioinformation. 2019;15(6):402–411. doi:10.6026/97320630015402
39. Haberska K, Vaz-Domínguez C, De Lacey AL, Dagys M, Reimann CT, Shleev S. Direct electron transfer reactions between human ceruloplasmin and electrodes. Bioelectrochemistry. 2009;76(1–2):34–41. doi:10.1016/j.bioelechem.2009.05.012
40. Stoj C, Kosman DJ. Cuprous oxidase activity of yeast Fet3p and human ceruloplasmin: implication for function. FEBS Lett. 2003;554(3):422–426. doi:10.1016/S0014-5793(03)01218-3
41. Musci G, Bonaccorsi di Patti MC, Calabrese L. Modulation of the redox state of the copper sites of human ceruloplasmin by chloride. J Protein Chem. 1995;14(7):611–619. doi:10.1007/BF01886887
42. Tian S, Jones SM, Jose A, Solomon EI. Chloride control of the mechanism of human serum ceruloplasmin (Cp) catalysis. J Am Chem Soc. 2019;141(27):10736–10743. doi:10.1021/jacs.9b03661
43. Saenko EL, Siverina OB, Basevich VV, Yaropolov AI. Study of ceruloplasmin oxidase activity. The effect of pH. Biochem Int. 1990;20(6):1049–1058.
44. Gulec S, Collins JF. Molecular mediators governing iron-copper interactions. Annu Rev Nutr. 2014;34:95–116. doi:10.1146/annurev-nutr-071812-161215
45. Arredondo M, Muñoz P, Mura CV, Nùñez MT. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am J Physiol Cell Physiol. 2003;284(6):C1525–1530. doi:10.1152/ajpcell.00480.2002
46. Zheng Y, Li XK, Wang Y, Cai L. The role of zinc, copper and iron in the pathogenesis of diabetes and diabetic complications: therapeutic effects by chelators. Hemoglobin. 2008;32(1–2):135–145. doi:10.1080/03630260701727077
47. Anderson GJ, Wang F. Essential but toxic: controlling the flux of iron in the body. Clin Exp Pharmacol Physiol. 2012;39(8):719–724. doi:10.1111/j.1440-1681.2011.05661.x
48. Tennant J, Stansfield M, Yamaji S, Srai SK, Sharp P. Effects of copper on the expression of metal transporters in human intestinal Caco-2 cells. FEBS Lett. 2002;527(1–3):239–244. doi:10.1016/S0014-5793(02)03253-2
49. Zheng G, Zhang J, Xu Y, et al. Involvement of CTR1 and ATP7A in lead (Pb)-induced copper (Cu) accumulation in choroidal epithelial cells. Toxicol Lett. 2014;225(1):110–118. doi:10.1016/j.toxlet.2013.11.034
50. Moriya M, Ho YH, Grana A, et al. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am J Physiol Cell Physiol. 2008;295(3):C708–721. doi:10.1152/ajpcell.00029.2008
51. Gulec S, Anderson GJ, Collins JF. Mechanistic and regulatory aspects of intestinal iron absorption. Am J Physiol Gastrointest Liver Physiol. 2014;307(4):G397–409. doi:10.1152/ajpgi.00348.2013
52. Lenartowicz M, Krzeptowski W. [Structure and function of ATP7A and ATP7B proteins–Cu-transporting ATPases]. Postepy Biochem. 2010;56(3):317–327. Polish.
53. de Romaña DL, Olivares M, Uauy R, Araya M. Risks and benefits of copper in light of new insights of copper homeostasis. J Trace Elem Med Biol. 2011;25(1):3–13. doi:10.1016/j.jtemb.2010.11.004
54. Bartee MY, Lutsenko S. Hepatic copper-transporting ATPase ATP7B: function and inactivation at the molecular and cellular level. Biometals. 2007;20(3–4):627–637. doi:10.1007/s10534-006-9074-3
55. De Filippis V, Vassiliev VB, Beltramini M, Fontana A, Salvato B, Gaitskhoki VS. Evidence for the molten globule state of human apo-ceruloplasmin. Biochim Biophys Acta. 1996;1297(2):119–123. doi:10.1016/S0167-4838(96)00139-2
56. Siotto M, Squitti R. Copper imbalance in Alzheimer’s disease: overview of the exchangeable copper component in plasma and the intriguing role albumin plays. Coord Chem Rev. 2018;371:86–95. doi:10.1016/j.ccr.2018.05.020
57. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. 2014;19(2):164–174.
58. Aigner E, Feldman A, Datz C. Obesity as an emerging risk factor for iron deficiency. Nutrients. 2014;6(9):3587–3600. doi:10.3390/nu6093587
59. Choi J, Masaratana P, Latunde-Dada GO, Arno M, Simpson RJ, McKie AT. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J Nutr. 2012;142(11):1929–1934. doi:10.3945/jn.112.160358
60. Chloupková M, Zhang AS, Enns CA. Stoichiometries of transferrin receptors 1 and 2 in human liver. Blood Cells Mol Dis. 2010;44(1):28–33. doi:10.1016/j.bcmd.2009.09.004
61. Anderson GJ, Frazer DM. Hepatic iron metabolism. Semin Liver Dis. 2005;25(4):420–432. doi:10.1055/s-2005-923314
62. De Domenico I, Ward DM, Di Patti MC, et al. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007;26(12):2823–2831. doi:10.1038/sj.emboj.7601735
63. Musci G, Polticelli F, Bonaccorsi di Patti MC. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J Biol Chem. 2014;5(2):204–215. doi:10.4331/wjbc.v5.i2.204
64. Tan MG, Kumarasinghe MP, Wang SM, Ooi LL, Aw SE, Hui KM. Modulation of iron-regulatory genes in human hepatocellular carcinoma and its physiological consequences. Exp Biol Med (Maywood). 2009;234(6):693–702. doi:10.3181/0807-RM-227
65. Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21(2):195–199. doi:10.1038/5979
66. Vasilyev VB. Looking for a partner: ceruloplasmin in protein-protein interactions. Biometals. 2019;32(2):195–210. doi:10.1007/s10534-019-00189-1
67. Altamura C, Squitti R, Pasqualetti P, et al. Ceruloplasmin/Transferrin system is related to clinical status in acute stroke. Stroke. 2009;40(4):1282–1288. doi:10.1161/STROKEAHA.108.536714
68. Goldstein IM, Kaplan HB, Edelson HS, Weissmann G. Ceruloplasmin: an acute phase reactant that scavenges oxygen-derived free radicals. Ann N Y Acad Sci. 1982;389:368–379. doi:10.1111/j.1749-6632.1982.tb22150.x
69. Ganini D, Canistro D, Jiang J, Stadler K, Mason RP, Kadiiska MB. Ceruloplasmin (ferroxidase) oxidizes hydroxylamine probes: deceptive implications for free radical detection. Free Radic Biol Med. 2012;53(7):1514–1521. doi:10.1016/j.freeradbiomed.2012.07.013
70. Cerone SI, Sansinanea AS, Streitenberger SA, Garcia MC, Auza NJ. Cytochrome c oxidase, Cu, Zn-superoxide dismutase, and ceruloplasmin activities in copper-deficient bovines. Biol Trace Elem Res. 2000;73(3):269–278. doi:10.1385/BTER:73:3:269
71. Inoue K, Akaike T, Miyamoto Y, et al. Nitrosothiol formation catalyzed by ceruloplasmin. Implication for cytoprotective mechanism in vivo. J Biol Chem. 1999;274(38):27069–27075. doi:10.1074/jbc.274.38.27069
72. Paradis M, Gagné J, Mateescu MA, Paquin J. The effects of nitric oxide-oxidase and putative glutathione-peroxidase activities of ceruloplasmin on the viability of cardiomyocytes exposed to hydrogen peroxide. Free Radic Biol Med. 2010;49(12):2019–2027. doi:10.1016/j.freeradbiomed.2010.09.030
73. Hellman NE, Kono S, Miyajima H, Gitlin JD. Biochemical analysis of a missense mutation in aceruloplasminemia. J Biol Chem. 2002;277(2):1375–1380. doi:10.1074/jbc.M109123200
74. Kono S, Miyajima H. Molecular and pathological basis of aceruloplasminemia. Biol Res. 2006;39(1):15–23. doi:10.4067/S0716-97602006000100003
75. Miyajima H, Kono S, Takahashi Y, Sugimoto M, Sakamoto M, Sakai N. Cerebellar ataxia associated with heteroallelic ceruloplasmin gene mutation. Neurology. 2001;57(12):2205–2210. doi:10.1212/WNL.57.12.2205
76. Borges MD, de Albuquerque DM, Lanaro C, Costa FF, Fertrin KY. Clinical relevance of heterozygosis for aceruloplasminemia. Am J Med Genet B Neuropsychiatr Genet. 2019;180(4):266–271. doi:10.1002/ajmg.b.32723
77. Corradini E, Buzzetti E, Dongiovanni P, et al. Ceruloplasmin gene variants are associated with hyperferritinemia and increased liver iron in patients with NAFLD. J Hepatol. 2021;75(3):506–513. doi:10.1016/j.jhep.2021.03.014
78. Pelucchi S, Ravasi G, Piperno A. Ceruloplasmin variants might have different effects in different iron overload disorders. J Hepatol. 2021;75(4):1003–1004. doi:10.1016/j.jhep.2021.05.005
79. Galicia-Garcia U, Benito-Vicente A, Jebari S, et al. Pathophysiology of Type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275. doi:10.3390/ijms21176275
80. Society CD. Guidelines for the prevention and control of type 2 diabetes in China (2017). Chin J Pract Internal Med. 2018;38(04):292–344.
81. Chacko SK, Cheluvappa R. Increased ceruloplasmin and fibrinogen in type 2 diabetes corresponds to decreased anti-oxidant activity in a preliminary tertiary South Indian hospital study. Exp Clin Endocrinol Diabetes. 2010;118(1):64–67. doi:10.1055/s-0029-1225647
82. Sarkar A, Dash S, Barik BK, et al. Copper and ceruloplasmin levels in relation to total thiols and GST in type 2 diabetes mellitus patients. Indian J Clin Biochem. 2010;25(1):74–76. doi:10.1007/s12291-010-0015-0
83. Sharma VK, Tumbapo A, Pant V, et al. Ceruloplasmin, a potential marker for glycemic status and its relationship with lipid profile in Type II diabetes mellitus. Asian J Med Sci. 2018;9(2):13–18. doi:10.3126/ajms.v9i2.19003
84. Rusticeanu M, Zimmer V, Schleithoff L, et al. Novel ceruloplasmin mutation causing aceruloplasminemia with hepatic iron overload and diabetes without neurological symptoms. Clin Genet. 2014;85(3):300–301. doi:10.1111/cge.12145
85. Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia. 1998;41(10):1241–1248. doi:10.1007/s001250051058
86. Odegaard AO, Jacobs DR
87. Fizelova M, Jauhiainen R, Kangas AJ, et al. Differential associations of inflammatory markers with insulin sensitivity and secretion: the Prospective METSIM Study. J Clin Endocrinol Metab. 2017;102(9):3600–3609. doi:10.1210/jc.2017-01057
88. Grossmann V, Schmitt VH, Zeller T, et al. Profile of the immune and inflammatory response in individuals with prediabetes and Type 2 diabetes. Diabetes Care. 2015;38(7):1356–1364. doi:10.2337/dc14-3008
89. Everett BM, Donath MY, Pradhan AD, et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol. 2018;71(21):2392–2401. doi:10.1016/j.jacc.2018.03.002
90. Tenenbaum A, Fisman EZ. Mirroring the CANTOS revolution: is anti-inflammatory therapy for diabetes just around the corner? Cardiovasc Diabetol. 2017;16(1):91. doi:10.1186/s12933-017-0573-z
91. Stienstra R, Joosten LA, Koenen T, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12(6):593–605. doi:10.1016/j.cmet.2010.11.011
92. Sokolov AV, Zakharova ET, Kostevich VA, Samygina VR, Vasilyev VB. Lactoferrin, myeloperoxidase, and ceruloplasmin: complementary gearwheels cranking physiological and pathological processes. Biometals. 2014;27(5):815–828. doi:10.1007/s10534-014-9755-2
93. Fardoun RZ. The use of vitamin E in type 2 diabetes mellitus. Clin Exp Hypertens. 2007;29(3):135–148. doi:10.1080/10641960701361601
94. Reddy VP, Zhu X, Perry G, Smith MA. Oxidative stress in diabetes and Alzheimer’s disease. J Alzheimers Dis. 2009;16(4):763–774. doi:10.3233/JAD-2009-1013
95. Hernández-Beltrán N, Moreno CB, Gutiérrez-Álvarez AM. Contribution of mitochondria to pain in diabetic neuropathy. Endocrinol Nutr. 2013;60(1):25–32. doi:10.1016/j.endonu.2012.03.005
96. Souza BM, Assmann TS, Kliemann LM, Gross JL, Canani LH, Crispim D. The role of uncoupling protein 2 (UCP2) on the development of type 2 diabetes mellitus and its chronic complications. Arq Bras Endocrinol Metabol. 2011;55(4):239–248. doi:10.1590/S0004-27302011000400001
97. Selvaraju V, Joshi M, Suresh S, Sanchez JA, Maulik N, Maulik G. Diabetes, oxidative stress, molecular mechanism, and cardiovascular disease–an overview. Toxicol Mech Methods. 2012;22(5):330–335. doi:10.3109/15376516.2012.666648
98. Islam KN, Takahashi M, Higashiyama S, Myint T, Uozumi N. Fragmentation of ceruloplasmin following non-enzymatic glycation reaction. J Biochem. 1995;118(5):1054–1060. doi:10.1093/jb/118.5.1054
99. Shukla N, Thompson CS, Angelini GD, Mikhailidis DP, Jeremy JY. Homocysteine enhances impairment of endothelium-dependent relaxation and guanosine cyclic monophosphate formation in aortae from diabetic rabbits. Diabetologia. 2002;45(9):1325–1331. doi:10.1007/s00125-002-0888-4
100. Jeppu AK, Kumar KA, Augusthy A. Plasma glucose and serum ceruloplasmin in metabolic syndrome and diabetes mellitus type 2. Recent Adv Biol Med. 2016;2(2016):651.
101. Srivatsan R, Das S, Gadde R, et al. Antioxidants and lipid peroxidation status in diabetic patients with and without complications. Arch Iran Med. 2009;12(2):121–127.
102. Li M, Fang F. Related analysis on type 2 diabetes and trace elements. Guangdong Trace Elements Sci. 2012;19(3):1–4.
103. Sanjeevi N, Freeland-Graves J, Beretvas SN, Sachdev PK. Trace element status in type 2 diabetes: a meta-analysis. J Clin Diagn Res. 2018;12(5):Oe01–oe08. doi:10.7860/JCDR/2018/35026.11541
104. Liu Q, Sun L, Tan Y, Wang G, Lin X, Cai L. Role of iron deficiency and overload in the pathogenesis of diabetes and diabetic complications. Curr Med Chem. 2009;16(1):113–129. doi:10.2174/092986709787002862
105. Lee EC, Ha E, Singh S, et al. Copper (II)-human amylin complex protects pancreatic cells from amylin toxicity. Phys Chem Chem Phys. 2013;15(30):12558–12571. doi:10.1039/c3cp44542a
106. Lei Y. Effect of Green Tea Polyphenols on Glucose Homeostasis and Its Mechanism in Ceruloplasmin Gene Knockout Mice. Hebei Medical University; 2017.
107. Arredondo M, Fuentes M, Jorquera D, et al. Cross-talk between body iron stores and diabetes: iron stores are associated with activity and microsatellite polymorphism of the heme oxygenase and type 2 diabetes. Biol Trace Elem Res. 2011;143(2):625–636. doi:10.1007/s12011-010-8895-7
108. Bonaccorsi di Patti MC, Cutone A, Polticelli F, et al. The ferroportin-ceruloplasmin system and the mammalian iron homeostasis machine: regulatory pathways and the role of lactoferrin. Biometals. 2018;31(3):399–414. doi:10.1007/s10534-018-0087-5
109. Lee MJ, Jung CH, Kang YM, et al. Serum ceruloplasmin level as a predictor for the progression of diabetic nephropathy in Korean men with Type 2 diabetes mellitus. Diabetes Metab J. 2015;39(3):230–239. doi:10.4093/dmj.2015.39.3.230
110. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond). 2013;124(3):139–152. doi:10.1042/CS20120198
111. Cooper GJ, Young AA, Gamble GD, et al. A copper (II)-selective chelator ameliorates left-ventricular hypertrophy in type 2 diabetic patients: a randomised placebo-controlled study. Diabetologia. 2009;52(4):715–722. doi:10.1007/s00125-009-1265-3
112. Adki KM, Kulkarni YA. Potential biomarkers in diabetic retinopathy. Curr Diabetes Rev. 2020;16(9):971–983. doi:10.2174/1573399816666200217092022
113. Chu J, Gao R, Zhao S, Lu G, Zhao D, Li J. 2016 Chinese guideline for the management of dyslipidemia in adults. Chin Circ J. 2016;31(10):937–953.
114. Li W, Zhou C. Advances in studies on hyperlipidemia, atherosclerosis and lipid metabolism. Chin J Pharmacol Toxicol. 2019;33(10):811.
115. Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017;3:17093. doi:10.1038/nrdp.2017.93
116. Rygiel K. Hypertriglyceridemia - common causes, prevention and treatment strategies. Curr Cardiol Rev. 2018;14(1):67–76. doi:10.2174/1573403X14666180123165542
117. Li T, Jiang S, Lu C, et al. Snapshots: endoplasmic reticulum stress in lipid metabolism and cardiovascular disease. Curr Issues Mol Biol. 2018;28:14–28. doi:10.21775/cimb.028.014
118. Cao SS, Luo KL, Shi L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol. 2016;231(2):288–294. doi:10.1002/jcp.25098
119. Sozen E, Ozer NK. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: an updated mini-review. Redox Biol. 2017;12:456–461. doi:10.1016/j.redox.2017.02.025
120. Lee SY, Hong IK, Kim BR, et al. Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice. Hepatology. 2015;62(1):135–146. doi:10.1002/hep.27804
121. Zhou W, Shen X, Wang L, Zhang Q, Lu Y. Influences of fenofibrate on oxidative stress and endoplasmic reticulum stress in livers of hyperlipidemic rats. J Pract Med. 2014;30(17):2718–2721.
122. Oe S, Miyagawa K, Honma Y, Harada M. Copper induces hepatocyte injury due to the endoplasmic reticulum stress in cultured cells and patients with Wilson disease. Exp Cell Res. 2016;347(1):192–200. doi:10.1016/j.yexcr.2016.08.003
123. Kono S, Suzuki H, Oda T, et al. Biochemical features of ceruloplasmin gene mutations linked to aceruloplasminemia. Neuromolecular Med. 2006;8(3):361–374. doi:10.1385/NMM:8:3:361
124. Shen Z. Correlation among blood lipids and main inflammatory factors levels in patients with coronary heart disease complicated hyperlipidemia. Chin J Cardiovasc Rehabil Med. 2017;26(4):388–390.
125. Chen S. Interconnectivity Between the Reaction System of Catalase and Hyperlipidemia. Jilin University; 2013.
126. Anila L, Vijayalakshmi N. Antioxidant action of flavonoids from Mangifera indica and Emblica officinalis in hypercholesterolemic rats. Food Chem. 2003;83(4):569–574. doi:10.1016/S0308-8146(03)00155-9
127. Zhang L, Liu X, Liang W, Zhong C. Study on oxidative stress level in patients with hyperlipidemia. Studies Trace Elements Health. 2016;33(01):9–10.
128. Samokyszyn VM, Miller DM, Reif DW, Aust SD. Inhibition of superoxide and ferritin-dependent lipid peroxidation by ceruloplasmin. J Biol Chem. 1989;264(1):21–26. doi:10.1016/S0021-9258(17)31218-8
129. Shukla N, Maher J, Masters J, Angelini GD, Jeremy JY. Does oxidative stress change ceruloplasmin from a protective to a vasculopathic factor? Atherosclerosis. 2006;187(2):238–250. doi:10.1016/j.atherosclerosis.2005.11.035
130. Wang B, Wang XP. Does ceruloplasmin defend against neurodegenerative diseases? Curr Neuropharmacol. 2019;17(6):539–549. doi:10.2174/1570159X16666180508113025
131. Commission N. The report on nutrition and chronic diseases in China (2015) release. Shanghai J Prevent Med. 2016;28(3):141.
132. Chen Y, Zhang Y, Kong Z, Yu J, Sun T, Zhang H. The prevalence of overweight and obesity in children and adolescents in China. Chin J Dis Control Prev. 2017;21(9):866–869, 878.
133. Tajik N, Golpaie A, Keshavarz SA, et al. Decreased plasma levels of ceruloplasmin after diet-induced weight loss in obese women. J Endocrinol Invest. 2012;35(6):566–569. doi:10.3275/7878
134. Moreno-Navarrete JM, Fernández-Real JM. The complement system is dysfunctional in metabolic disease: evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin Cell Dev Biol. 2019;85:164–172. doi:10.1016/j.semcdb.2017.10.025
135. Ziakas A, Gavrilidis S, Souliou E, et al. Ceruloplasmin is a better predictor of the long-term prognosis compared with fibrinogen, CRP, and IL-6 in patients with severe unstable angina. Angiology. 2009;60(1):50–59. doi:10.1177/0003319708314249
136. Ruggiero C, Ehrenshaft M, Cleland E, Stadler K. High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am J Physiol Endocrinol Metab. 2011;300(6):E1047–1058. doi:10.1152/ajpendo.00666.2010
137. Raffaella C, Francesca B, Italia F, Marina P, Giovanna L, Susanna I. Alterations in hepatic mitochondrial compartment in a model of obesity and insulin resistance. Obesity. 2008;16(5):958–964. doi:10.1038/oby.2008.10
138. Guo Y, Chen P, Xiao W. Mitochondrial dysfunction and exercise regulation in adipose tissue during obesity. Chin J Biochem Mol Biol. 2020;36(10):1145–1150.
139. Curtis JM, Grimsrud PA, Wright WS, et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59(5):1132–1142.
140. Yuan K, Lin N, Chen H. Relationship between intestinal flora and pathogenesis of obesity. Med Recapitulate. 2018;24(21):4166–4171.
141. Barra NG, Anhê FF, Cavallari JF, Singh AM, Chan DY, Schertzer JD. Micronutrients impact the gut microbiota and blood glucose. J Endocrinol. 2021;250(2):R1–r21. doi:10.1530/JOE-21-0081
142. Mayneris-Perxachs J, Cardellini M, Hoyles L, et al. Iron status influences non-alcoholic fatty liver disease in obesity through the gut microbiome. Microbiome. 2021;9(1):104. doi:10.1186/s40168-021-01052-7
143. Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev. 2011;32(4):550–570. doi:10.1210/er.2010-0030
144. Lehr S, Hartwig S, Lamers D, et al. Identification and validation of novel adipokines released from primary human adipocytes. Mol Cell Proteomics. 2012;11(1):
145. Safavi SM, Ziaei R, Maracy MR. Association of serum ceruloplasmin level with obesity: some components of metabolic syndrome and high-sensitive C-reactive protein in Iran. J Obes. 2012;2012:951093. doi:10.1155/2012/951093
146. Göçmen AY, Sahin E, Semiz E, Gümuşlü S. Is elevated serum ceruloplasmin level associated with increased risk of coronary artery disease? Can J Cardiol. 2008;24(3):209–212. doi:10.1016/S0828-282X(08)70586-5
147. Mori T, Sasaki J, Kawaguchi H, et al. Serum glycoproteins and severity of coronary atherosclerosis. Am Heart J. 1995;129(2):234–238. doi:10.1016/0002-8703(95)90003-9
148. Reunanen A, Knekt P, Aaran RK. Serum ceruloplasmin level and the risk of myocardial infarction and stroke. Am J Epidemiol. 1992;136(9):1082–1090. doi:10.1093/oxfordjournals.aje.a116573
149. Mänttäri M, Manninen V, Hurrunen JK, et al. Serum ferritin and ceruloplasmin as coronary risk factors. Eur Heart J. 1994;15(12):1599–1603. doi:10.1093/oxfordjournals.eurheartj.a060440
150. Enbergs A, Dorszewski A, Luft M, et al. Failure to confirm ferritin and caeruloplasmin as risk factors for the angiographic extent of coronary arteriosclerosis. Coron Artery Dis. 1998;9(2–3):119–124.
151. Klipstein-Grobusch K, Grobbee DE, Koster JF, et al. Serum caeruloplasmin as a coronary risk factor in the elderly: the Rotterdam Study. Br J Nutr. 1999;81(2):139–144. doi:10.1017/S0007114599000276
152. Engström G, Stavenow L, Hedblad B, et al. Inflammation-sensitive plasma proteins and incidence of myocardial infarction in men with low cardiovascular risk. Arterioscler Thromb Vasc Biol. 2003;23(12):2247–2251. doi:10.1161/01.ATV.0000102924.11767.8D
153. Engström G, Hedblad B, Stavenow L, et al. Fatality of future coronary events is related to inflammation-sensitive plasma proteins: a population-based prospective cohort study. Circulation. 2004;110(1):27–31. doi:10.1161/01.CIR.0000133277.88655.00
154. Verma VK, Ramesh V, Tewari S, Gupta RK, Sinha N, Pandey CM. Role of bilirubin, vitamin C and ceruloplasmin as antioxidants in coronary artery disease [CAD]. Indian J Clin Biochem. 2005;20(2):68–74. doi:10.1007/BF02867403
155. Brunetti ND, Pellegrino PL, Correale M, De Gennaro L, Cuculo A, Di Biase M. Acute phase proteins and systolic dysfunction in subjects with acute myocardial infarction. J Thromb Thrombolysis. 2008;26(3):196–202. doi:10.1007/s11239-007-0088-7
156. Deepa M, Pasupathi P, Sankar KB, Rani P, Kumar SP. Free radicals and antioxidant status in acute myocardial infarction patients with and without diabetes mellitus. Bangladesh Med Res Counc Bull. 2009;35(3):95–100. doi:10.3329/bmrcb.v35i3.2999
157. Kumar A, Nagtilak S, Sivakanesan R, Gunasekera S. Cardiovascular risk factors in elderly normolipidemic acute myocardial infarct patients–a case controlled study from India. Southeast Asian J Trop Med Public Health. 2009;40(3):581–592.
158. Tang WH, Wu Y, Nicholls SJ, et al. Subclinical myocardial necrosis and cardiovascular risk in stable patients undergoing elective cardiac evaluation. Arterioscler Thromb Vasc Biol. 2010;30(3):634–640. doi:10.1161/ATVBAHA.109.201210
159. Tang WH, Wu Y, Hartiala J, et al. Clinical and genetic association of serum ceruloplasmin with cardiovascular risk. Arterioscler Thromb Vasc Biol. 2012;32(2):516–522. doi:10.1161/ATVBAHA.111.237040
160. Xu Y, Lin H, Zhou Y, Cheng G, Xu G. Ceruloplasmin and the extent of heart failure in ischemic and nonischemic cardiomyopathy patients. Mediators Inflamm. 2013;2013:348145. doi:10.1155/2013/348145
161. Grammer TB, Kleber ME, Silbernagel G, et al. Copper, ceruloplasmin, and long-term cardiovascular and total mortality (the Ludwigshafen Risk and Cardiovascular Health Study). Free Radic Res. 2014;48(6):706–715. doi:10.3109/10715762.2014.901510
162. Daybanyrova LV, Shevchenko OP. Clinical significance levels of c-reactive protein and ceruloplasmin in patients with ischemic heart disease. Wiad Lek. 2015;68(4):517–519.
163. Bao X, Borné Y, Johnson L, et al. Comparing the inflammatory profiles for incidence of diabetes mellitus and cardiovascular diseases: a prospective study exploring the “common soil” hypothesis. Cardiovasc Diabetol. 2018;17(1):87. doi:10.1186/s12933-018-0733-9
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.