Back to Journals » OncoTargets and Therapy » Volume 8
Molecular diagnostics of a single drug-resistant multiple myeloma case using targeted next-generation sequencing
Authors Ikeda H, Ishiguro K, Igarashi T, Aoki Y, Hayashi T, Ishida T, Sasaki Y, Tokino T, Shinomura Y
Received 13 April 2015
Accepted for publication 17 August 2015
Published 5 October 2015 Volume 2015:8 Pages 2805—2815
DOI https://doi.org/10.2147/OTT.S86515
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Daniele Santini
Hiroshi Ikeda,1 Kazuya Ishiguro,1 Tetsuyuki Igarashi,1 Yuka Aoki,1 Toshiaki Hayashi,1 Tadao Ishida,1 Yasushi Sasaki,1,2 Takashi Tokino,2 Yasuhisa Shinomura1
1Department of Gastroenterology, Rheumatology and Clinical Immunology, 2Medical Genome Sciences, Research Institute for Frontier Medicine, Sapporo Medical University, Sapporo, Japan
Abstract: A 69-year-old man was diagnosed with IgG λ-type multiple myeloma (MM), Stage II in October 2010. He was treated with one cycle of high-dose dexamethasone. After three cycles of bortezomib, the patient exhibited slow elevations in the free light-chain levels and developed a significant new increase of serum M protein. Bone marrow cytogenetic analysis revealed a complex karyotype characteristic of malignant plasma cells. To better understand the molecular pathogenesis of this patient, we sequenced for mutations in the entire coding regions of 409 cancer-related genes using a semiconductor-based sequencing platform. Sequencing analysis revealed eight nonsynonymous somatic mutations in addition to several copy number variants, including CCND1 and RB1. These alterations may play roles in the pathobiology of this disease. This targeted next-generation sequencing can allow for the prediction of drug resistance and facilitate improvements in the treatment of MM patients.
Keywords: multiple myeloma, drug resistance, genome-wide sequencing, semiconductor sequencer, target therapy
Introduction
Multiple myeloma (MM) is characterized by malignant plasma cell proliferation in the bone marrow (BM) associated with monoclonal protein in the serum and/or urine.1,2 Hematopoietic stem cell transplantation and novel agents such as bortezomib, thalidomide, and lenalidomide have improved the survival of MM patients.3,4 However, most patients eventually relapse even after the achievement of a complete therapeutic response. Improvements in molecular profiling technologies have provided new insight into the basic molecular events underlying the development of MM as well as the mechanisms of anticancer drug resistance. The transition from the long-established one-size-fits-all approach to new strategies based on individual genetic profiles provides an opportunity to transform current diagnostics into individual prognostic and even predictive classifications.
In MM, there are two distinct genetic subtypes based on copy number alterations and translocations. Approximately half of all MM cases are hyperdiploid, which is characterized by multiple trisomies of chromosomes 3, 5, 7, 9 11, 15, 19, and 21 and a lower prevalence of primary translocations involving the immunoglobulin heavy chain (IgH) locus at 14q32.5,6 The remaining cases form the nonhyperdiploid group, and chromosomes 8, 13, 14, and 16 are frequently lost. Nonhyperdiploid myeloma is strongly associated with translocations of IgH alleles with various partner chromosomes. Copy number alterations in chromosomal regions, such as 1q, 6q, 8p, and 16q, occur in both subtypes. Overall, nonhyperdiploid MM is associated with worse survival compared with hyperdiploid MM.
Methods for determining DNA content and, ultimately, ploidy in MM include conventional cytogenetics, fluorescence in situ hybridization (FISH), comparative genomic hybridization, and, recently, massively parallel whole genome sequencing. Because unique mutations have been observed in individual human cancer samples, the identification and characterization of the molecular alterations of individual cancer patients is a critical step toward the development of more effective personalized therapies. For example, next-generation sequencing (NGS) technologies have revolutionized cancer genomics research by providing a comprehensive method of detecting genomic alterations associated with somatic cancer.7–9 In this study, we sequenced all exons of 409 cancer-related genes in matched tumor and normal DNA samples from a multidrug-resistant myeloma patient using a next-generation semiconductor sequencing protocol.
Case report
A 69-year-old male presented in October 2010 with back pain. Physical examination and magnetic resonance imaging revealed large focal lesions in the fourth thoracic vertebra (Figure 1A) and first lumbar vertebra (Figure 1B). Laboratory evaluation revealed a white blood cell count of 2.5×103/μL with no atypical cells, a red blood cell count of 3.67×106/μL, a hemoglobin level of 11.6 g/dL, and a platelet count of 122×106/μL. The serum total protein level was 10.7 g/dL, the albumin level was 3.4 g/dL, the serum β2 microglobulin level was 4.2 mg/dL, and the serum calcium level was 8.9 mg/dL. The concentrations of IgG, IgA, and IgM were 6,284, 34, and 25 mg/dL, respectively. The monoclonal protein IgG was increased, and serum immunofixation revealed the production of IgG with λ light-chain restriction (data not shown). The proliferation of plasma cells (more than 10% among all nucleated cells) was also detected in BM aspirates. When BM biopsy was performed, the infiltration of plasma cells expressing IgG λ monoclonal protein was identified by pathological investigation, and the patient was diagnosed with MM (Stage II according to the International Staging System). Chromosome analysis at this time using conventional Giemsa banding of BM-derived metaphase spreads revealed a normal karyotype (46, XX) in all analyzed cells. The patient was then treated with one cycle of high-dose dexamethasone, followed by three cycles of bortezomib plus dexamethasone. He achieved complete response according to the International Myeloma Working Group uniform response criteria. His symptoms were also significantly improved.
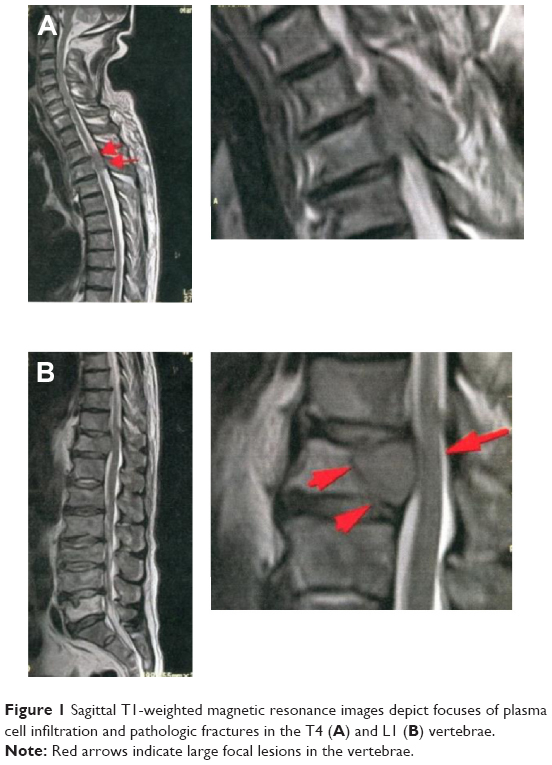 | Figure 1 Sagittal T1-weighted magnetic resonance images depict focuses of plasma cell infiltration and pathologic fractures in the T4 (A) and L1 (B) vertebrae. |
In August 2012, the serum concentrations of IgG and free light chain (FLC) gradually increased, suggesting the worsening of his MM. His complete blood count was as follows: 1.8×103/μL white blood cells, 3.61×106/μL red blood cells, 11.6 g/dL hemoglobin, and 55×106/μL platelets. BM analysis showed complex aberrations often observed in this patient (Figures 2A and 2B) and an elevated plasma cell percentage (59.8%). His karyotype was 39, XY, del(1)(p22p36), −3, −6, der(8)t(6:8)(p11.1:p23), −10, t(11:14)(q13:q32), −12, add(13)(q22), add(16)(q22), −17, add(18)(p11), −19, −20, +mar [4]/46, XY [20]. The chromosomal translocation t(11:14)(q13:q32), which generates the IgH/CCND1 fusion gene, was also identified in our case by FISH (Figure 2C).
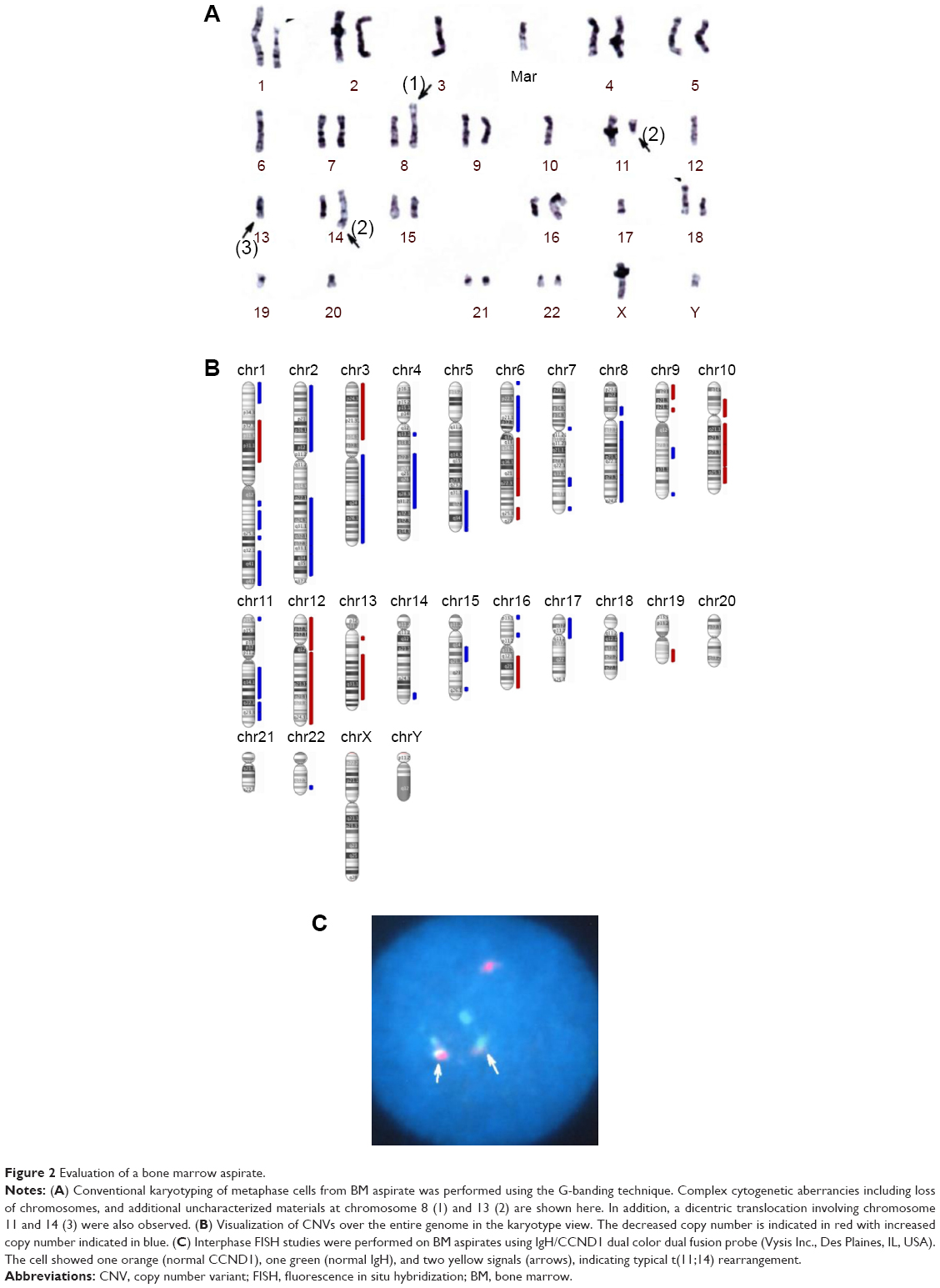 | Figure 2 Evaluation of a bone marrow aspirate. |
The patient was started on a bortezomib, cyclophosphamide, and dexamethasone regimen. No serious complications occurred during the course of the treatment, and a partial response was observed with a decrease in the serum FLC value. After seven cycles of this regimen, however, his condition progressively deteriorated, with increases in serum lambda immunoglobulin light chain and LDH, a deterioration of renal function, and the appearance of circulating plasma cells in the peripheral blood (up to 5% of the total peripheral leukocyte population). He was admitted for combination chemotherapy with combination chemotherapy with bortezomib, cyclophosphamide, lenalidomide, and dexamethasone, but his response was poor (Figure 3), and an increase in myeloma cells was detected by BM biopsy. Unfortunately, the patient has since passed away, and his family did not choose to perform postmortem examination.
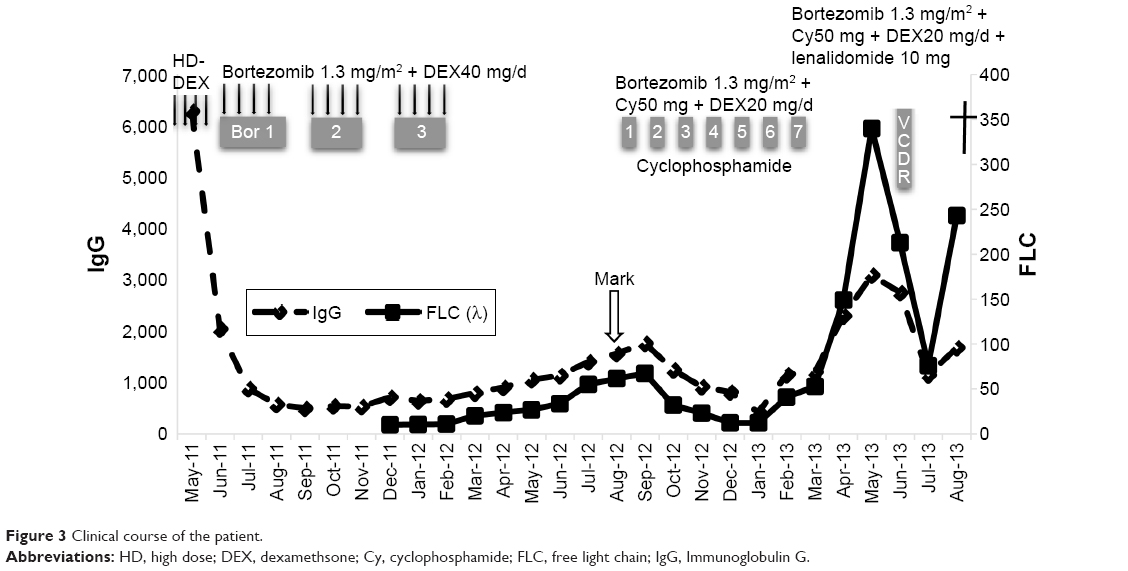 | Figure 3 Clinical course of the patient. |
The patient provided consent for use of his medical record and samples for clinical and research purposes, and the examination was performed in accordance with the Declaration of Helsinki. The sequence study was approved by the Institutional Review Boards of Sapporo Medical University. Retrospectively, to better understand the molecular pathogenesis in this patient, we sequenced 409 cancer-related genes in matched tumor and nontumor DNA samples at relapse in August 2012 using an Ion Torrent PGM (Life Technologies, Carlsbad, CA, USA). DNA was extracted from magnetic bead–enriched BM CD138 positive tumor cells from the patient, and CD138 negative cells were used as matched nontumor cells. DNA (40 ng) was used for multiplex polymerase chain reaction (PCR) amplification with an Ion Ampliseq Comprehensive Cancer Panel (Life Technologies), enabling the targeted coverage of all exons of 409 cancer-related genes frequently cited and mutated (covered regions =95.4% of total). The 15,992 amplicons obtained represented more than 1.2 Mb of target sequence. Library preparation and sequencing with an Ion Torrent PGM was performed as previously described.9 The mean read depths were 125× (tumor) and 152× (normal). Alignment to the human genome build 19 and variant calling were performed by Ion Reporter Software 4.0. Mutations were also validated by conventional Sanger sequencing. We identified eight nonsynonymous somatic mutations (6.49 mutations/Mb; Table 1). We included missense mutations in seven genes (SYNE1, IKBKB, ERBB3, MYH11, CYLD, TP53, and CDH2) and a frameshift mutation in EGFR. Changes in relative copy number were also assessed from the sequencing data, and we identified 133 copy number variant (CNV) regions, including 87 gain and 46 loss regions (Figure 2B and Table S1). Importantly, we found a gain in the copy number of CCND1, a gene encoding cyclin D1. To check the contamination of tumor cells in the CD138 negative subset, we compared CD138 negative DNA of this patient and peripheral blood DNA from two healthy donors. We found single nucleotide variants in the CD138 negative DNA of this patient; however, all variants had previously been reported in the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/) (Tables 2 and 3). Therefore, we can rule out tumor cell contamination in the CD138 negative subset.
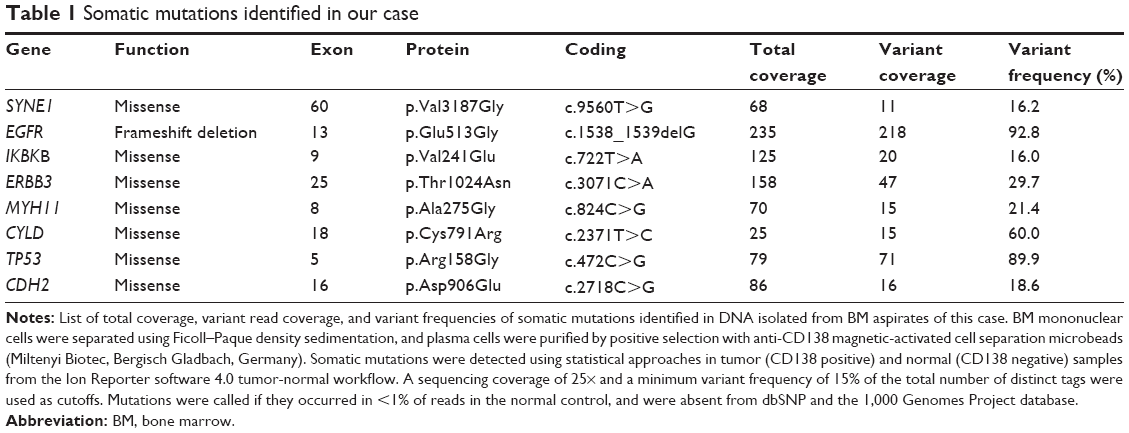 | Table 1 Somatic mutations identified in our case |
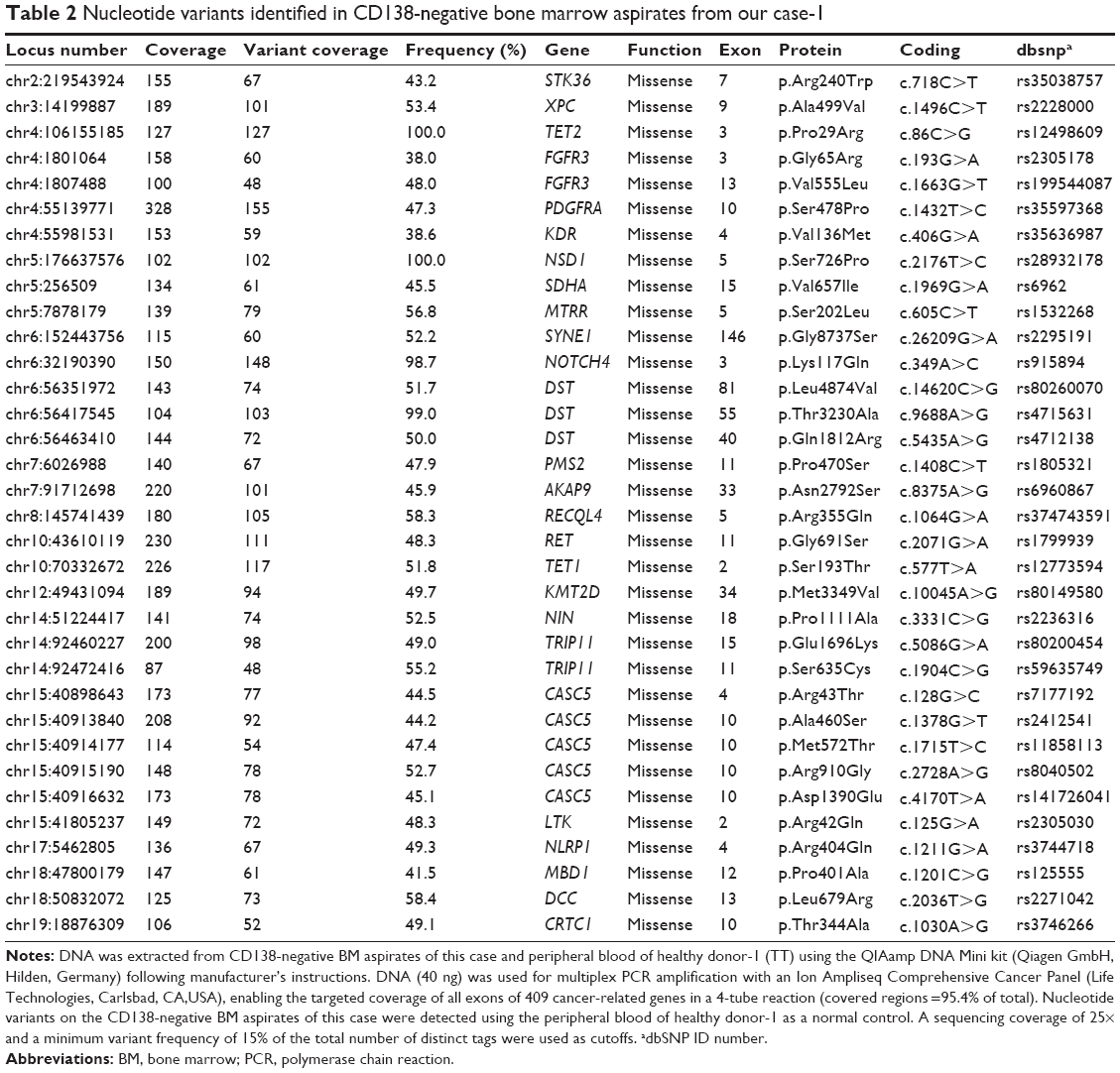 | Table 2 Nucleotide variants identified in CD138-negative bone marrow aspirates from our case-1 |
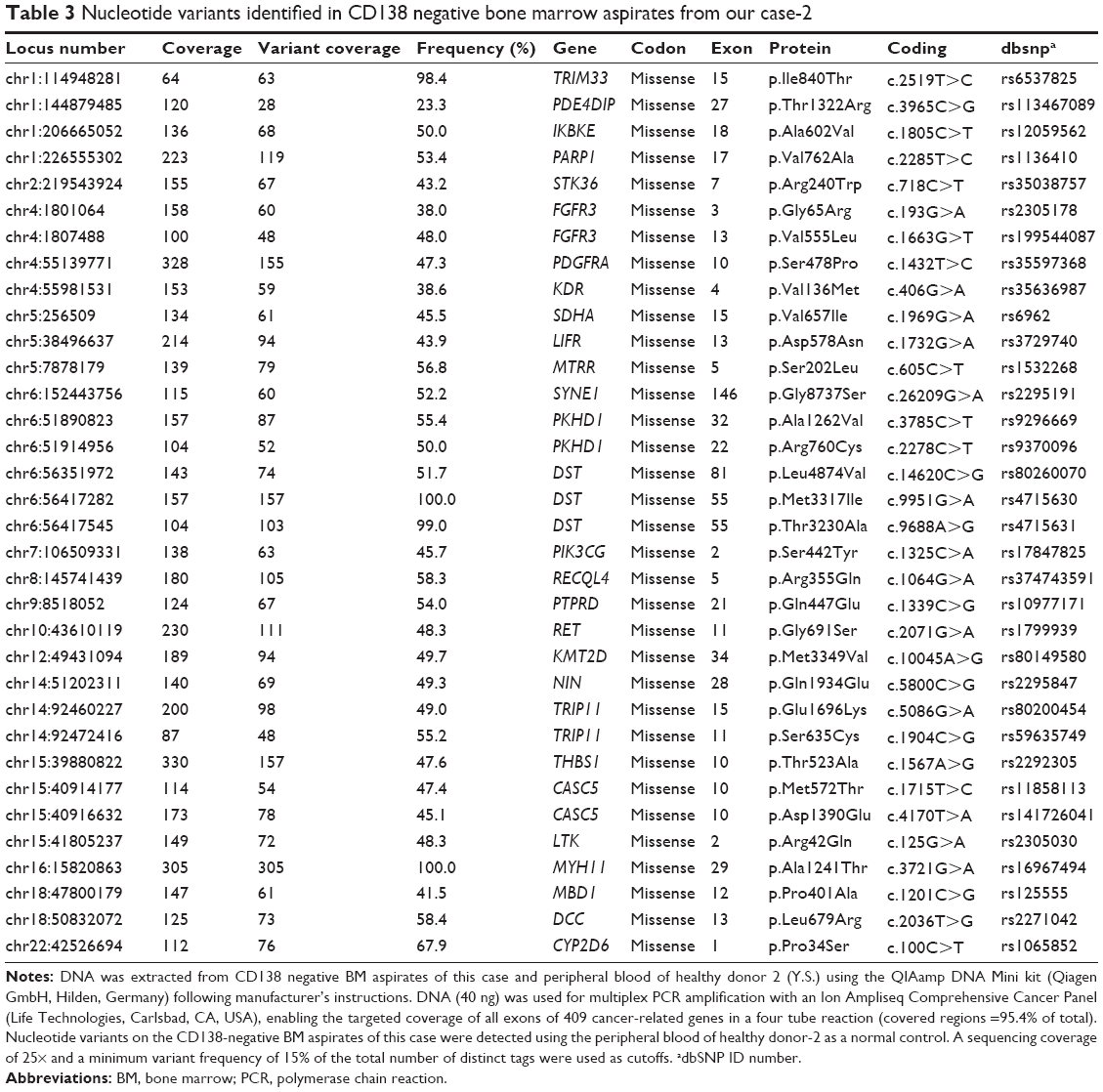 | Table 3 Nucleotide variants identified in CD138 negative bone marrow aspirates from our case-2 |
Discussion
MM is a plasma cell malignancy characterized by a heterogeneous clinical course. Treatments for MM have remarkably improved in recent years, due in part to the introduction of novel therapies such as bortezomib, thalidomide, and lenalidomide. Despite these advancements, the prognosis of patients with relapse and refractory MM remains poor, and novel therapies are needed. Alternatively, the identification of novel targets or signaling pathways regulating myeloma cell proliferation would improve the clinical outcome and survival of refractory MM patients. Several pathways related to drug resistance and cell survival, such as Notch1, Akt, and NF-κB, are activated to protect MM cells from death.10–12 Here, we describe the characterization of genetic abnormalities found in myeloma cells in a patient with refractory MM.
In nonhyperdiploid MM, the IgH gene (14q32) commonly fuses with FGFR3 (4p16), MMSET (4p16.3), CCND3 (6p21), CCND1 (11q13), and MAF (16q23), resulting in the direct or indirect dysregulation of cyclin D.13 Among the nonhyperdiploid myelomas, the hypodiploid subtype (≦44 chromosomes) has the most aggressive clinical phenotype, but the genetic differences between the groups have not been completely defined. Cytogenetic analysis revealed that this patient had a hypodiploid karyotype with 39 chromosomes and complex chromosomal abnormalities, including t(11:14)(q13:q32). CCND1 expression is generally related to copy number aberrations. Although we did not analyze CCND1 mRNA expression, CNV analysis revealed a gain of 11q13–q21, suggesting the presence of cyclin D1 dysregulation.
Several NGS platforms are available for the sequencing of targeted genomic regions to analyze a variety of disease-associated changes, such as point mutations, insertions, deletions, and CNVs. CNV analysis of the sequencing data revealed that this patient had diverse DNA copy number alterations, including large and regional gains and losses (Figure 2B and Table S1). Additionally, we detected eight somatic mutations among 409 cancer-related genes (Table 1). We considered gene sets based on existing insights into the biology of this MM patient. It has been proposed that activation of the NF-kB pathway is important in the pathogenesis of MM, as well as in resistance to chemotherapy. We observed two point mutations and three CNVs affecting NF-κB pathway genes, including IKBKB, CYLD, IKBKE, CD79B, and SYK (Tables 1 and S1). Although additional experiments are required to establish the functional significance of genetic alterations of these genes in MM cells, NF-κB pathway activation may be involved in the molecular pathogenesis of this patient’s disease.
Alterations in the tumor suppressor retinoblastoma (RB) and p53 or their respective pathways are frequently observed in human cancers.14 In MM, a deletion or mutation of p53 (17p13) or RB1 (13q14.2) is considered to be predictive of poor prognosis. We found a monoallelic chromosome 17 deletion and a missense mutation in the DNA-binding domain of p53 (Arg158Gly), suggesting the abrogation of p53 transcription. Moreover, the deletion of the CNV in 13q14.2 was detected in this patient, resulting in the inactivation of two key regulators of the cell cycle, RB1 and p53.
A recent study has described an increased rate of mutations in receptor tyrosine kinases (RTKs) and their associated signaling effectors, pointing to a more potent role of this pathway in MM than was previously appreciated.15 We found two mutations in RTKs, including a missense mutation in ERBB3 and a truncating mutation in EGFR, and have suggested a role of aberrant RTK signaling in the development or progression of MM in this patient. In addition, few studies have addressed the functional roles of the remaining three mutated genes (SYNE1, MYH11, and CDH2) in myeloma, and further investigations are required. SYNE1 is frequently silenced by DNA methylation in primary cancers of the colon and lung,16,17 suggesting that a loss of SYNE1 function may be a genetic event that promotes tumor progression. MYH11 is a member of the myosin family, and inversion at the MYH11 locus is found in acute myeloid leukemia.18 Additionally, MYH11 mutations have been shown to occur in human colorectal cancers with microsatellite instability.19 N-cadherin, encoded by the CDH2 gene, is a transmembrane protein and plays an important role in cell adhesion. Recently, circulating N-cadherin levels was reported to be a negative prognostic factor in patients with MM.20 In this study, we performed comprehensive genomic analyses using PCR target enrichment and semiconductor-based sequencing of matched tumor and normal DNA samples obtained from an individual with refractory MM. We detected several genetic alterations that may have been associated with the poor prognosis and poor response to chemotherapy of this patient. Although its value should be further confirmed in larger samples, targeted NGS is considered a valuable tool for high-throughput genetic testing in clinical research.
Acknowledgments
This work was supported in part by the Scientific Support Programs for Cancer Research Grant-in-Aid Scientific Research on Innovative Areas Ministry of Education, Culture, Sports, Science, and Technology. This is the same research abstract number 4424 AACR Annual Meeting Philadelphia 2015.
Disclosure
The authors report no conflicts of interest in this work.
References
Ikeda H, Hideshima T, Fulciniti M, et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin Cancer Res. 2009;15:4028–4037. | ||
Görgün G, Calabrese E, Hideshima T, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115:5202–5213. | ||
Anderson KC. Therapeutic advances in relapsed or refractory multiple myeloma. J Natl Compr Canc Netw. 2013;11:676–679. | ||
Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197–2208. | ||
Chinen Y. Phosphoinositide protein kinase PDPK1 is a crucial cell signaling mediator in multiple myeloma. Cancer Res. 2014;74:7418–7429. | ||
Görgün G, Calabrese E, Soydan E, et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood. 2010;116:3227–3237. | ||
Damerla RR, Chatterjee B, Li Y, Francis RJ, Fatakia SN, Lo CW. Ion Torrent sequencing for conducting genome-wide scans for mutation mapping analysis. Mamm Genome. 2014;3–4:120–128. | ||
Singh RR, Patel KP, Routbort MJ, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013;5:607–622. | ||
Chapman MA. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. | ||
Nefedova Y, Cheng P, Alsina M, et al. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103:3503–3510. | ||
Hideshima T, Catley L, Raje N, et al. Inhibition of Akt induces significant downregulation of survivin and cytotoxicity in human multiple myeloma cells. Br J Haematol. 2007;138:783–791. | ||
Markovina S, Callander NS. Bortezomib-resistant nuclear factor-kappa B activity in multiple myeloma cells. Mol Cancer Res. 2008;6:1356–1364. | ||
Jimenez-Zepeda VH, Braggio E, Fonseca R, et al. Dissecting karyotypic patterns in non-hyperdiploid multiple myeloma: an overview on the karyotypic evolution. Clin Lymphoma Myeloma Leuk. 2013;13:552–558. | ||
Fonseca R, Blood E, Rue M, Harrington D, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101:4569–4575. | ||
Leich E, Weiβbach S, Klein HU, et al. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion- and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013;3:102. | ||
Tessema M, Willink R, Do K, et al. Promoter methylation of genes in and around the candidate lung cancer susceptibility locus 6q23–25. Cancer Res. 2008;68:1707–1714. | ||
Schuebel KE, Chen W, Cope L, Glöckner SC, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3(9):1709–1723. | ||
Castilla LH, Garrett L, Adya N, et al. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat Genet. 1999;23(2):144–146. | ||
Alhopuro P, Phichith D, Tuupanen S, et al. Unregulated smooth-muscle myosin in human intestinal neoplasia. Proc Natl Acad Sci U S A. 2008;105(14):5513–5518. | ||
Vandyke K, Chow AW, Williams SA, et al. Circulating N-cadherin levels are a negative prognostic indicator in patients with multiple myeloma. Br J Haematol. 2013;161(4):499–507. |
Supplementary material
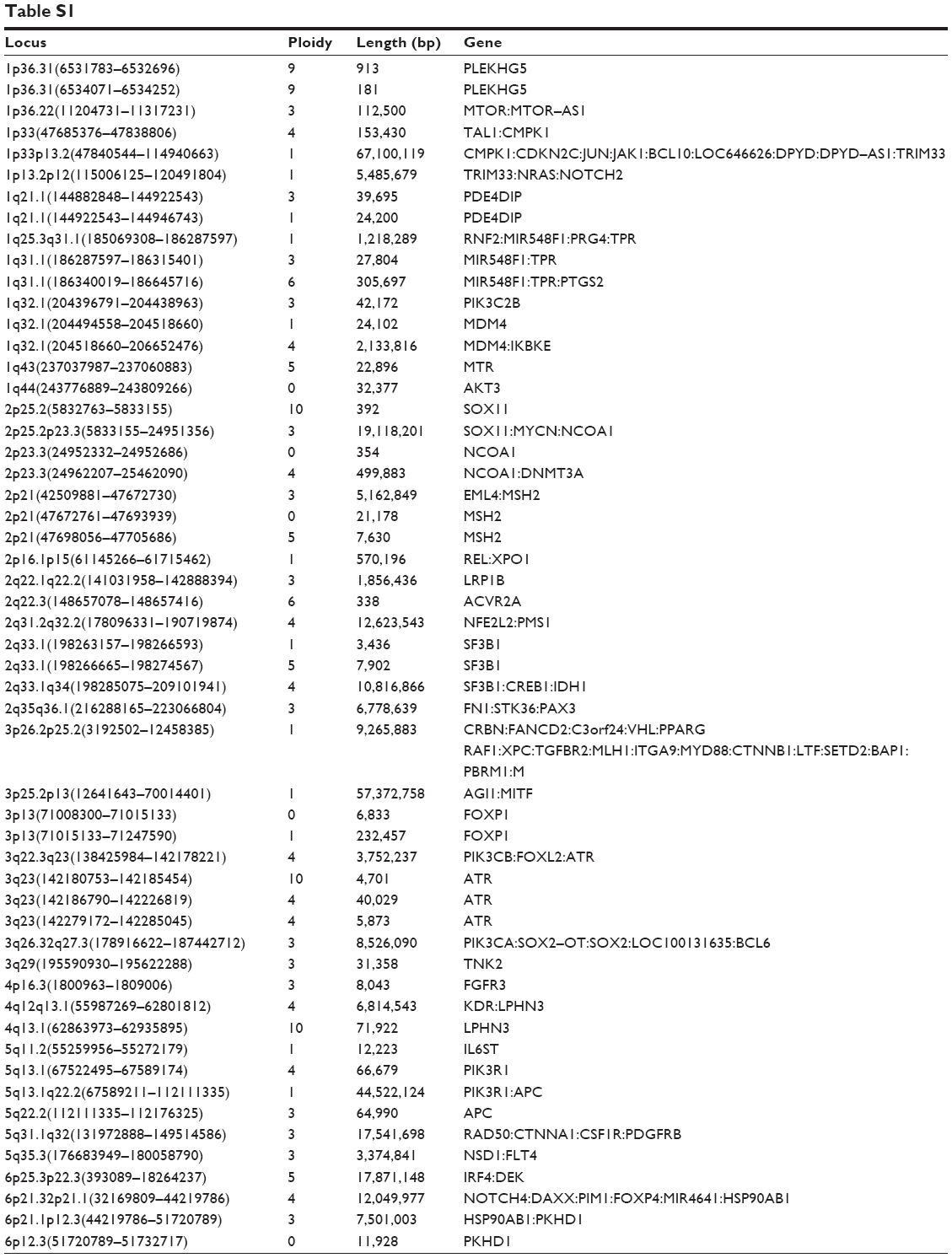 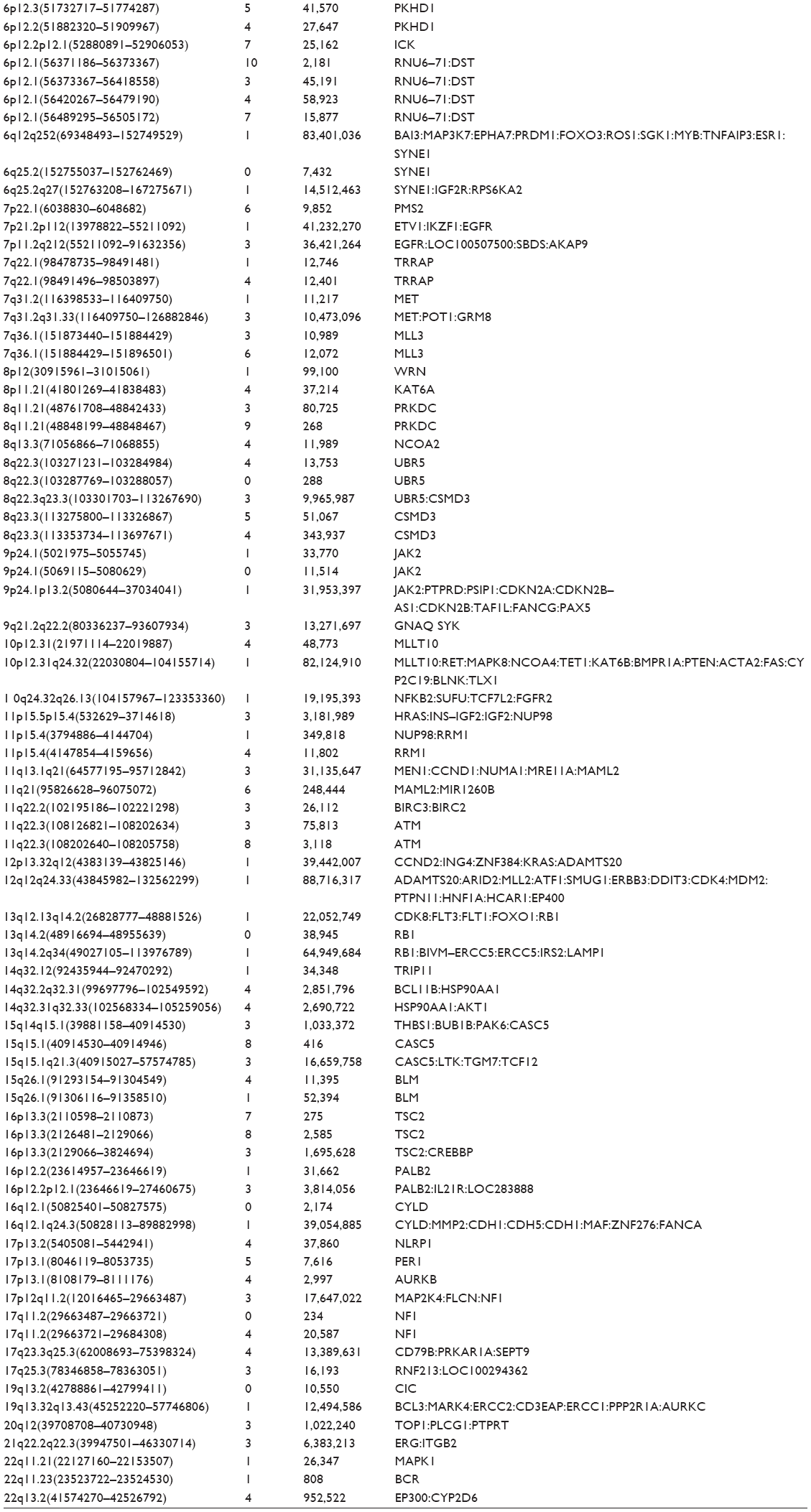 | Table S1 |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.