Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 12
Molecular dynamics study to improve the substrate adsorption of Saccharomycopsis fibuligera R64 alpha-amylase by designing a new surface binding site
Authors Baroroh U ![]() , Yusuf M
, Yusuf M ![]() , Rachman SD, Ishmayana S
, Rachman SD, Ishmayana S ![]() , Hasan K
, Hasan K ![]() , Subroto T
, Subroto T ![]()
Received 13 December 2018
Accepted for publication 19 March 2019
Published 7 June 2019 Volume 2019:12 Pages 1—13
DOI https://doi.org/10.2147/AABC.S198110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Umi Baroroh,1,* Muhammad Yusuf,2,3,* Saadah Diana Rachman,2 Safri Ishmayana,2 Khomaini Hasan,4 Toto Subroto2,3
1Biotechnology Master Program, Postgraduate School, Universitas Padjadjaran, Bandung 40132, West Java, Indonesia; 2Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Sumedang 45363, West Java, Indonesia; 3Research Center for Molecular Biotechnology and Bioinformatics, Universitas Padjadjaran, Bandung 40133, West Java, Indonesia; 4Faculty of Medicine, Universitas Jenderal Achmad Yani, Cimahi, West Java, Indonesia
*These authors contributed equally to this work
Background: Carbohydrate binding module (CBM) and surface binding site (SBS) are two important parts of amylase which respond to the raw starch digestion. They are related to the enzyme ability to adsorb and to catalyze the starch hydrolysis. However, starch processing is still expensive due to the high temperature in the gelatinization step. Therefore, direct starch digestion is more favorable. One of the solutions is to use α-amylase with high starch adsorptivity, which is expected to be capable of digesting starch below the gelatinization temperature. In Indonesia, Saccharomycopsis fibuligera R64 α-amylase (Sfamy R64) is one of the enzymes with the highest activity on starch. However, its raw starch adsorptivity was low. The aim of this study was to propose an in-silico model of Sfamy R64 mutant by introducing a new SBS using molecular dynamics (MD) simulation.
Methods: The structural behavior of Sfamy R64 and positive control were studied using MD simulation. Furthermore, the mutants of Sfamy R64 were designed to have a stable SBS which mimics the positive control. The substrate affinity in all systems was evaluated using the molecular mechanics generalized Born surface area (MM/GBSA) method.
Results: The stability of a new SBS constructed by seven substitutions and a loop insertion was improved throughout MD simulation. The substrate was consistently bound to the SBS over 55 ns of simulation, as compared to 14 ns in wild-type. The structural behavior of SBS in mutant and positive control was similar. The interaction energies of the positive control, wild-type, and mutant were −17.6, −5.2, and −8.2 kcal/mol, respectively.
Conclusion: The enhanced substrate binding in the mutant, due to the existence of a new SBS, suggests the potential of improving starch adsorptivity of Sfamy R64. This result should be useful in developing an enzyme with better substrate adsorption based on the rational computer-aided molecular design approach.
Keywords: surface binding site, α-amylase, Sfamy R64, starch adsorptivity, molecular dynamics simulation
Introduction
Carbohydrates can attach to the enzyme through the catalytic and the non-catalytic site such as Carbohydrate Binding Module (CBM) and Surface Binding Site (SBS).1 CBM is a group of well-known and categorized module based on sequence similarity in the CAZy database.2 In some enzymes, CBM is a contiguous amino acid sequences to the catalytic module via linkers. The sequence of CBM fold independently and mostly has a β-sandwich motif.3 Currently, there are 84 defined families of CBMs (
However, the non-catalytic carbohydrate binding domain is not always appeared as CBM. Many studies on the structures of glycoside hydrolases (GHs) showed that carbohydrates bind to a non-catalytic surface region of the catalytic module. This surface is also called a secondary binding site, which is not homologous by sequence, but they consist of aromatic residues.6
Several functions of SBSs in GHs are: (1) to bring the substrate closer to the enzyme, (2) to help the catalytic action by assisting catalysis by bringing the substrates into the active site cavity, (3) to disturb the structure of substrate before catalysis, (4) to hold the substrate on the surface of enzyme for the next reactions, (5) to activate the enzyme allosterically, (6) to release the product of reaction, and (7) to attach the GH to the outer membrane of the host cells.7–9
Starch granules possess a crystalline and amorphous form which is rigid and difficult to be degraded. One of the strategies for improving catalytic efficiency is by incorporating the SBS(s) in various enzymes. α-Amylase is a crucial enzyme for starch digesting process. α-Amylase, or 1,4-α-D-glucan glucanohydrolase with EC 3.2.1.1, hydrolyze the 1,4-α-glycosidic bonds of starch or glycogen randomly, producing various lengths of oligosaccharides.10
In the starch processing industry, α-amylase is utilized to hydrolyze the starch granules. Due to the low solubility of starch granules in cold water,11 a heating process at 105°C is required to open the crystalline structure of starch, thus make it more accessible to be digested by the enzyme.12 But, this process requires high energy and the high cost of production.13 A starch digesting process at low temperature is more preferred.12,14,15 Shiau et al15 found that the hydrolysis performance of amylolytic enzyme to the raw starch was related to the starch adsorptivity properties. CBM and SBS have been suggested to be responsible for the adsorption capacity of α-amylase.16
Saccharomycopsis fibuligera R64 is found in Indonesia. It produced α-amylase and glucoamylase and showed the highest amylolytic activity among 136 local isolates.17 However, the raw starch adsorption of α-amylase from S. fibuligera R64 (Sfamy R64) was low. Therefore, the structure of Sfamy R64 is predicted to lack of CBM and/or SBS. It is known that Sfamy R64 has an optimum temperature and pH of 50 °C and 5.0, respectively.18
The approach of Computer-aided Molecular Design (CAMD) should be used to design a modified protein with desired properties. CAMD has been utilized to develop Fungamyl™, an amylase-like enzyme with high thermostability and also works at the low pH.19 Using a similar concept, CAMD can be applied to design a Sfamy R64 mutant with better starch adsorption without compromising its amylolytic activity.
A raw starch adsorbing enzyme which shares high sequence similarity with Sfamy R64 is Aspergillus niger α-amylase (71% homology). This enzyme possessed one SBS in the C domain, consists of tyrosine and tryptophan as the main residues. Its substrate-bound structure has been crystallized and resolved at 1.8 Å.20 Therefore, the positive control for our design is an A. niger α-amylase.
The aim of this study was to propose an in-silico model of Sfamy R64 mutant by introducing a new SBS on its surface using a CAMD approach. The dynamical behavior of a new mutant of Sfamy R64 was studied using MD simulation. The binding affinity of the substrate on the SBS was computed using MM/GBSA method.
Material and methods
Preparation of model
The model structure of Sfamy R64 and mutants were prepared by homology modeling method using MODELLER 9.15,21,22 as reported previously.23 The selection for the template is a crucial step in homology modeling. The structure of A. niger α-amylase with PDB ID 2GUY was used as a template for all models based on the sequence similarity (71% homology) and the structure quality (1.59 Å). The model mutants of Sfamy R64 were built by changing and inserting some amino acid to mimic the positive control. The model structures were evaluated by the Ramachandran plot using PROCHECK and the Z-score using ProSA-web.24,25 The calcium ion (Ca2+) was added to all models located in the A/B domain as a cofactor. The structure of A. niger α-amylase with PDB ID 2GVY was used as a positive control. Maltose which bound to the C domain of A. niger α-amylase, or SBS, was used in all model systems studied by MD simulation.
Table 1 shows the five MD systems prepared in this study. The cysteine-type and the protonation state of histidine were manually annotated to their chemical environment. The parameters of the substrate (maltose) were calculated using the AM1-BCC method by antechamber program.26 The solvent system of a box TIP3P water was added to the solute with the least distance 10 Å between protein and the edge of the box. The sodium ion was used to neutralize the system.
 | Table 1 The list of the MD system |
Molecular dynamics simulation
AMBER14 was utilized to perform minimization and MD simulation.27 First, minimization was done by using 1,000 and 4,000 steps of steepest descent and conjugate gradient, respectively. The MD system was gradually heated to 323 K over 60 ps in the NVT ensemble. Harmonic restraint of 5 kcal/molÅ2 on the complex was used in the heating stage. Furthermore, 1 ns of NPT equilibration was done. In this stage, harmonic restraints gradually reduced by 1 kcal/molÅ2 until it reached zero. Finally, production run in NPT ensemble was done for 100 ns. The time step at the production run was 2 fs since the SHAKE algorithm was used. Langevin thermostat was used to control the temperature. The collision frequency parameter was set to 1 ps−1. Berendsen barostat was used to control the pressure. The parameter of coupling constant and target pressure was set to 1 ps and 1 bar, respectively. The non-bonded cutoff value was set to 9 Å. Particle Mesh Ewald was activated to treat the long-range electrostatics. AmberTools was used to analyze the MD trajectories.
Binding energy calculation
MMPBSA.py program was used to calculate the pairwise interaction energy using a single trajectory method.28 Binding free energy (ΔGbind) between an amylase (A) and maltose (M) was calculated based on the MM/GBSA method:
(1) 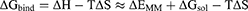
(2) 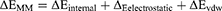
(3) 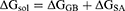
In these equations, ΔH is the enthalpy, and T is the temperature (K). ΔEMM is the molecular mechanical (MM) energy change in the gas phase, which consists of ΔEinternal (internal energy), ΔEelectrostatic (Coulomb electrostatic term), and ΔEvdw (van der Waals interaction term). ΔGsol is the solvation free energy, consists of ΔGGB (electrostatic solvation energy or polar contribution calculated by GB method) and ΔGSA (nonelectrostatic solvation component or nonpolar contribution). The interval step and salt concentration used in the binding energy calculation was 1 ns and 150 mM, respectively.
Results
Based on its molecular shape, there are three types of SBS, including a flat surface type,29 which found in our positive control. In this study, three different SBS were prepared, namely MT1, MT2, and MT3. The quality of all mutant models was assessed by Ramachandran plot and Z-score analysis. More than 90% of the residue was located in the most favored region of all models, and none of the residues were located in the disallowed region (Figure 1). Also, the Z-score of all mutants were also located in the range of X-ray structure quality and had a similar score with the template (Figure 2). These results indicated a good quality of the model structure.24,25 The behavior of maltose, the simplest form of amylase substrate, was observed using MD simulations. The initial coordinate of maltose was taken from the positive control. MD systems of MT1-MT3 were superimposed to the PDB ID 2GVY to have similar binding configurations of the substrate.
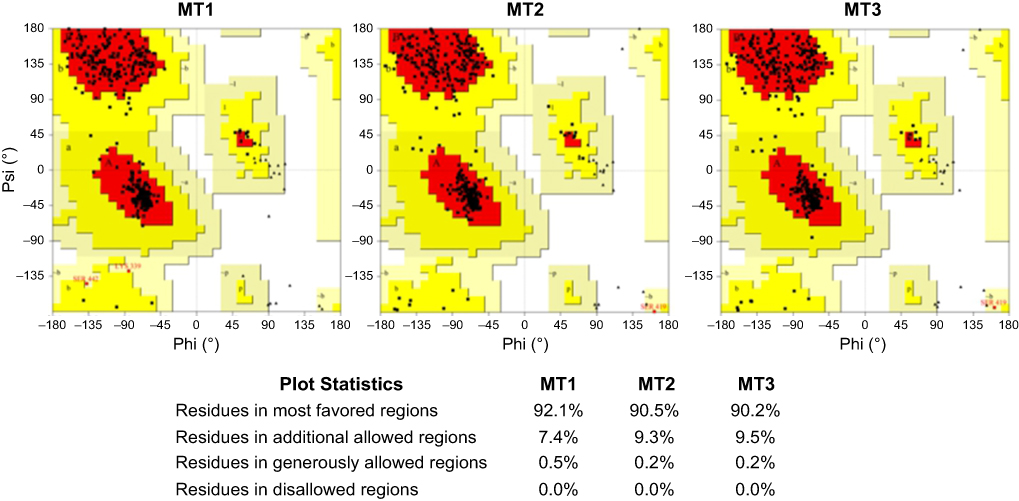 | Figure 1 Ramachandran plot of MT1–MT3. Abbreviations: MT1, mutant 1; MT2, mutant 2; MT3, mutant 3. |
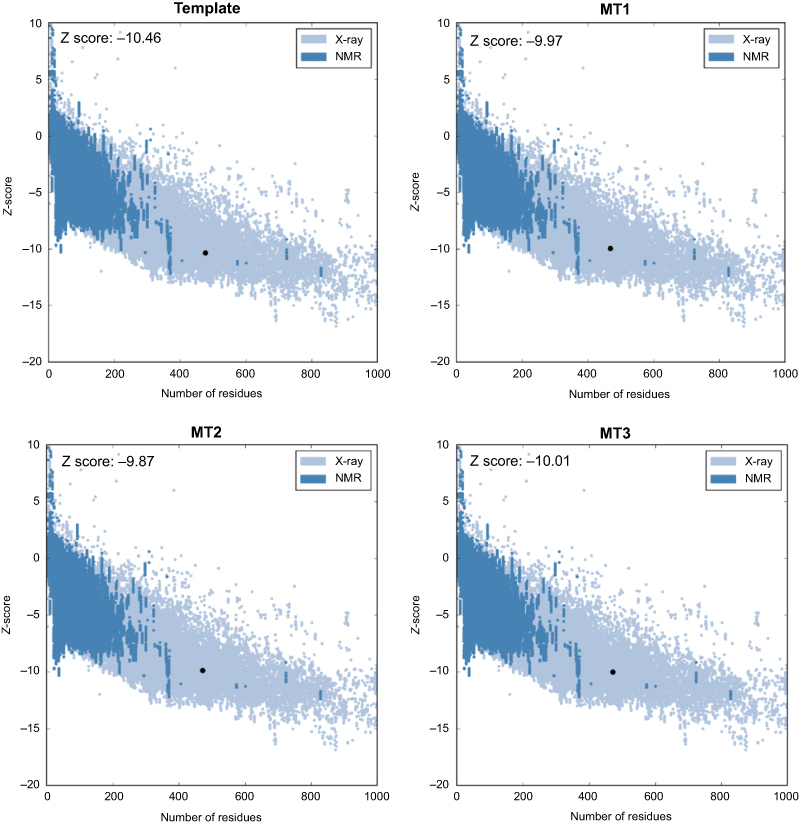 | Figure 2 Z-score of template and MT1–MT3. Abbreviations: MT1, mutant 1; MT2, mutant 2; MT3, mutant 3; NMR, nuclear magnetic resonance. |
Table 1 shows the five MD systems investigated in this study, including positive control, wild-type Sfamy R64, and three mutants. The root mean square deviation (RMSD) calculated for the protein backbone atom and the substrate over 100 ns of production trajectory is provided in Figure 3. The RMSD profile of protein backbone atom (Figure 3A) indicates that the positive control has the lowest RMSD which followed by MT2 with average RMSD values of 1.1 Å and 1.3 Å, respectively. The rest systems have similar values of average RMSD (±1.5 Å). Interestingly, the deviation of maltose in all MD systems was different (Figure 3B). In the positive control, maltose bound to the SBS over 72 ns, while that of Sfamy R64 (without SBS) was only 14 ns. The maltose binding in three mutants was better than the wild-type, which are 17 ns, 23 ns, and 55 ns in MT1, MT2, and MT3, respectively. Therefore, the importance of SBS in substrate binding is suggested.
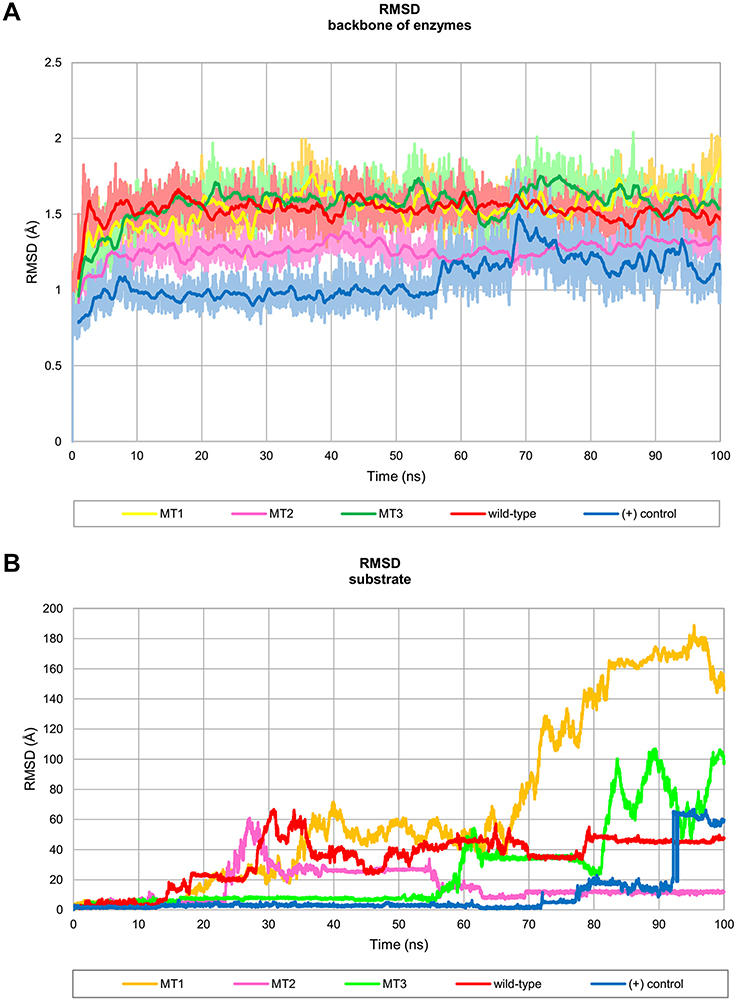 | Figure 3 Profile of RMSD throughout 100 ns of simulation. (A) RMSD backbone of the enzyme which presented in 100 frames moving average and (B) RMSD of substrate binding. Abbreviations: RMSD, root mean square deviation; MT1, mutant 1; MT2, mutant 2; MT3, mutant 3; (+) control, A. niger α-amylase. |
The root mean square fluctuation (RMSF) profile of all MD systems are shown in Figure 4. The high fluctuations in the terminals were expected because there were not restrained and the high fluctuations in the catalytic site region were observed, ie, residue number 25, 130, and 155, due to the absence of substrate at the catalytic site. Hence, the loop in the catalytic site was transformed into closed conformation (Figure 5). The other high fluctuations, ie, around residues 307, 375, 415, and 430, was a natural character of the loop structure. However, these residues were located far away from the SBS region.
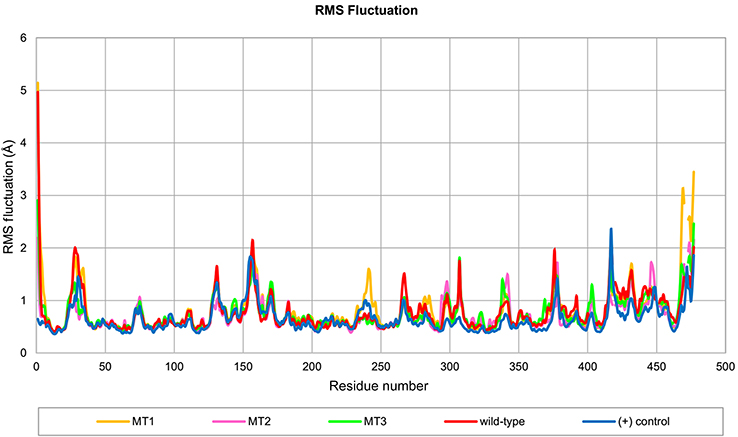 | Figure 4 Profile of RMS fluctuation throughout 100 ns of MD simulation. Abbreviations: RMS, root mean square; MD, molecular dynamics; MT1, mutant 1; MT2, mutant 2; MT3, mutant 3; (+) control, A. niger α-amylase. |
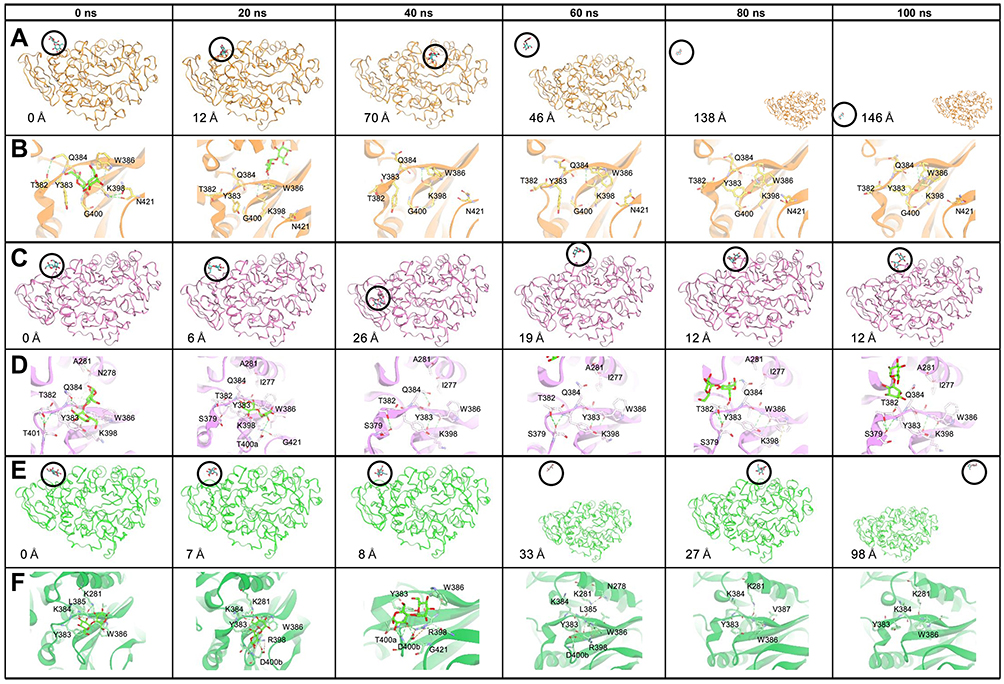 | Figure 5 Time evolution snapshot every 20 ns and interaction around the substrate in initial position. (A), (C), and (E) show the substrate position along the simulation and the distance of the substrate from the initial position. (B), (D), and (F) show the interaction that occurred around the substrate. The yellow, pink, and green color show the MT1, MT2, and MT3, respectively. The substrate is visualized in the green stick, and the hydrogen bond in the green dashed line. Abbreviations: MT1, mutant 1; MT2, mutant 2; MT3, mutant 3. |
The calculated binding energy between maltose and SBS using the MM/GBSA method in all mutants were better than the wild-type (Table 3), indicating that SBS is important for substrate binding. The calculated binding energy of all systems was generally in agreement with the design of SBS. A variation in the calculated binding energy of mutants is caused by the mobility of maltose throughout the simulation. Since the MM/GBSA calculation computed all interactions that formed between maltose and protein surface during the 100 ns of simulation, then MT2 showed lower binding energy than the MT3. Figure 3B shows that at the end of the simulation, maltose in MT1 system was deviated up to 180 Å from the initial position, farther than that of MT2 and MT3 (10 Å and 100 Å, respectively). Therefore, the binding energy of MT2 was the lowest, which followed by MT3 and MT1. However, SBS in MT3 successfully held the maltose longer than MT2 (Figure 3A). Since maltose deviated from the SBS of MT3 at 55 ns, then a separate binding energy calculation was done on the 50 ns trajectory, showing MT3 with the lowest binding energy among all mutant systems (Table 3). Nevertheless, the motion of maltose away from SBS, as also occurred in the positive control system, showed that substrate adsorption in SBS was temporary. This result is in agreement with the role of SBS in leading the substrate to enter the catalytic site.
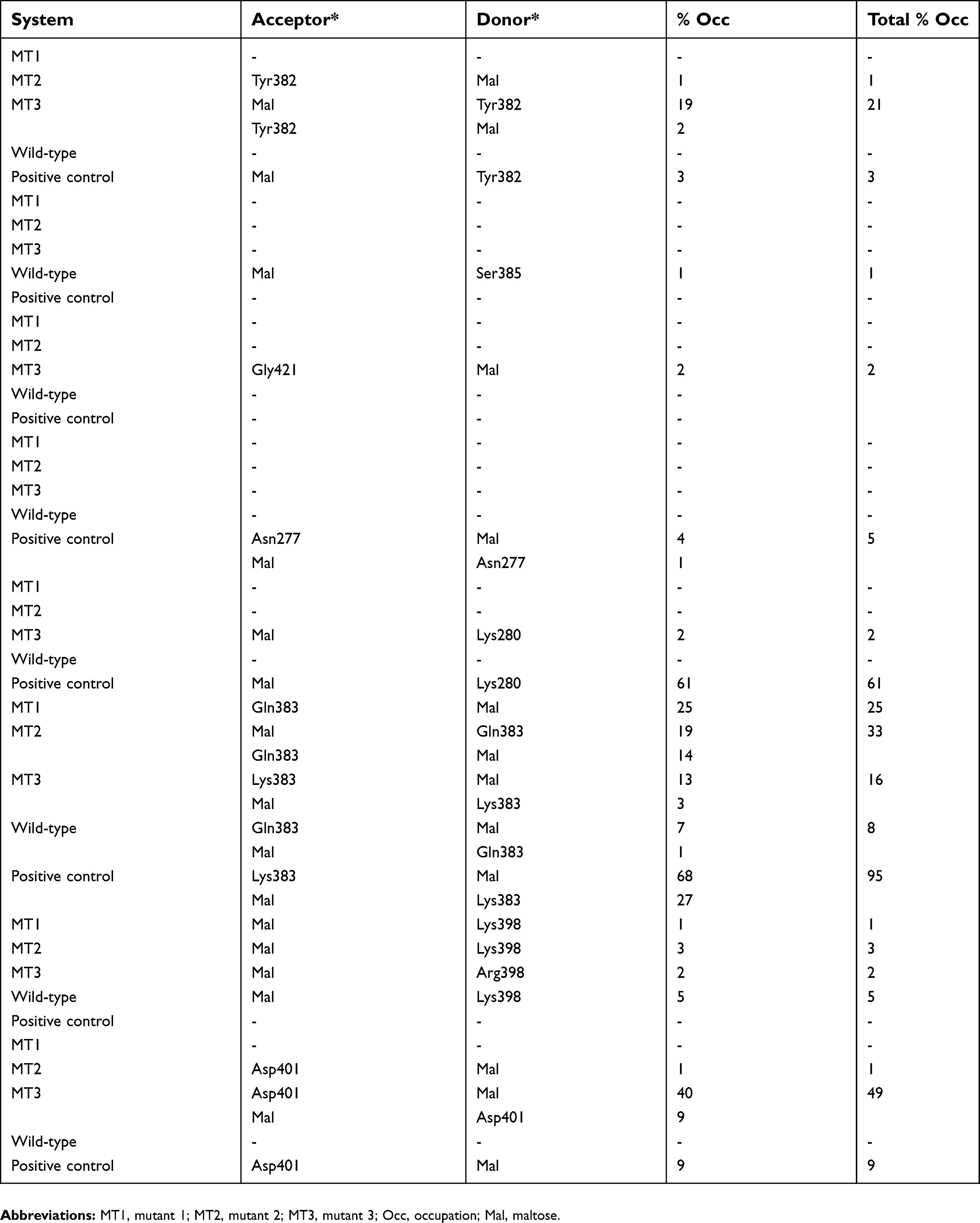 | Table 2 Hydrogen bond formation between substrate and enzyme throughout 100 ns of MD simulations |
 | Table 3 Computed binding energy between enzyme and substrate |
The explanation on the rationale behind the modifications of all three mutants is discussed below.
Designing SBS in MT1
MT1 is an extended MD simulation that has been done by Yusuf et al23. In our systems, the timescale is prolonged to 100 ns. In this SBS, serine at 383 and 386 position was replaced by aromatic residues, tyrosine, and tryptophan, respectively, to mimic the characteristic of positive control. Our results showed that the maltose could bind longer than the wild-type, although the time evolution snapshots at every 20 ns (Figure 5) showed that the substrate was moving away at the 20 ns for 12 Å from the initial coordinate. Furthermore, it was getting away for 146 Å at the end of the simulation. Hydrogen bonds around the SBS and substrate were only occurred at the beginning of simulation with Q384 and G400 (Figure 5B). No hydrogen bond was formed by these two aromatic residues (≥1% of occupancy) (Table 2). Interestingly, the pairwise decomposition of interaction energies between the substrate and these two aromatic residues were lower than the wild-type (Figure 6). It was indicated that the hydrophobic interaction dominated their forces during simulation. The missing hydrogen bond and the loose substrate binding were suggested due to the absence of stabilizing residues at the SBS. The conformation of SBS residues in MT1 was also different from that of positive control. However, this conformation was similar to that of positive control from 60 ns until 100 ns (Figure 7). A bulky asparagine at position 421 was predicted to change the conformation of W386 to deviate from its position in the positive control. After the substrate went away from the SBS, the orientation of N421 was changed, thus providing more space to W386 to have similar conformation with the control positive. Hence, N421 needed to be substituted with the smallest residue, ie, glycine. Also, an additional loop under the SBS that found in the positive control is required to stabilize the conformation. These observations were then guiding us to develop the other mutants to optimize the conformation of SBS residues.
 | Figure 6 Pairwise decomposition energy between maltose and side chain of residues around the mutation points in all systems.Residue number according to PDB ID 2GVY. Abbreviations: MT1, mutant 1; MT2, mutant 2; MT3, mutant 3; (+) control, A. niger α-amylase. |
 | Figure 7 Time evolution snapshot every 20 ns at the two aromatic residues of SBS. (A) positive control, (B) MT1, (C) MT2, and (D) MT3. Abbreviations: SBS, substrate binding site; MT1, mutant 1; MT2, mutant 2; MT3, mutant 3. |
Substrate-stabilizing SBS in MT2 by adding an extra loop and avoiding steric hindrances
MT2 was built to handle the MT1 problem. In this system, the substrate could bind longer until 23 ns. Time evolution snapshots at every 20 ns (Figure 5) revealed that the substrate remained close to the SBS at 20 ns, although it was departed for 6 Å from the initial position. It was indicated that the stacking interaction still occurred at a considerable distance. MT2 increased the chances of hydrogen bond formation by the contribution of an additional loop. Pairwise decomposition of interaction energies between substrate and MT2 was lower than wild-type and MT1 (Figure 6). The hydrogen bond between the substrate with Y383 was also formed in MT2. It was indicated that the additional loop could help the proper orientation of Y383, indicating its importance to support the SBS (Figure 7). Whereas the conformation and orientation of W386 and its surrounding residues were still different from the positive control, especially at the top region of SBS (Figure 7C). It is predicted that SBS requires supporting hydrophobic forces to maintain its stability. In the positive control, lysines above the SBS (K281 and K384 in Sfamy R64 numbering) contributed to the formation of a hydrogen bond with the substrate (Table 2). At the same position, Sfamy R64 was occupied with alanine, not lysine. Therefore, the movement of the substrate away from the SBS of MT2 in 23 ns might be due to the less of hydrophobic interaction above the SBS residues, and also the extra cavity provided by the smaller residue of A281. In addition, the rigidity of R397 below the SBS residues in positive control system might also play an important role to stabilize the aromatic residues by hydrophobic interaction. It is noted that at the same position, Sfamy R64 occupies a more flexible lysine. Based on these observations, another mutant, namely MT3, was designed to improve the stability of SBS of Sfamy R64.
The most stable SBS of Sfamy R64 in MT3
Finally, the last mutant was designed to overcome the problems that appeared in MT2, particularly to provide supporting hydrophobic forces to the stability of SBS. The RMSD of the substrate in MT3 showed that it could bind to the SBS until 55 ns. Time evolution snapshot every 20 ns (Figure 5) revealed that substrate binding in MT3 was better than the two previous mutants. After introducing the A281K, a new hydrogen bond with the substrate was formed, as also indicated in the pairwise decomposition energy involving K281 (Figure 6). Moreover, Q384K in MT3 formed a hydrogen bond with the substrate, and also stabilized K281 through hydrophobic forces. Also, K398R mutation showed to enhance the stability of SBS residues from below, due to the rigidity of arginine. It is observed that the role of additional loop 400–401 in MT3 to be more efficient than in MT2, indicated from the higher number of hydrogen bond formed with the substrate (Table 2). This observation corresponds well with the pairwise decomposition energy (Figure 6), where Y383 in MT3 contributed to the lower binding energy with the substrate as compared with other mutants.
In general, the time-dependent conformations of Y383 and W386 in MT3 is similar to that of the positive control (Figure 7). The modifications of residues at the top and bottom of the SBS have successfully changed the orientation of Y383 and W386. It is noted that all modifications surrounding the SBS are within the distance of 5 Å and positioned at the surface of the enzyme. The summary of structural modifications in MT3 is presented in Figure 8.
 | Figure 8 The summary of SBS modification in MT3. The substrate is shown in green, the two key SBSs are shown in gray, and the supporting hydrophobic region, supporting loop, and supporting residues are shown in the blue, red, and yellow circles, respectively. Abbreviations: SBS, substrate binding site; MT3, mutant 3. |
From our results, it is concluded that designing an SBS in amylase is not only by introducing the aromatic residue(s) or by increasing hydrogen bond formation, but also considering the stabilizing residues around the SBS.
Discussion
Samaeidaryan29 have characterized the SBS in GH family using computational study. From a total of 119 crystal structure from the protein data bank of GHs containing carbohydrate bound SBS, one or two aromatic residues mostly existed in a flat surface. The crystal structure of A. niger amylase possesses one SBS with two aromatic residues, which is absent in Sfamy R64 structure (based on the superimposition of these two structures). Previously, we have done a preliminary computational study to introduce SBS in the structure of Sfamy R64.23 The results showed that the binding of maltose was only until 17 ns, due to the unstable SBS as compared to the positive control. Therefore, this study was conducted to stabilize the SBS in a longer timescale. The sequence of Sfamy R64 consists of two glycosylation sites at position N127 and N198.30 Schwanniomyces occidentalis SWA2 amylase which shares high sequence identity with Sfamy R64 also has two possible glycosylation sites located at position N134 and N229. Mutation analysis in that position revealed that N229 is the single glycosylated residue and play an important role in the activity, stability, levels, and kinetics.31 Sequence alignment of N299 and N198 for SWA2 and Sfamy R64, respectively shown that these two sequences are in the same position. It was indicated that N198 is the glycosylation site for Sfamy R64. Nevertheless, in our simulation, the glycosylation site was deleted since its location far away from the SBS. In addition, deglycosylation in A. oryzae did not affect its stability.32
Mutation analysis on SBS has been done in some studies to investigate the importance of this site. SusG α-amylase has one SBS with tryptophan and tyrosine as main residues. The activity extremely loss up to 56% for insoluble cornstarch when SBS was removed.33 Barley AMY1 has two SBSs; mutation study was done in SBS1 and SBS2. A single mutation on SBS2 decreased the activity about 10 fold (Kd=1.4 mg/mL) as compared to the wild-type and when both of SBSs were mutated into alanine residue, the activity completely loss for barley starch granule (Kd>100 mg/mL).9 Our finding by introducing an SBS in Sfamy R64 also indicates that the substrate binding increased when compared to the wild-type. In this study, to only substitute SBS residues is not enough to adsorb the substrate. Further analysis of the structure of the enzyme is important to make the SBS is working well and not disturbing the stability of the enzyme structure. Computer-aided molecular design should be useful to predict the best conformation of the enzyme-substrate complex.
Amalia et al34 have mutated Sfamy R64 with Y375W to improve the substrate binding on raw starch. The results showed that the mutation was unsatisfied to improve the ability of the enzyme to bind substrate. In our simulation on the wild-type Sfamy R64, the fluctuation of residue number 375 was high. Therefore, mutating this residue by tryptophan alone could not stabilize the substrate binding. Moreover, the addition of a new disulfide bridge between A and C domain of Sfamy R64 resulted in similar activity and pH optimum with the wild-type.35 Chemical modification of Sfamy R64 to improve the stability also has been done by introducing the nonpolar group. As a result, the enzyme stability was increased. In another study, the addition of a cross-linking agent to the enzyme had protected the calcium ion. Moreover, the use of polyethylene glycol for hydrophilization had caused resistance towards tryptic digestion.30
Conclusion
A computational model of SBS in MT3 improved the maltose binding as compared to the wild-type. The SBS was introduced by S383Y/S386W/N421G/S278N/A281K/Q384K/K398R and insertions of G400-S401insTDGS. Mutations were determined by considering the surrounding residues of SBS to improve the stability of the binding site. It is predicted that the MT3 could have improved the starch adsorptivity of Sfamy R64.
Acknowledgments
We want to thank Universitas Padjadjaran for their support of this work by the Internal Grant through Riset Kompetensi Dosen Unpad Funding (No. 2362/UN6.D/KS/2018) and the Academic Leadership Grant. Also, we acknowledge computing services supported by a PPTI Grant (No. 05/E/KPT/2018) from Ristekdikti. We would also like to thank Professor Soetijoso Soemitro for his enlightening discussions about enzyme engineering.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cockburn D, Nielsen MM, Christiansen C, et al. Surface binding sites in amylase have distinct roles in recognition of starch structure motifs and degradation. Int J Biol Macromol. 2015;75:338–345. doi:10.1016/j.ijbiomac.2015.01.054
2. Davies GJ, Gloster TM, Henrissat B. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr Opin Struct Biol. 2005;15:637–645. doi:10.1016/j.sbi.2005.10.008
3. Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. 2004;781:769–781. doi:10.1042/BJ20040892
4. Barchiesi J, Hedin N, Casati DFG, Ballicora MA, Busi MV. Functional demonstrations of starch binding domains present in Ostreococcus tauri starch synthases isoforms. BMC Res Notes. 2015;8:1–12. doi:10.1186/s13104-015-1598-6
5. Gilbert HJ, Knox JP, Boraston AB. Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules. Curr Opin Struct Biol. 2013;23:1–9. doi:10.1016/j.sbi.2013.05.005
6. Cuyvers S, Dornez E, Delcour JA, Courtin CM. Occurrence and functional significance of secondary carbohydrate binding sites in glycoside hydrolases. Crit Rev Biotechnol. 2012;32:93–107. doi:10.3109/07388551.2011.561537
7. Ye Z, Miyake H, Tatsumi M, Nishimura S, Nitta Y. Two additional carbohydrate-binding sites of β-amylase from Bacillus cereus var. mycoides are involved in hydrolysis and raw starch-binding. J Biochem. 2004;135(3):355–363. doi:10.1093/jb/mvh043
8. Sevcik J, Hostinova E, Solovicova A, Gasperik J, Dauter Z, Wilson KS. Structure of the complex of a yeast glucoamylase with acarbose reveals the presence of a raw starch binding site on the catalytic domain. FEBS J. 2006;273:2161–2171. doi:10.1111/j.1742-4658.2006.05230.x
9. Nielsen MM, Bozonnet S, Seo E-S, et al. Two secondary carbohydrate binding sites on the surface of barley α-amylase 1 have distinct functions and display synergy in hydrolysis of starch granules. Biochemistry. 2009;48(32):7686–7697. doi:10.1021/bi900795a
10. Janecek S, Balaz S. α-Amylases and approaches leading to their enhanced stability. FEBS Lett. 1992;304(1):1–3.
11. Hamilton LM, Kelly CT, Fogarty WM. Purification and properties of the raw starch-degrading α-amylase of Bacillus sp. IMD 434. Biotechnol Lett. 1999;21:111–115. doi:10.1023/A:1005413816101
12. Singh H, Soni SK. Production of starch-gel digesting amyloglucosidase by Aspergillus oryzae HS-3 in solid state fermentation. Process Biochem. 2001;37:453–459. doi:10.1016/S0032-9592(01)00238-2
13. Sun H, Zhao P, Ge X, et al. Recent advances in microbial raw starch degrading enzymes. Appl Biochem Biotechnol. 2010;160(4):988–1003. doi:10.1007/s12010-009-8579-y
14. Kelly CT, Mctigue MA, Doyle EM, Fogarty WM. The raw starch-degrading alkaline amylase of Bacillus sp IMD. J Ind Microbiol. 1995;15:9–10. doi:10.1007/BF01569973
15. Shiau J, Hung H, Jeang C. Improving the thermostability of raw-starch-digesting amylase from a Cytophaga sp. by site-directed mutagenesis. Appl Environ Microbiol. 2003;69(4):2383–2385. doi:10.1128/AEM.69.4.2383-2385.2003
16. Cockburn D, Wilkens C, Ruzanski C, et al. Analysis of surface binding sites (SBSs) in carbohydrate active enzymes with focus on glycoside hydrolase families 13 and 77 – a mini-review. Biologia (Bratisl). 2014;69:705–712. doi:10.2478/s11756-014-0373-9
17. Ismaya WT, Hasan K, Subroto T, Natalia D, Soemitro S.Chromatography as the major tool in the identification and the structure-function relationship study of amylolytic enzymes from Saccharomycopsis fibuligera R64. In: de Azevedo Calderon, editor. Chromatography - The most versatile method of chemical analysis. Leonardo de Azevedo Calderon. London: Intech Open; 2012:271-294. doi:10.5772/51325.
18. Hasan K, Ismaya WT, Kardi I, et al. Proteolysis of α-amylase from Saccharomycopsis fibuligera: characterization of digestion products. Biologia. 2008;63(6):1044–1050. doi:10.2478/s11756-008-0167-z
19.
20. Zagar VA, Dijkstra BW. Monoclinic crystal form of Aspergillus niger α-amylase in complex with maltose at 1.8 Å resolution. Protein Struct Commun. 2006;62:716–721. doi:10.1107/S1744309106024729
21. Fiser A, Šali A. MODELLER: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi:10.1016/S0076-6879(03)74020-8
22. Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi:10.1006/jmbi.1993.1626
23. Yusuf M, Baroroh U, Hasan K, Diana S, Ishmayana S, Subroto T. Computational model of the effect of a surface-binding site on the Saccharomycopsis fibuligera R64 α-amylase to the substrate adsorption. Bioinform Biol Insights. 2017;11:1–8. doi:10.1177/1177932217738764
24. Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystalogr. 1993;26(2):283–291. doi:10.1107/S0021889892009944
25. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi:10.1093/nar/gkm290
26. Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi:10.1002/jcc.10128
27. Case DA, Darden T, Iii TEC, et al. Amber14. San Francisco: University of California; 2014.
28. Miller BR, Mcgee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA. py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8:3314–3321. doi:10.1021/ct300418h
29. Samaeidaryan S. Characterization of surface binding sites in glycoside hydrolases: a computational study. J Mol Recognit. 2017;e2624. doi:10.1002/jmr.2624
30. Ismaya WT, Hasan K, Kardi I, et al. Chemical modification of Saccharomycopsis fibuligera R64 α-amylase to improve its stability against thermal, chelator, and proteolytic inactivation. Appl Biochem Biotechnol. 2013;170:44–57. doi:10.1007/s12010-013-0164-8
31. Yanez E, Carmona TA, Tiemblo M, Jime A, Ya E. Expression of the Schwanniomyces occidentalis SWA2 amylase in Saccharomyces cerevisiae: role of N-glycosylation on activity, stability and secretion. Biochem J. 1998;71:65–71. doi:10.1042/bj3290065
32. Eriksen SH, Jensen B, Olsen J. Effect of N-linked glycosylation on secretion, activity, and stability of α-amylase from Aspergillus oryzae. Curr Microbiol. 1998;37:117–122.
33. Koropatkin NM, Smith TJ. SusG: a unique cell-membrane-associated α-amylase from a prominent human gut symbiont targets complex starch molecules. Struct Des. 2010;18(2):200–215. doi:10.1016/j.str.2009.12.010
34. Amalia R, Ismaya WT, Puspasari F, et al. Heterologous expression of α-amylase from Saccharomycopsis fibuligera R64 and its Tyr401Trp mutant in Pichia pastoris. Microbiol Indones. 2016;10(1):23–29. doi:10.5454/mi.10.1.4
35. Natalia D, Vidilaseris K, Ismaya WT, et al. Effect of introducing a disulphide bond between the A and C domains on the activity and stability of Saccharomycopsis fibuligera R64 α-amylase. J Biotechnol. 2014;195:8–14.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.