Back to Journals » OncoTargets and Therapy » Volume 10
Molecular alterations and clinical prognostic factors for cholangiocarcinoma in Thai population
Authors Trachu N, Sirachainan E, Larbcharoensub N, Rattanadech W, Detarkom S, Monnamo N, Kamprerasart K, MunTham D, Sukasem C, Reungwetwattana T
Received 14 June 2017
Accepted for publication 4 September 2017
Published 11 October 2017 Volume 2017:10 Pages 4955—4968
DOI https://doi.org/10.2147/OTT.S143982
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
N Trachu,1,2 E Sirachainan,3 N Larbcharoensub,4 W Rattanadech,3 S Detarkom,3 N Monnamo,1 K Kamprerasart,4 D MunTham,5 C Sukasem,6,7 T Reungwetwattana3
1Research Center, Faculty of Medicine Ramathibodi Hospital, 2Molecular Medicine Program, Multidisciplinary Unit, Faculty of Science, 3Division of Medical Oncology, Department of Medicine, 4Division of Anatomical Pathology, Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, 5Section for Mathematic, Faculty of Science and Technology, Rajamangala University of Technology Suvarnabhumi, 6Division of Pharmacogenomics and Personalized Medicine, Department of Pathology, 7Laboratory for Pharmacogenomics, Somdech Phra Debaratana Medical Center (SDMC), Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Abstract: This study explores genomic alterations in cholangiocarcinoma (CCC) tissues in Thai patients. We identified and reviewed the records of patients who had been diagnosed with CCC and for whom sufficient tumor samples for DNA and RNA extraction were available in our database. The specimens were explored for EGFR, KRAS, BRAF, and PIK3CA mutations and ROS1 translocation in 81 samples. Immunohistochemistry staining for HER2, ALK, and Ki-67 expression was tested in 74 samples. Prevalence of EGFR, KRAS, and PIK3CA mutations in this study was 21%, 12%, and 16%, respectively. No BRAF V600 mutation or ROS1 translocation was found. Patients with T790M mutation had a significantly longer overall survival (18.84 months) than those with the other types of EGFR mutations (4.08 months; hazard ratio [HR]: 0.26, P=0.038) and also had a significantly lower median Ki-67 (22.5% vs 80%, P=0.025). Furthermore, patients with PIK3CA mutations had a significantly longer median progression-free survival (15.87 vs 7.01 months; HR: 0.46, P=0.043). Strongly positive HER2 expression was found in only 1 patient, whereas ALK expression was not found. The presence of EGFR and/or PIK3CA mutations implies that targeted drugs may provide a feasible CCC treatment in the future.
Keywords: cholangiocarcinoma, targeted therapy, gene alterations
Introduction
Biliary tract cancer (BTC) consists of both cholangiocarcinoma (CCC) and gallbladder cancer. Approximately 90% of BTCs are adenocarcinoma arising from the epithelial lining of the gallbladder and intrahepatic and extrahepatic bile duct. CCC is classified, according to its anatomical location in biliary tract, into 3 subtypes: intrahepatic, perihilar, and distal extrahepatic.
BTC shows differences in etiology, prevalence, and molecular alterations between Caucasian and Asian populations. BTC is relatively rare in Europe and the USA. Age-adjusted rates of CCC are reportedly lowest in non-Hispanic white people and black people (both 2.1 per 100,000) and highest in Hispanic and Asian populations (2.8–3.3 per 100,000).1,2 The highest rates are found in Eastern Asia especially the northeast of Thailand (85 per 100,000),3 whereas rates are low in South, Central, and Western Asia, as well as in Northern and Eastern Europe.4 The National Cancer Institute in Thailand estimated that liver and bile duct cancers were the most common cancers in men, with an estimated 8,000 new cases per year; the highest incidence was found in Khon Kaen, north-eastern region of Thailand.5
The etiology of CCC in Asian countries is liver fluke infestation especially Opisthorchis viverrini and Clonorchis sinensis. It induced chronic inflammation leading to oxidative DNA damage of the biliary epithelium and malignant transformation. C. sinensis infestation is common in rural area of Korea and China, whereas O. viverrini infestation is highly prevalent in the northeast of Thailand.6
Thailand has the highest incidence of intrahepatic CCC in the world, perhaps related to a tradition of eating raw fish, which may be contaminated with O. viverrini.7
Although surgery is the only curative treatment for CCC, the resection rate is quite low and variable, as most patients present with advanced disease. Median survival after CCC resection is 10–40 months. Patients with locally advanced or metastatic BTC have poor prognoses, with 5-year survival rates of 5%–10%.8 The response rate (RR) of first-line systemic chemotherapy (gemcitabine- or 5-fluorouracil [5-FU]-based regimens) is 10%–40%.9 Patients who received single-agent gemcitabine had median overall survival (mOS) of 6.5–11.5 months,10,11 whereas patients treated with 5-FU/leucovorin (LV) had mOS of 6–6.5 months.12–14 For patients who received combined 5-FU and cisplatin, mOS was 9.5–10 months.15–17 The other gemcitabine combination regimen was studied in a Phase II trial that showed an RR of 9%–36% and mOS of 11–15.4 months. In addition, a Phase III randomized study (ABC-02) showed longer overall survival (OS) from gemcitabine/cisplatin over gemcitabine alone and led to this combined regimen becoming a standard first-line treatment.18 However, survival of patients with advanced disease remains poor with the current treatments.19
In the era of individualized medicine and targeted therapy, the molecular pathogenesis of CCC is worthy of study. Established mutations and amplification of known oncogenes had been shown in previous studies, including various molecular differences between Caucasian and Asian populations; for example, 8%–22% of Caucasians with CCC showed BRAF mutations, whereas no BRAF mutations were seen in Asian patients, who had higher rates of KRAS and PIK3CA mutations and ROS1 gene rearrangements; however, EGFR mutation rates were similar in Asian and Caucasian patients (14%–17%).20–38
In our previous pilot study by Detarkom,39 we found that some clinical prognostic factors affected survival, including staging, Eastern Cooperative Oncology Group (ECOG) performance status, surgical resection, and carbohydrate antigen 19-9 (CA19-9) pretreatment level. The study showed a trend of better OS in a patient with strong ALK expression, but due to the shortage of tissue sample, fluorescent in situ hybridization (FISH) for ALK could not be performed. Therefore, in this study, we explored clinical factors more extensively to predict prognosis and further studied genomic alteration in CCC in Thai patients with an aim to develop new treatment for this lethal disease.
Materials and methods
Study cohort, data, and clinical characteristics
This study used a computerized search of the tumor registry database of Ramathibodi Hospital for patients treated from November 2007 to December 2013. The data were accessed using the International Classification of Disease-10 (ICD-10) and the database from the tumor bank of the Pathology Department. We selected patients who had been diagnosed with BTC, and from whom adequate tumor tissue for extracting DNA was available. We fully reviewed their medical records, with particular regard to the natural history of their disease including clinical and tumor characteristics (age, sex, smoking status, staging, tumor type, presenting symptoms, CA19-9 and carcinoma embryonic antigen [CEA] levels at diagnosis, viral hepatitis B surface antigen [HBsAg], and anti-hepatitis C virus [HCV] status, patient’s birthplace, resection procedure, lymph node status, and medical treatment).
This study was approved by the ethics committee of Ramathibodi Hospital, Mahidol University, Bangkok, Thailand (EC approval number 11-56-03). As data collection and further analyses were performed without disclosure of the identity and private information of patients, informed consents for the review of medical records and the use of archived tissue samples were not required by the ethics committee of Ramathibodi Hospital.
We categorized tumor into 2 groups (intrahepatic and hilar/extrahepatic/gallbladder) by Bismuth classification that defines intrahepatic as a tumor that is located in intrahepatic bile duct, hilar type is located from common hepatic duct to position of cystic duct, extrahepatic is located at common bile duct to ampulla of Vater, and gallbladder type is tumor located at gallbladder and cystic duct.
Age at diagnosis was divided into 4 ranges: ≤50, 51–60, 61–70, or ≥71 years. Staging of disease was performed by using American Joint Committee on Cancer (AJCC) TNM staging system (seventh edition, 2010) according to the diagnosis.
Performance status was evaluated by ECOG scale and criteria. ECOG criteria was defined as follows: 0= fully active, able to carry on all pre-disease performance without restriction; 1= restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature, eg, light house work, office work; 2= ambulatory and capable of all self-care but unable to carry out any work activities. Up and about more than 50% of waking hours; 3= capable of only limited self-care, confined to bed or chair more than 50% of waking hours; 4= completely disabled, cannot carry on any self-care. Totally confined to bed or chair; 5= death. We grouped patients into 2 groups as ECOG 0–1 and ECOG 2–4 for analyzing the data.
Presenting symptoms were categorized into 2 categories: asymptomatic or symptomatic with any symptoms (gastrointestinal-related symptoms and other symptoms that were not related to gastrointestine, eg, weight loss, back pain, and dyspnea).
Surgery was categorized into curative, diagnostic, or palliative procedure. Two categories of systemic chemotherapy were gemcitabine-based regimen (gemcitabine single agent, gemcitabine with cisplatin, and gemcitabine with carboplatin) or non-gemcitabine-based regimen (capecitabine, or 5-FU with LV). Lymph node status was obtained by pathological report from surgical specimen. HCV infection was evaluated by enzyme immunoassay to detect HCV antibodies. Hepatitis B virus (HBV) infection was evaluated by enzyme-linked immunosorbent assay (ELISA) to detect HBsAg. Blood for CA19-9&CEA was collected at the time of first visit. Regarding hometown, patients from northern and north-eastern part of Thailand were analyzed compared with those who came from central, western, and southern part of Thailand, because north and north-eastern part of Thailand has highest incident rate of CCC. The result of treatment was assessed at the time after complete treatment with surgery, radiation therapy, or chemotherapy by clinical or radiological examination with Response Evaluation Criteria In Solid Tumors (RECIST) criteria.
Molecular alterations study: formalin-fixed, paraffin-embedded (FFPE) tissue blocks from our archives, with tumor cellularity ≥50%, were routinely prepared by a pathologist at the Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University. This study was approved by the research ethics committee of Faculty of Medicine Ramathibodi Hospital, Mahidol University (approval ID 11-56-03).
We randomly selected 81 FFPE tissue blocks from the years 2010–2013 and prepared them for ALK, HER2, and Ki-67 immunohistochemistry (IHC) staining. Molecular testing for mutations in KRAS, BRAF, EGFR, and PIK3CA from DNA extractions and RNA extractions was performed for FISH (only for ALK+ samples), and ROS1 translocation tests.
Molecular testing for PIK3CA, BRAF, KRAS, and EGFR mutations
Paraffin-embedded tissue was dissolved in xylene and followed by 2 washes with 100% ethanol to remove residual xylene. Tissue was digested at 56°C for 1 hour, then at 90°C 1 hour, with 180 μL of ATL buffer and 20 μL of proteinase K. After digestion, 200 μL of alkaline lysis (AL) buffer was added. The solution was transferred into a spin column and washed with the wash buffers provided in the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The DNA was eluted in 30 μL of ATE buffer and was ready for use in amplification reactions or for storage at −20°C.
PIK3CA, BRAF, KRAS, and EGFR mutation testing by amplification-refractory mutation system-based quantitative polymerase chain reaction (ARMS-qPCR)
DNA samples were subjected to PIK3CA, BRAF, KRAS, and EGFR mutant analysis using AmoyDx PIK3CA 5 Mutations, BRAF V600 Mutations, KRAS 7 Mutations, and EGFR 29 Mutations Detection Kits (Amoy Diagnostics, Xiamen, China). These kits employ ARMS-real-time (RT) PCR technology to detect 5 common mutations in the PIK3CA gene, V600 mutation in the BRAF gene, 7 mutations in KRAS codons 12 and 13, and 29 mutations in EGFR gene (Table S1). The experiments and analyses were performed according to the manufacturer’s instructions, using Bio-Rad CFX96 RT-PCR (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Sanger sequencing for EGFR mutations
We assembled 10 ng DNA with 10 μL AmpliTaq Gold® PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) and 10 μM forward and reverse EGFR-specific primers (Table S2). PCRs were amplified, and the PCR product was then purified. Sequencing reactions were performed using chain-terminating dideoxynucleotides (BigDye® 1.0; Thermo Fisher Scientific) and loaded into an automated ABI 310 sequencer. Eventually, the data were analyzed by Sequencer 3.1.1 Software (Thermo Fisher Scientific).
Droplet digital PCR (ddPCR) for EGFR mutations
ddPCR reagents were ordered from Bio-Rad Laboratories Inc. Combined nucleic DNA and primer/probe mixes for EGFR T790M were custom made by Thermo Fisher Scientific. PCRs were performed from a DNA template, 1× ddPCR Mastermix (Bio-Rad Laboratories Inc.), TaqMan probe, and 20× custom primers made specifically for each assay. Each ddPCR mix was loaded into the wells of a droplet generator cartridge (Bio-Rad Laboratories Inc.). The target DNA and background DNA were randomly distributed in droplets, which were transferred to a 96-well PCR plate. The plate was sealed and subjected to the PCR protocol. The 96-well PCR plate was loaded into the QX-100 droplet reader (Bio-Rad Laboratories Inc.). Data were read and analyzed by QuantaSoft analysis software (Bio-Rad Laboratories Inc.).
ROS1 translocation testing by RT-PCR
Paraffin was removed from FFPE tissue sections by treatment with xylene. Samples were incubated at 56°C for 15 min; then, at 80°C for15 min with lysis buffer, which contained proteinase K, treat lysate with DNase was then mixed with buffer red blood cell and ethanol. The solution was applied to an RNeasy MinElute spin column. RNA was eluted into 14 μL of RNase-free water and was ready for use or for storage at −80°C.
We evaluated FFPE tumor samples for ROS1 fusion using the AmoyDxROS1 Gene Fusions Detection Kit (Amoy Diagnostics). About 50 ng/mL of RNA OD260/OD280 value (1.9–2.0 of RNA) sample was used for reverse transcription and RT-PCR of 4 reactions of ROS1 Fusion Gene Detection Kit according to the manufacturer’s instruction. This kit detected 14 ROS1 gene fusions with various spliced genes and exons. We analyzed reaction sample reference gene Ct value ≤20. If sample 6-carboxyfluorescein (FAM) Ct value was <30, the sample was considered positive for one of the variants detected by reaction mixture.
IHC staining
All IHC staining was performed on 4 μm-thick FFPE tissue sections. The slides were deparaffinized, and antigen retrieval was performed. We identified ALK, HER2, and Ki-67 expression by IHC using D5F3, HER2/neu, and Ki-67 antibodies, respectively, on the sections. For ALK, FFPE tissues were sectioned at 4 μm thickness and stained with anti-ALK rabbit monoclonal antibody (clone D5F3; 1:20 dilution; Ventana Medical Systems, Tucson, AZ, USA), using the Optiview DAB IHC Detection Kit and Optiview Amplification Kit with the Ventana Benchmark XT Stainer (Ventana Medical Systems) according to the manufacturer’s protocol. Immunoreactivity was scored as follows: 0, no staining; 1+, faint cytoplasmic staining; 2+, moderate cytoplasmic staining; 3+, strong granular cytoplasmic staining in ≥10% of tumor cells. Immunoreactivity was evaluated as positive or negative according to the manufacturer’s protocol. If the specimen had positive IHC staining for ALK, we then performed FISH to confirm ALK rearrangement.
Tissue sections were IHC stained for HER2 on BenchMark XT IHC/ISH staining module (Ventana Medical Systems), using the technical protocol XT UltraView DAB V3 by incubation with anti-HER2/neu (4B5) rabbit monoclonal primary antibody. Antigen detection was carried out using UltraView Universal DAB IHC Detection Kit (Ventana Medical Systems). IHC staining was assessed and scored by a pathologist. HER2 expression scores of 0 and 1+ were considered to be HER2 negative, and 2+ and 3+ as HER2 positive. A standard criterion for HER2 scoring was utilized.40
Tissue sections were IHC stained for Ki-67 on a BenchMark XT IHC/ISH staining module (Ventana Medical Systems) using the technical protocol XT Ultra View DAB V3 by incubation with Confirm anti-Ki-67 (30-9) rabbit monoclonal primary antibody. Antigen detection was carried out using UltraView Universal DAB IHC Detection Kit. IHC staining was assessed and scored by a pathologist. The labeling index of the Ki-67 in each tumor was estimated as a percentage of positive cells out of 100–1,000 counted tumor cells.
Statistical analysis
Statistical analyses were performed using STATA software v.13 (StataCorp LP, College Station, TX, USA). OS and progression-free survival (PFS) were calculated and censored on January 31, 2015. OS and PFS curves were drawn using the Kaplan–Meier method. The log-rank test was used to compare survival rates by each variable. Univariate analysis of OS and PFS used the Cox proportional hazards regression model. Significantly, prognostic factors were included in subsequent multivariate analyses. P≤0.05 was considered significant. The correlation between molecular alteration and survival was compared by log-rank test. Mean Ki-67 in each molecular alteration group was compared by the Mann–Whitney U test.
Results
EGFR mutation
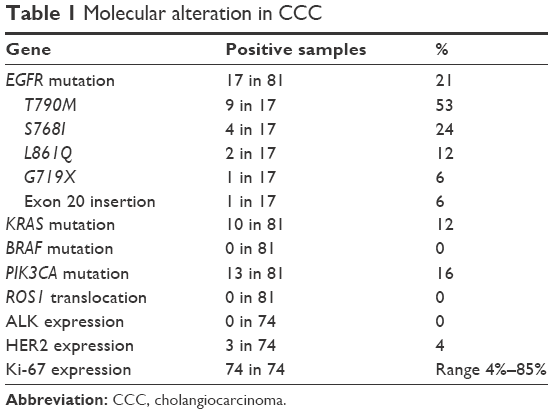
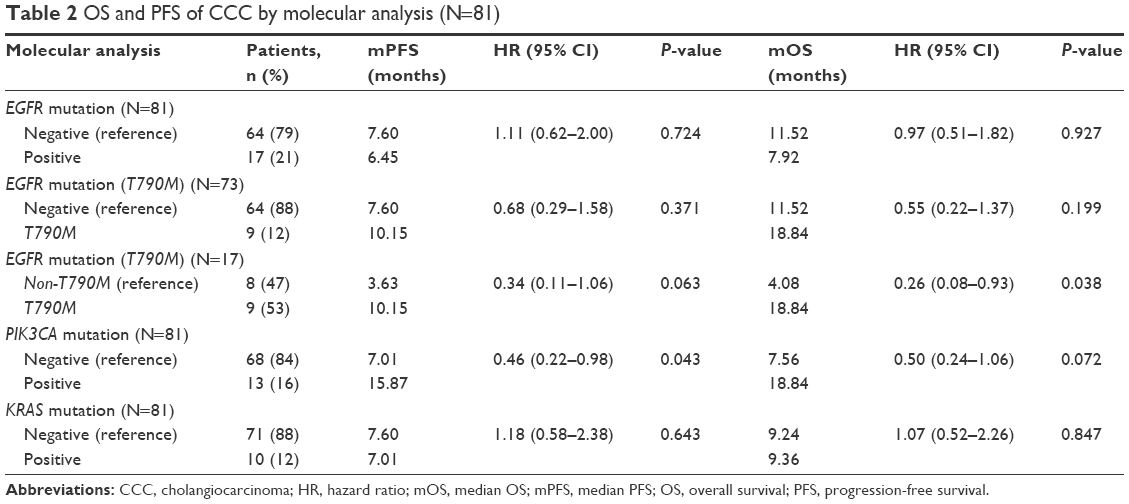
We found that 17 out of 81 samples (21%) were positive for EGFR mutations, comprising 9 with T790M, 4 with S768I, 2 with L861Q, 1 with G719X, and 1 with insertion (Table 1). The EGFR+ group showed a nonsignificant trend of longer mOS than the EGFR− group (7.92 vs 11.52 months, P=0.927) (Table 2 and Figure 1A). Median PFS (mPFS) was comparable between the EGFRMut+ and EGFRMut− groups (6.45 vs 7.60 months, P=0.724; Table 2 and Figure 2A). Patients with T790MMut+ mutations group showed a nonsignificant trend of longer mOS (18.84 vs 11.52 months, P=0.199) and mPFS (10.15 vs 7.60 months, P=0.371; Table 2 and Figures 1B and 2B) than the EGFRMut− group. However, patients with T790M mutations had a significant longer mOS (18.84 months) than the non-T790M mutation group (4.08 months, P=0.038; Table 2 and Figure 1C); the T790M group also had a trend of longer mPFS (10.15 vs 3.63 month, 0.063; Table 2 and Figure 2C).
 | Table 1 Molecular alteration in CCC |
 | Table 2 OS and PFS of CCC by molecular analysis (N=81) |
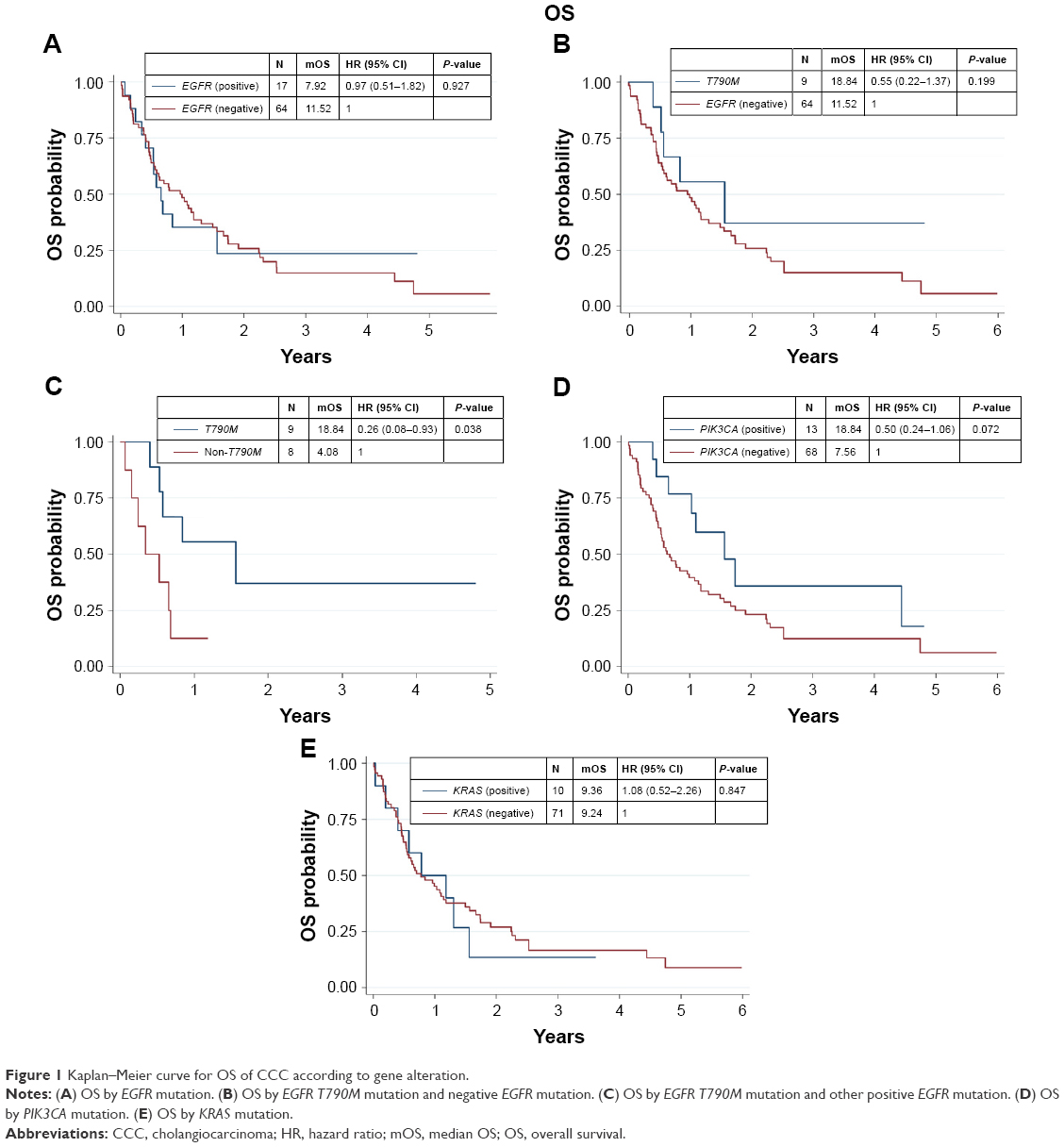 | Figure 1 Kaplan–Meier curve for OS of CCC according to gene alteration. |
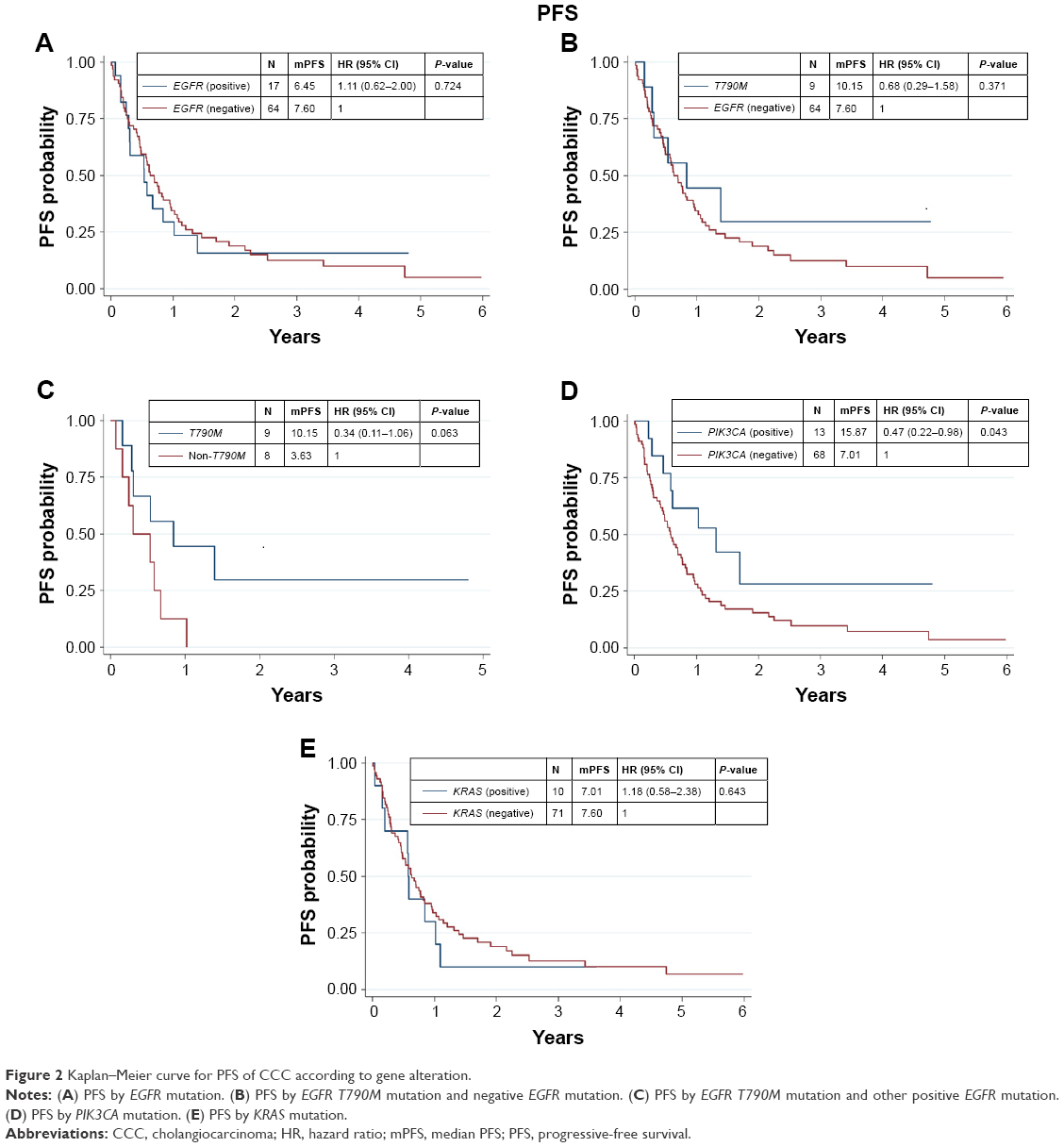 | Figure 2 Kaplan–Meier curve for PFS of CCC according to gene alteration. |
We further validated the positive results of EGFRMut+ mutation results by the Sanger direct sequencing method which is a gold standard method for mutation analysis, and by ddPCR, which is a highly sensitive method for detecting low percentages of gene mutations (0.1%–5% of gene frequency) compared with ARMS-qPCR (1%–5% of frequency).41 We found 100% concordance of EGFR T790M mutation by ddPCR, compared with qRT-PCR, but only 5 of 9 (55.55%) T790M mutations were detected by direct sequencing (Table 3 and Figure 3). We did not confirm exon 20 insertion and L861Q in ddPCR, because we did not have enough DNA. However, we confirmed L861Q with Sanger direct sequencing, but unfortunately we did not find it.
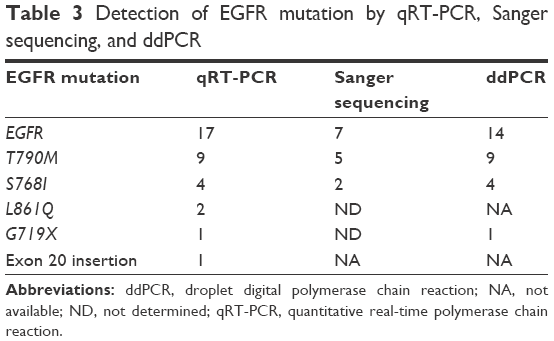 | Table 3 Detection of EGFR mutation by qRT-PCR, Sanger sequencing, and ddPCR |
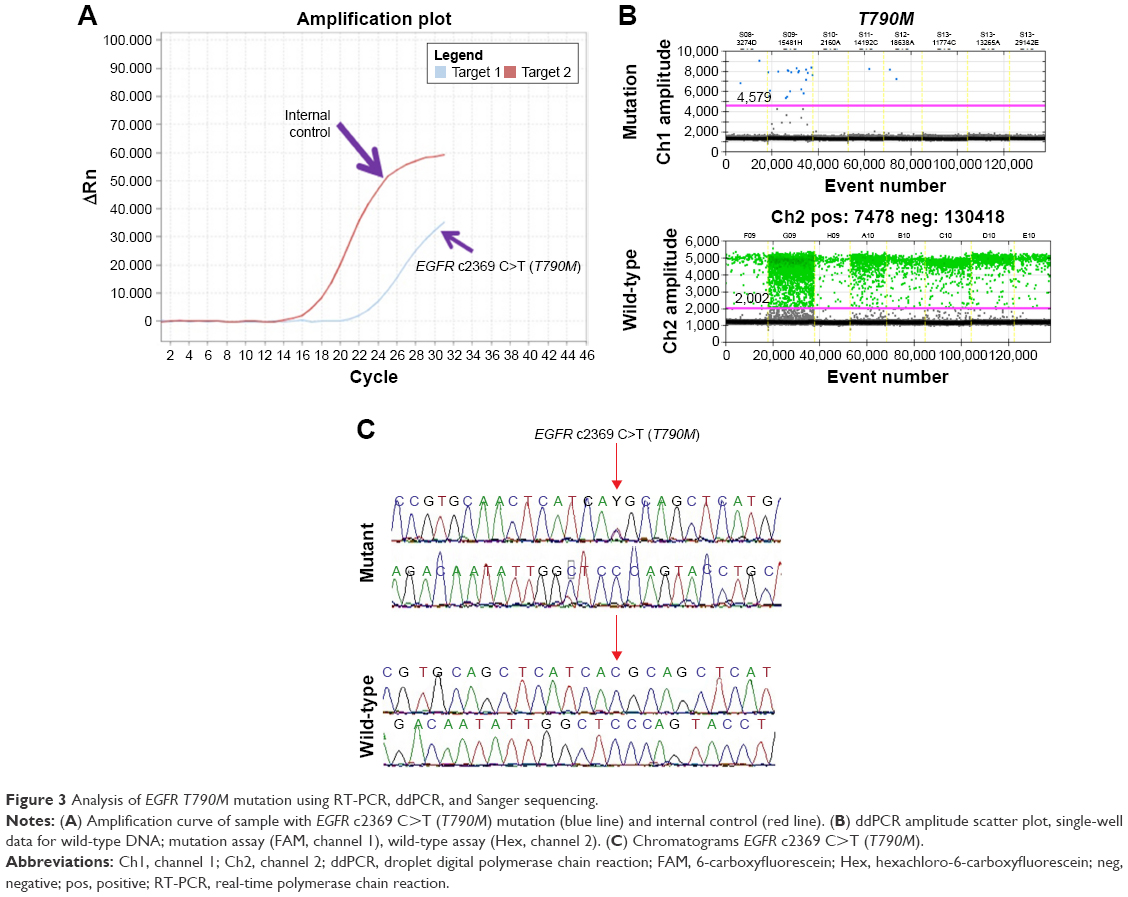 | Figure 3 Analysis of EGFR T790M mutation using RT-PCR, ddPCR, and Sanger sequencing. |
PIK3CA mutation
In 81 samples, 13 (16%) were positive for PIK3CA mutations (PIK3CAMut+), which comprised 6 with E545K, 3 with E542K, 3 with H1047R, and 1 with double mutations of E542K and E545K (Table 1). The PIK3CAMut+ had a trend of longer mOS than the PIK3CAMut− group (18.84 vs 7.56 months, P=0.072) and significantly longer mPFS than the PIK3CAMut− group (15.87 vs 7.01 months, P=0.043; Table 2 and Figures 1D and 2D).
KRAS mutation, BRAF mutation, and ROS1 translocation
We found that 10 out of the 81 samples (12%) were positive for KRAS mutation (KRASMut+), comprising 2 with G12Asp, 1 with G12Ala, 3 with 12V, 1 with G12S, 1 with G13Asp, and 1 with double mutations of G12Asp and G12 (Table 1). The KRASMut+ and KRASMut− groups did not significantly differ in mOS or mPFS at P<0.05 (Table 2 and Figures 1E and 2E). We detected no BRAF mutation or ROS1 translocation in the 81 samples (Table 1).
Clinical characteristics and prognostic factors based on significant gene alterations
The clinical characteristics and clinical data categorized based on each molecular alterations (EGFR, EGFR T790M, PIK3CA, and KRAS) of CCC patients were similar as listed in Table 4, except staging that was significantly different based on KRAS mutation and CEA baseline level at cut point of 15 μg/L was significantly different based on PIK3CA mutation.
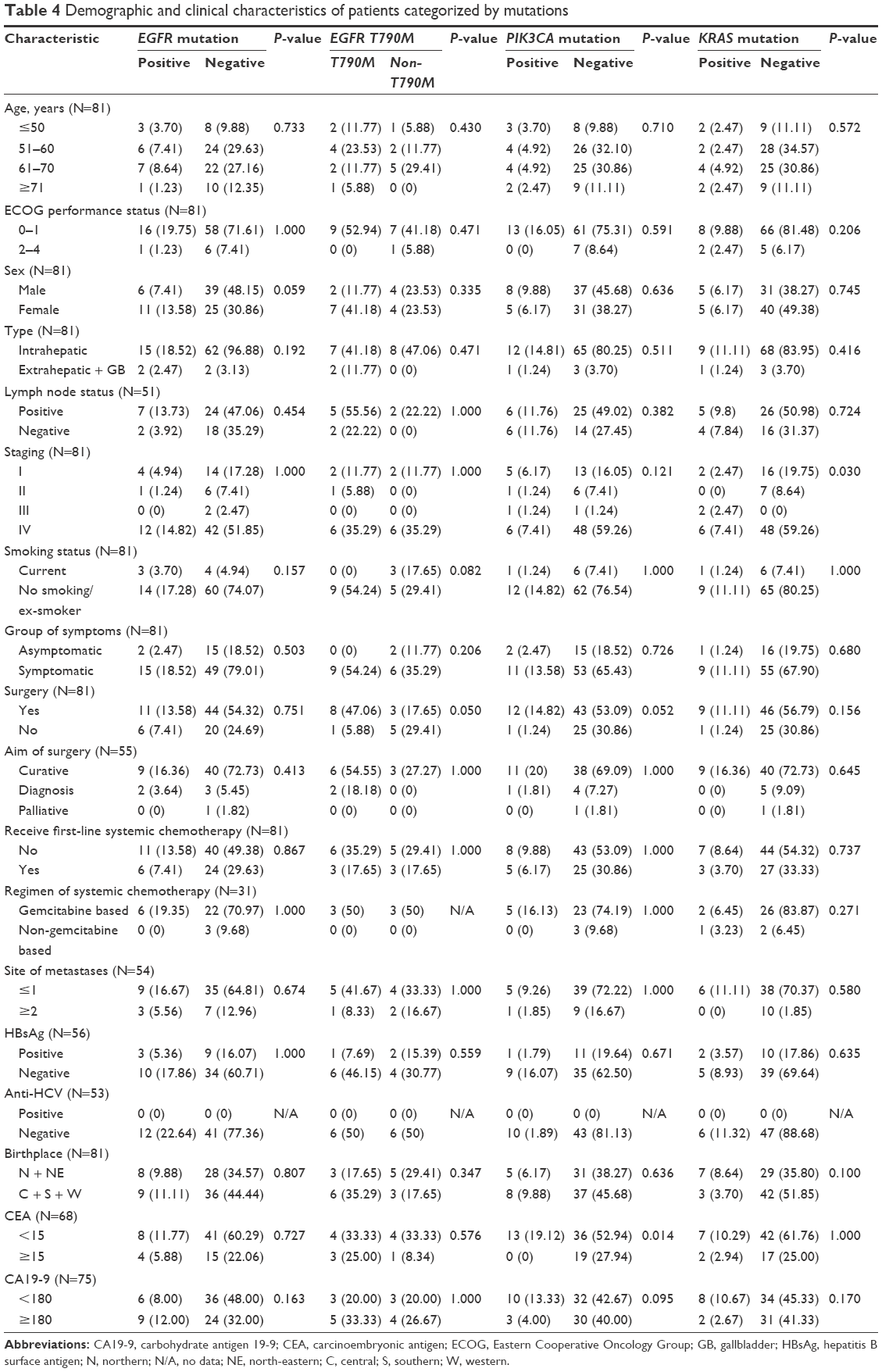 | Table 4 Demographic and clinical characteristics of patients categorized by mutations |
IHC
Not all 81 patients were tested for ALK expression due to the exhaustion of tumor tissue. We tested 74 samples for ALK expression, all of which showed negative ALK expression (Table 1). We found strongly positive staining (IHC 2+ and 3+) for HER2 expression in 3 out of 74 (4%) samples. Another 9 samples had IHC 1+ staining for HER2 (Table 1). Median Ki-67 was 42.5% (interquartile range: 4%–85%). The EGFRMut+ and EGFRMut− groups did not significantly differ in median Ki-67 (65% vs 40%, P=0.121). However, T790M samples had a significantly lower median Ki-67 than did samples without T790M mutations (22.5% vs 80%, P=0.025). The PIK3CAMut+ group had lower median Ki-67 but not significantly so (25% and 47.5%, P=0.273). The KRASMut+ and KRASMut− groups did not significantly differ in median Ki-67 (22.5% vs 45%, P=0.260; Table 5).
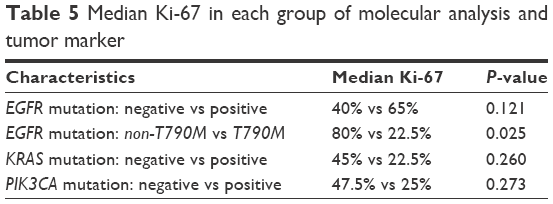 | Table 5 Median Ki-67 in each group of molecular analysis and tumor marker |
Discussion
CCC is a lethal malignancy that usually presents at an advanced stage. Due to the difficulty of early diagnosis and limited effective treatment, CCC has a poor prognosis. Few new treatments seem to be in development or in trials, because of the lack of clinical data and oncogenic data. However, as most relevant studies show differences in clinical characteristics and genetic alterations in Western and Asian populations, we have therefore investigated the clinical data and genomic alterations in Thai patients.
In our study, we found 21% of Thai patients with CCC had EGFRMut+ tumor tissues, which was higher than that seen in other populations investigated in previous studies.32,42–44 Types of EGFR mutation included 1 case in exon 18 (G719X), 13 cases in exon 20 (9 of T790M, 4 of S768I), and 3 cases in exon 21 (2 of L861Q and 1 of insertion). Consistent with a Taiwan study,43 no exon 19 mutation was identified, but contrast to Italian study,42 which found an exon 19 substitution (K757R) in 1 out of 40 samples. Interestingly, we found a high rate of T790M mutation (50% of EGFR mutation), and this group had significant longer OS and a trend of longer PFS than did the non-T790M group. However, the study by Chang et al43 reported EGFR mutation to be the strongest independent predictor of shorter OS. In addition to longer survival, patients with the T790M mutation in our study also had a significant lower median Ki-67 compared with the non-T790M group – interestingly, as we know that Ki-67 indicates cell proliferation.
To our knowledge, no study has previously reported this correlation between T790M mutation and clinicopathological factors or prognosis in patients with CCC.
Approximately 12% of patients had KRAS mutations, most of which were located in codon 12 and only 1 in codon 13. One of the previous studies reported 16% of KRAS mutation in codon 12 in Thai population.45 Other populations have shown a reported 13%–50% prevalence of KRAS mutations in BTCs.22,36,37,43,46 BRAF mutation was not detected in this study, which was similar to the report by Xu et al36 in a Chinese population, whereas Tannapfel et al22 demonstrated 22% (15/69) had positive BRAF mutations but found no significant correlation with the other clinicopathological factors and patient survival. We could not find the ROS1 translocation in our Thai CCC cohort, although a 2011 study found 8.7% of ROS1 translocations in an Asian population.35
PIK3CA mutation was rare in a previous study; Riener et al47 reportedly found it in only 1 out of 11 (9%) intrahepatic CCC samples and 1 out of 23 (4%) in gallbladder carcinoma samples. Another study from China showed that 32.4% of patients with CCC had PIK3CA mutations.36 However, our study is the first to report the prevalence of PIK3CA mutation in Thai patients with CCC. Interestingly, the PIK3CAMut+ group had significantly longer mPFS than did the PIK3CAMut− group. However, no previous study had analyzed patient survival in this group, although Xu et al36 found that PIK3CAMut− CCC was detected at a more advanced stage and in more aggressive forms.
We identified HER2 expression in only 3 out of 74 samples (4%), whereas Nakazawa et al26 identified HER2 overexpression in 15.7% of gallbladder carcinoma samples and 5.1% of extrahepatic bile duct carcinoma samples. We found strongly positive (3+) HER2 in 1 sample, from a patient who was diagnosed with early-stage disease and underwent curative surgery; 4 months later, he developed metastatic disease at lung, liver, adrenal gland, and lymph nodes. He received palliative chemotherapy, but unfortunately the disease was very aggressive, and his OS was 6 months after his diagnosis. This may imply that high HER2 expression is associated with more aggressive tumors.
IHC for Ki-67 showed a median Ki-67 of 42.5%. A previous study showed that high Ki-67 expression was correlated with advanced stage disease and could be used as a prognostic biomarker for CCC.48 We also found low median Ki-67 (22.5%) in the T790M mutation group, which correlated with longer survival.
We have validated our study’s results by both Sanger direct sequencing technique and ddPCR technique; we found 100% concordance between ARMS-RT-PCR and ddPCR. This result was also similar to that of Zhang et al41 in a Chinese population. However, we found only 55.6% concordance between Sanger direct sequencing technique and RT-PCR technique. Previous studies found that RT-PCR was significantly more sensitive in detecting mutation than Sanger DNA sequencing. Each technique has different limitations in detecting solid tumor mutations; for example, Sanger direct sequencing technique can detect 15%–20% mutant alleles, whereas ddPCR can detect low abundance mutations, present in only 0.02% of alleles.49,50 The ddPCR is a new technology to detect invasive genotyping of cfDNA in plasma, which can thus help detect acquired resistance in lung cancer.51–53
Our study had some limitations. First, because of the retrospective nature of the study, some data had been missing. Second, archival tissue specimens may suffer DNA damage, which can affect genomic alteration testing.
Conclusion
Cancer genetics may guide direct therapeutic decision in CCC, including the use of targeted drugs. EGFR mutations and PIK3CA mutations may imply and guide targeted therapy for CCC in the future, but due to the small sample size of this study, thus, we need to further explore in the larger population. Our study also suggested ARMS-RT-PCR or ddPCR for detecting low percentage of gene mutations.
Acknowledgments
The abstract of this paper (abstract no 2383) was presented at the European Cancer Congress 2015 as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in the European Journal of Cancer: http://www.ejcancer.com/article/S0959-8049(16)31299-0/abstract. This study was supported by grants from Ramathibodi Comprehensive Cancer Center, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Thailand. This work is supported by The Thailand Research Fund and Medical Research Council-UK (Newton Fund). Project no DBG 598006.
Disclosure
The authors report no conflicts of interest in this work.
References
Tyson GL, El-Serag HB. Risk factors for cholangiocarcinoma. Hepatology. 2011;54(1):173–184. | ||
Shaib YH, El-Serag HB, Davila JA, Morgan R, McGlynn KA. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case-control study. Gastroenterology. 2005;128(3):620–626. | ||
Oh JK, Weiderpass E. Infection and cancer: global distribution and burden of diseases. Ann Glob Health. 2014;80(5):384–392. | ||
Jemal ABF, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Petcharin S, Pattarawin A. Cancer incidence and leading sites. In Cancer In Thailand, Vol V; 2001–2003. Ministry of Public Health, Bangkok; 2010. | ||
Shin HR, Oh JK, Masuyer E, et al. Epidemiology of cholangiocarcinoma: an update focusing on risk factors. Cancer Sci. 2010;101(3):579–585. | ||
Sripa B, Kaewkes S, Sithithaworn P, et al. Liver fluke induces cholangiocarcinoma. PLoS Med. 2007;4(7):e201. | ||
Anderson CD, Pinson CW, Berlin J, Chari RS. Diagnosis and treatment of cholangiocarcinoma. Oncologist. 2004;9(1):43–57. | ||
Butthongkomvong K, Sirachainan E, Jhankumpha S, Kumdang S, Sukhontharot OU. Treatment outcome of palliative chemotherapy in inoperable cholangiocarcinoma in Thailand. Asian Pac J Cancer Prev. 2013;14(6):3565–3568. | ||
Raderer M, Hejna MH, Valencak JB, et al. Two consecutive phase II studies of 5-fluorouracil/leucovorin/mitomycin C and of gemcitabine in patients with advanced biliary cancer. Oncology. 1999;56(3):177–180. | ||
Penz M, Kornek GV, Raderer M, et al. Phase II trial of two-weekly gemcitabine in patients with advanced biliary tract cancer. Ann Oncol. 2001;12(2):183–186. | ||
Glimelius B, Hoffman K, Sjödén PO, et al. Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann Oncol. 1996;7(6):593–600. | ||
Falkson G, MacIntyre JM, Moertel CG. Eastern cooperative oncology group experience with chemotherapy for inoperable gallbladder and bile duct cancer. Cancer. 1984;54(6):965–969. | ||
Choi CW, Choi IK, Seo JH, et al. Effects of 5-fluorouracil and leucovorin in the treatment of pancreatic-biliary tract adenocarcinomas. Am J Clin Oncol. 2000;23(4):425–428. | ||
Thongprasert S. The role of chemotherapy in cholangiocarcinoma. Ann Oncol. 2005;16(suppl 2):ii93–ii96. | ||
Ducreux M, Rougier P, Fandi A, et al. Effective treatment of advanced biliary tract carcinoma using 5-fluorouracil continuous infusion with cisplatin. Ann Oncol. 1998;9(6):653–656. | ||
Taïeb J, Mitry E, Boige V, et al. Optimization of 5-fluorouracil (5-FU)/cisplatin combination chemotherapy with a new schedule of leucovorin, 5-FU and cisplatin (LV5FU2-P regimen) in patients with biliary tract carcinoma. Ann Oncol. 2002;13(8):1192–1196. | ||
Valle J, Wasan H, Palmer DH, et al; ABC-02 Trial Investigators. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;2010(362):1273–1281. | ||
Valle JW. Advances in the treatment of metastatic or unresectable biliary tract cancer. Ann Oncol. 2010;21(suppl 7):vii345–vii348. | ||
Terada T, Ashida K, Endo K, et al. c-erbB-2 protein is expressed in hepatolithiasis and cholangiocarcinoma. Histopathology. 1998;33(4):325–331. | ||
Tannapfel A, Benicke M, Kataline A. Frequency of p16INK4A alterations and k-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut. 2000;47(5):721–727. | ||
Tannapfel A, Sommerer F, Benicke M, et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 2003;52(5):706–712. | ||
Aishima SI, Taguchi KI, Sugimachi K, Shimada M, Sugimachi K, Tsuneyoshi M. c-erbB-2 and c-Met expression relates to cholangiocarcinogenesis and progression of intrahepatic cholangiocarcinoma. Histopathology. 2002;40(3):269–278. | ||
Endo K, Yoon BI, Pairojkul C, Demetris AJ, Sirica AE. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology. 2002;36(2):439–450. | ||
Rashid A, Ueki T, Gao Y-T, et al. K-ras mutation, p53 overexpression, and microsatellite instability in biliary tract cancers: a population-based Study in China. Clin Cancer Res. 2002;8(10):3156–3163. | ||
Nakazawa K, Dobashi Y, Suzuki S, Fujii H, Takeda Y, Ooi A. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol. 2005;206(3):356–365. | ||
Ooi A, Suzuki S, Nakazawa K, et al. Gene amplification of Myc and its coamplification with ERBB2 and EGFR in gallbladder adenocarcinoma. Anticancer Res. 2009;29(1):19–26. | ||
Kawamoto T, Krishnamurthy S, Tarco E, et al. HER receptor family: novel candidate for targeted therapy for gallbladder and extrahepatic bile duct cancer. Gastrointest Cancer Res. 2007;1(6):221–227. | ||
Kim HJ, Yoo TW, Park DI, et al. Gene amplification and protein overexpression of HER-2/neu in human extrahepatic cholangiocarcinoma as detected by chromogenic in situ hybridization and immunohistochemistry: its prognostic implication in node-positive patients. Ann Oncol. 2007;18(5):892–897. | ||
Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98(2):418–425. | ||
Harder J, Waiz O, Otto F, et al. EGFR and HER2 expression in advanced biliary tract cancer. World J Gastroenterol. 2009;15(36):4511–4517. | ||
Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol. 2010;28(21):3531–3540. | ||
Pignochino Y, Sarotto I, Peraldo-Neia C, et al. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer. 2010;10:631. | ||
Shafizadeh N, Grenert JP, Sahai V, Kakar S. Epidermal growth factor receptor and HER-2/neu status by immunohistochemistry and fluorescence in situ hybridization in adenocarcinomas of the biliary tree and gallbladder. Hum Pathol. 2010;41(4):485–492. | ||
Gu TL, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6(1):e15640. | ||
Xu RF, Sun JP, Zhang SR, et al. KRAS and PIK3CA but not BRAF genes are frequently mutated in Chinese cholangiocarcinoma patients. Biomed Pharmacother. 2011;65(1):22–26. | ||
O’Dell MR, Huang JL, Whitney-Miller CL, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72(6):1557–1567. | ||
Dai R, Li J, Fu J, et al. The tyrosine kinase c-Met contributes to the pro-tumorigenic function of the p38 kinase in human bile duct cholangiocarcinoma cells. J Biol Chem. 2012;287(47):39812–39823. | ||
Detarkom S. A Pilot Study of Molecular Alterations and the Clinical Prognostic Factors of Cholangiocarcinoma in Thai Population. Ramathibodi Hospital, Mahidol. Eur J Cancer. 2015;51(2):e9. | ||
Wolff AC, Hammond ME, Hicks DG, et al; American Society of Clinical Oncology; College of American Pathologists. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. | ||
Zhang B, Chun-Wei X, Yun S, et al. Comparison of droplet digital PCR and conventional quantitative PCR for measuring EGFR gene mutation. Exp Ther Med. 2015;9(4):1383–1388. | ||
Leone F, Cavalloni G, Pignochino Y, et al. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin Cancer Res. 2006;12(6):1680–1685. | ||
Chang YT, Chang MC, Huang KW, Tung CC, Hsu C, Wong JM. Clinicopathological and prognostic significances of EGFR, KRAS and BRAF mutations in biliary tract carcinomas in Taiwan. J Gastroenterol Hepatol. 2014;29(5):1119–1125. | ||
Gwak GY, Yoon JH, Shin CM, et al. Detection of response-predicting mutations in the kinase domain of the epidermal growth factor receptor gene in cholangiocarcinomas. J Cancer Res Clin Oncol. 2005;131(10):649–652. | ||
Wattanasirichaigoon S, Tasanakhajorn U, Jesadapatarakul S. The incidence of K-ras codon 12 mutations in cholangiocarcinoma detected by polymerase chain reaction technique. J Med Assoc Thai. 1998;81(5):316–323. | ||
Voss JS, Holtegaard LM, Kerr SE, et al. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol. 2013;44(7):1216–1222. | ||
Riener MO, Bawohl M, Clavien PA, Jochum W. Rare PIK3CA hotspot mutations in carcinomas of the biliary tract. Genes Chromosomes Cancer. 2008;47(5):363–367. | ||
Zhao W, Zhang B, Guo X, et al. Expression of Ki-67, Bax and p73 in patients with hilar cholangiocarcinoma. Cancer Biomark. 2014;14(4):197–202. | ||
Zhang H, Zheng X, Ji T, et al. Comparative screening of K-ras mutations in colorectal cancer and lung cancer patients using a novel real-time PCR with ADx-K-ras kit and Sanger DNA sequencing. Cell Biochem Biophys. 2012;62(3):415–420. | ||
Bárbara A, Esther C, Ana SG, et al. A comparison of EGFR mutation testing methods in lung carcinoma: direct sequencing, real-time PCR and immunohistochemistry. PLoS One. 2012;7(8):e43842. | ||
Xu Q, Zhu Y, Bai Y, et al. Detection of epidermal growth factor receptor mutation in lung cancer by droplet digital polymerase chain reaction. Onco Targets Ther. 2015;8:1533–1541. | ||
Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20(6):1698–1705. | ||
Hudecova I. Digital PCR analysis of circulating nucleic acids. Clin Biochem. 2015;48(15):948–956. |
Supplementary materials
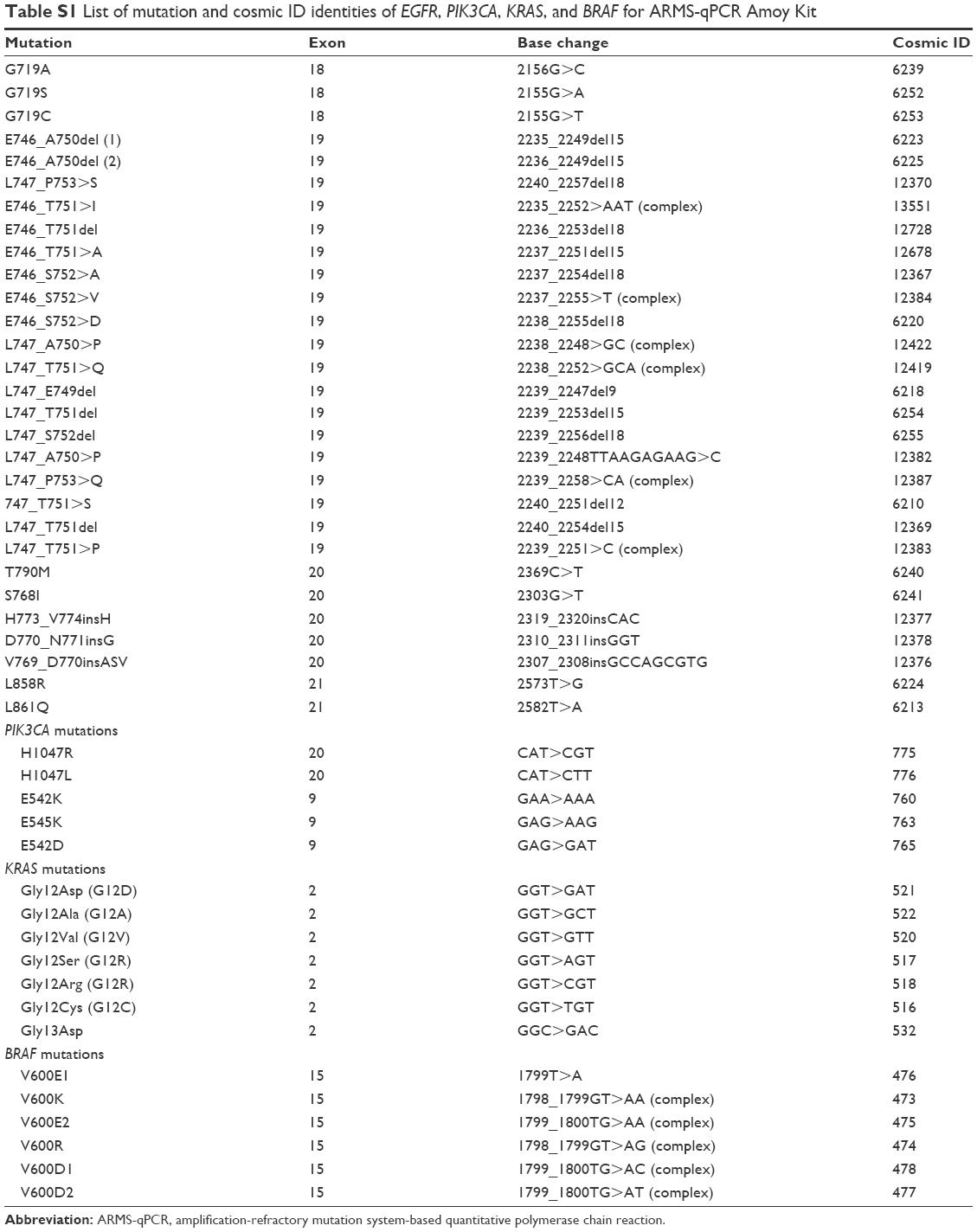 | Table S1 List of mutation and cosmic ID identities of EGFR, PIK3CA, KRAS, and BRAF for ARMS-qPCR Amoy Kit |
 | Table S2 List of primers for direct sequencing EGFR (Ref seq: NG_007726.3) |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.