Back to Journals » International Journal of General Medicine » Volume 13
Molecular Characterization and Disease-Related Morbidities of β-Thalassemia Patients from the Northeastern Part of Iraq
Authors Amin S ![]() , Jalal S
, Jalal S ![]() , Ali K, Rasool L
, Ali K, Rasool L ![]() , Osman T, Ali O, M-Saeed A
, Osman T, Ali O, M-Saeed A
Received 2 September 2020
Accepted for publication 13 November 2020
Published 9 December 2020 Volume 2020:13 Pages 1453—1467
DOI https://doi.org/10.2147/IJGM.S277947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Shaema Amin,1 Sana Jalal,2 Kosar Ali,3 Luqman Rasool,4 Tara Osman,4 Omed Ali,5 Abdalhamid M-Saeed4
1Hiwa Hematology/Oncology Sulaymaniyah Cancer Center, Sulaymaniyah, Iraq; 2Department of Pathology, College of Medicine, University of Sulaymaniyah, Sulaymaniyah, Iraq; 3Department of Medicine, College of Medicine, University of Sulaymaniyah, Sulaymaniyah, Iraq; 4Thalassemia and Congenital Blood Disorders Center, Sulaymaniyah, Iraq; 5Otolaryngology Head and Neck Surgery Center, Sulaymaniyah Teaching Hospital, Sulaymaniyah, Iraq
Correspondence: Shaema Amin
Hiwa Hematology/Oncology Sulaymaniyah Cancer Center, Sulaymaniyah, Iraq
Tel +964-7701499546
Email [email protected]
Background: β-thalassemia is a significant problem in the northeastern part of Iraq, and has imposed a huge burden on the health authorities.
Objective: To identify the molecular characterization and morbidity prevalence in transfusion-dependent thalassemia (TDT) and non-transfusion dependent thalassemia (NTDT) phenotypes in northeastern Iraq.
Patients and Methods: This is a cross-sectional study conducted on 242 β-thalassemia patients from 162 families. Reverse hybridization technique and direct gene sequencing were used to characterize β-thalassemia mutations, and medical records of the patients were reviewed with a well-designed questionnaire.
Results: A total of 22 β-globin mutations arranged in 53 different genotypes were identified: IVS-II-1 (G> A) (35.7%), followed by IVS-I-6 (T> C) (18.0%), and codon 8/9 (+G) (8.5%) were the most frequent. Among disease-related morbidities, bone disease amounted to (66.9%), followed by endocrinopathies (32.2%), hepatobiliary complications (28.9%), and pulmonary hypertension (9.9%), whereas thrombosis, extramedullary hemopoiesis, and leg ulcers were less frequent.
Conclusion: The overall complications rate was 78.9%, with a growing probability of complications with advanced age, with evidently higher rates in patients with β0β0 and β0β+ genotypes that explain the role of underlying genetic defects in the pathophysiology of disease complications.
Keywords: transfusion dependent thalassemia, non-transfusion dependent thalassemia, HBB mutations, morbidities, risk factors, Iraq
Introduction
Beta-thalassemia (β-thal) represents a group of recessively inherited disorders of hemoglobin synthesis that imposed a significant health burden in the tropical belt area, particularly in under-resourced countries, including Iraq.1 The underlying pathophysiology of the disease is multifactorial and stems from a reduced or absent generation of the β-globin chain of the hemoglobin causing α: β chain imbalance, intracellular accumulation of free α-chains, and subsequent red cell destruction ending in ineffective erythropoiesis; the hallmark of β-thalassemia.2
Despite the significant progress and availability of different treatment modalities in clinical practice, yet there are multiple serious morbidities which arise from chronic anemia, and progressive iron accumulation in different organs as a consequence of repeated transfusion therapy as well as enhanced iron absorption from the gut.3,4
β-thalassemia is a major public health concern in Iraq, including the Kurdistan Region with a carrier rate of 4.1% in Sulaymaniyah (northeastern Iraq),5 and an estimated prevalence rate of 37.1/100,000 in Iraq.6 The present study aimed to identify the molecular characterization, and morbidity prevalence in transfusion-dependent thalassemia (TDT) and non-transfusion dependent thalassemia (NTDT) phenotypes in northeastern Iraq.
Patients and Methods
Patients
A cross-sectional study on 242 β-thalassemia patients from 162 families was conducted at Sulaymaniyah Thalassemia Center between July 2018-August 2019. The medical records of the patients were reviewed. The inclusion criteria of NTDT were; age at initial presentation ≥2 years, Hb level maintain between 7 and 10 g/dL, and intermittent need for blood transfusion for NTDT, while for TDT; age at initial presentation <2 years, Hb levels 6–7 g/dl, and regular transfusion requirement.7–9
Ethical Approval
All participants provided informed consent, while the parent or legal guardian of participants under the legal age of majority in Iraq provided with written informed consent. The present study was commenced after obtaining approval from the ethical committee at the College of Medicine, Sulaymaniyah University, Kurdistan Region of Iraq (approval No. 61 on April 16, 2018). All methods were performed in accordance with the Helsinki Declaration.
Clinical Evaluation
Full medical information regarding family history of thalassemia, patient’s age, age at the diagnosis and first transfusion, transfusion history (frequency of transfusion regimen within the last year), bone fracture, any documented thrombotic event, DM, radiological evidence of EMH, parent consanguinity and rate of parenthood were retrieved from patients’ electronic records. Details regarding the utilization of hydroxyurea treatment and iron-chelating drugs within the last year were also recorded. Also, thorough physical examination was performed including evaluation of thalassemia facies, height to judge growth retardation (height> 2 SD below 3rd percentile for the patient age and gender),10 and leg ulceration, the size of the liver and spleen (non-splenectomized) was measured, with screening for gall bladder calculus by ultrasound examination.
Laboratory Findings
Hematological Investigations
Five milliliters of blood was collected from each patient; 2.5 mL was transferred to an EDTA containing tube and the remaining 2.5 mL to a plain tube. The EDTA blood samples were used for routine measurement of complete blood count with a fully automated hematology analyzer (Swelab, Spånga, Sweden), and the estimation of HbA2 and HbF by high-performance liquid chromatography (HPLC) [D-10, Bio-Rad Laboratories, Hercules, CA, USA]. Then, the remaining blood in EDTA tubes was centrifuged; a buffy coat layer was removed for DNA extraction immediately or frozen at −20 for later DNA processing. These investigations were performed just before the next blood transfusion.
Biochemical and Other Investigations
Serum was used to estimate ferritin levels by Enzyme-Linked Immunosorbent Assay (ELISA) (Biokit-USA), liver function tests (LFT) (alanine transaminase; ALT), blood glucose (mg/dL), virological tests; Hepatitis B surface antigen (HBsAg), anti-hepatitis C virus (HCV) antibodies and Human Immune Deficiency Virus (HIV) antibody 1 and 2 (Plasmatic Laboratory Product-UK). Thyroid function test (Thyroid-stimulating hormone TSH and free T4), using the enzyme immunoassay (TOSOH-Japan) was estimated in patients ≥ 10 years annually according to the Thalassemia International Federation (TIF) guidelines.11 Subclinical hypothyroidism was diagnosed based on high TSH>4.7 μIU/mL combined with normal free T4>0.8 ng/dl, while patients with high TSH>4.7 μIU/mL and low free T4 <0.8 ng/dl were diagnosed as overt hypothyroidism.12
Molecular Investigations
Genomic DNA was extracted from peripheral blood (buffy coat) by phenol-chloroform method,13 then the extracted DNA was amplified by a multiplex polymerase reaction (PCR) with subsequent reverse hybridization to characterize 22 common Mediterranean β-thalassemia mutations using β-globin StripAssay MED® Kit (Vienna Labordiagnostica GmbH, Vienna, Austria). All steps were performed in line with the manufacturer’s instructions. The mutations include: −101 (C> T), −87 (C> G); −30 (T> A); codon 5 (-CT); codon 6 (G> A); codon 6 (A> T); codon 6 (-A); codon 8 (-AA); codon 8/9 (+G); codon 15 (G> A); codon 27 (G> T); IVS I-1 (G> A); IVS I-5 (G> C); IVS I-6 -(T>C); IVS I-110 (G> A); IVS I-116 (T> G), IVS -I-130 (G> C); codon 39 (C> T); codon 44 (-C); IVS II-1 (G> A); IVS II-745 (C> G); IVS II-848 (C> A). Direct sequencing of the entire β-globin gene (HBB) was performed for 24 patients when reverse hybridization did not reveal the underlying β-globin alleles at Kariminejad-Najmabadi Pathology and Genetic center in Tehran, Iran using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit and the ABI Prism 377 DNA Automatic sequencer (Perkin Elmer, Foster City, CA). Screening for -α,3,7 -α,2,4 and the –Med-1 α-thal deletions and α-gene triplication by gap PCR,14 were also conducted for the above 24 uncharacterized samples.
Bone Mineral Density Assessment
Patients aged ≥10 years were screened for osteoporosis annually by measuring bone mineral density (BMD) using Dual Energy X-ray Absorptiometry (DEXA) scan as recommended by Thalassemia International Federation (TIF).11 According to World Health Organization criteria, patients with low BMD and a Z-score of ≤ −2.5 SD below the mean for the age and sex-matched control (in patients up to the age of 20 years) and T-score of ≤ −2.5 SD (in those aged 20 years or more) were considered as osteoporosis based on determining the BMD (g/cm2) at the lumbar spine (L1-L4) and femoral neck.15,16
Echocardiography Assessment
Screening for PHT was performed by periodic echocardiography evaluation for all patients at their regular visiting dates. Patients detected with pulmonary artery systolic pressure (PASP) ≥25 mmHg combined with exertional dyspnea at rest and no evidence of left-side heart failure were diagnosed as pulmonary hypertension.17
Statistical Analysis
SPSS program (version 21) was applied for data analysis (IBM SPSS Statistical Package for the Social Sciences). For quantitative variables, the difference in the means was assessed using an independent t-test and while comparing the means among several groups was performed using the ANOVA test. A Chi-square test was used to compare the categorical data among different groups of patients. Tukey’s HSD (Honestly Significant Difference) was used as a Post-Hoc test for pair comparison amongst groups. P -values of ≤ 0.05 were used as a cutoff point for the significance of statistical tests.
Results
Demographic Data, Clinical Characteristics, and Conventional Management of β-Thalassemia Patients
A total of 242 β-thal patients from 162 families were recruited; including 159 non-transfusion dependent thalassemia (NTDT) and 83 transfusions dependent thalassemia (TDT) patients. The mean age was 17 ± 10.2 years, with a range of (1.4–54 years). They were 129 males (53.3%) and 113 females (46.7%), with a male: female ratio (1.1:1). The mean age at diagnosis and initiation of transfusion was 5 ± 6.3 years and 4 ± 5.8 years, respectively. One hundred twenty-three (50.8%) had splenomegaly, with a mean spleen size of 13.8 ± 3.5cm, 34.3% of which underwent splenectomy. On the other hand, 87 thalassemia patients (36%) had hepatomegaly, with a mean hepatic size of 13.8 ± 2.5cm (Table 1).
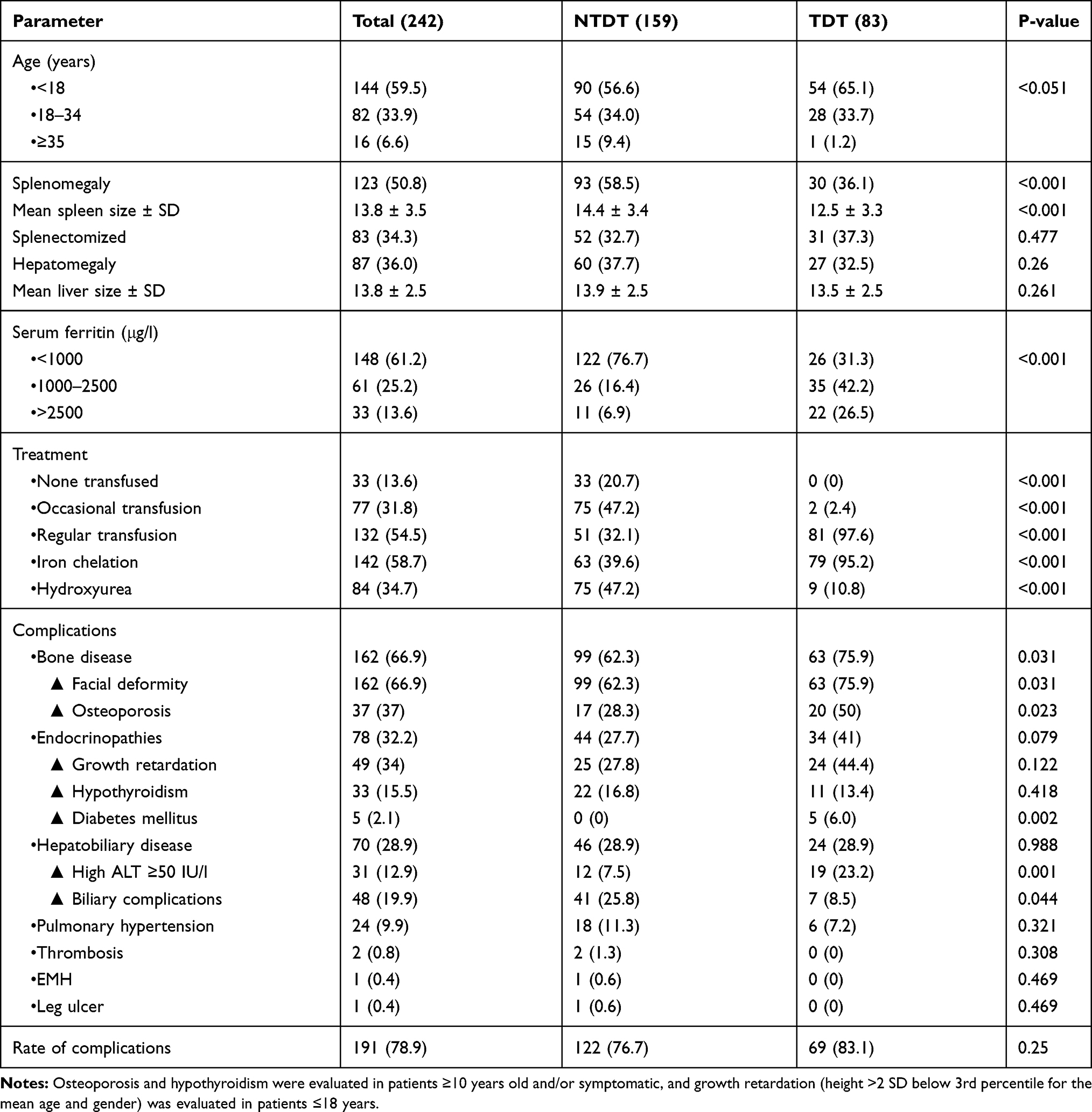 |
Table 1 Demographic and Disease Characteristics of 242 β-Thalassemia Patients |
As for thalassemia patients’ management, 132 patients (54.5%) were on regular transfusion therapy (> 3 times/year), while 77 (31.8%) patients received occasional transfusion regimen (0–3/year); during a severe illness, pregnancy, or surgery, whereas 13.6% had never been transfused. In addition, 142 patients (58.7%) received iron-chelating therapy for at least one-year duration, and Deferasirox and Desferoxamine were used in 83.9% and 16.1%, respectively. In the current study, 84 (34.7%) used hydroxyurea therapy and had revealed a potentially lower annual transfusion rate (3 ± 5.3), a lower mean s.ferritin (695.2 ± 1039.5) and hence, a significantly lower requirement for iron chelation therapy, in 18.3% as opposed to those patients without hydroxyurea therapy, p-value <0.001 (Table 2).
 |
Table 2 Impact of Hydroxyurea Therapy on Disease Characteristics |
Disease-Associated Morbidities
Bone disease (facial deformity and osteoporosis) was the most recurrent morbidity, reported in 162 patients (66.9%), followed by endocrinopathies [growth retardation, hypothyroidism, and diabetes mellitus (DM)] in 78 patients (32.2%), hepatobiliary complications [abnormal liver function tests (LFT); ALT≥50 IU/l and biliary complications; cholelithiasis and cholecystectomy] in 28.9%, and pulmonary hypertension (PHT) in 9.9% (Table 1). Other complications; thrombotic events, leg ulcers, and extramedullary hemopoiesis (EMH) were less frequent. No patient with heart failure was identified. Moreover, the rate of developing the above-mentioned disease-related morbidities increased with age, and this was particularly evident in TDT patients (Figure 1).
 |
Figure 1 Frequency of disease-related morbidities among 242 β-thalassemia patients at different age intervals. |
Disease Characteristics and Morbidities in TDT and NTDT Patients
Patients with TDT were diagnosed and transfused at an earlier age (0.8 ± 0.5 years), and (0.75 ± 0.5 years), respectively in comparison to NTDT patients (7.3 ± 6.9 years), and (6.1 ± 6.6 years), respectively p-value <0.001. Further, the vast majority of TDT patients enrolled in this study received regular blood transfusions and iron chelation therapy, 97.6%, and 95.2%, respectively, while in contrast, 32.1% of NTDT patients required regular transfusions, and in fact, 20.7% had not been transfused (Figure 2). On the other hand, less than half of NTDT patients required occasional blood transfusions, though they had received hydroxyurea therapy. Additionally, of 83 patients who underwent splenectomy, 62.7% were NTDT patients. Furthermore, s.ferritin level≥ 1000 µg/l was more frequently detected in TDT patients, p-value <0.001 (Table 1).
 |
Figure 2 Clinical management of 242 β-thalassemia patients. |
Regarding disease-related morbidities, TDT patients reported a significantly higher frequency of bone complications (both facial deformity and osteoporosis) as opposed to NTDT (Table 1), and (Figure 3). Likewise, abnormal LFT and growth retardation were more frequent in patients with TDT, and DM was only detected in this phenotype. In contrast, biliary complications, PHT, and hypothyroidism were more prevalent among NTDT patients, with thrombosis, EMH, and leg ulcers only encountered in this group. The reported complications rate in this study was 78.9%, with a slightly higher rate in TDT in comparison to NTDT patients, 83.1% vs 76.7%, respectively (Table 1).
 |
Figure 3 Distribution of disease-related morbidities among 242 β-thalassemia patients. |
Furthermore, osteoporosis and abnormal LFT were less frequently observed among patients who used hydroxyurea therapy, on the other hand, high ALT ≥50 IU/l was more frequently reported among patients with ferritin levels ≥1000 μg/l, patients on regular transfusion and iron chelation therapy, p-value <0.001. Moreover, pulmonary hypertension and biliary complications were significantly reported among splenectomized patients, p-value 0.033. Additionally, the later complication was significantly documented among patients on occasional transfusion therapy and patients ≥18 years old, p-value <0.001 (Table 3).
 |
Table 3 Univariate Analysis of Disease-Related Morbidities Among 242 β-Thalassemia Patients |
Laboratory Results
Hemoglobin (Hb) level at the time of the study ranged between 4.1–13.8 g/dl with a mean of 8.8 ± 1.3 g/dl. The mean MCV and MCH were 74.4 ± 9.7 fl and 25.6 ± 3.8 pg, respectively. Hb F at first presentation ranged between 4.6% −99.8% with a mean of 65.7 ± 34.8%, while mean Hb A2 was 3.2 ± 2.3% with a range of 0.2% – 11.2%. Also, the s.ferritin level ranged between 27–9882 μg/l with a mean of 1250.5 ± 1472.5 μg/l. Moreover, 31 patients (12.8%) had been diagnosed with abnormal LFT, over 90% of which had s. ferritin ≥ 1000 µg/l and received iron chelation therapy, 12 (38.7%) had hepatomegaly and 6 (19.4%) were HCV positive. The mean TSH was 3.5± 2µIU/mL, with a range of (0.4–15.3), with no significant difference between TDT and NTDT patients. Lastly, Hb, MCV, MCH, ALT, s.ferritin, and HCV infection differed significantly between TDT and NTDT patients (Table 4).
 |
Table 4 Laboratory Investigations of 242 β-Thalassemia Patients in the Current Study |
β-Thalassemia Mutations and Genotypes of Patients
Among 484 alleles from 242 β-thalassemia patients, a total of 22 β-globin alleles were identified, 12 of which were β0, 9 were β+, with a single dominant β-thalassemia like mutation codon 127 (A >G). The three most frequent β-thal mutations were: IVS-II-1 (G >A), followed by IVS-I-6 (T >C), and codon 8/9; 35.7%, 18.0%, and 8.5%, respectively. Other mutations were less frequent (Table 5). Fourteen β-thal mutations were determined by reverse hybridization, while the remaining 8 mutations were identified by direct sequencing: IVS-I-128, CAP +1 (A> C), codon 36/37, codon 25/26, codon 82/82, codon 127, +20/IVS-II-745, and IVS-II-850.
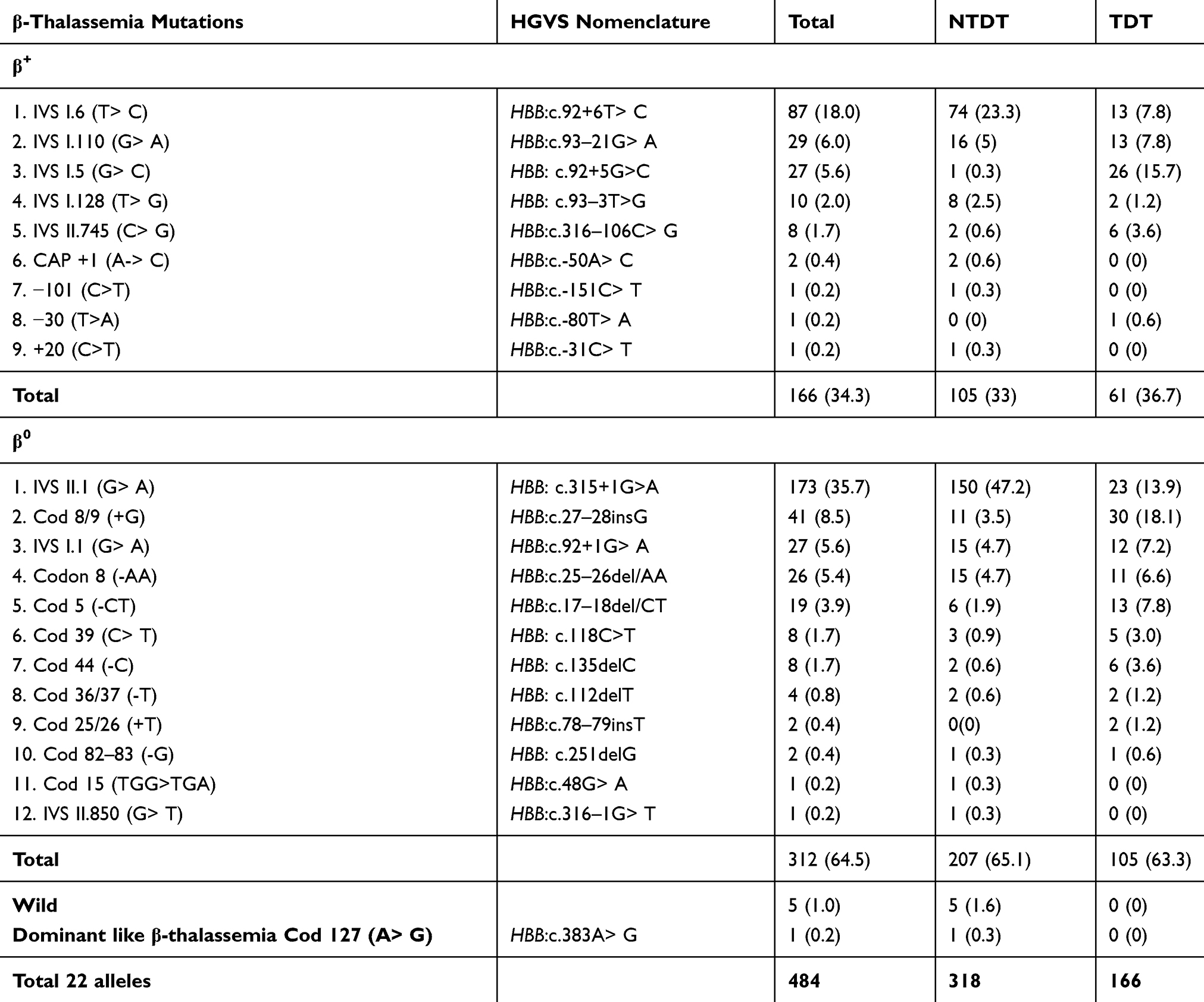 |
Table 5 The Reported β-Globin Gene Mutations in the Current Study |
In the current study, 53 various genotypes were identified; homozygous IVS-II-1, homozygous IVS-I-6, and compound heterozygous IVS-II-1/codon 8/9 were the most frequent (Table 6). The commonest genotype among NTDT patients was homozygous IVS-II-1, as opposed to homozygous codon 8/9 in TDT patients. Homozygous mutations were determined in (76.3%) patients, of which, 62.9% were the result of the consanguineous marriage, p-value <0.001, and parent consanguinity rate was determined in 52.5%.
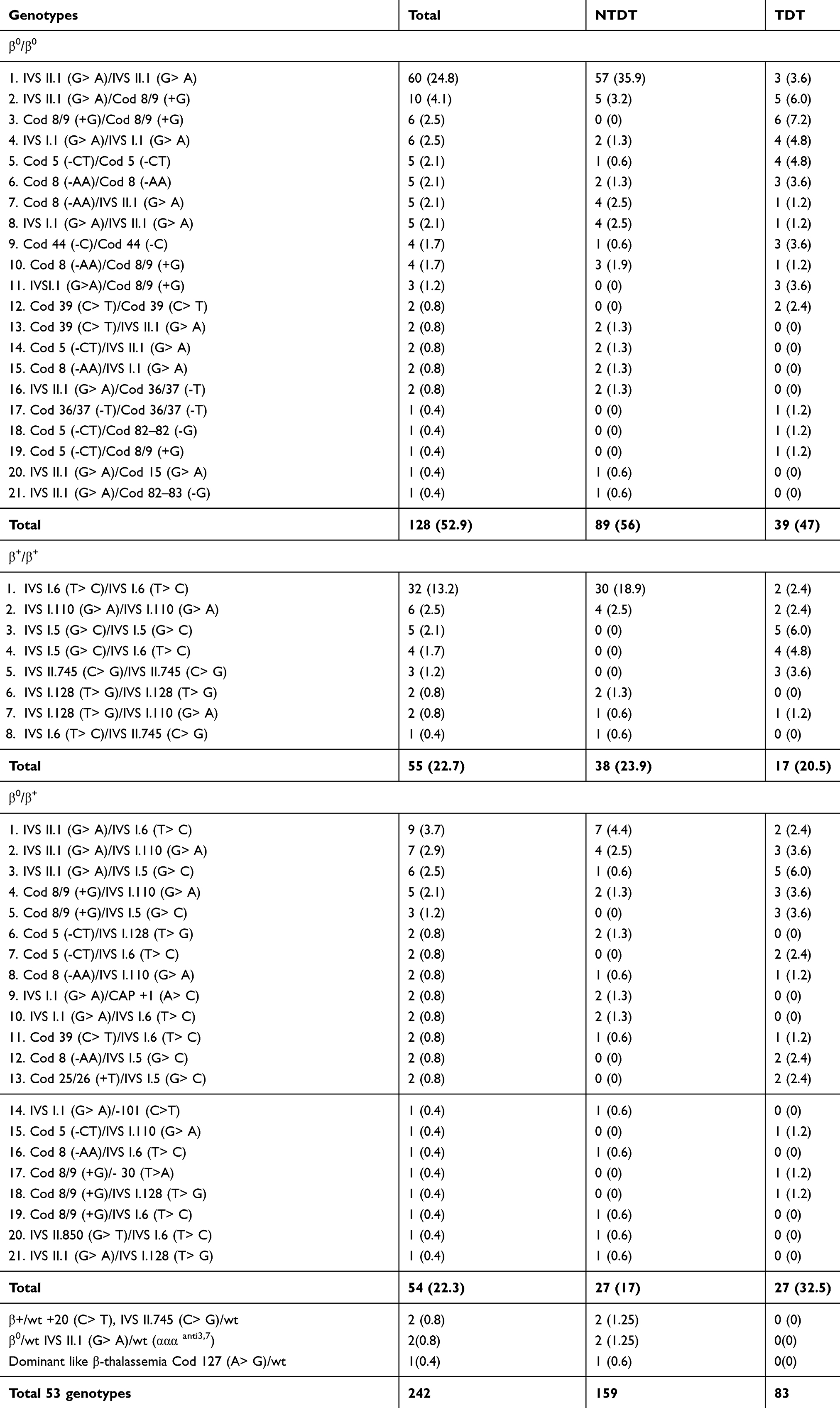 |
Table 6 Genotypes of 242 Thalassemia Patients in the Current Study |
Clinical Characteristics of the Studied Genotypes
Patients with β0β0 and β0β+ genotypes were diagnosed earlier and had an earlier onset of blood transfusion in comparison to the β+β+ genotype. Their HbA2 level was the lowest and Hb F was the highest. Generally, the above genotypes had a higher frequency of disease morbidities, though not at significant levels when compared with β+β+ genotype (apart from facial deformity) (Table 7).
 |
Table 7 Genotype Correlation with Different Parameters |
Discussion
Our study has highlighted the molecular defects of TDT and NTDT and explained the differential clinical picture, proportion of disease complications, and management options in both disease phenotypes.
IVS-II-1 (G> A) (the Mediterranean β 0 thal mutation) was the most prevalent mutation detected in this study (35.7%) with the highest frequency (47.2%) among NTDT patients in consistence with earlier studies from central Iraq,18 and Iran.19,20 In contrast, studies from other Kurdish provinces-Iraq,21,22 Turkey,23 Lebanon,9 and Egypt24 revealed the IVS-I-6 (Mediterranean β+ thal mutation), which is the second common β-thal mutation reported in this study and among NTDT patients, as the most frequent mutation.
The third most recurrent mutation, codon 8/9 (+G) (β0 Asian Indian mutation), was more frequent in TDT patients in agreement with an earlier small-sized study from Sulaymaniyah.25 However, IVS-II-1 is shown to be the most common β-thal mutation in TDT patients in earlier studies from Iraq,26–28 Iran,19 and Kuwait.29 Further, IVS-I-110 (Mediterranean β+ thal mutation) was the most frequent mutation in studies from Turkey,30 Egypt,31 and Lebanon.32 The discrepancy in the prevalence of frequent mutations is attributed to different study populations and different sample sizes.
Although β+ thal mutations (β+/β+ or β0/β+) have contributed to 40.9% of NTDT genotypes included in this study, NTDT had an evidently less severe phenotype regarding the age of diagnosis, frequency of transfusion, and the frequency of most of the disease-related complications. This would highly propose the coinheritance of disease modifiers such as single nucleotide polymorphism in the three major quantitative trait loci (QTLs) to induce Hb F synthesis and/or inheritance of α-thalassemia to modify the unbalance between α: β globin chains and subsequently less ineffective erythropoiesis.33
Conventional management of TDT patients at our center complies with clinical management guidelines of Thalassemia International Federation,34 which comprised regular transfusion to maintain Hb>9–10 g/dl and iron chelation therapy to prevent iron-related toxicity. Likewise, our figure of splenectomy (37.3%) is within the range reported from Iraq and neighboring countries (11.3%-95%),6,30,35,36 though, the indications of splenectomy needs to be revisited and restricted to selected cases to avert the risk of hypercoagulability and overwhelming infection post-splenectomy.37
As for NTDT, the transfusion regimen implemented was individually tailored to meet patients’ demands, and only 32.1% of NTDT received regular blood transfusions, 39.6% required iron chelation therapy while 32.7% were splenectomized. These figures are lower in comparison to figures reported by the “Optimal Care Study,”38 while we have reported a higher proportion of NTDT patients (47.2%) receiving hydroxyurea.38,39 Interestingly, our data on hydroxyurea therapy in thalassemia patients is supporting previous encouraging results from optimal care study and a single report evaluating 6 years of hydroxyurea therapy in NTDT.40 Eighty-four β-thal patients (including 9 TDT) used this Hb F inducer and revealed a potentially lower s.ferritin, annual transfusion frequency, and chelation therapy requirement (Table 2), illustrating the effect of hydroxyurea in improving α: β globin chain imbalance and eventually more effective erythropoiesis.
Disease complications were encountered in 78.9% of the enrolled patients and the rate was more frequent among TDT patients. The discrepancy in the multimorbidity profile of TDT and NTDT has been attributed to the difference in the underlying genotype and clinical management lines (transfusion frequency, chelation, more frequent splenectomy and the use of fetal hemoglobin inducing therapy).38,41
Bone abnormalities, the most prevalent morbidity documented in this study, were detected at a higher frequency among TDT patients, while it was reported to be more profound in NTDT,38,42 as a result of chronic anemia, enhanced ineffective erythropoiesis, and consequent bone marrow expansion. This variation might be attributed to the more frequent use of hydroxyurea in just less than half of our NTDT patients, with further support by iron chelation therapy (39.6% of NTDT patients) to lower the rate of bone complications to 62.3% vs 75.9% in TDT, p-value = 0.031 (Table 1), in consistence with previous studies.38,41 However, this high prevalence of bone complications warrants close follow-up with early and appropriate initiation of therapy.
Endocrine abnormalities are among the most common complications of β-thalassemia.43 A lower prevalence of endocrine diseases was reported among NTDT patients which might be related to the lower extent of blood transfusion, slower iron accumulation rate, and hepatic predominance of iron-loading,44 in agreement with our figures (Figure 3).
Unlike previous reports,28,35 hypothyroidism (97.1% subclinical) was diagnosed at a higher frequency in NTDT compared to TDT (Figure 3). As for DM, it was also observed in well transfused and regularly chelated TDT patients,34 suggesting the role of other factors in the pathogenesis including individual sensitivity to iron and chronic anemia. Five TDT patients were identified with DM in this study; two had s.ferritin ≥1000 μg/l.
Among different body organs susceptible to damage in β-thalassemia patients, the liver represents a major target and iron overload is considered as the most important single cause, while HCV infection is the second acting in synergy, particularly to increase the risk of hepatocellular carcinoma. Drug toxicity (chelation therapy) is an added risk factor.45,46
The total frequency of hepatobiliary complications in this study was the same for TDT and NTDT patients, at 28.9% (Table 1). However, abnormal LFT was more prevalent among TDT, explained by a significantly higher frequency of raised s.ferritin ≥1000 μg/l, and HCV infection together with regular iron chelation therapy requirements,45,47 whereas biliary complications were more common in NTDT patients (Figure 3).
The higher frequency of biliary complications observed in NTDT had been supported by earlier studies,35,41 and explained by the underlying chronic hemolytic anemia.48 Furthermore, it was significantly reported among splenectomized patients and patients aged ≥18 years old (Table 3), in agreement with earlier results.38,48,49 Furthermore, 75 (31%) β-thal patients enrolled had hepatomegaly with normal LFT, half of them used iron chelation therapy, 25.3% were positive for HCV infection and 22.7% had s.ferritin ≥1000 μg/l. In view of the above findings, a monthly follow-up of hepatic transaminases is justified for the early detection of hepatic complications.
Another potential complication reported in this study was PHT, a disease progression complication in the absence or inappropriate blood transfusion, its prevalence was higher in NTDT patients in consistent with other studies.35,50 Chronic thromboembolism has been also implicated in the pathophysiology of this morbidity, in particular patients who underwent splenectomy,51 in accordance with our result (Table 3). Besides, this study had revealed that the rate of PHT was frequently observed in patients who used chelation therapy in contrast to other reports,38,50 where iron chelation had reduced the incidence of PHT. This finding can be attributed to the small sample size of patients (24) with PHT.
Despite compliance with the management guidelines for thalassemia patients, we have reported a high complication rate, with an increased probability of complications with advanced age (Figure 1). Additionally, since access to blood for transfusion therapy in limited-resource countries is a challenge and poses a considerable health burden,51 a preventive program for hemoglobinopathies based on the concept of premarital screening, counseling, and the prenatal diagnosis was the only viable option in Sulaymaniyah/Iraq to minimize the birth of affected fetuses. Sulaymaniyah Hemoglobinopathies Preventive Program started in 2008, and over the first 5 years, the program has succeeded to reduce the birth of affected fetuses by hemoglobinopathies by 65%, and can possibly serve as a prototype for other regional programs.52
Limitations of the current study are using s.ferritin to estimate the iron overload instead of liver R2 magnetic imaging, and measuring PASP by Doppler Echocardiography to determine PHT instead of right heart catheterization. Furthermore, genetic modifiers including coinheritance of α-thalassemia and QTL polymorphism were not performed due to the lack of financial support.
Conclusions
The current study, the largest from Iraq and Kurdistan region on β-thalassemia patients, revealed that β0 was the most frequent β-thal mutation, a result that is rather distinct from earlier reports of Iraq and nearby countries. Additionally, using hydroxyurea therapy among NTDT patients resulted in potentially lower serum ferritin, transfusion, and chelation therapy requirements. Furthermore, the high complications rate in limited-resource countries predestines timely medical interference to curtail these complications before reaching the point of irreversibility.
Moreover, the role of genetic modifiers of disease severity needs to be addressed in future studies for a better understanding of disease pathophysiology. In the end, a feasible regional preventive program is the key to reduce the number of affected births in thalassemia high prevalence settings.
Abbreviations
β-thal, Beta-thalassemia; TDT, Transfusion Dependent Thalassemia; NTDT, Non-transfusion Dependent Thalassemia; DM, Diabetes Mellitus; EMH, Extra Medullary Hemopoiesis; EDTA, Ethylene Diamine Tetra Acetic acid; HPLC, High-Performance Liquid Chromatography; ELISA, Enzyme-Linked Immunosorbent Assay; LFT, Liver Function Test; ALT, Alanine Transaminase; TSH, Thyroid Stimulating Hormone; PCR, Polymerase Chain Reaction; BMD, Bone Mineral Density; DEXA, Dual Energy X-ray Absorptiometry; TIF, Thalassemia International Federation; PHT, Pulmonary Hypertension; PASP, Pulmonary Artery Systolic Pressure; s.ferritin, Serum Ferritin; Hb, Hemoglobin; MCV, Mean Corpuscular Volume; MCH, Mean Corpuscular Hemoglobin; HCV, Hepatitis C Virus; QTL, Quantitative Trait Loci.
Data Sharing Statement
All data related to this study are included within this article and can be requested from the corresponding author on reasonable request.
Acknowledgments
The authors would like to thank all enrolled thalassemia patients and medical staff of Thalassemia and Congenital Blood Disorders Center in Sulaymaniyah, for their great cooperation throughout this work. Special thanks to Dr. Sherko Saeed F. Zmnako for his advice regarding the arrangement of the references by EndNote Software.
Disclosure
The authors declare no conflicts of interest.
References
1. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704–712.
2. Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26:S12–S15. doi:10.1016/S0268-960X(12)70005-X
3. Rund D. Thalassemia 2016: modern medicine battles an ancient disease. Am J Hematol. 2016;91(1):15–21. doi:10.1002/ajh.24231
4. Nemeth E. Hepcidin in β-thalassemia. Ann N Y Acad Sci. 2010;1202(1):31–35. doi:10.1111/j.1749-6632.2010.05585.x
5. Jalal SD, Al-Allawi NA, Faraj AH, Ahmed NH. Prevalence of haemoglobinopathies in Sulaimani–Iraq. Duhok Med J. 2008;2(1):71–79.
6. Kadhim KA, Baldawi KH, Lami FH. Prevalence, incidence, trend, and complications of Thalassemia in Iraq. Hemoglobin. 2017;41(3):164–168. doi:10.1080/03630269.2017.1354877
7. Cappellini MD, Musallam KM, Cesaretti C, Taher A Thalassemia intermedia. Paper presented at: Disorders of Erythropoiesis, Erythrocytes and Iron Metabolism. Genoa, Italy: Forum Service Editore 2009.
8. Borgna-Pignatti C, Marsella M, Zanforlin N. The natural history of thalassemia intermedia. Ann N Y Acad Sci. 2010;1202(1):214–220. doi:10.1111/j.1749-6632.2010.05550.x
9. Qatanani M, Taher A, Koussa S, et al. β-Thalassaemia intermedia in Lebanon. Eur J Haematol. 2000;64(4):237–244. doi:10.1034/j.1600-0609.2000.90087.x
10. Najafipour F, Sarisorkhabi R, Bahrami A, et al. Evaluation of endocrine disorders in patients with thalassemia major. Iranian J Endocrinol Metabol. 2008;10(1):35–43.
11. Taher A, Vichinsky E, Musallam K, Cappellini M-D, Viprakasit V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT). Nicosia, Cyprus: Thalassaemia International Federation,; 2013.
12. Baskin HJ, Cobin RH, Duick DS, et al. American association of clinical endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. Endocrine Pract. 2002;8(6):457–469. doi:10.4158/1934-2403-8.6.457
13. Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2001.
14. Oron-Karni V, Filon D, Oppenheim A, Rund D. Rapid detection of the common mediterranean α-globin deletions/rearrangements using PCR. Am J Hematol. 1998;58(4):306–310.
15. World Health O. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: Report of a WHO study group [meeting held in Rome from 22 to 25 June 1992]. Geneva: World Health Organization; 1994.
16. Kanis JA. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group. Osteoporosis Int. 1994;4(6):368–381.
17. Greiner S, Jud A, Aurich M, et al. Reliability of noninvasive assessment of systolic pulmonary artery pressure by doppler echocardiography compared to right heart catheterization: analysis in a large patient population. J Am Heart Assoc. 2014;3(4):e001103. doi:10.1161/JAHA.114.001103
18. Al-Allawi NAS, Puehringer H, Raheem RA, Oberkanins C. Genetic modifiers in β-thalassemia intermedia: a study on 102 Iraqi Arab patients. Genet Test Mol Biomarkers. 2015;19(5):242–247. doi:10.1089/gtmb.2014.0310
19. Maryami F, Azarkeivan A, Fallah MS, Zeinali S, Large Cohort A. Study of genotype and phenotype correlations of beta- thalassemia in Iranian population. Int J Hematol Oncol Stem Cell Res. 2015;9(4):198–202.
20. Rahimi Z, Muniz A, Parsian A. Detection of responsible mutations for beta thalassemia in the Kermanshah Province of Iran using PCR-based techniques. Mol Biol Rep. 2010;37(1):149–154. doi:10.1007/s11033-009-9560-0
21. Al-Allawi NAS, Jalal SD, Mohammad AM, Omer SQ, Markous RSD. Thalassemia intermedia in Northern Iraq: a single center experience. Biomed Res Int. 2014;2014:9.
22. Shamoon RP, Al-Allawi NAS, Cappellini MD, Di Pierro E, Brancaleoni V, Granata F. Molecular basis of β-Thalassemia Intermedia in Erbil Province of Iraqi Kurdistan. Hemoglobin. 2015;39(3):178–183. doi:10.3109/03630269.2015.1032415
23. ALTAY Ç, GÜRGEY A. β-Thalassemia Intermedia in Turkey. Ann N Y Acad Sci. 1990;612(1):81–89. doi:10.1111/j.1749-6632.1990.tb24293.x
24. Elmezayen AD, Kotb SM, Sadek NA, Abdalla EM. β-Globin mutations in Egyptian patients with β-thalassemia. Lab Med. 2015;46(1):8–13. doi:10.1309/LM1AYKG6VE8MLPHG
25. Saud A, Hasan F, Al A, et al. Molecular and biochemical study on β-thalassemia patients in Iraq. Current Res Microbiol Biotechnol. 2014;1:160–165.
26. Al-Allawi NAS, Hassan KMA, Sheikha AK, Nerweiy FF, Dawood RS, Jubrael J. Thalassemia mutations among transfusion-dependent thalassemia major patients in Northern Iraq. Mol Biol Int. 2010;2010.
27. Saleh K, Kakey E. Some Molecular Characterization of Βeta-Thalassemia Major in Koya City. 2018.
28. Omer WA. Molecular characterization of beta-thalassemia mutations in Baghdad. Iraqi J Commun Med. 2010;23(2):90–95.
29. Adekile AD, Gu LH, Baysal E, et al. Molecular characterization of α-thalassemia determinants, β-thalassemia alleles, and βs haplotypes among Kuwaiti Arabs. Acta Haematol. 1994;92(4):176–181. doi:10.1159/000204216
30. Aydinok Y, Oymak Y, Atabay B, et al. A national registry of thalassemia in Turkey: demographic and disease characteristics of patients, achievements, and challenges in prevention. Turk j Haematol. 2018;35(1):12–18. doi:10.4274/tjh.2017.0039
31. Al-Akhras A, Badr M, El-Safy U, et al. Impact of genotype on endocrinal complications in β-thalassemia patients. Biomed Rep. 2016;4(6):728–736. doi:10.3892/br.2016.646
32. Zahed L, Qatanani M, Nabulsi M, Taher A. β-Thalassemia mutations and haplotype analysis in Lebanon. Hemoglobin. 2000;24(4):269–276. doi:10.3109/03630260008993133
33. Rujito L, Sasongko TH. Genetic background of β thalassemia modifier: recent update. J Biomed Transl Res. 2018;4:12–21. doi:10.14710/jbtr.v4i1.2541
34. Cappellini M-D, Cohen A, Porter J, Taher A, Viprakasit V. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). Nicosia, Cyprus: Thalassaemia International Federation; 2014.
35. Isma’eel H, Shamseddeen W, Taher A, et al. Ventricular late potentials among thalassemia patients. Int J Cardiol. 2009;132(3):453–455. doi:10.1016/j.ijcard.2007.08.103
36. Hashemieh M, Azarkeivan A, Radfar M, et al. Prevalence of osteoporosis among thalassemia patients from zafar adult thalassemia clinic, Iran. Iranian J Blood Cancer. 2014;6(3):143–148.
37. Taher AT, Musallam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thrombosis Haemostasis. 2010;8(10):2152–2158. doi:10.1111/j.1538-7836.2010.03940.x
38. Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892. doi:10.1182/blood-2009-09-243154
39. Rafsanjani KA, Mafi N, Tafreshi RI. Complications of β-thalassemia intermedia in Iran During 1996–2010 (Single-Center Study). Pediatr Hematol Oncol. 2011;28(6):497–508. doi:10.3109/08880018.2011.572144
40. Karimi M, Darzi H, Yavarian M. Hematologic and clinical responses of thalassemia intermedia patients to hydroxyurea during 6 years of therapy in Iran. J Pediatr Hematol Oncol. 2005;27(7):380–385. doi:10.1097/01.mph.0000174386.13109.28
41. Sleiman J, Tarhini A, Bou-Fakhredin R, Saliba AN, Cappellini MD, Taher AT. Non-transfusion-dependent thalassemia: an update on complications and management. Int J Mol Sci. 2018;19(1):182. doi:10.3390/ijms19010182
42. Cappellini MD, Musallam KM, Taher AT. Insight onto the Pathophysiology and Clinical Complications of Thalassemia Intermedia. Hemoglobin. 2009;33(sup1):S145–S159. doi:10.3109/03630260903351528
43. Skordis N, Sanctis V, Soliman A. Endocrine Disease. In: Cappellini CA, Porter J, et al., editors. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT).
44. Baldini M, Marcon A, Cassin R, et al. Beta-Thalassaemia intermedia: evaluation of endocrine and bone complications. Biomed Res Int. 2014;2014:5. doi:10.1155/2014/174581
45. Lai ME, Origa R, Danjou F, et al. Natural history of hepatitis C in thalassemia major: a long-term prospective study. Eur J Haematol. 2013;90(6):501–507. doi:10.1111/ejh.12086
46. Moukhadder HM, Halawi R, Cappellini MD, Taher AT. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: a comprehensive review. Cancer. 2017;123(5):751–758. doi:10.1002/cncr.30462
47. Triantos C, Kourakli A, Kalafateli M, et al. Hepatitis C in patients with β-thalassemia major. A single-centre experience. Ann Hematol. 2013;92(6):739–746. doi:10.1007/s00277-013-1692-6
48. Musallam KM, Taher AT, Rachmilewitz EA. beta-thalassemia intermedia: a clinical perspective. Cold Spring Harb Perspect Med. 2012;2(7):a013482. doi:10.1101/cshperspect.a013482
49. Cappellini MD, Motta I. New therapeutic targets in transfusion-dependent and -independent thalassemia. Hematology. 2017;2017(1):278–283. doi:10.1182/asheducation-2017.1.278
50. Derchi G, Galanello R, Bina P, et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of B-thalassemia patients using right heart catheterization. Circulation. 2014;129(3):338–345. doi:10.1161/CIRCULATIONAHA.113.002124
51. Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet (London, England). 2018;391(10116):155–167. doi:10.1016/S0140-6736(17)31822-6
52. Al-Allawi NA, Jalal SD, Ahmed NH, Faraj AH, Shalli A, Hamamy H. The first five years of a preventive programme for haemoglobinopathies in Northeastern Iraq. J Med Screen. 2013;20(4):171–176. doi:10.1177/0969141313508105
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.