Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 8
Molecular dynamics simulations: advances and applications
Authors Hospital A, Goñi JR, Orozco M, Gelpi J
Received 27 July 2015
Accepted for publication 9 October 2015
Published 19 November 2015 Volume 2015:8 Pages 37—47
DOI https://doi.org/10.2147/AABC.S70333
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Adam Hospital,1 Josep Ramon Goñi,2,3 Modesto Orozco,1–4 Josep L Gelpí2–4
1Institute for Research in Biomedicine, The Barcelona Institute of Science and Technology, 2Joint BSC-IRB Research Program in Computational Biology, 3Barcelona Supercomputing Center, 4Department of Biochemistry and Molecular Biology, University of Barcelona, Barcelona, Spain
Abstract: Molecular dynamics simulations have evolved into a mature technique that can be used effectively to understand macromolecular structure-to-function relationships. Present simulation times are close to biologically relevant ones. Information gathered about the dynamic properties of macromolecules is rich enough to shift the usual paradigm of structural bioinformatics from studying single structures to analyze conformational ensembles. Here, we describe the foundations of molecular dynamics and the improvements made in the direction of getting such ensemble. Specific application of the technique to three main issues (allosteric regulation, docking, and structure refinement) is discussed.
Keywords: molecular dynamics, allostery, docking, conformational ensembles, structure prediction, refinement
Introduction
The study of the macromolecular structure is a key point in the understanding of biology. Biological function is based on molecular interactions, and these are a consequence of macromolecular structures. Since initial structure determinations in the 50s, both in the protein and in the nucleic acid worlds, the increase in the knowledge of how macromolecular structures are built has been continuous. At present, protein data bank (PDB)1 holds more than 110,000 entries, including more than 100,000 proteins, 2,800 nucleic acids, alone or forming complexes, and approximately 20,000 small molecules complexed to macromolecules. Molecular recognition rules as defined by such structural knowledge powers the understanding of basic biological phenomena, like enzyme mechanisms and regulation, transport across membranes, the building of large structures like ribosomes, or viral capsids, or how DNA is read and transcription is controlled. The study and prediction of protein–protein interaction networks is one of the growing fields in modern systems biology. On a more practical note, protein three-dimensional (3D) structures are the basis for structure-based drug design. The simple visual analysis of 3D structures of protein or nucleic acids, as obtained from the experiments, has driven large number of successful studies in biochemistry. However, despite their enormous utility, structures stored at the PDB provide only a partial view of 3D structure. Both protein and nucleic acids are flexible entities, and dynamics can play a key role in their functionality. Proteins undergo significant conformational changes while performing their function. As a rule, any complex made by any protein implies some structural rearrangement. This can be easily checked just by comparing a series of PDB entries that just differ in a small ligand bound to a given protein. Figure 1 shows a superimposition of experimental acetylcholinesterase structures. There are no changes in the overall fold, just small rearrangements in the structure; however, these differences are large enough to fool ligand-docking algorithms. Larger conformational changes are also present in the known protein structures.2–6 Conformational changes are a common part of an enzymes’ catalytic cycle.7 For instance, loop or domain closures contribute to isolate the active site from solvent and, in so doing, alter the chemical environment around substrates, or trigger the catalytic event by bringing essential partners together.8–10 Also, allostery is the most common enzyme regulation strategy. Allosteric regulation is entirely based on the possibility of a given protein to coexist in two or more conformations of comparable stability. Binding to ligands (allosteric regulators), or simply protein concentration, or crowding, may switch stabilities among conformations and trigger the shape transition. Additionally, some features of protein function can be understood only when dynamic properties are taken into account. For instance, diffusion of small substrates through heme-dependent enzyme molecules requires the transient appearance of channels in the protein structure.11–18 Also, cavities have transient phenomena that in some cases can only be revealed or analyzed following its dynamics.19,20
 | Figure 1 Structure variability within a protein family. |
In the case of nucleic acids, conformational changes are even more complex. Standard B-DNA has a relatively simple structure in comparison with protein or complex RNAs; however, it is an extremely plastic molecule that undergoes large conformational changes to adapt to its interaction partners. Binding of transcription factors to DNA, for example, is not only dependent on DNA sequence recognition, but also a direct consequence of the ability of the DNA molecule to adapt to the protein surface.21,22
The traditional approach to understand conformation influence on macromolecular function is to cumulate experimental structures covering the conformational space. This has led to the generation of crystal structures for macromolecules in several environments, or macromolecules complexed with different molecules, and contributes to the enormous redundancy seen in the PDB. Examples of this approach are the 87 structures for CK2 homologues (Figure 2A), where a common fold is maintained, and different degrees of conformational variation are clearly visible, especially in loop regions. A single experiment could generate conformational ensembles as those taken from nuclear magnetic resonance experiments (Figure 2B). In the latter case, the source of the variability found is rather a consequence of the lack of experimental data in some specific regions of the structure. Indeed, the study of PDB as source for molecular flexibility has been exploited in some extent.18,23–26 Taking such “experimental ensembles” a partial view of the macromolecule flexibility can be obtained, although PDB composition is necessarily biased. Theoretical techniques appear as the most convenient way to obtain a picture of macromolecular dynamic properties. Recent advances in the performance of simulation algorithms, including specific strategies to increase the conformational sampling, have popularized the concept of “conformational ensemble”, as the alternative to the analysis of PDB’s single structures. Ensembles can be analyzed to derive thermodynamic properties of the system, like entropy or free energy.27 If properly built, ensembles can also be used to reconstruct complex conformational transitions or even folding events.27–29 Ensembles are also a better reference to reproduce experimental results, as experiments measure averages of properties over a real ensemble.30,31
 | Figure 2 Experimental ensembles. |
Molecular dynamics simulation
Molecular dynamics (MD) simulation, first developed in the late 70s,32,33 has advanced from simulating several hundreds of atoms to systems with biological relevance, including entire proteins in solution with explicit solvent representations, membrane embedded proteins, or large macromolecular complexes like nucleosomes34,35 or ribosomes.36,37 Simulation of systems having ~50,000–100,000 atoms are now routine, and simulations of approximately 500,000 atoms are common when the appropriate computer facilities are available. This remarkable improvement is in large part a consequence of the use of high performance computing (HPC), and the simplicity of the basic MD algorithm (Figure 3). An initial model of the system is obtained from either experimental structures or comparative modeling data. The simulated system could be represented at different levels of detail. Atomistic representation is the one that leads to the best reproduction of the actual systems. However, coarse-grained representations are becoming very popular when large systems or long simulations are required (see Orozco et al38 for a review of such strategies). Solvent representation is a key issue in system definition. Several approaches have been assayed39–47 but, again, the most effective is the simplest one, the explicit representation of solvent molecules, although at the expense of increasing the size of the simulated systems. Explicit solvent is able to recover most of the solvation effects of real solvent including those from entropic origin like the hydrophobic effect. Once the system is built, forces acting on every atom are obtained by deriving equations, the force-fields, where potential energy is deduced from the molecular structure.48–53 Force-fields are complex equations, but they are easy to calculate. The simplicity of the force-field representation of molecular features: springs for bond length and angles, periodic functions for bond rotations and Lennard–Jones potentials, and the Coulomb’s law for van der Waals and electrostatic interactions, respectively, assures that energy and force calculations are extremely fast even for large systems. Force-fields currently used in atomistic molecular simulations differ in the way they are parameterized. Parameters are not necessarily interchangeable, and not all force-fields allow to represent all molecule types, but simulations conducted using modern force-fields are normally equivalent.54,55 Once the forces acting on individual atoms are obtained, classical Newton’s law of motion is used to calculate accelerations and velocities and to update the atom positions. As integration of movement is done numerically, to avoid instability, a time step shorter than the fastest movements in the molecule should be used. This ranks normally between 1 and 2 fs for atomistic simulations, and is the major bottleneck of the simulation procedure. Microsecond-long simulations, barely scratching the time scales of biological processes, require iterating over this calculation cycle 109 times. This is one of the strengths of coarse-grained strategies. As a more simplified representation of the system is used, much larger time steps are possible, and therefore the effective length of the simulations is dramatically extended. Of course, this can be obtained at the expense of the accuracy of the simulation ensemble. Algorithmic advances, that include fine-tuning of energy calculations, parallelization, or the use of graphical processing units (GPUs), have largely improved the performance of MD simulations.
 | Figure 3 Molecular dynamics basic algorithm. |
The present generation of computers takes benefit of parallelism and accelerators to speed-up the process. The most popular simulation codes (AMBER,56 CHARMM,57 GROMACS,58 or NAMD59) have long been compatible with the messaging passing interface (MPI). When a large number of computer cores can be used simultaneously, MPI can greatly reduce the computation time. To benefit the locality of interactions, the general strategy is to distribute the system to simulate among processors. This strategy is called spatial decomposition. Only a small fragment of the system has to be simulated in each processor. The most efficient division is not based in the list of particles, but in their position in space. Each processor deals with a region of space irrespective of which particles are present there. Communication between processors is also reduced, as only those simulating neighboring regions have to share information (see Larsson et al60 for a review). As stated, the use of accelerators, mainly GPU, has become a major breakthrough in simulation codes. Originally designed to handle computer graphics, GPUs have evolved into general-purpose, fully programmable, high-performance processors and represent a major technical improvement to perform atomistic MD. Most major MD codes have already been prepared for GPUs, and even MD codes written specifically to be used on GPUs have been developed (ACEMD61). Simulation on GPUs alone or combined with MPI is, at present, the default strategy for high-throughput MD simulations. Remarkably, while simulations have been the most popular use of HPC in life sciences, the increasing power and sophistication of GPUs is leading to a greater use of personal workstations with a comparable performance.
Strategies to improve ensemble generation
Pure computational brute force, just making longer simulations, is not enough to extend the conformational sampling in biomolecular systems. The complex shape of the free energy landscape makes most of the simulations explore just a small region around the energy minimum closest to the initial conformation. With the availability of the present HPC systems, an obvious strategy is to perform a series of parallel simulations with several starting conformations. Although this could be efficient, it requires a specific knowledge of the system to simulate, and cannot be applied as a general strategy. This approach is particularly useful when several crystal structures are available (for instance in the case of allosterically regulated enzymes). A second problem that appears when collections of parallel simulations are calculated, is the generation of a usable ensemble out of the trajectories obtained. Recently the Markov state model (MSM) theory has been used to this end.62–65 MSM theory discretizes the conformational ensemble in a collection of states, and constructs a matrix with the transition probabilities among them. The analysis of such a matrix would allow reconstruction of the global behavior of the system. Since the transition rates converge more rapidly than the population of the involved states, this approach has the advantage that the collection of simulations is not required to be especially long. This approach has been used mainly in the study of folding processes,28,66 but also in the kinetic characterization of the formation of ligand–protein complexes.67 Other approaches have been designed to increase the sampling space in single simulations, like metadynamics,68–71 for instance, where already visited conformations are penalized; weighted ensembles, where additional simulations are started when new conformational spaces are visited;72 or accelerated MD where energy barriers are artificially reduced.73–75 However, even with a perfect sampling, MD simulations cannot surmount barriers in the energy landscape higher than the total energy added to the system. The obtained ensemble with a single simulation is limited to those states that are accessible at the simulation temperature. Simulations at high temperatures were common in the origins of MD, but they lead to unrealistic trajectories, and hence should be combined to room temperature runs. This approach, called simulated annealing, has been largely replaced by replica exchange methods.76,77 Such methods launch parallel simulations in different conditions. The most common variation is simulation temperature. The sampling ability of the simulation increases with temperature. Higher temperature simulations can surmount energy barriers and explore new regions of the ensemble. Periodically, energies of the different simulations are compared and structures are swapped according to its energy rank. The resulting simulation has sampled a larger conformational space, due to high temperature simulations, and retains the ability to represent the low-temperature states of the system. The main difference with the simulated annealing approach is that a realistic ensemble of the system is obtained and thermodynamic information can be derived from simulations. The idea has been extended to other simulation strategies. Most remarkably, replicas based on differences in the Hamiltonian replica exchange, including alchemical free energy calculations78 or constant-pH simulations,79–83 are becoming popular.
Tools to popularize MD
Preparation for simulation implies the following of a series of operations that are far from being just routine. First, the initial structure comes from the experiment. Expected issues include nonstructured or missing regions or residues, nonstandard ligands, or even structures bearing errors in the interpretation of experimental data. When a single system is simulated, all the effort in the preparation of the system is worth, as it assures the quality of the simulation result. Such setup is usually done manually, with a considerable human effort. A standard procedure to set up a system implies a number of well-known procedures: fixing structure errors; ionization of titrable amino acids; addition of structural water molecules, counter ions, and solvent; and energy minimization and equilibration of the system at the desired temperature. An expert modeler normally carries out these procedures using a set of helper programs. Such an expert has the necessary knowledge to surmount specific problems that may arise. For instance, the workflow used in the MoDEL project84 was programmed to run automatically, but a nonnegligible fraction of over 1,500 proteins prepared failed at some point of the process. With this scenario, for newcomers to MD simulation, even a single system setup could represent an unaffordable problem. Even worse, nonexpert users tend to blindly use default procedures leading easily to artifactual trajectories, which are hard to distinguish from the correct ones. This strongly contributes to the lack of popularity of biomolecular simulations among the bioinformatics or the biochemical community. MD simulations have been restricted to those research groups bearing the necessary expertise. Solving this issue requires an automatic setup of the simulation system. We would be looking for a clever black box for the nonexperts, but also for a robust software suite that can account for a large set of unrelated protein structures. All major MD codes56–59 come with a set of accompanying programs, which perform most steps of the preparation. Additionally, a number of initiatives, combining those tools with a user-friendly interface, have come into the scene to address this problem. CHARMM-Gui85 and CHARMMing,86 for CHARMM, or Guimacs,87 Gromita,88 and jSimMacs,89 for GROMACS, provide automatic setup functionality. VMD90 provides a number of plug-ins that allow to launch simulations with NAMD. Most of these tools provide a friendly environment to prepare systems for simulation without the need of a deep knowledge of the underlying operations, thus facilitating the access to the field for the newcomers. Unfortunately, due to the lack of a standard for the representation of molecular simulation data, most helper applications are restricted to a single MD package, and data is not easily interchangeable. Besides, although most use some kind of embedded scripting language, automation of procedures is not a straightforward task. Lessons learned in the preparation of the MoDEL database, by our group, leaded to the generation of a new set of tools, MDMoby and MDWeb91 that try to cover both aspects of the problem. On one hand, MDMoby provides a full set of web services, covering all setup, simulation, and analysis operations. The modular nature of such collection of web services allows incorporating them as a tool kit to the design of complex setup protocols and to run them programmatically. In turn, MDWeb, a web-based interface, provides a user-friendly bench where user can check for the quality of the input structure, tailor their own setup protocols, or use a collection of predefined ones.
Application: understanding allostery
Most regulation phenomena in proteins are explained within conformational transitions. The concept of allostery that translates conformational dynamics in functional implications has been analyzed since the early times of protein biochemistry.92–94 Conformation shift involved in allostery spans from small rearrangements to large quaternary shifts as those originally accounted by the Monod model. In any case, there is a general agreement that conformational shifts involved in allosteric transitions are simple in terms of collective movements.95 For this reason, molecular simulations could be a natural tool to understand allostery. However, the ability of free atomistic simulation algorithms to follow a complete transition path is limited. Most of the traditional reports of simulations in this field use simplified frameworks, like discrete MD96 or Go-Models,97 or even popular nonsimulation equivalents like elastic network models,98–101 and seek to find the transition path between known experimental structures. With full atom representations, it is usual to trick the algorithm by using targeted,102–104 or supervised MD105 where the simulation is artificially driven to the desired conformation. In this case, the analysis of the path could give insight into the energetics and details of the allosteric transition. For those cases where allosteric regulation is known to occur, but one of the ends is unknown, long simulations (alone106,107 or with enhanced conformational sampling) are required.108,109 The direct use of conformational ensembles without any conditioning is still out of the routine possibilities of present MD simulations; however, specific cases with well-defined collective motions could be feasible. Figure 4 shows an example where only 50 ns of simulation allowed for a conformational shift in Bacillus stearothermophilus lactate dehydrogenase (PDB code: 1LDN). This enzyme is known to exist in two states: a fully active, tetrameric state and a less active, dimeric one. Fructose-1,6-bisphosphate is a known allosteric regulator that allows the tetrameric state to be formed.110,111 The construction of stable dimeric state by site-directed mutagenesis allowed for the analysis of the less active form in more detail and confirmed that a significant conformational shift occurs when the tetrameric state is formed.112,113 The simulation of a B. stearothermophilus lactate dehydrogenase dimer protein starting with the conformation of the tetrameric one, as obtained from the experimental structure, reveals a significant intersubunit movement compatible with the experimental behavior of the enzyme (Figure 4). In this case, no computational bias was introduced; however, protein setup was done mimicking experimental conditions where the conformational shift is known to occur. The use of experimental restrains (but not necessarily the target structure) is being exploited to guide the simulation.31,114 Alternatively, free long simulations may be replaced by shorter simulations and analyzed with MSMs to provide quantitative insight.63–65 However target structure should be at some point explored to allow reconstructing the full process. The power of molecular simulations to uncover allosteric regulations is not in any doubt; however, there is still a long way until it could be routinely applied to all cases.
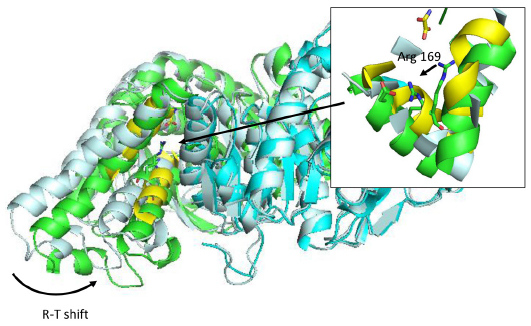 | Figure 4 R-T transition on Bacillus stearothermophilus lactate dehydrogenase after 50 ns simulation in explicit solvent. |
Application: molecular docking and drug design
One of the most practical application of the concept of molecular recognition are docking strategies, either small molecule or protein docking. To understand how a ligand, typically a substrate or a regulator, binds to its macromolecular counterpart is a key issue in the understanding of function itself, and it is the basis of structurally driven drug design. The recognition process is by nature dynamic.115 Molecules are flexible entities, and the recognition process itself implies structural rearrangements, and this shape adjustment is part of the binding process not only from the structural point of view, but also from the energetics. Although this is a generally well-accepted idea, docking algorithms are far from considering dynamic effects as a routine. Most docking or virtual screening codes work on rigid structures as obtained from the PDB. Figure 5 shows a traditional cross-docking experiment where a collection of acetylcholinesterase ligands are docked back in the same set of receptor structures. Protein structures correspond to the ones shown in Figure 1. In this experiment, all receptor structures correspond to the same protein, but crystallized with a different ligand; and all ligands are known to bind the receptor in the same place and pose. In these conditions, the experiment just measures the impact of small receptor rearrangements caused by ligand binding, on the docking efficiency. The usual result, as the one shown in Figure 5, is that even though the protein does not change, a different PDB structure implies poorer docking results. Even docking of a ligand back on its original PDB structure (diagonal results in Figure 5) tends to fail due to the usual overcompression of structures derived from X-ray crystallography. This problem is especially relevant in protein–protein docking where considerable differences are found between bound and unbound structures. For ligand-docking methods, ligand flexibility could be largely recovered by using conformer families.116–118 In the case of protein flexibility, solutions are not so extended. Most of them use algorithms to select from a limited alternative set of protein conformations, either precomputed or simulated.119–121 It is also possible to introduce flexibility a posteriori as a refinement process.122 It is still not clear whether structural adjustment comes from selection of available conformations or it is induced by the binding process itself,123,124 but in any case the use of structural ensembles instead of single structures can be expected to improve the binding prediction.125–127 The concept of “ensemble docking” usually requires the selection of a representative set of snapshots coming from a simulation and uses them as targets in normal docking procedures.128–130 Integration of docking and simulation in a single calculation is less popular, but some examples have been reported.16,67,121,131
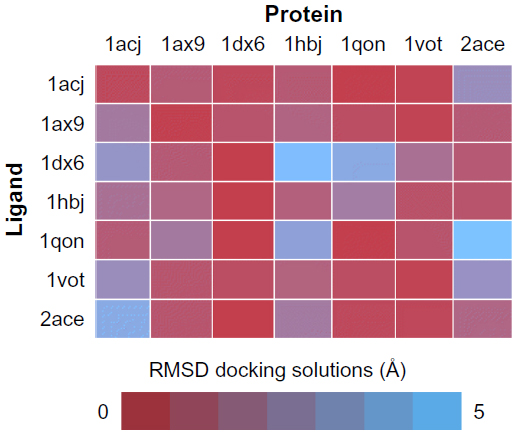 | Figure 5 Cross-docking experiment with selected acetylcholinesterase structures from PDB. |
The use of simulations for the improvement of virtual screening or docking processes has a clear advantage. However, due to the speed requirements of docking, most methods based on traditional atomistic simulations are too slow to be considered, when used in a real scenario. Coarse-grained methods or any sort of accelerated MD could be a way to take benefit of simulation in a near future.
Application: refining structure predictions
Structure prediction has been one of the most ancient problems addressed in structural bioinformatics. MD, including the longest simulations performed, has been extensively used for ab initio protein structure prediction,132–136 aiming to simulate protein folding from scratch, although this is not the preferred strategy to obtain theoretical model of protein structure. Instead, template-based modeling is the most efficient technique.137–144 In template-based modeling, one or several 3D structures of protein showing a reasonable degree of similarity to the protein of interest are taken as templates. Irrespective of the modeling algorithm, the end result is a model bearing the new amino acid sequence and a structure averaging the used templates. In most cases, the last step of the prediction procedure implies relaxation of the structure using normally molecular mechanics. In others, restrained simulations are used throughout all the process.144 The use of MD simulations looks like an obvious step in refining such models.145 Simulation would allow the structure to adapt to the new sequence, and in theory give a more realistic model. Although this point is reasonable as a concept, MD simulations require systems to be close to their equilibrium (native) conformation. Otherwise, significant and difficult to detect artifacts may occur. Critical assessment of protein structure prediction contests,139 where prediction algorithms face problems with known but nonpublic 3D structures, provide an excellent dataset to test this issue. Applying different MD approaches to the refinement of such predictions has led to a number of conclusions. The most naive approach, a single simulation starting from the predicted conformation, tends to deviate significantly from the desired structure.145–147 Apparently, inaccuracies in the model have a large impact in the quality of the simulation results. Instead, results clearly indicate that deviation from the original structure is directly correlated with the loss of quality of the model. A second conclusion is that the ensemble of structures taken from the simulations is a closer representation of the target structure, thus indicating that the native and original structures both lie within the conformational space of the simulation.146
Conclusion
MD simulations have already more than 40 years of history. However, it was not until the recent years that MD has achieved time scales that begin to be compatible with biological processes. At present, when routine simulations are approaching the microsecond scale, conformational changes, or ligand binding can be effectively simulated. The improvement of the computational equipment, especially the use of GPUs, and the improvements made in the optimization of MD algorithms, including coarse-grained ones, allow us to move from the analysis of single structures, the basis of the molecular modeling as we know it, to the analysis of conformational ensembles. Conformational ensembles are a much better representation of real macromolecules, as they account for flexibility and dynamic properties (including all thermodynamic information) and ease the match with experimental results. Although the shift in concept is clear, and the technology is coming along, there is still a long way until biomolecular simulations, the generation of conformational ensembles, would become a routine. Tools exist that make the setup of a macromolecular system much easier, and even allow the nonexperts to enter the simulation world. However, lack of representation standards, much less optimized analysis tools, and even the difficulties in simply storing and transmitting the huge amount of trajectory data that is generated are still issues that remain to be solved. In any case, MD is already a valuable tool in helping to understand biology.
Disclosure
The authors report no conflicts of interest in this work.
References
Berman HM, Battistuz T, Bhat TN, et al. The protein data bank. Acta Crystallogr D Biol Cryst. 2002;58:899–907. | |
Flores S, Echols N, Milburn D, et al. The database of macromolecular motions: new features added at the decade mark. Nucleic Acids Res. 2006;34(Database issue):D296–D301. | |
Bhabha G, Biel JT, Fraser JS. Keep on moving: discovering and perturbing the conformational dynamics of enzymes. Acc Chem Res. 2015; 48(2):423–430. | |
Cockrell GM, Kantrowitz ER. ViewMotions rainbow: a new method to illustrate molecular motions in proteins. J Mol Graph Model. 2013;40:48–53. | |
Gerstein M, Krebs W. A database of macromolecular motions. Nucleic Acids Res. 1998;26(18):4280–4290. | |
Goh CS, Milburn D, Gerstein M. Conformational changes associated with protein-protein interactions. Curr Opin Struct Biol. 2004; 14(1):104–109. | |
Kokkinidis M, Glykos NM, Fadouloglou VE. Protein flexibility and enzymatic catalysis. Adv Protein Chem Struct Biol. 2012;87:181–218. | |
Stevens RC, Lipscomb WN. Allosteric control of quaternary states in E. coli aspartate transcarbamylase. Biochem Biophys Res Commun. 1990;171(3):1312–1318. | |
Shinoda T, Arai K, Shigematsu-Iida M, et al. Distinct conformation-mediated functions of an active site loop in the catalytic reactions of NAD-dependent D-lactate dehydrogenase and formate dehydrogenase. J Biol Chem. 2005;280(17):17068–17075. | |
Karplus M. Role of conformation transitions in adenylate kinase. Proc Natl Acad Sci U S A. 2010;107(17):E71; author reply E72. | |
Kalko SG, Gelpi JL, Fita I, Orozco M. Theoretical study of the mechanisms of substrate recognition by catalase. J Am Chem Soc. 2001;123(39):9665–9672. | |
Jakopitsch C, Droghetti E, Schmuckenschlager F, Furtmuller PG, Smulevich G, Obinger C. Role of the main access channel of catalase-peroxidase in catalysis. J Biol Chem. 2005;280(51):42411–42422. | |
Bidon-Chanal A, Marti MA, Crespo A, et al. Ligand-induced dynamical regulation of NO conversion in Mycobacterium tuberculosis truncated hemoglobin-N. Proteins. 2006;64(2):457–464. | |
Hara I, Ichise N, Kojima K, et al. Relationship between the size of the bottleneck 15 A from iron in the main channel and the reactivity of catalase corresponding to the molecular size of substrates. Biochemistry. 2007;46(1):11–22. | |
Bidon-Chanal A, Marti MA, Estrin DA, Luque FJ. Dynamical regulation of ligand migration by a gate-opening molecular switch in truncated hemoglobin-N from Mycobacterium tuberculosis. J Am Chem Soc. 2007;129(21):6782–6788. | |
Guallar V, Lu C, Borrelli K, Egawa T, Yeh SR. Ligand migration in the truncated hemoglobin-II from Mycobacterium tuberculosis: the role of G8 tryptophan. J Biol Chem. 2009;284(5):3106–3116. | |
Daigle R, Guertin M, Lague P. Structural characterization of the tunnels of Mycobacterium tuberculosis truncated hemoglobin N from molecular dynamics simulations. Proteins. 2009;75(3):735–747. | |
Ahalawat N, Murarka RK. Conformational changes and allosteric communications in human serum albumin due to ligand binding. J Biomol Struct Dyn. 2015:1–13. | |
Barbany M, Meyer T, Hospital A, et al. Molecular dynamics study of naturally existing cavity couplings in proteins. PLoS One. 2015;10(3):e0119978. | |
Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc Natl Acad Sci U S A. 2015;112(9):2734–2739. | |
Bouvier B, Zakrzewska K, Lavery R. Protein-DNA recognition triggered by a DNA conformational switch. Angew Chem Int Ed Engl. 2011;50(29):6516–6518. | |
Zakrzewska K, Lavery R. Towards a molecular view of transcriptional control. Curr Opin Struct Biol. 2012;22(2):160–167. | |
Perez A, Noy A, Lankas F, Luque FJ, Orozco M. The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 2004;32(20):6144–6151. | |
Leo-Macias A, Lopez-Romero P, Lupyan D, Zerbino D, Ortiz AR. An analysis of core deformations in protein superfamilies. Biophys J. 2005;88(2):1291–1299. | |
Velazquez-Muriel JA, Rueda M, Cuesta I, Pascual-Montano A, Orozco M, Carazo JM. Comparison of molecular dynamics and superfamily spaces of protein domain deformation. BMC Struct Biol. 2009;9:6. | |
Micheletti C. Comparing proteins by their internal dynamics: exploring structure-function relationships beyond static structural alignments. Phys Life Rev. 2013;10(1):1–26. | |
Baxa MC, Haddadian EJ, Jumper JM, Freed KF, Sosnick TR. Loss of conformational entropy in protein folding calculated using realistic ensembles and its implications for NMR-based calculations. Proc Natl Acad Sci U S A. 2014;111(43):15396–15401. | |
Noe F, Schuette C, Vanden-Eijnden E, Reich L, Weikl TR. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc Natl Acad Sci U S A. 2009;106(45):19011–19016. | |
Naganathan AN, Orozco M. The native ensemble and folding of a protein molten-globule: functional consequence of downhill folding. J Am Chem Soc. 2011;133(31):12154–12161. | |
Candotti M, Esteban-Martin S, Orozco M. Atomistic insights on protein-urea structural organization from MD simulations of a chemically denatured protein ensemble determined by NMR. FEBS J. 2012;279:531–531. | |
Bryn Fenwick R, Esteban-Martin S, Salvatella X. Understanding biomolecular motion, recognition, and allostery by use of conformational ensembles. Eur Biophys J. 2011;40(12):1339–1355. | |
McCammon JA, Gelin BR, Karplus M. Dynamics of folded proteins. Nature. 1977;267(5612):585–590. | |
Warshel A, Levitt M. Theoretical studies of enzymic reactions – dielectric, electrostatic and steric stabilization of carbonium-ion in reaction of lysozyme. J Mol Biol. 1976;103(2):227–249. | |
Roccatano D, Barthel A, Zacharias M. Structural flexibility of the nucleosome core particle at atomic resolution studied by molecular dynamics simulation. Biopolymers. 2007;85(5–6):407–421. | |
Sharma S, Ding F, Dokholyan NV. Multiscale modeling of nucleosome dynamics. Biophys J. 2007;92(5):1457–1470. | |
Tinoco I Jr, Wen JD. Simulation and analysis of single-ribosome translation. Phys Biol. 2009;6(2):025006. | |
Brandman R, Brandman Y, Pande VS. A-site residues move independently from P-site residues in all-atom molecular dynamics simulations of the 70S bacterial ribosome. PLoS One. 2012; 7(1):e29377. | |
Orozco M, Orellana L, Hospital A, et al. Coarse-grained representation of protein flexibility. Foundations, successes, and shortcomings. In: Christov C, editor. Advances in Proteins Chemistry and Structural Biology, Vol 85: Computational Chemistry Methods in Structural Biology. Waltham, MA: Academic Press; 2011:183–215. | |
Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35(2):133–152. | |
Roux B, Simonson T. Implicit solvent models. Biophys Chem. 1999;78(1–2):1–20. | |
Orozco M, Luque FJ. Theoretical methods for the description of the solvent effect in biomolecular systems. Chem Rev. 2000; 100(11):4187–4226. | |
Bashford D, Case DA. Generalized born models of macromolecular solvation effects. Ann Rev Phys Chem. 2000;51:129–152. | |
Haberthuer U, Caflisch A. FACTS: fast analytical continuum treatment of solvation. J Comput Chem. 2008;29(5):701–715. | |
Luchko T, Gusarov S, Roe DR, et al. Three-dimensional molecular theory of solvation coupled with molecular dynamics in Amber. J Chem Theory Comput. 2010;6(3):607–624. | |
Vorobjev YN. Advances in implicit models of water solvent to compute conformational free energy and molecular dynamics of proteins at constant pH. Adv Protein Chem Struct Biol. 2011;85:281–322. | |
Kleinjung J, Fraternali F. Design and application of implicit solvent models in biomolecular simulations. Curr Opin Struct Biol. 2014;25:126–134. | |
Anandakrishnan R, Drozdetski A, Walker RC, Onufriev AV. Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations. Biophys J. 2015;108(5):1153–1164. | |
Hermans J, Berendsen HJC, Vangunsteren WF, Postma JPM. A consistent empirical potential for water-protein interactions. Biopolymers. 1984;23(8):1513–1518. | |
Mackerell AD, Wiorkiewiczkuczera J, Karplus M. An all-atom empirical energy function for the simulation of nucleic-acids. J Am Chem Soc. 1995;117(48):11946–11975. | |
Ott KH, Meyer B. Parametrization of GROMOS force field for oligosaccharides and assessment of efficiency of molecular dynamics simulations. J Comput Chem. 1996;17(8):1068–1084. | |
MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. | |
Cornell WD, Cieplak P, Bayly CI, et al. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc. 1995;117(19):5179–5197. | |
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105(28):6474–6487. | |
Rueda M, Ferrer-Costa C, Meyer T, et al. A consensus view of protein dynamics. Proc Natl Acad Sci U S A. 2007;104(3):796–801. | |
Perez A, Lankas F, Luque FJ, Orozco M. Towards a molecular dynamics consensus view of B-DNA flexibility. Nucleic Acids Res. 2008; 36(7):2379–2394. | |
Case DA, Darden TA, Cheatham TE I, et al. AMBER 12. San Francisco, CA: University of California; 2012. | |
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. | |
Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. | |
Nelson MT, Humphrey W, Gursoy A, et al. NAMD: a parallel, object oriented molecular dynamics program. Int J Supercomput Appl High Perform Comput. 1996;10(4):251–268. | |
Larsson P, Hess B, Lindahl E. Algorithm improvements for molecular dynamics simulations. Wiley Interdiscip Rev Comput Mol Sci. 2011; 1(1):93–108. | |
Harvey MJ, Giupponi G, De Fabritiis G. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput. 2009;5(6):1632–1639. | |
Pan AC, Roux B. Building Markov state models along pathways to determine free energies and rates of transitions. J Chem Phys. 2008; 129(6):064107. | |
Bowman GR, Huang X, Pande VS. Using generalized ensemble simulations and Markov state models to identify conformational states. Methods. 2009;49(2):197–201. | |
Chodera JD, Noe F. Markov state models of biomolecular conformational dynamics. Curr Opin Struct Biol. 2014;25:135–144. | |
Da LT, Sheong FK, Silva DA, Huang X. Application of Markov state models to simulate long timescale dynamics of biological macromolecules. Adv Exp Med Biol. 2014;805:29–66. | |
Voelz VA, Bowman GR, Beauchamp K, Pande VS. Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1-39). J Am Chem Soc. 2010;132(5):1526–1528. | |
Buch I, Giorgino T, De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2011;108(25):10184–10189. | |
Micheletti C, Laio A, Parrinello M. Reconstructing the density of states by history-dependent metadynamics. Phys Rev Lett. 2004; 92(17):170601. | |
Laio A, Rodriguez-Fortea A, Gervasio FL, Ceccarelli M, Parrinello M. Assessing the accuracy of metadynamics. J Phys Chem B. 2005; 109(14):6714–6721. | |
Raiteri P, Laio A, Gervasio FL, Micheletti C, Parrinello M. Efficient reconstruction of complex free energy landscapes by multiple walkers metadynamics. J Phys Chem B. 2006;110(8):3533–3539. | |
Barducci A, Bonomi M, Parrinello M. Metadynamics. Wiley Interdiscip Rev Comput Mol Sci. 2011;1(5):826–843. | |
Dickson A, Brooks CL. WExplore: hierarchical exploration of high-dimensional spaces using the weighted ensemble algorithm. J Phys Chem B. 2014;118(13):3532–3542. | |
Abdul-Wahid B, Feng H, Rajan D, et al. AWE-WQ: fast-forwarding molecular dynamics using the accelerated weighted ensemble. J Chem Inf Model. 2014;54(10):3033–3043. | |
Doshi U, Hamelberg D. Towards fast, rigorous and efficient conformational sampling of biomolecules: advances in accelerated molecular dynamics. Biochim Biophys Acta. 2015;1850(5):878–888. | |
Miao Y, Feixas F, Eun C, McCammon JA. Accelerated molecular dynamics simulations of protein folding. J Comput Chem. 2015; 36(20):1536–1549. | |
Nymeyer H, Gnanakaran S, Garcia AE. Atomic simulations of protein folding, using the replica exchange algorithm. Methods Enzymol. 2004;383:119–149. | |
Kubitzki MB, de Groot BL. Molecular dynamics simulations using temperature-enhanced essential dynamics replica exchange. Biophys J. 2007;92(12):4262–4270. | |
Meng Y, Dashti DS, Roitberg AE. Computing alchemical free energy differences with Hamiltonian replica exchange molecular dynamics (H-REMD) simulations. J Chem Theory Comput. 2011; 7(9):2721–2727. | |
Burgi R, Kollman PA, Van Gunsteren WF. Simulating proteins at constant pH: an approach combining molecular dynamics and Monte Carlo simulation. Proteins. 2002;47(4):469–480. | |
Stern HA. Molecular simulation with variable protonation states at constant pH. J Chem Phys. 2007;126(16):164112. | |
Baptista AM, Martel PJ, Petersen SB. Simulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titration. Proteins. 1997;27(4):523–544. | |
Goh GB, Hulbert BS, Zhou H, Brooks CL. Constant pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins. 2014;82(7):1319–1331. | |
Itoh SG, Damjanovic A, Brooks BR. pH replica-exchange method based on discrete protonation states. Proteins. 2011;79(12):3420–3436. | |
Meyer T, D’Abramo M, Hospital A, et al. MoDEL (molecular dynamics extended library): a database of atomistic molecular dynamics trajectories. Structure. 2010;18(11):1399–1409. | |
Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. | |
Miller BT, Singh RP, Klauda JB, Hodoscek M, Brooks BR, Woodcock HL 3rd. CHARMMing: a new, flexible web portal for CHARMM. J Chem Inf Model. 2008;48(9):1920–1929. | |
Kota P. GUIMACS – a Java based front end for GROMACS. In Silico Biol. 2007;7(1):95–99. | |
Sellis D, Vlachakis D, Vlassi M. Gromita: a fully integrated graphical user interface to gromacs 4. Bioinform Biol Insights. 2009;3:99–102. | |
Roopra S, Knapp B, Omasits U, Schreiner W. jSimMacs for GROMACS: a Java application for advanced molecular dynamics simulations with remote access capability. J Chem Inf Model. 2009; 49(10):2412–2417. | |
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14(1):33–38. | |
Hospital A, Andrio P, Fenollosa C, Cicin-Sain D, Orozco M, Gelpi JL. MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. Bioinformatics. 2012;28(9):1278–1279. | |
Monod J, Changeux JP, Jacob F. Allosteric proteins and cellular control systems. J Mol Biol. 1963;6(4):306–329. | |
Monod J, Wyman J, Changeux JP. On nature of allosteric transitions – a plausible model. J Mol Biol. 1965;12(1):88–118. | |
Koshland DE, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5(1):365–385. | |
Henzler-Wildman KA, Thai V, Lei M, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450(7171):838–844. | |
Sfriso P, Emperador A, Orellana L, Hospital A, Lluis Gelpi J, Orozco M. Finding conformational transition pathways from discrete molecular dynamics simulations. J Chem Theory Comput. 2012; 8(11):4707–4718. | |
Sfriso P, Hospital A, Emperador A, Orozco M. Exploration of conformational transition pathways from coarse-grained simulations. Bioinformatics. 2013;29(16):1980–1986. | |
Kim MK, Jernigan RL, Chirikjian GS. Efficient generation of feasible pathways for protein conformational transitions. Biophys J. 2002;83(3):1620–1630. | |
Bahar I, Rader AJ. Coarse-grained normal mode analysis in structural biology. Curr Opin Struct Biol. 2005;15(5):586–592. | |
Orellana L, Rueda M, Ferrer-Costa C, Lopez-Blanco JR, Chacon P, Orozco M. Approaching elastic network models to molecular dynamics flexibility. J Chem Theory Comput. 2010;6(9):2910–2923. | |
Orellana L, Orozco M. Understanding protein dynamics with coarse-grained models: from structures to disease. FEBS J. 2012;279:528–528. | |
Schlitter J, Engels M, Kruger P, Jacoby E, Wollmer A. Targeted molecular-dynamics simulation of conformational change – application to the T-R transition in insulin. Mol Simul. 1993;10(2–6):291–308. | |
Kruger P, Verheyden S, Declerck PJ, Engelborghs Y. Extending the capabilities of targeted molecular dynamics: simulation of a large conformational transition in plasminogen activator inhibitor 1. Protein Sci. 2001;10(4):798–808. | |
Perdih A, Kotnik M, Hodoscek M, Solmajer T. Targeted molecular dynamics simulation studies of binding and conformational changes in E. coli MurD. Proteins. 2007;68(1):243–254. | |
Deganutti G, Cuzzolin A, Ciancetta A, Moro S. Understanding allosteric interactions in G protein-coupled receptors using supervised molecular dynamics: a prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg Med Chem. 2015;23(14):4065–4071. | |
Arkhipov A, Shan Y, Das R, et al. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152(3):557–569. | |
Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol. 2009;19(2):120–127. | |
Anderson JS, Mustafi SM, Hernández G, LeMaster DM. Statistical allosteric coupling to the active site indole ring flip equilibria in the FK506-binding domain. Biophys Chem. 2014;192:41–48. | |
Boczek EE, Reefschläger LG, Dehling M, et al. Conformational processing of oncogenic v-Src kinase by the molecular chaperone Hsp90. Proc Natl Acad Sci U S A. 2015;112(25):E3189–E3198. | |
Wigley DB, Muirhead H, Gamblin SJ, Holbrook JJ. Crystallization of a ternary complex of lactate-dehydrogenase from Bacillus stearothermophilus. J Mol Biol. 1988;204(4):1041–1043. | |
Wigley DB, Gamblin SJ, Turkenburg JP, et al. Structure of a ternary complex of an allosteric lactate-dehydrogenase from Bacillus stearothermophilus at 2.5 A resolution. J Mol Biol. 1992;223(1):317–335. | |
Cameron AD, Roper DI, Moreton KM, Muirhead H, Holbrook JJ, Wigley DB. Allosteric activation in Bacillus stearothermophilus lactate-dehydrogenase investigated by an x-ray crystallographic analysis of a mutant designed to prevent tetramerization of the enzyme. J Mol Biol. 1994;238(4):615–625. | |
Jackson RM, Gelpi JL, Cortes A, et al. Construction of a stable dimer of Bacillus stearothermophilus lactate dehydrogenase. Biochemistry. 1992;31(35):8307–8314. | |
Esteban-Martin S, Bryn Fenwick R, Salvatella X. Refinement of ensembles describing unstructured proteins using NMR residual dipolar couplings. J Am Chem Soc. 2010;132(13):4626–4632. | |
Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci U S A. 1958;44(2):98–104. | |
Hawkins PCD, Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model. 2012;52(11):2919–2936. | |
Ishikawa Y. A script for automated 3-dimentional structure generation and conformer search from 2-dimentional chemical drawing. Bioinformation. 2013;9(19):988–992. | |
Klett J, Cortes-Cabrera A, Gil-Redondo R, Gago F, Morreale A. ALFA: Automatic ligand flexibility assignment. J Chem Inf Model. 2014;54(1):314–323. | |
Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. | |
Nabuurs SB, Wagener M, De Vlieg J. A flexible approach to induced fit docking. J Med Chem. 2007;50(26):6507–6518. | |
Borrelli KW, Vitalis A, Alcantara R, Guallar V. PELE: protein energy landscape exploration. A novel Monte Carlo based technique. J Chem Theory Comput. 2005;1(6):1304–1311. | |
Emperador A, Solernou A, Sfriso P, et al. Efficient relaxation of protein-protein interfaces by discrete molecular dynamics simulations. J Chem Theory Comput. 2013;9(2):1222–1229. | |
Csermely P, Palotai R, Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci. 2010;35(10):539–546. | |
Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci U S A. 2005;102(52):18908–18913. | |
Fukunishi Y. Structural ensemble in computational drug screening. Exp Opin Drug Metab Toxicol. 2010;6(7):835–849. | |
Ivetac A, McCammon JA. A molecular dynamics ensemble-based approach for the mapping of druggable binding sites. Methods Mol Biol. 2011;819:3–12. | |
Okamoto Y, Kokubo H, Tanaka T. Ligand docking simulations by generalized-ensemble algorithms. In: KarabenchevaChristova T, editor. Dynamics of Proteins and Nucleic Acids. Vol 92. Waltham, MA: Academic Press; 2013:63–91. | |
Fenollosa C, Oton M, Andrio P, Cortes J, Orozco M, Goni JR. SEABED: Small molEcule activity scanner weB servicE baseD. Bioinformatics. 2015;31(5):773–775. | |
Maria Novoa E, Ribas de Pouplana L, Barril X, Orozco M. Ensemble docking from homology models. J Chem Theory Comput. 2010; 6(8):2547–2557. | |
Osguthorpe DJ, Sherman W, Hagler AT. Exploring protein flexibility: incorporating structural ensembles from crystal structures and simulation into virtual screening protocols. J Phys Chem B. 2012; 116(23):6952–6959. | |
Chaudhuri R, Carrillo O, Laughton CA, Orozco M. Application of drug-perturbed essential dynamics/molecular dynamics (ED/MD) to virtual screening and rational drug design. J Chem Theory Comput. 2012;8(7):2204–2214. | |
Dorn M, E Silva MB, Buriol LS, Lamb LC. Three-dimensional protein structure prediction: methods and computational strategies. Comput Biol Chem. 2014;53:251–276. | |
Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334(6055):517–520. | |
Piana S, Lindorff-Larsen K, Shaw DE. Protein folding kinetics and thermodynamics from atomistic simulation. Proc Natl Acad Sci U S A. 2012;109(44):17845–17850. | |
Piana S, Lindorff-Larsen K, Shaw DE. Atomic-level description of ubiquitin folding. Proc Natl Acad Sci U S A. 2013;110(15):5915–5920. | |
Bonneau R, Baker D. Ab initio protein structure prediction: progress and prospects. Ann Rev Biophys Biomol Struct. 2001;30:173–189. | |
Lance BK, Deane CM, Wood GR. Exploring the potential of template-based modelling. Bioinformatics. 2010;26(15):1849–1856. | |
Joo K, Lee J, Lee S, Seo JH, Lee SJ, Lee J. High accuracy template based modeling by global optimization. Proteins. 2007;69:83–89. | |
Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr Opin Struct Biol. 2005;15(3):285–289. | |
Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010; 5(4):725–738. | |
Zhang Y. Protein structure prediction: when is it useful? Curr Opin Struct Biol. 2009;19(2):145–155. | |
Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19(8):1015–1018. | |
Misura KMS, Baker D. Progress and challenges in high-resolution refinement of protein structure models. Proteins. 2005;59(1):15–29. | |
Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. | |
Raval A, Piana S, Eastwood MP, Dror RO, Shaw DE. Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins. 2012;80(8):2071–2079. | |
Mirjalili V, Feig M. Protein structure refinement through structure selection and averaging from molecular dynamics ensembles. J Chem Theory Comput. 2013;9(2):1294–1303. | |
Chen J, Brooks CL III. Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins. 2007;67(4):922–930. | |
Pronk S, Pall S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.