Back to Journals » The Application of Clinical Genetics » Volume 7
Molecular analysis of beta-globin gene mutations among Thai beta-thalassemia children: results from a single center study
Authors Boonyawat B ![]() , Monsereenusorn C
, Monsereenusorn C ![]() , Traivaree C
, Traivaree C ![]()
Received 22 August 2014
Accepted for publication 7 October 2014
Published 10 December 2014 Volume 2014:7 Pages 253—258
DOI https://doi.org/10.2147/TACG.S73058
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Martin Maurer
Boonchai Boonyawat,1 Chalinee Monsereenusorn,2 Chanchai Traivaree2
1Division of Genetics, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand; 2Division of Hematology/Oncology, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand
Background: Beta-thalassemia is one of the most common genetic disorders in Thailand. Clinical phenotype ranges from silent carrier to clinically manifested conditions including severe beta-thalassemia major and mild beta-thalassemia intermedia.
Objective: This study aimed to characterize the spectrum of beta-globin gene mutations in pediatric patients who were followed-up in Phramongkutklao Hospital.
Patients and methods: Eighty unrelated beta-thalassemia patients were enrolled in this study including 57 with beta-thalassemia/hemoglobin E, eight with homozygous beta-thalassemia, and 15 with heterozygous beta-thalassemia. Mutation analysis was performed by multiplex amplification refractory mutation system (M-ARMS), direct DNA sequencing of beta-globin gene, and gap polymerase chain reaction for 3.4 kb deletion detection, respectively.
Results: A total of 13 different beta-thalassemia mutations were identified among 88 alleles. The most common mutation was codon 41/42 (-TCTT) (37.5%), followed by codon 17 (A>T) (26.1%), IVS-I-5 (G>C) (8%), IVS-II-654 (C>T) (6.8%), IVS-I-1 (G>T) (4.5%), and codon 71/72 (+A) (2.3%), and all these six common mutations (85.2%) were detected by M-ARMS. Six uncommon mutations (10.2%) were identified by DNA sequencing including 4.5% for codon 35 (C>A) and 1.1% initiation codon mutation (ATG>AGG), codon 15 (G>A), codon 19 (A>G), codon 27/28 (+C), and codon 123/124/125 (-ACCCCACC), respectively. The 3.4 kb deletion was detected at 4.5%. The most common genotype of beta-thalassemia major patients was codon 41/42 (-TCTT)/codon 26 (G>A) or betaE accounting for 40%.
Conclusion: All of the beta-thalassemia alleles have been characterized by a combination of techniques including M-ARMS, DNA sequencing, and gap polymerase chain reaction for 3.4 kb deletion detection. Thirteen mutations account for 100% of the beta-thalassemia genes among the pediatric patients in our study.
Keywords: mutation analysis, beta-globin gene, Thai children
Introduction
Thalassemia is the most common inherited blood disorder in Southeast Asia and is caused by reduced or absent synthesis of the globin chains of hemoglobin (Hb) leading to imbalance of the globin chains.1,2 Beta-thalassemia is one of the major types of thalassemia and is caused by a mutation in the beta-globin gene (HBB) on chromosome 11. The clinical and hematological spectrum of beta-thalassemia ranges from silent carrier to clinically manifested conditions including severe transfusion dependent beta-thalassemia major and beta-thalassemia intermedia (TI).3,4 In Thailand, both beta-thalassemia and hemoglobin E (HbE) represent one of the most common forms distributed in all regions especially in Northeast Thailand. Most of the severe beta-thalassemia results from the interaction of beta-thalassemia and HbE.5,6
The molecular basis of thalassemia has been studied worldwide. More than 300 different beta-globin gene mutations have been characterized. Most of the beta-thalassemia mutations are caused by point mutations, small deletions or insertions within the coding regions and the exon-intron junctions. The types of the mutation are typically ethnic specific.7–9 In Thailand, the prevalence of beta-thalassemia carriers varies from 3%–9%.10 To date, more than 30 different mutations have been identified.11–13 The heterogeneity of the mutations makes it difficult to identify the mutation in some beta-thalassemia patients. Various DNA analysis techniques such as dot blot analysis, reverse dot blot, allele specific amplification using amplification refractory mutation system (ARMS) or direct DNA sequencing have been widely used to identify beta-globin gene mutations.14–17
This study aimed to characterize the beta-globin gene mutations in 80 pediatric patients who carry beta-thalassemia mutations and were followed-up in Phramongkutklao Hospital, a tertiary care center for thalassemia patients from all regions, especially central Thailand.
Patients and methods
Patient selection
Eighty unrelated beta-thalassemia patients who attended the Hematology Clinic at the Department of Pediatrics, Phramongkutklao Hospital, Bangkok, Thailand, from January 2013 to December 2013 were enrolled in our study. The study protocol was approved by the Institutional Review Board of Phramongkutklao Hospital, Phramongkutklao College of Medicine, Thailand. Sixty-five patients had clinically manifested beta-thalassemia including 57 with beta-thalassemia/HbE and eight with homozygous or compound heterozygous beta-thalassemia. Fifteen patients had heterozygous beta-thalassemia. All patients were diagnosed at 18 years of age or less. Patients with homozygous beta-thalassemia and beta-thalassemia/HbE were clinically classified into severe transfusion dependent thalassemia major and mild TI based on criteria such as age at presentation, average Hb level at the steady state and transfusion frequency history, as previously described.18
Mutation analysis
After informed consent was obtained, a total of 80 peripheral blood ethylenediaminetetraacetic acid (EDTA) samples from all individuals were collected. Genomic DNA was extracted from peripheral blood lymphocytes using an AxyPrep™ blood genomic DNA miniprep kit according to manufacturer’s protocol. The beta-globin gene mutations were first characterized using two sets of allele specific polymerase chain reaction (PCR) or multiplex amplification refractory mutation system (M-ARMS) to detect seven common mutations in Chinese and Southeast Asian populations including codon 41/42 (-TCTT), codon 17 (A>T), nucleotide -28 (A>G), IVS-II-654 (C>T), codon 71/72 (+A), IVS-I-1 (G>T) and IVS-I-5 (G>C) as previously described.15,17 These mutations have been found to be responsible for 80%–90% of beta-thalassemia alleles in this region.19,20 Unknown beta-thalassemia genes were further characterized by direct DNA sequencing of all coding regions and exon-intron boundaries to detect uncommon point mutations and small rearrangements in the beta-globin gene according to protocols previously described elsewhere.16 Beta-thalassemia alleles that remained uncharacterized by the above M-ARMS and DNA sequencing methods were subsequently screened by gap-PCR to detect 3.4 kb deletion of the entire beta-globin gene, previously reported in Thai populations.21
Results
A total of 88 beta-thalassemia alleles from eight homozygous or compound heterozygous beta-thalassemia patients, 57 beta-thalassemia/HbE patients, and 15 heterozygous beta-thalassemia individuals were included in our study. All 80 subjects were from unrelated families and 93.8% (75/80) of subjects lived in Bangkok and other provinces in central Thailand. Of these 65 patients who had clinically manifested beta-thalassemia, 92.3% (60/65) and 7.7% (5/65) of patients presented with beta-thalassemia major and TI, respectively. In the 60 beta-thalassemia major patients, 86.7% (52/60) of patients had beta-thalassemia/HbE whereas only 13.3% (8/60) of patients had homozygous or compound heterozygous beta-thalassemia. Alternatively, all patients with homozygous or compound heterozygous beta-thalassemia and 91.2% (52/57) of beta-thalassemia/HbE patients in this study presented with severe transfusion dependent beta-thalassemia major. Concerning genotype of beta-thalassemia major patients, the most common genotype was codon 41/42 (-TCTT)/codon 26 (G>A) or betaE accounting for 40%, and the second most common was codon 17 (A>T)/betaE accounting for 18.3% of all beta-thalassemia major genotypes (Table 1 and Figure 1).
 | Table 1 Genotype of 65 clinically manifested beta-thalassemia patients |
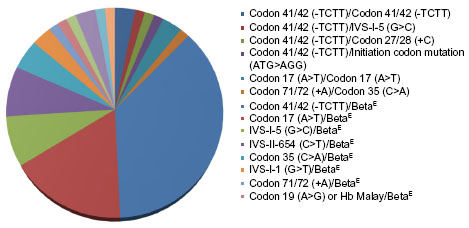 | Figure 1 Type of genotype in beta-thalassemia patients. |
To elucidate the molecular basis of these 88 beta-thalassemia alleles, we first characterized each patient’s DNA by two sets of M-ARMS. Seventy-five alleles (85.2%) were identified by this method including 33 alleles (37.5%) of codon 41/42 (-TCTT), 23 alleles (26.1%) of codon 17 (A>T), seven alleles (8%) of IVS-I-5 (G>C), six alleles (6.8%) of IVS-II-654 (C>T), four alleles (4.5%) of IVS-I-1 (G>T) and two alleles (2.3%) of codon 71/72 (+A) (Table 2). Interestingly, nucleotide -28 (A>G) was not identified in our study. In 13 alleles (14.8%), neither set revealed beta-thalassemia mutation. Thus, direct DNA sequencing of all three coding exons and flanking exon-intron junctions in the beta-globin gene was the next step to characterize these 13 alleles. Six uncommon mutations were detected in nine alleles (10.2%) including four alleles (4.5%) of codon 35 (C>A) and one allele (1.1%) for initiation codon mutation (ATG>AGG), codon 15 (G>A), codon 19 (A>G), codon 27/28 (+C) and codon 123/124/125 (-ACCCCACC), respectively (Figure 2). The remaining alleles, uncharacterized by M-ARMS and DNA sequencing, were further identified by gap-PCR to detect 3.4 kb deletion and this deletion was found in all four remaining alleles (4.5%).
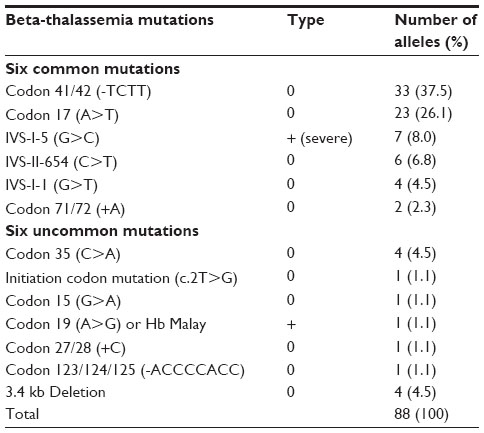 | Table 2 The frequency of beta-thalassemia mutations in 88 alleles |
 | Figure 2 Six uncommon mutations identified by direct DNA sequencing. |
In all, 100% of our 88 beta-thalassemia alleles were characterized by a combination of these techniques including two sets of M-ARMS, direct DNA sequencing and 3.4 kb deletion detection gap-PCR. Excluding the betaE-globin gene, 13 different beta-thalassemia mutations were encountered in the present study. The 4 bp deletion (-TCTT) in codons 41/42 was the most common mutation identified in our study and accounted for 37.5% of the alleles. The codon 17 (A>T) was the second most common and accounted for 26.1% of the alleles. Together, these two common mutations accounted for more than half (63.6%) of affected alleles. All other common mutations except nucleotide -28 (A>G) accounted for 21.6% of the alleles.
Discussion
This study provided useful information regarding the frequency distribution of beta-thalassemia mutations among pediatric patients especially in central Thailand since more than 90% of patients lived in Bangkok and other provinces in this region. In all, 92% of children who had clinically manifested beta-thalassemia had the beta-thalassemia major phenotype and more than 85% of pediatric patients with beta-thalassemia major had compound heterozygous beta-thalassemia and HbE, which is highly prevalent in Thailand.5,6,8,10 The frequency of the betaE-globin gene was not included in the analysis of beta-thalassemia mutation frequency since HbE can be easily detected by Hb electrophoresis.
Several methods can determine beta-thalassemia mutations.14–17 This study revealed that M-ARMS detected seven common mutations in Chinese and Southeast Asian populations and was able to detect 85.2% of the alleles as in previous reports.19,20 Since M-ARMS can detect only a given set of mutations specific to the primers employed, direct DNA sequencing is the next step to identify various point mutations and small rearrangements in the beta-globin gene. Six less frequent mutations were identified in 10.2% of the alleles. The disadvantage of DNA sequencing is that large deletions of the gene are undetectable. Thus, gap-PCR is the final step to detect 3.4 kb deletion, previously reported in Thai populations.21 A combination of these techniques could identify beta-thalassemia mutations in all 88 alleles (100%) of pediatric patients in our study.
Excluding the betaE-globin gene, 13 different beta-thalassemia mutations were detected in 80 unrelated patients. The two most common mutations detected were codon 41/42 (-TCTT) and codon 17 (A>T), accounting for 37.5% and 26.1% of the affected alleles, respectively. All other common mutations detected by M-ARMS accounted for 21.6% of the alleles. In comparison with the previous study of beta-thalassemia mutations in Thailand, this revealed that the frequency of beta-globin gene mutations identified in our study was different. First, the nucleotide -28 (A>G) was not identified in our study. The nucleotide -28 (A>G) is a beta+-thalassemia mutation causing the mild beta-thalassemia phenotype; thus, this mutation has been observed only in TI patients.22 This present study demonstrated that TI was the minority group (less than 10%) among pediatric patients with symptomatic beta-thalassemia. This may explain why this mutation was not observed in our study. Second, the frequency of other common mutations including codon 41/42 (-TCTT), codon 17 (A>T), IVS-II-654 (C>T), codon 71/72 (+A), IVS-I-1 (G>T) and IVS-I-5 (G>C) in our study was similar to other studies in central Thailand.8,10 However, allele frequency of this present study was different from studies in other parts of Thailand. Codon 41/42 (-TCTT) was the most common mutation identified in our population, who mainly lived in central Thailand. This frameshift mutation is also the most common mutation found in the other parts of Thailand, People’s Republic of China, and Southeast Asia.20 While codon 17 (A>T) was the second most common mutated allele in central, northern, and northeast Thailand, IVS-I-5 (G>C) was the second most common in southern Thailand.23 IVS-I-5 (G>C) was previously reported as the most common mutation among Thai Muslim patients in southern Thailand.24 Additionally, codon 71/72 (+A) accounted for 2.3% in the present study while this mutation accounted for 6% in north16 and 13.1% in northeast13 Thailand indicating a higher frequency of this mutation in this area.
Six uncommon mutations including codon 35 (C>A), codon 15 (G>A), codon 19 (A>G) or Hb Malay, codon 27/28 (+C), initiation codon mutation (ATG>AGG) and codon 123/124/125 (-ACCCCACC) were identified by direct DNA sequencing. Although no new mutation was detected, two rare mutations in Thailand, initiation codon mutation and 8 bp deletion in exon 3, were found in our study. Initiation codon mutation (ATG>AGG) was first described in a Chinese beta-thalassemia patient in 1990.25 In Thailand, this mutation was reported once in 2005 in two siblings with beta-thalassemia/HbE.26 Although this mutation has seriously affected beta-globin chain synthesis and most likely causes beta0-thalassemia phenotype, all three reported patients had the mild phenotype, which can be explained by co-inheritance with heterozygous 3.7 kb deletion alpha-thal-2 genotype among Chinese patient and the association with C>T polymorphism at -158(G)gamma-globin gene in the two Thai siblings. Our patient’s phenotype was transfusion dependent beta-thalassemia major, diagnosed at 9 months of age, whereas the genotype was compound heterozygous between codon 41/42 (-TCTT) and initiation codon mutation. Both mutated alleles cause the beta0-thalassemia phenotype. Eight-base deletion in exon 3 or codon 123/124/125 (-ACCCCACC) was first characterized in northeast Thai children with the severe beta-thalassemia/HbE phenotype.27 This deletion results in the synthesis of 135 amino acids betaX chain (beta-Khon Kaen), which are highly unstable and degraded soon after translation. Extensive alteration of amino acids at the codon 123 to 131 caused dominant inclusion body beta-thalassemia traits as in our patient.
Beta-thalassemia/HbE is a major thalassemia problem in Thailand and can be associated with various clinical phenotypes ranging from mild thalassemia intermedia to severe transfusion dependent thalassemia major.5,6,8,10 As in our study, 85% of beta-thalassemia major patients and 100% of TI patients had compound heterozygous beta-thalassemia and HbE. In beta-thalassemia major, the most common encountered genotype was codon 41/42 (-TCTT)/betaE (40%), followed by codon 17 (A>T)/betaE (18.3%), IVS-I-5 (G>C)/betaE (8.3%) and IVS-II-654 (C>T)/betaE (8.3%), which made up for 75% of all detected genotypes. Excluding the betaE-thalassemia mutation, all beta-thalassemia mutations in our symptomatic patients were classified as beta0 mutation except for codon 19 (A>G) or Hb Malay, classified as beta+ mutation and identified only in one patient.28
Interestingly, our patient with codon 19 (A>G)/betaE had transfusion dependent thalassemia major manifestation instead of mild TI. On the other hand, compound heterozygotes among four beta0 mutations including codon 41/42 (-TCTT), codon 17 (A>T), 3.4 kb deletion, IVSI-1 (G>T) and betaE genotype manifested as TI instead of beta-thalassemia major phenotype. This phenomenon can be explained by several genetic and nongenetic factors, which may play roles in determining the variability of the disease.5,22,29,30 However, the additional genetic analysis (genetic modifiers) was not performed in our study.
The concept of pre-implantation genetic diagnosis is to allow transfer of embryos to the uterus in assisted reproduction procedures. This technique is rapid and suitable as a noninvasive clinical tool for identifying genetic disorders for the purpose of reducing selective miscarriages such as thalassemia major.31,32 Our study revealed different beta-thalassemia mutations maybe clinically applied to preimplantation genetic protocol, permitting molecular genetic analysis to amplify a specific region on the beta-globin gene for a couple.
In conclusion, the present study demonstrates the heterogeneity of molecular defects causing beta-thalassemia in Thai children. All of the beta-thalassemia alleles have been characterized by a combination of techniques including M-ARMS, direct DNA sequencing, and gap-PCR for 3.4 kb deletion detection. Thirteen mutations accounted for 100% of the beta-thalassemia genes in our study. The frequency obtained should represent the frequency of beta-globin gene mutations among pediatric patients, who mainly lived in central Thailand.
Acknowledgment
This study was approved by and received funding from the Phramongkutklao College of Medicine.
Disclosure
The authors declare no conflicts of interest in this work.
References
Weatherall DJ. The thalassemia syndromes. Tex Rep Biol Med. 1980;40:323–333. | |
Vichinsky EP. Changing patterns of thalassemia worldwide. Ann N Y Acad Sci. 2005;1054:18–24. | |
Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61–76. | |
Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005; 353(11):1135–1146. | |
Fucharoen S, Winichagoon P. Haemoglobinopathies in southeast Asia. Indian J Med Res. 2011;134:498–506. | |
Fucharoen S, Ketvichit P, Pootrakul P, Siritanaratkul N, Piankijagum A, Wasi P. Clinical manifestation of beta-thalassemia/hemoglobin E disease. J Pediatr Hematol Oncol. 2000;22(6):552–557. | |
Akhavan-Niaki H, Derakhshandeh-Peykar P, Banihashemi A, et al. A comprehensive molecular characterization of beta thalassemia in a highly heterogeneous population. Blood Cells Mol Dis. 2011;47(1):29–32. | |
Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia: molecular biology and clinical medicine. Hemoglobin. 1997;21(4): 299–319. | |
Giardine B, van Baal S, Kaimakis P, et al. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum Mutat. 2007;28(2):206. | |
Wasi P, Pootrakul S, Pootrakul P, Pravatmuang P, Winichagoon P, Fucharoen S. Thalassemia in Thailand. Ann N Y Acad Sci. 1980;344: 352–363. | |
Fukumaki Y, Fucharoen S, Fucharoen G, et al. Molecular heterogeneity of beta-thalassemia in Thailand. Southeast Asian J Trop Med Public Health. 1992;23 Suppl 2:14–21. | |
Thein SL, Winichagoon P, Hesketh C, et al. The molecular basis of beta-thalassemia in Thailand: application to prenatal diagnosis. Am J Hum Genet. 1990;47(3):369–375. | |
Fucharoen S, Fucharoen G, Sriroongrueng W, et al. Molecular basis of beta-thalassemia in Thailand: analysis of beta-thalassemia mutations using the polymerase chain reaction. Hum Genet. 1989;84(1):41–46. | |
Mirasena S, Shimbhu D, Sanguansermsri M, Sanguansermsri T. Detection of beta-thalassemia mutations using a multiplex amplification refractory mutation system assay. Hemoglobin. 2008;32(4):403–409. | |
Bhardwaj U, Zhang YH, Lorey F, McCabe LL, McCabe ER. Molecular genetic confirmatory testing from newborn screening samples for the common African-American, Asian Indian, Southeast Asian, and Chinese beta-thalassemia mutations. Am J Hematol. 2005;78(4):249–255. | |
Sirichotiyakul S, Saetung R, Sanguansermsri T. Analysis of beta-thalassemia mutations in northern Thailand using an automated fluorescence DNA sequencing technique. Hemoglobin. 2003;27(2): 89–95. | |
Fucharoen S, Fucharoen G, Ratanasiri T, Jetsrisuparb A, Fukumaki Y. A simple non radioactive method for detecting beta-thalassemia/hbe disease: application to prenatal diagnosis. Southeast Asian J Trop Med Public Health. 1995;26 Suppl 1:278–281. | |
Ho PJ, Hall GW, Luo LY, Weatherall DJ, Thein SL. Beta-thalassaemia intermedia: is it possible consistently to predict phenotype from genotype? Br J Haematol. 1998;100(1):70–78. | |
Old JM, Khan SN, Verma I, et al. A multi-center study in order to further define the molecular basis of beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2001;25(4):397–407. | |
Kazazian HH Jr, Dowling CE, Waber PG, Huang S, Lo WH. The spectrum of beta-thalassemia genes in China and Southeast Asia. Blood. 1986;68(4):964–966. | |
Sanguansermsri T, Pape M, Laig M, Hundrieser J, Flatz G. Beta zero-thalassemia in a Thai family is caused by a 3.4 kb deletion including the entire beta-globin gene. Hemoglobin. 1990;14(2):157–168. | |
Nuntakarn L, Fucharoen S, Fucharoen G, Sanchaisuriya K, Jetsrisuparb A, Wiangnon S. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast Thailand. Blood Cells Mol Dis. 2009;42(1):32–35. | |
Nopparatana C, Panich V, Saechan V, et al. The spectrum of beta-thalassemia mutations in southern Thailand. Southeast Asian J Trop Med Public Health. 1995;26 Suppl 1:229–234. | |
Laosombat V, Nopparatana C, Wongchanchailert M, Wiriyasateinkul A. Molecular basis of beta-thalassemia in Thai Muslim patients in the the south of Thailand. Southeast Asian J Trop Med Public Health. 1997; 28 Suppl 3:104–105. | |
Lam VM, Xie SS, Tam JW, Woo YK, Gu YL, Li AM. A new single nucleotide change at the initiation codon (ATG----AGG) identified in amplified genomic DNA of a Chinese beta-thalassemic patient. Blood. 1990;75(5):1207–1208. | |
Viprakasit V, Chinchang W, Suwanthol L, Tanphaichitr VS. Common origin of a rare beta-globin initiation codon mutation (ATG-->AGG) in Asians. Clin Lab Haematol. Dec 2005;27(6):409–415. | |
Fucharoen G, Fuchareon S, Jetsrisuparb A, Fukumaki Y. Eight-base deletion in exon 3 of the beta-globin gene produced a novel variant (beta khon kaen) with an inclusion body beta-thalassemia trait. Blood. 1991;78(2):537–539. | |
Yang KG, Kutlar F, George E, et al. Molecular characterization of beta-globin gene mutations in Malay patients with Hb E-beta-thalassaemia and thalassaemia major. Br J Haematol. 1989;72(1):73–80. | |
Olivieri NF, Pakbaz Z, Vichinsky E. HbE/beta-thalassemia: basis of marked clinical diversity. Hematol Oncol Clin North Am. 2010;24(6): 1055–1070. | |
Rund D, Fucharoen S. Genetic modifiers in hemoglobinopathies. Cur Mol Med. 2008;8(7):600–608. | |
Nasri NW, Jamal AR, Abdullah NC, Razi ZR, Mokhtar NM. Preimplantation genetic diagnosis for beta-thalassemia using single-cell DNA analysis for codons 17 and 26 of beta-globin gene. Arch Med Res. 2009;40(1):1–9. | |
Hung CC, Chen SU, Lin SY, et al. Preimplantation genetic diagnosis of beta-thalassemia using real-time polymerase chain reaction with fluorescence resonance energy transfer hybridization probes. Anal Biochem. 2010;400(1):69–77. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.