Back to Journals » Journal of Inflammation Research » Volume 15
Modulation of Inflammation-Related Lipid Mediator Pathways by Celastrol During Human Macrophage Polarization
Authors Zhang K, Jordan PM, Pace S, Hofstetter RK ![]() , Werner M, Chen X, Werz O
, Werner M, Chen X, Werz O ![]()
Received 4 January 2022
Accepted for publication 25 March 2022
Published 2 June 2022 Volume 2022:15 Pages 3285—3304
DOI https://doi.org/10.2147/JIR.S356964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Kehong Zhang,1,2 Paul Mike Jordan,1 Simona Pace,1 Robert K Hofstetter,1 Markus Werner,1 Xinchun Chen,2 Oliver Werz1
1Department of Pharmaceutical/Medicinal Chemistry, Institute of Pharmacy, Friedrich Schiller University Jena, Jena, D-07743, Germany; 2Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, Department of Pathogen Biology, Shenzhen University School of Medicine, Shenzhen, 518000, People’s Republic of China
Correspondence: Oliver Werz, Email [email protected]
Background and Purpose: Celastrol (CS) is a major active ingredient of the Chinese/Asian herb Tripterygium wilfordii that is frequently used as phytomedicine to treat inflammation and autoimmune diseases. We showed before that short-term exposure to CS (1 μM) favorably impacts the biosynthesis of inflammation-related lipid mediators (LM) in human polarized macrophages by modulating the activities of different lipoxygenases (LOXs). However, whether CS regulates the expression of LOXs and other related LM-biosynthetic enzymes during macrophage polarization is unknown. Here, we investigated how CS affects LM-biosynthetic enzyme expression on the protein level and studied concomitant LM signature profiles during polarization of human monocyte-derived macrophages (MDM) towards M1- and M2-like phenotypes.
Methods and Results: We used LM metabololipidomics to study the long-term effects of CS on LM profile signatures after manipulation of human monocyte-derived macrophages (MDM) during polarization. Exposure of MDM to low concentrations of CS (ie, 0.2 μM) during polarization to an inflammatory M1 phenotype potently suppressed the formation of pro-inflammatory cyclooxygenase (COX)- and 5-LOX-derived LM, especially prostaglandin (PG)E2. Notably, gene and enzyme expression of COX-2 and microsomal PGE2 synthase (mPGES)-1 as well as M1 markers were strongly decreased by CS during M1-MDM polarization, along with impaired activation of nuclear factor-κB and p38 mitogen-activated protein kinase. During IL-4-induced M2 polarization, CS decreased the capacity of the resulting M2-MDM to generate pro-inflammatory COX and 5-LOX products as well but it also reduced the formation of 12/15-LOX products and specialized pro-resolving mediators, without affecting the levels of liberated fatty acid substrates.
Conclusion: Depending on the timing and concentration, CS not only favorably affects LOX activities in macrophages but also the expression of LM-biosynthetic enzymes during macrophage polarization connected to changes of inflammation-related LM which might be of relevance for potential application of CS to treat inflammatory disorders.
Keywords: inflammation, macrophages, lipoxygenase, cyclooxygenase, lipid mediators, specialized pro-resolving mediators, celastrol
Graphical Abstract:

Introduction
Inflammation is an essential immune response to pathogens, tissue injury, and other harmful events, in order to remove the stimulus and to accomplish tissue repair and regeneration.1 Prolonged inflammatory processes can lead to persistent inflammation and tissue damage, and ultimately to chronic diseases.1–4 Numerous anti-inflammatory drugs, either steroidal or nonsteroidal, have long been applied to treat inflammatory disorders, however, the inevitable and various side-effects are limiting their therapeutic use.3,5–9 In this respect, the discovery of anti-inflammatory agents from natural sources with better tolerability is continuously popular and accepted for modern pharmaceutical therapies.10–13
Aberrant biosynthesis of lipid mediators (LMs) is causative for many chronic inflammatory diseases.2,14–16 LMs are generated from polyunsaturated fatty acids (PUFA) in complex networks via cyclooxygenases (COX), lipoxygenases (LOX), and cytochrome P450 enzymes as key players, in conjunction with additional LM-biosynthetic enzymes.17–19 These LMs include, on the one hand, pro-inflammatory eicosanoids that initiate and maintain inflammation but also specialized pro-resolving mediators (SPMs) promoting the resolution of inflammation and the return to homeostasis on the other.2,16,18 Pro-inflammatory eicosanoids such as COX-derived prostaglandins (PGs) and 5-LOX-derived leukotrienes (LTs) are derived from arachidonic acid (AA) that is liberated from membrane phospholipids, mainly by cytosolic phospholipase (cPL)A2 upon cell stimulation.20 Thus, 5-LOX and the inducible COX-2 isoform are major targets to treat pathological inflammation by suppressing the biosynthesis of pro-inflammatory LMs.21 However, uncontrolled inflammation is now widely appreciated to be promoted also by a deficiency of SPMs including lipoxins (LX), maresins (MaR), protectins (PD), and resolvins (Rv) that all counter-regulate excessive and persistent inflammation and promote its resolution.22–24 Therefore, pharmacological strategies capable of shifting from formation of pro-inflammatory PGs and LTs to inflammation-resolving SPMs may constitute novel and innovative approaches for intervention with chronic inflammatory diseases.24,25
Plant-derived pentacyclic triterpenes from the lupane, oleanane, and ursane groups possess substantial pharmacological relevance with multi-target properties such as anti-inflammatory, wound healing, anti-bacterial, anti-viral, hepatoprotective, and anti-tumoral effects, combined with low toxicity.26–28 Major representatives with marked anti-inflammatory features encompass betulin/betulinic acid, lupeol, boswellic acids, ursolic acid, glycyrrhetinic acid, and oleanolic acid. The pentacyclic triterpenoid quinone methide celastrol (CS) is one major active component contained in the Chinese traditional medicinal plant Tripterygium wilfordii Hook F (TwHF) and has been examined for the treatment of various inflammation-associated diseases.29–32 Like other pentacyclic triterpene acids, CS possesses anti-inflammatory, anti-tumor, anti-oxidant, and immunomodulatory activities, and is now being increasingly recognized as a promising clinical candidate for the therapy of autoimmune disease, in particular rheumatoid arthritis (RA).30,33,34 The compound has been studied in-depth for interference with pro-inflammatory cytokines and chemokines, and modulation of cellular inflammatory responses.35–37 But still, the exact mechanisms underlying the beneficial actions of CS in the treatment of inflammatory disease are not entirely clear. Compared to other pentacyclic triterpene acids, CS contains an α,β-unsaturated carbonyl as part of the quinone methide, conferring it susceptible to conjugate addition due to the highly electrophilic carbon C-6,38 and thus CS may differ in its pharmacological profile and target interactions.
With respect to interference with LM biosynthesis, CS is a multi-enzyme inhibitor targeting sPLA2-IIA, in addition to 5-LOX and COX-2, supporting its potential as an anti-inflammatory drug.39,40 Moreover, we recently reported that CS at 1 µM promotes SPM biosynthesis by activation of 15-LOX-1 as a key enzyme during short-term (3 h) treatment in human monocyte-derived macrophages (MDM).40 In mice, CS impaired zymosan-induced LT formation along with elevated levels of SPM and other 12-/15-LOX-derived LM in peritoneal exudates, spleen, and plasma in vivo.40 But how CS impacts the LM networks on the protein expression level under long-term treatment has not been elucidated yet. Here we employed human MDM that were polarized towards M1- and M2-like subtypes in order to acquire phenotypic LM profiles,41 and we aimed at elucidating how CS affects LM-biosynthetic enzyme expression on the protein level and concomitant LM signature profiles during macrophage polarization. Our results show for the first time that CS considerably impacts the expression of LM-biosynthetic enzymes during macrophage polarization with implications for the respective LM signature profiles and thus for the occurrence of the macrophage phenotype.
Materials and Methods
Materials
Celastrol (CS; item number 70950) was supplied from Biomol GmbH (Hamburg, Germany). Deuterated and non-deuterated LM standards for ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS-MS) were purchased from Cayman Chemicals (Ann Arbor, MI). All other chemicals and reagents were obtained from Sigma-Aldrich (Steinheim, Germany), unless stated otherwise.
Isolation of Cells from Human Blood
Human leukocyte concentrates from freshly withdrawn blood (16 IU heparin/mL) from healthy adult male and female volunteers were obtained from the Department of Transfusion Medicine at the University Hospital of Jena, Germany. The research was conducted in accordance with the Declaration of Helsinki. The research protocols have been approved by the local ethical committee and were performed in accordance with guidelines and regulations; informed consent was obtained. Peripheral blood mononuclear cells (PBMC) were separated by dextran sedimentation, followed by density gradient centrifugation on lymphocyte separation medium (C-44010, Promocell, Heidelberg, Germany). PBMC from the intermediate fraction were washed with PBS pH 5.9/0.9% NaCl (1:1, v/v) and resuspended in PBS pH 7.4 and 1 mM CaCl2. PBMC were finally seeded in RPMI 1640 (Thermo Fisher Scientific, Schwerte, Germany) containing 10% (v/v) heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin in cell culture flasks (Greiner Bio-one, Frickenhausen, Germany) for 1.5 h at 37°C and 5% CO2 for adherence of monocytes.
Differentiation and Polarization of Human MDM and Incubation for LM Formation
For differentiation of monocytes to macrophages, M0GM-CSF and M0M-CSF were generated by incubating freshly isolated blood monocytes with 20 ng/mL granulocyte macrophage-colony stimulating factor (GM-CSF) or M-CSF (Cell Guidance Systems Ltd., Cambridge, UK), respectively, in RPMI 1640 supplemented with 10% FCS, 2 mmol/L L-glutamine, and 100 U/mL penicillin-streptomycin for 6 days. M1-MDM were obtained by incubation of M0GM-CSF with 100 ng/mL lipopolysaccharide (LPS) and 20 ng/mL interferon (IFN)γ (Peprotech, Hamburg, Germany) for 48 h, while M2-MDM were obtained by incubation of M0M-CSF with 20 ng/mL interleukin (IL)-4 (Peprotech) for 48 h. To assess the effects of CS on LM pathways during macrophage polarization, M0GM-CSF or M0M-CSF (2 × 106/mL, each) were pre-treated with CS (0.2 µM) or DMSO (0.1%) as a vehicle for 15 minutes before the addition of polarizing agents. Cell supernatants from treated M1-MDMs and M2-MDMs were carefully removed and cells were further incubated in 1 mL PBS containing 1 mM CaCl2 and kept at 37°C with or without 1% Staphylococcus aureus 6850-conditioned medium (SACM) for another 90 minutes to generate LM.42 The reaction was stopped by transferring supernatants (1 mL) into 2 mL ice-cold MeOH, and deuterated LM standards (200 nM d8-5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2, and 10 µM d8-AA; Cayman Chemical/Biomol GmbH, Hamburg, Germany) were added. Then, samples were processed for LM analysis by solid phase extraction and UPLC-MS-MS as described below.
Lipid Mediator Metabololipidomics by UPLC-MS-MS
Samples were kept at −20°C for at least 60 minutes to allow for protein precipitation. After centrifugation (1,200×g, 4°C, 10 minutes), acidified H2O (8 mL, final pH=3.5) was added and samples were subjected to solid phase extraction (Sep-Pak® Vac 6cc 500 mg/6 mL C18; Waters, Milford, MA). Briefly, columns were equilibrated with 6 mL methanol and 2 mL H2O before sample loading, washed with 6 mL H2O and 6 mL n-hexane, before eluting LMs with 6 mL methyl formate. The eluent was evaporated (TurboVap LV, Biotage, Uppsala, Sweden) and the residue resuspended in 100 µL methanol/water (50/50, v/v) for UPLC-MS-MS analysis using an Acquity™ UPLC (Waters, Milford, MA) and a QTRAP 5500 Mass Spectrometer (ABSciex, Darmstadt, Germany) equipped with a Turbo V™ Source and electrospray ionization. LM were separated on an ACQUITY UPLC® BEH C18 column (1.7 µm, 2.1 mm × 100 mm; Waters, Eschborn, Germany) at 50°C with a flow rate of 0.3 mL/min and a mobile phase consisting of methanol-water-acetic acid (starting at 42:58:0.01, v/v/v) that was ramped to 86:14:0.01 over 12.5 minutes and then to 98:2:0.01 for 3 minutes.43 The QTRAP 5500 was operated in negative mode using scheduled multiple reaction monitoring (MRM) coupled with information-dependent acquisition. The scheduled MRM window was 60 seconds, optimized LM parameters were adopted,43 and the curtain gas pressure was set to 35 psi. The retention time and at least six diagnostic ions for each LM were confirmed by means of an external standard (Cayman Chemical/Biomol GmbH, Hamburg, Germany). Quantification was achieved by calibration curves for each LM. Linear calibration curves were obtained for each LM and gave r2 values of 0.99.43
Determination of Cell Viability by MTT Assay
M0GM-CSF or M0M-CSF (2×105/mL) in a 96-well plate were pre-incubated with 0.1% vehicle (DMSO), CS, or 1% Triton X-100 that was used as a positive control, for 15 minutes. Then, 100 ng/mL LPS and 20 ng/mL IFNγ or 20 ng/mL IL-4 (for M1 and M2 polarization, respectively) were added. After 48 hours, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/mL, 20 µL; Sigma-Aldrich, Munich, Germany) solution was added in the darkness for 2–3 hours at 37°C and 5% CO2, and formazan product was solubilized with sodium dodecyl sulfate (SDS, 10% in 20 mM HCl). The absorbance was measured at 570 nm using a Multiskan Spectrum microplate reader (Thermo Fisher Scientific, Schwerte, Germany).
Analysis of LM-Biosynthetic Enzyme Expression by SDS-PAGE and Western Blot
M0GM-CSF and M0M-CSF MDM (2×106/mL) were pre-treated with 0.2 µM CS or 0.1% DMSO as a vehicle for 15 minutes prior to addition of 100 ng/mL LPS plus 20 ng/mL IFNγ or 20 ng/mL IL-4 (for M1- or M2-MDM polarization, respectively) or vehicle for 48 hours. Then, cells were lysed as previously described42 and the protein concentration was determined by DC-protein assay kit (Bio-Rad Laboratories GmbH, Munich, Germany). Lysates were mixed with 4×SDS loading buffer (50 mM Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 1% (v/v) β-mercaptoethanol, 12.5 mM EDTA, 0.02% (w/v) bromophenol blue) followed by heating at 95°C for at least 5 minutes. Each sample was adjusted to the same amount of protein and separated on 8% (for cPLA2α and COX-2), 10% (for 15-LOX-1, 15-LOX-2, LTA4H, phospho-p38 MAPK, p38 MAPK, phospho-NF-κB p65, NF-κB p65), 16% (for COX-1, mPGES-1, 5-LOX, and FLAP) SDS-PAGE gels and then blotted onto nitrocellulose membranes (Amersham Protran Supported 0.45 μm nitrocellulose, GE Healthcare, Freiburg, Germany). The membranes were incubated with the following primary antibodies: rabbit polyclonal anti-cPLA2α, 1:1,000 (2832S; Cell Signaling Technology); rabbit monoclonal anti-COX-2, 1:1,000 (12282S; Cell Signaling Technology); mouse monoclonal anti-15-LOX-1, 1:500 (ab119774; Abcam, Cambridge, UK); rabbit polyclonal anti-15-LOX-2, 1:500 (ab23691; Abcam); rabbit monoclonal anti-LTA4H, 1:1,000 (ab133512; Abcam); rabbit polyclonal anti-COX-1, 1:1,000 (4841S; Cell Signaling Technology); rabbit polyclonal anti-mPGES-1, 1:5,000 (kindly provided by Dr. Per-Johan Jakobsson, Karolinska Institute, Stockholm, Sweden); rabbit polyclonal anti-5-LOX, 1:1,000 (to a peptide corresponding to the C-terminal 12 amino acids of 5-LOX: CSPDRIPNSVAI; kindly provided by Dr. M. E. Newcomer, Louisiana State University, Baton Rouge, LA); rabbit polyclonal anti-FLAP, 1:1,000 (ab85227; Abcam); rabbit monoclonal anti-p38 MAPK, 1:1,000 (8690S; Cell Signaling); rabbit polyclonal anti-phospho-p38 MAPK (Thr180/Tyr182), 1:1,000 (9211S; Cell Signaling); rabbit monoclonal anti-NF-κB p65 (C22B4), 1:1,000 (4764S; Cell Signaling); mouse monoclonal anti-phospho-NF-κB p65 (Ser536), 1:1,000 (13346S; Cell Signaling); mouse monoclonal anti-β-actin, 1:1,000 (3700S; Cell Signaling); rabbit monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and 1:1,000 (5174S; Cell Signaling). Immunoreactive bands were stained with IRDye 800CW Goat anti-Mouse IgG (H+L), 1:10,000 (926–32210, LI-COR Biosciences, Lincoln, NE), IRDye 800CW Goat anti-Rabbit IgG (H+L), 1:15,000 (926 32211, LI-COR Biosciences), and/or IRDye 680LT Goat anti-Mouse IgG (H+L), 1:40,000 (926–68020, LI-COR Biosciences), and visualized by an Odyssey infrared imager (LI-COR Biosciences). Data from densitometric analysis were background corrected.
Real Time PCR
M0GM-CSF (106/mL) were pre-treated with 0.2 µM CS or 0.1% DMSO as a vehicle for 15 minutes prior to exposure to 100 ng/mL LPS and 20 ng/mL IFNγ. After 0, 6, 24, or 48 hours, total RNA was isolated from the cells using E.Z.N.A.® Total RNA Kit I (Omega Bio-tek, Inc. via VWR, Dresden, Germany) according to the manufacturer´s protocol and then quantified by NanoVue (GE Healthcare; Spekol, Analytic Jena, Germany). Reverse transcription reaction was performed with 0.5–1.5 µg RNA in a 20 µL reaction using a High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems™ via Thermo Fisher Scientific, Schwerte, Germany) according to the manufacturer’s instructions. The qPCR reaction was performed in a qTOWER3 G touch Instrument (Analytik Jena) using the PerfeCTa™ SYBR® Green SuperMix, ROX™ kit (Quantabio via VWR, Dresden, Germany) containing optimized concentrations of MgCl2, dNTPs, AccuStart Taq DNA Polymerase, SYBR Green I dye, ROX Reference Dye, and stabilizers. For qPCR, a 25 µL reaction mix containing PerfeCTa SYBR Green SuperMix, ROX (2X), diluted cDNA template, and 0.5 µM of forward and reverse primer (listed in Table 1) was used. Reaction mixes were prewarmed for 10 minutes at 95°C, followed by 40 cycles of denaturation for 15 seconds at 95°C, annealing for 30 seconds at 60°C, and extension for 30 seconds at 72°C. Data were collected and processed with qPCRsoft 4.1 software (Analytik Jena). The 2−ΔΔCT method was used to calculate relative gene expression levels.44 GAPDH was used as a reference gene. As quality controls, negative control measurements, melting curves analysis, and determination of qPCR efficiency by LinRegPCR 2021.1 software (developed by Dr. J. M. Ruijter, Dept. Medical Biology Amsterdam, Academic Medical Centre, University of Amsterdam)45 were performed.
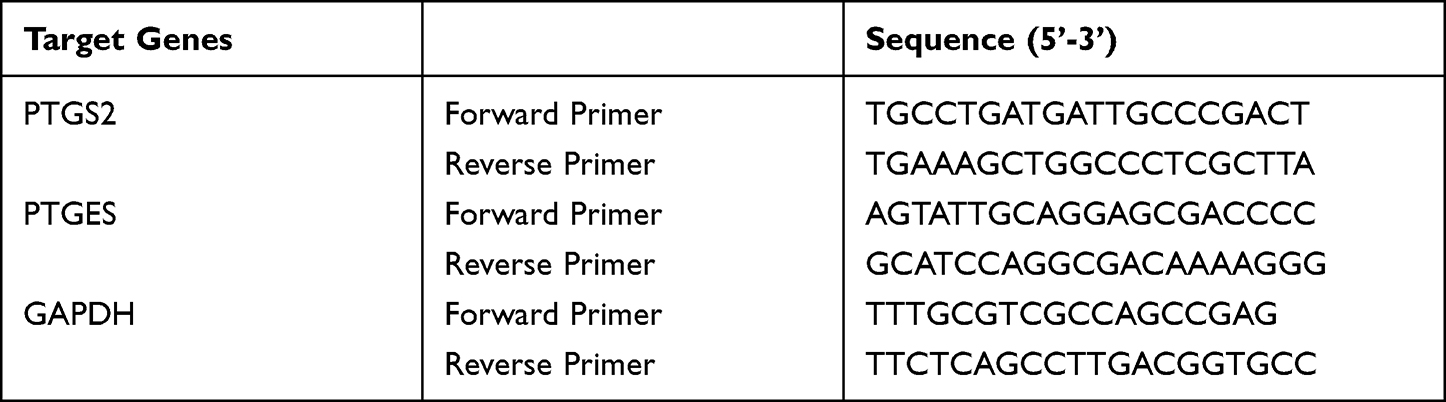 |
Table 1 Prime Sequences for qPCR Assays |
Flow Cytometry
M0M-CSF (2×106/mL) were treated with CS (0.2 µM) or vehicle (0.1% DMSO) for 48 hours. Then, cells were stained in PBS pH 7.4 containing 0.5% bovine serum albumin, 2 mM EDTA, and 0.1% sodium azide by Zombie Aqua™ Fixable Viability Kit (Biolegend, San Diego, CA) for 5 minutes at 4°C to determine cell viability. Non-specific antibody binding was blocked by using mouse serum (10 minutes at 4°C) prior to staining by the following fluorochrome-labeled antibodies (20 minutes, 4°C): FITC anti-human CD14 (clone M5E2, #555397, BD Biosciences, San Jose, CA), APC-H7 anti-human CD80 (clone L307.4, #561134, BD Biosciences), PE-Cy7 anti-human CD54 (clone HA58, #353115, Biolegend), PE anti-human CD163 (clone GHI/61, #556018, BD Biosciences), and APC anti-human CD206 (clone 19.2, #550889, BD Biosciences) to determine M1 and M2 surface marker expression using a LSRFortessaTM cell analyzer (BD Biosciences), and data were analyzed using FlowJo X Software (BD Biosciences).
Statistical Analysis
Results are expressed as mean±SEM of each independent experiment, where n represents the indicated numbers from separate donors performed on different days. Statistical analysis and graphs were made by using GraphPad Prism 9 software (San Diego, CA). Paired t-test was used to analyze experiments for comparison of two groups; while one-way ANOVA or multiple paired t-tests were applied for multiple comparisons as indicated. A p-value ≤0.05 is a criterion for statistical significance.
Results
Impact of CS on MDM Viability and Expression of M1- and M2-Phenotypic Surface Markers During Polarization
Macrophages exhibit high plasticity where inflammatory M1-like cells strongly express COX-2 and 5-LOX generating mainly PGs/TX and LTs, while anti-inflammatory M2-like phenotypes express abundant 15-LOX-1 and produce substantial amounts of SPMs.41,43,46 In order to study how CS affects these LM networks and connected LM formation during macrophage polarization, we employed human MDMs that were either polarized for 48 hours towards M1- and M2-like phenotypes or left untreated for 48 hours, in the presence and absence of CS, with subsequent stimulation by bacterial exotoxins (using SACM) to evoke several LM-biosynthetic pathways.42
Since CS (at ≥3 µM) can be cytotoxic for innate immune cells,40 we first determined its impact on the viability of MDMs during polarization. MDMs, differentiated with GM-CSF (M0GM-CSF), were polarized to M1-MDMs by LPS and IFNγ, while MDMs, differentiated with M-CSF (M0M-CSF), were polarized to M2-MDMs by IL-4. MTT assays after 48 hours of polarization showed that CS up to 0.3 µM caused only minor detrimental effects but, at ≥1 µM CS, the viability of both M1- and M2-MDMs was significantly reduced; the EC50 values were determined at 3.5 µM and 2.7 µM for M1- and M2-MDM, respectively (Figure 1A and B). Thus, we limited the concentration of CS to 0.2 µM for subsequent experiments, in order to exclude cytotoxicity as a trigger for potential modulatory effects on LM networks.
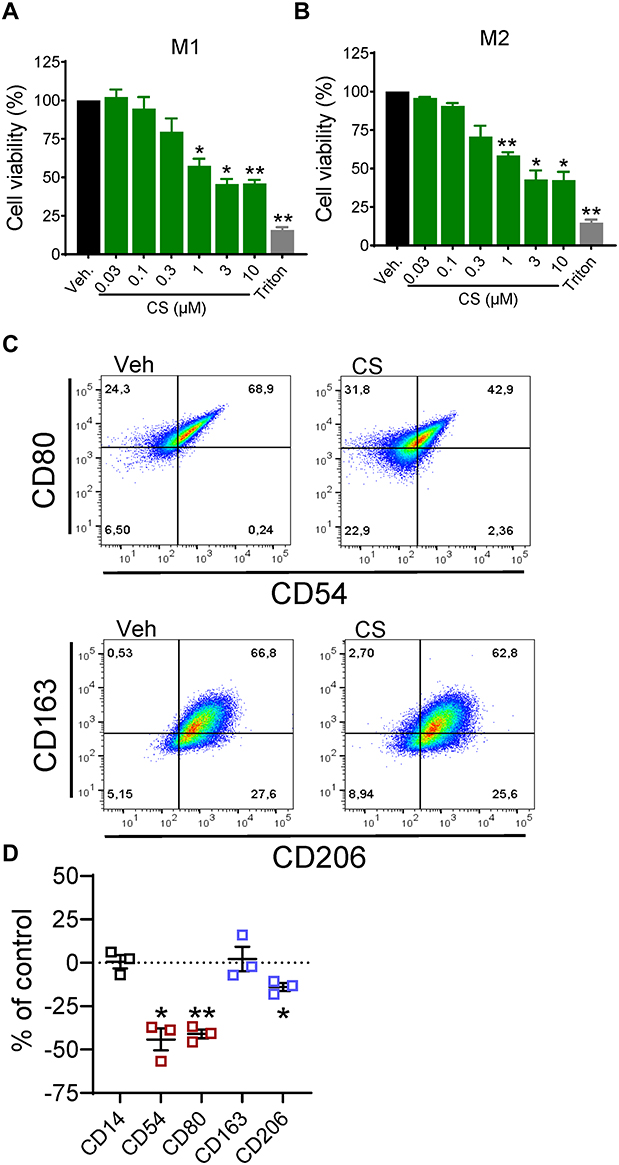 |
Figure 1 Impact of celastrol (CS) on cell viability and polarization of human MDM. (A and B) Effects of CS on cell viability. M0GM-CSF (A) and M0M-CSF (B) were pre-treated with CS at the indicated concentrations, 0.1% DMSO as vehicle, or 1% Triton X-100 as positive control, for 15 minutes before adding the polarizing agents (LPS/IFNγ for M1-, IL-4 for M2-MDM). After 48 hours, cell viability was assessed by MTT assay. Values are means±SEM, n=3, expressed as a percentage of vehicle control (=100%); * p<0.05, ** p<0.01 vs control group, one-way ANOVA for multiple comparisons with Dunnett’s correction. (C and D) Effects of CS on the expression of macrophage phenotype surface markers. M0M-CSF were treated with 0.2 µM CS or 0.1% DMSO (as vehicle) for 48 hours. (C) Expression of surface markers CD54 and CD80 (M1-like) as well as CD163 and CD206 (M2-like) among living CD14+ cells was analyzed by flow cytometry; shown are representative pseudocolor dot plots of the M1-like and M2-like surface markers. (D) Mean fluorescence intensity (MFI) of each marker in (C) was determined. The change of the MFI from CS-treated MDM against the MFI of vehicle-treated cells was calculated and is given in % of control in scatter dot plots as single values and means±SEM, n=3. * p<0.05, ** p<0.01 CS vs control group, ratio paired t-test. |
We then assessed whether CS could affect macrophage polarization, as suggested by others before.37,47 We studied the expression of CD54 and CD80 as markers for human M1-MDMs and CD163 and CD206 as markers for M2-MDMs41 in the presence or absence of 0.2 µM CS. We found a significant reduction of the two M1 markers CD54 and CD80 by CS after 48 hours with a minor impact on M2 markers, where only CD206 was slightly reduced but not CD163 (Figure 1C and D). These data indicate that CS impairs the polarization towards pro-inflammatory M1-like macrophages, while marginally affecting M2-like markers.
Impact of CS on LM Signature Profiles in Human M1-Like MDM
In order to study how CS affects LM signature profiles of M1-like MDMs, we used M0GM-CSF that were pre-incubated (15 minutes) with 0.2 µM CS and then polarized for 48 hours with LPS/IFNγ towards M1-MDMs or left unpolarized. Afterwards, LM biosynthesis was assessed in unstimulated or SACM-challenged MDMs upon incubation for another 90 minutes. In agreement with results from previous studies,40–43 polarization to M1-MDMs led to high amounts of COX products (PGE2, PGD2, PGF2α, TXB2, 11-HEPE, and 11-HETE), formed even in the absence SACM as stimulus, especially PGE2 (Figure 2, Table 2). Also, the capacity to generate 5-LOX products (LTB4, t-LTB4, 5-HETE, 5-HEPE) and to release the PUFAs AA, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) was strongly upregulated during polarization and further elevated by subsequent SACM-challenge (Figure 2, Table 2). A similar pattern was observed for generation of 12/15-LOX products (Figure 2), although the overall amounts of these LMs were comparably low (Table 2).
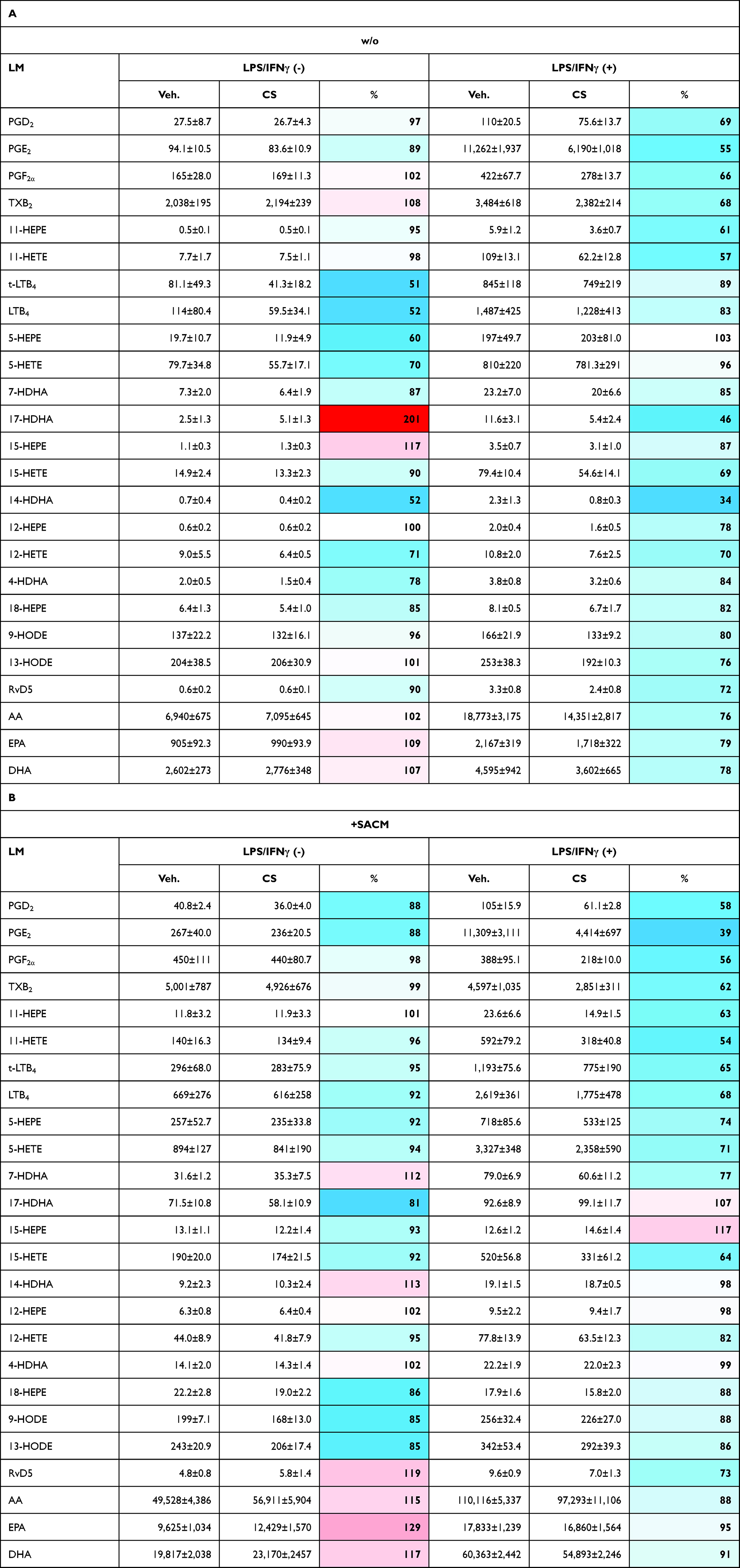 |
Table 2 Effects of Celastrol (CS) on LM Pathways During Human M1-MDM Polarization |
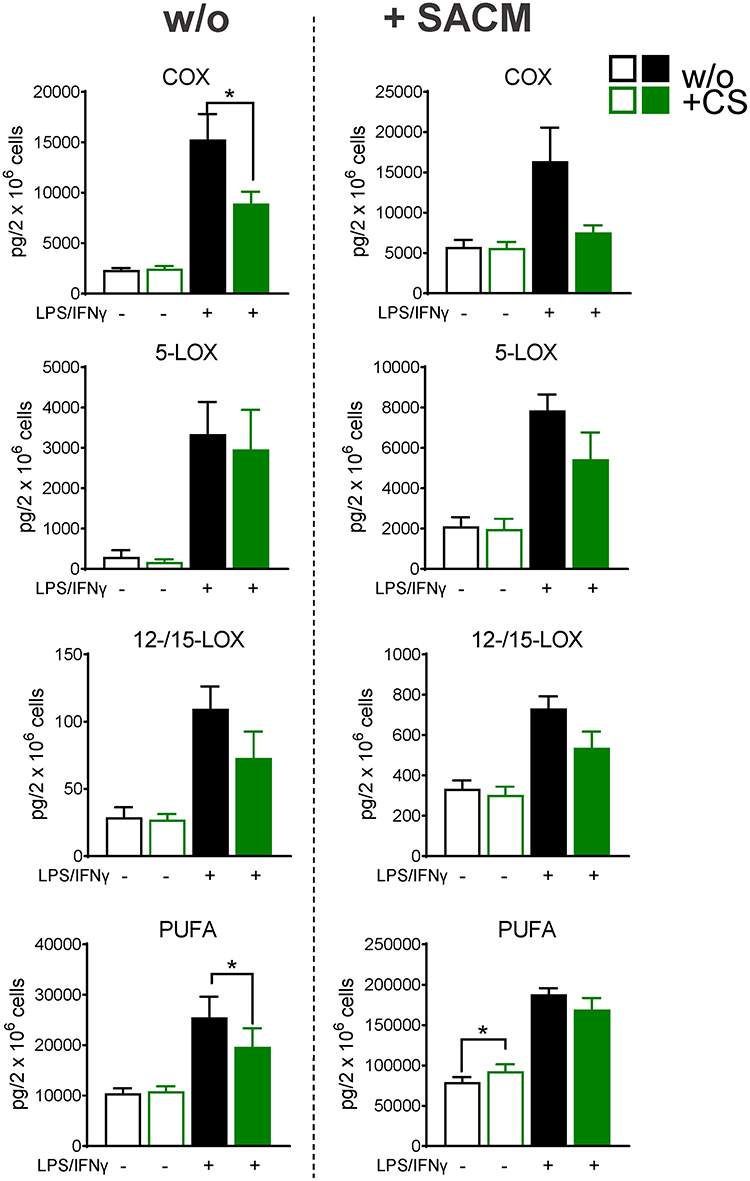 |
Figure 2 Celastrol (CS) modulates LM pathways during polarization towards human M1-MDM. M0GM-CSF were pre-treated with 0.2 µM CS or 0.1% DMSO (as vehicle) for 15 minutes before adding the polarizing agents LPS/IFNγ or vehicle. After 48 hours, cells were incubated in 1 mL PBS pH 7.4 plus 1 mM CaCl2 without or with 1% S. aureus-conditioned medium (SACM) for 90 minutes. Produced LM were analyzed in cell supernatants by UPLC-MS-MS. The sum of COX products (PGE2, PGD2, PGF2α, TXB2), 5-LOX products (LTB4, t-LTB4, 5-HETE, 5-HEPE), 12/15-LOX products (17-HDHA, 15-HETE, 15-HEPE, 14-HDHA, 12-HETE, 12-HEPE), and PUFA (AA, EPA, DHA) are shown as pg/2× 106 cells of CS-treated and vehicle-treated cells, in the resting state or upon challenge with SACM. Data are means±SEM, n=4 and were log-transformed for statistical analysis, * p<0.05, CS vs control group, one-way ANOVA for multiple comparisons with Sidak’s correction. |
CS (0.2 µM) suppressed the strong formation of COX products in M1-MDMs by about 50% regardless of SACM-challenge, while in unpolarized MDMs, CS was without marked effects after 48 hours incubation (Figure 2, Table 2A and B). Most striking (>60%) inhibition was evident for PGE2 (Table 2B). Formation of 5-LOX products (LTB4, t-LTB4, 5-HETE, 5-HEPE) was less affected by CS, with major effects in unpolarized/unstimulated cells and in SACM-activated M1-MDMs (Figure 2, Table 2B). A similar pattern was found for modulation of 12/15-LOX products (14-HDHA, 12-HETE, 12-HEPE, 17-HDHA, 15-HETE, and 15-HEPE) by CS, although it should be noted that unstimulated cells produced rather low amounts of several 12/15-LOX products (eg, 15-HEPE, 14-HDHA, 12-HEPE; Table 2A and B) due to minute protein levels of the 12- and 15-LOXs in M1-MDMs.41,43 Suppression of 12/15-LOX products in SACM-activated M1-MDMs by CS was evident only for the substantially formed 15-HETE (Table 2B) that can be generated in M1-MDMs as 15(R)-HETE by COX enzymes.43 Liberation of PUFA, regardless of the experimental conditions, was not significantly altered by CS (Figure 2, Table 2A and B). Together, CS efficiently repressed the strong COX product formation acquired during M1 polarization, especially of PGE2, with a tendency of reduced 5-LOX and 12/15-LOX product levels in M1-MDMs when elicited by SACM.
Impact of CS on LM Profiles in Anti-Inflammatory M2-Like MDM
We then assessed the effects of CS on LM formation in M2-like MDMs using M0M-CSF that were pre-incubated (15 minutes) with 0.2 µM CS, polarized for 48 hours towards M2-MDMs or left untreated (= unpolarized), and LM biosynthesis was then assessed in unstimulated or SACM-challenged cells after 90 minutes incubation. Except TXB2, only relatively low amounts of COX, 5-LOX, and 12/15-LOX products were formed in unpolarized M0M-CSF or M2-MDMs (cultured for 48 hours) that received no SACM (Table 3, Figure 3A). However, challenge with SACM strikingly induced formation of essentially all LMs regardless of polarization. As reported before,40–43 M2-MDMs produced much higher amounts of 12/15-LOX products and SPMs compared to M1-MDMs, especially upon challenge with SACM (Table 3B).
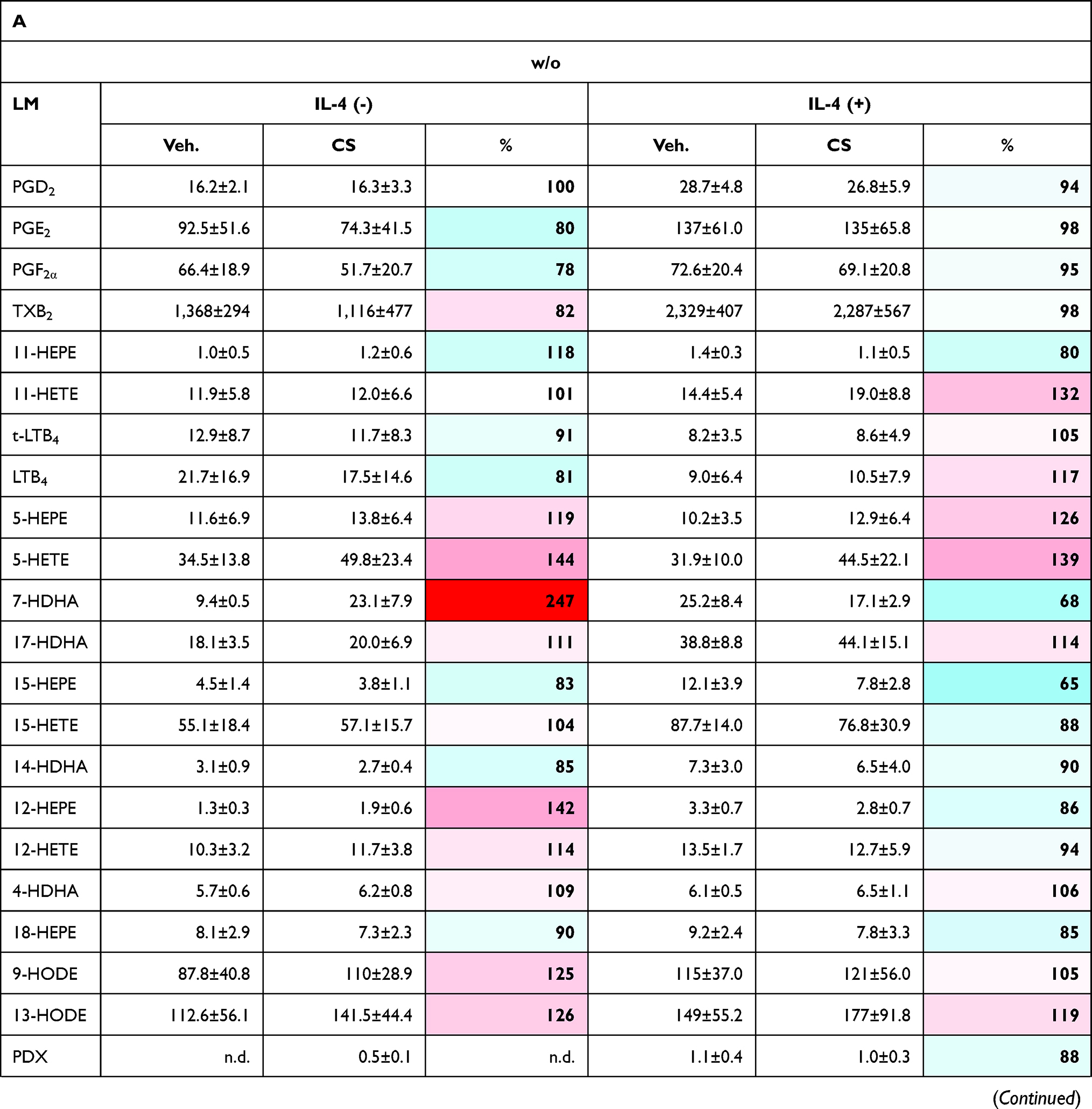 | 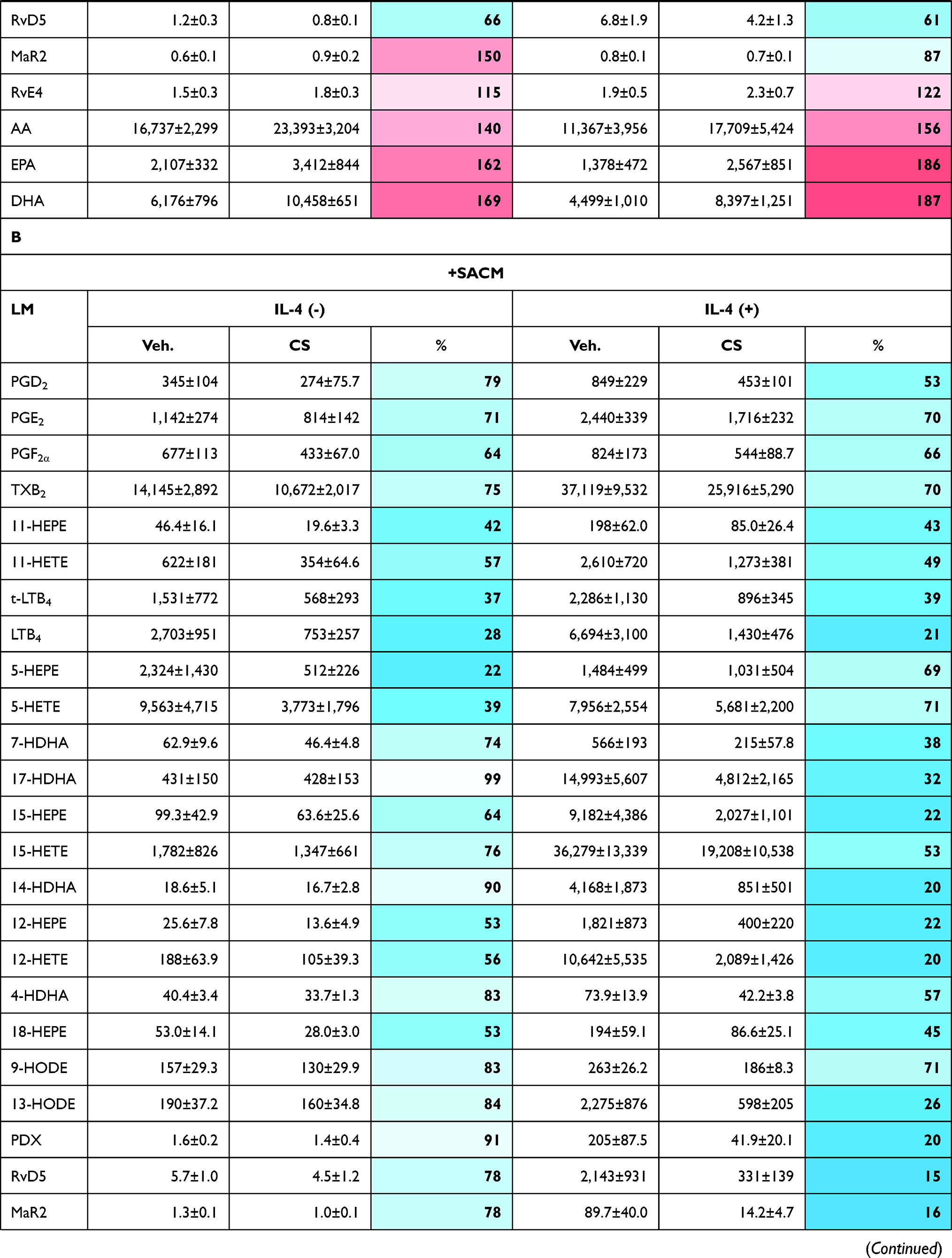 | 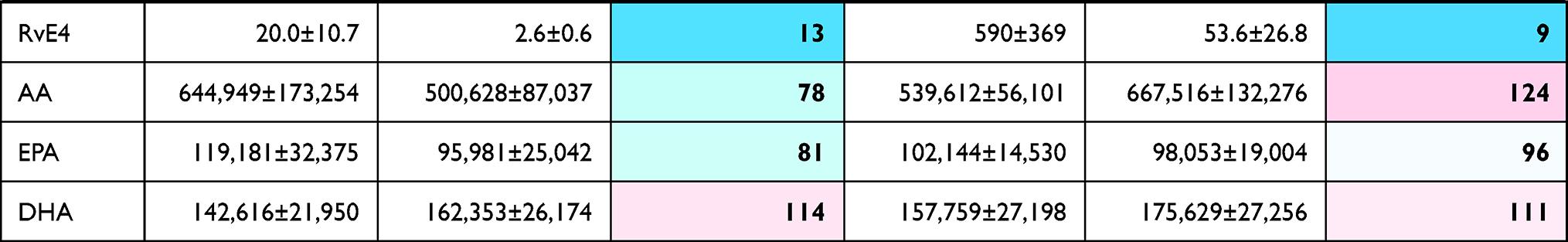 |
Table 3 Effects of Celastrol (CS) on LM Pathways During Human M2-MDM Polarization |
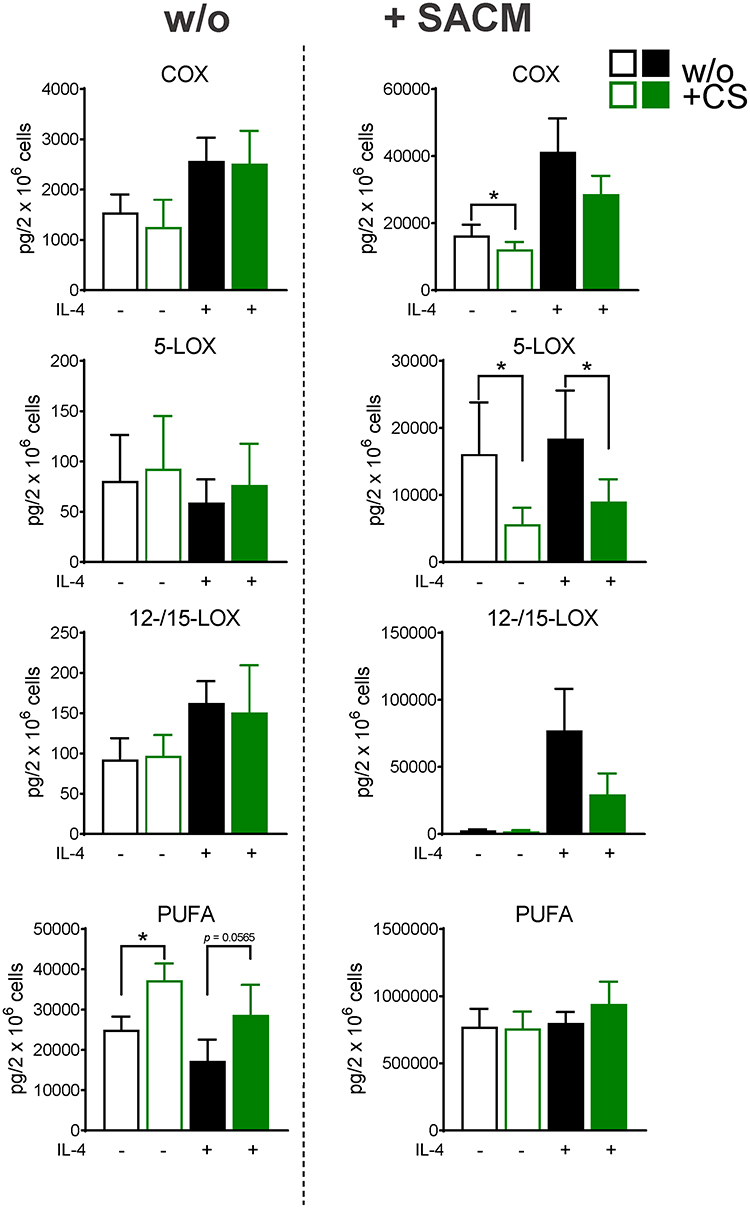 |
Figure 3 Celastrol (CS) modulates LM pathways during polarization towards human M2-MDM. M0M-CSF were pre-treated with 0.2 µM CS or 0.1% DMSO (as vehicle) for 15 minutes before adding the polarizing agent IL-4 or vehicle. After 48 hours, cells were incubated in 1 mL PBS pH 7.4 plus 1 mM CaCl2 without or with 1% S. aureus-conditioned medium (SACM) for 90 minutes. Produced LM were analyzed in cell supernatants by UPLC-MS-MS. The sum of COX products (PGE2, PGD2, PGF2α, TXB2), 5-LOX products (LTB4, t-LTB4, 5-HETE, 5-HEPE), 12/15-LOX products (17-HDHA, 15-HETE, 15-HEPE, 14-HDHA, 12-HETE, 12-HEPE), and PUFA (AA, EPA, DHA) are shown as pg/2×106 cells of CS-treated and vehicle-treated cells, in the resting state or upon challenge with SACM. Data are means±SEM, n=4 and were log-transformed for statistical analysis, * p<0.05, CS vs control group, one-way ANOVA for multiple comparisons with Sidak’s correction. |
In the presence of CS, unpolarized M0M-CSF or M2-MDMs (that generated low amounts of LMs) were hardly and inconsistently affected, without significant modulation of LM biosynthesis, albeit levels of free PUFAs were elevated (Table 3A, Figure 3). However, in the case of SACM stimulation, treatment of MDMs (unpolarized or M2 cells) with CS for 48 hours suppressed the subsequent COX and 5-LOX product formation, most prominently for 5-LOX products in unpolarized M0M-CSF. Also, a tendency for reduced 12/15-LOX product formation was obvious in SACM-challenged cells due to CS, and formation of the detectable SPM, that is, PDX, RvD5, MaR2, and RvE4 as well as their monohydroxylated precursors 17-HDHA, 14-HDHA, and 15-HEPE, were suppressed by CS (Table 3B). Note that, despite the suppressive effects of CS on generation of essentially all COX/LOX products upon challenge with SACM, the levels of free PUFAs remained mainly unaffected by CS (Figure 3). Taken together, incubation of M0M-CSF in the presence of CS with or without polarization agent IL-4 strongly suppresses the subsequent capacity to generate massive COX and 5-LOX products as well as SPMs elicited by SACM-challenge.
Effect of CS on the Expression of LM-Biosynthetic Enzymes in Human MDM
Since CS markedly affected LM pathways during MDMs culture and polarization with mainly suppressive outcome, it appeared reasonable that CS may compromise the expression level of the respective LM-biosynthetic enzymes during the incubations. Thus, we assessed the protein levels of the enzymes in M1- and M2-MDMs by Western Blot which are relevant for the biosynthesis of those LMs that were affected by CS, namely cPLA2, 5-LOX, FLAP, LTA4H, 15-LOX-1 (only in M2-MDMs), mPGES-1 (only in M1-MDMs), COX-1, COX-2, and 15-LOX-2. M0GM-CSF and M0M-CSF were pretreated (15 minutes) with 0.2 µM CS and then kept for 48 hours with and without polarizing agents (LPS/IFNγ for M1; IL-4 for M2). In the absence of LPS/IFNγ, CS did not affect protein levels of any of the LM-biosynthetic enzymes in M0GM-CSF within 48 hours (Figure 4A and B). However, the upregulated expression of COX-2 obtained during polarization of M0GM-CSF towards M1-MDMs, was significantly reduced by CS, with a concomitant decrease of mPGES-1 protein, but without alteration of the levels of other enzymes (ie, cPLA2, 5-LOX, FLAP, LTA4H, 15-LOX-2, COX-1) (Figure 4A and B). In analogy to M0GM-CSF the protein levels after 48 hours culture of M0M-CSF were not significantly affected by CS (Figure 4C and D). When M0M-CSF were polarized towards M2-MDMs with IL-4 for 48 hours, the presence of CS caused a tendency for reduced protein levels of most of the enzymes, except for LTA4H, being most pronounced and significant for 15-LOX-1 and -2 (Figure 4C and D). Conclusively, CS clearly impairs COX-2 and mPGES-1 protein during M1 polarization with minor inhibitory effects on cPLA2 and 5-LOX but markedly on 15-LOX-1 and -2 during polarization towards M2, which fits well to the strong suppression of COX product formation, especially of PGE2, in M1- and overall decreased LM formation in M2-MDMs after SACM-challenge, respectively.
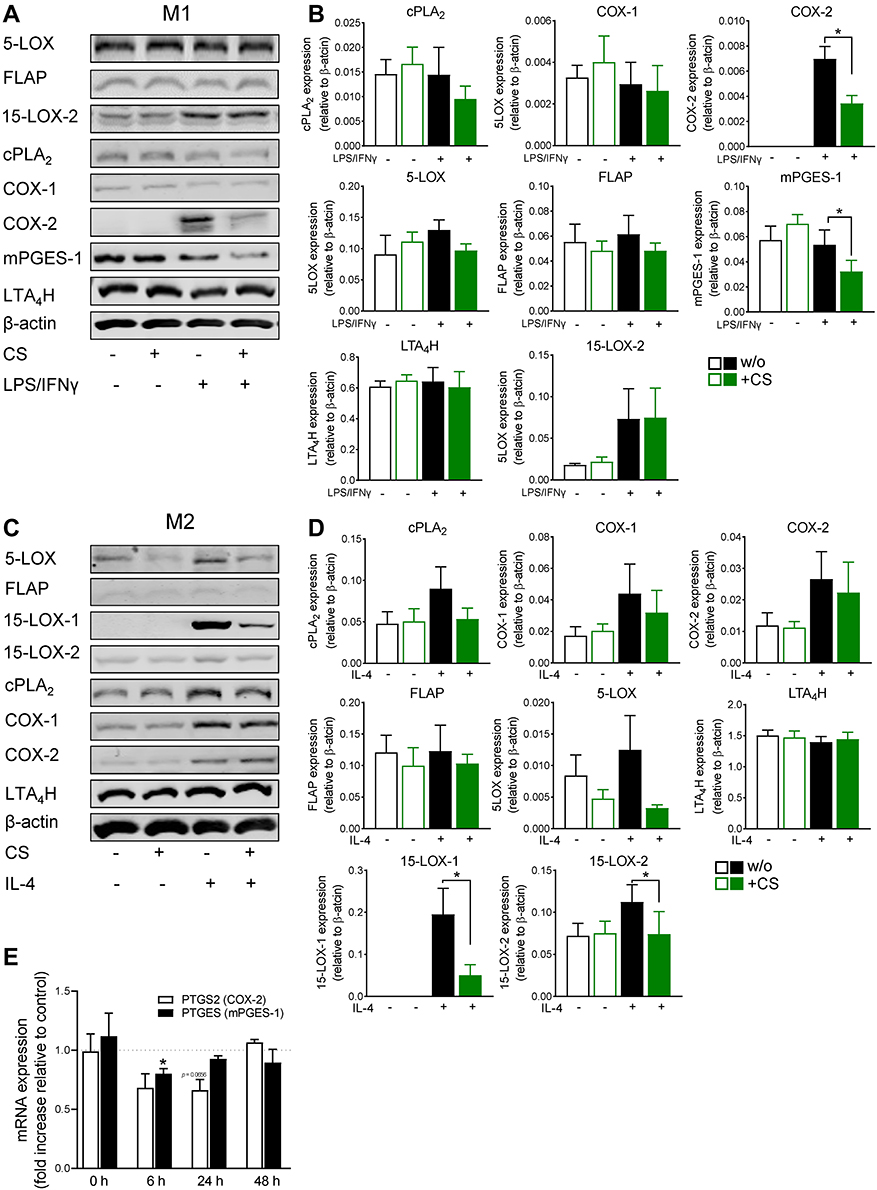 |
Figure 4 Celastrol (CS) modulates the expression of LM-biosynthetic enzymes during polarization of human MDM. (A and B) M0GM-CSF and (C and D) M0M-CSF were pre-treated with 0.2 µM CS or 0.1% DMSO (as vehicle) for 15 minutes before adding the polarizing agents LPS/IFNγ or IL-4, respectively, or vehicle. After 48 hours, cells were harvested, lysates were prepared, and proteins were analyzed by SDS-PAGE and Western blotting. Immunoreactive protein bands of LM-biosynthetic enzymes are shown for (A) M1-MDM and (C) M2-MDM. Immunoreactive bands of LM-biosynthetic enzymes were analyzed by densitometry for proteins derived from (B) M1-MDM and (D) M2-MDM, normalized to β-actin. Data are shown as means±SEM from n=4 separate donors. Densitometric ratios were used for statistical analysis. * p<0.05, CS vs control group, one-way ANOVA for multiple comparisons with Sidak’s correction. (E) Effects of CS on PTGS2 and PTGES mRNA levels during polarization towards human M1-MDM. M0GM-CSF were pretreated with 0.2 µM CS or 0.1% DMSO (as vehicle) for 15 minutes and polarized for 0, 6, 24, and 48 hours towards M1-MDM using LPS/IFNγ. RNA was isolated, transformed in cDNA by reverse transcription, and amplified by qPCR for quantification. Data are expressed as fold increase to DMSO control for n=3 separate donors. Statistics: * expression ratio is significantly different from 1, p<0.05, CS vs control group; multiple paired t-test with Holm-Sidak correction. |
Since COX-2 and mPGES-1 are known to be upregulated during M1 macrophage polarization48 and because CS efficiently impaired COX-2 and mPGES-1 protein levels in M1-MDMs, we investigated if CS would affect these enzymes also on the mRNA level. Analysis by qPCR showed that CS downregulated both PTGS2 (COX-2) and PTGES (mPGES-1) during M1-MDM polarization, after 6 and 24 hours (Figure 4E). These data suggest that CS interferes with COX-2, and to a lesser extent with mPGES-1, at the transcription level, with respective consequences for COX-2/mPGES-1 protein and COX-2 product formation.
Effects of CS on the Activation of NF-κB and p38 MAPK in Human MDMs
LPS-stimulated phosphorylation (and thus activation) of NF-κB and p38 MAPK is crucial for COX-2 expression in macrophages.49,50 Accordingly, the phosphorylation of NF-κB p65 was increased upon LPS/IFNγ-treatment of M0GM-CSF after 3 and 6 hours. This was clearly reduced when 0.2 µM CS was included during the incubation (Figure 5A). Phosphorylation of p38 MAPK was slightly elevated by LPS/IFNγ-treatment after 6 hours (but not yet after 3 hours), and again, CS impaired this stimulatory effect (Figure 5B). Together, these data suggest that CS may block the phosphorylation of both NF-κB p65 and p38 MAPK during M1-polarization of human MDMs which may result in impaired expression of COX-2.
 |
Figure 5 Effects of celastrol (CS) on the activation of NF-κB and p38 MAPK in human M1-MDM. M0GM-CSF were pretreated for 15 minutes with 0.2 µM CS or 0.1% DMSO (as vehicle) prior to polarization to M1-MDM using LPS/IFNγ for 3 or 6 hours. Protein phosphorylation of (A) NF-κB p65 and (B) p38 MAPK was analyzed by SDS-PAGE and Western blotting using phospho-specific antibodies of cell lysates; the respective unphosphorylated proteins were used for normalization. Representative Western blots of n=3 independent experiments are shown; data are means±SEM. Densitometric ratios were used for statistical analysis. *p<0.05, CS-treated vs DMSO-treated, one-way ANOVA for multiple comparisons with Sidak’s correction. |
Discussion
Here we show that CS from anti-rheumatic Tripterygium wilfordii modulates the biosynthetic pathways of pro-inflammatory and anti-inflammatory LMs which are acquired during macrophage polarization. When human MDMs were polarized to the M1 phenotype, CS markedly suppressed the capacity for exotoxin-elicited formation of pro-inflammatory COX products, especially of PGE2, and to a minor extent also of 5-LOX-derived LM such as LTB4. Accordingly, CS markedly decreased the protein levels of COX-2 and mPGES-1 with moderate suppression of 5-LOX during M1-MDM polarization, in parallel to impaired expression of M1 phenotypic surface markers. Similarly, during polarization of MDMs towards the M2 phenotype, CS suppressed the capacity of exotoxin-challenged cells to generate COX and 5-LOX products but also SPM formation was considerably inhibited along with impaired 15-LOX-1 and -2 protein levels. Since pro-inflammatory PG and LT are typical features of M1 macrophages,41 lowering these LM by CS confirms impaired M1 polarization. Thus, our data suggest mainly anti-inflammatory properties of CS by suppressing pro-inflammatory LM pathways in M1-like macrophages, but they imply also an impact of CS on inflammation resolution by impairing the acquired capacity for pronounced SPM generation in M2 macrophages by down-regulating 15-LOX-1/2.
CS was recently shown to promote a switch from LT biosynthesis to formation of SPM and other 12/15-LOX-derived LM in short-term (3 hours) incubations when given to polarized MDMs that either were in a resting state or stimulated by SACM.40 These effects were traced back to direct interference of CS with 5-LOX and 15-LOX-1. Also, after short-term (2.5 hours) treatment of mice, CS given intraperitoneally impaired zymosan-induced LT formation and simultaneously elevated the levels of SPM and related 12-/15-LOX-derived LM in peritoneal exudates, spleen, and plasma.40 Under these short-term conditions, CS appears to act as a direct enzyme modulator, inhibiting 5-LOX activity with reduced LTB4 levels but promoting 15-LOX-1 activation yielding elevated SPM concentrations. Such LM class switch from pro-inflammatory to pro-resolving LOX-derived LM is considered as an innovative pharmacological strategy for actively promoting the resolution of inflammation.23–25 In the present study, where CS was evaluated in long-term incubations during macrophage polarization, the compound affected the protein level of the LM biosynthetic enzymes with consequent changes in the LM profiles upon subsequent cell stimulation. CS-containing Tripterygium wilfordii glycosides (TWG) also efficiently suppressed agonist-induced formation of 5-LOX products during short-term incubations of M1-MDMs and neutrophils while pronounced SPM formation and 12/15-LOX products were evident in M2-MDMs.51 Note that in analogy to our present data with CS, during 48 hours M1-MDM polarization these TWG decreased the capacity to generate 5-LOX and COX products and impaired COX-2 and mPGES-1 protein levels as well as M1 markers.51
Our results suggest that the suppression of the COX-2 and mPGES-1 protein levels by CS during M1 polarization is due to inhibition of the NF-κB and, at least to some extent, of the p38 MAPK pathways. COX-2 and mPGES-1 are upregulated during M1 macrophage polarization48 and NF-κB and p38 MAPK are known to be required for induction of COX-2 expression in macrophages by LPS,49,50 the agent used together with IFNγ to trigger M1-MDM polarization. Previous studies demonstrated that CS could control macrophage polarization through modulating the cross-talk among LPS-stimulated MAPKs (ie, p38 MAPK, ERK1/2, JNK) and nuclear translocation of NF-κB p65 and other transcription factor-related axes.37,52 The α,β-unsaturated carbonyl of the quinone methide of CS confers its position C-6 highly electrophilic and thus susceptible for conjugation to cysteine thiols, whereby CS targets the early virus-encoded protein Tat53 and proteostasis.38 In fact, NF-κB contains redox-regulated cysteine residues and covalently modification of the thiol moieties (eg, by S-nitrosylation at Cys-6254) results in the inhibition of NF-κB DNA recognition and binding.55 Possibly, CS binds these crucial thiols of the reactive cysteine residues in NF-κB, thereby suppressing its signaling activity. Macrophage polarization towards functionally opposite phenotypes is eventually caused by the activation of different signaling pathways, transcription factors, and cytokine secretion, which are of importance for the progression and resolution of inflammatory responses of various human diseases.56–58 Canonically activated macrophages (M1-like) often exhibit antibacterial and antitumor functions, and are characterized by a high production of various pro-inflammatory cytokines as well as PGs and LTs.41,46,56 On the other hand, alternatively activated macrophages (M2-like) are regarded as anti-inflammatory phenotypes involved in immunosuppression and tissue repair, producing anti-inflammatory cytokines and numerous SPMs.41,46,56,59 Our current data suggest that CS may govern the balance of macrophage polarization suppressing the occurrence of a M1-like phenotype since the characteristic upregulation of COX-2 and mPGES-1 as well as CD54 and CD80 were prevented by CS during M1 polarization. This is supported by findings in mice, where CS blocked M1 polarization in diet-induced obese animals,37 and when loaded in nanomicelles, reduced the expression of the M1 biomarkers TNF-α, IL-1β, IL-6, and inducible nitric oxide synthase.47 In our hands, CS did not or hardly affected the M2-like markers CD163 and CD206, but impaired the expression levels of the 15-LOX-1 and 15-LOX-2 that are key enzymes in SPM biosynthesis,60,61 and where at least 15-LOX-1 protein is strongly upregulated in human M2-MDMs.41 Similarly, in diet-induced obese mice, the expression of the M2 biomarkers arginase-1 and IL-10 were only marginally altered by CS, while M1 markers were strikingly impaired.47 Another study demonstrated that CS protects against acute ischemic stroke-induced brain injury by promoting microglia/macrophage M2 polarization.62 Our results show that M2 polarization in the presence of CS impaired the capacity to produce SPMs upon exotoxin challenge of M2-MDMs, which implies rather detrimental consequences for inflammation resolution. Moreover, CS significantly impaired the viability of M1- and M2-MDMs during long-term (48 hour) incubations at concentrations ≥1 µM, which is in agreement with the well-known toxicity of CS63 and thus further questions the pharmacotherapeutic safety of this natural product. It is interesting that a variety of nanotechnology-based CS formulations have been developed that were able to reduce the toxicity and/or improved bioavailability.64 Future investigations of nanotechnology-based CS formulations in our experimental systems of macrophage polarization and LM biosynthesis might reveal if cytotoxicity and SPM-impairing effects may be circumvented. Indeed, drug delivery by nanofluids improved the efficacy of isoniazid65 or of bromocriptine,66 and encapsulation of a cytotoxic indirubin derivative into polymer-based nanoparticles reduced its detrimental impact on monocyte viability.67
Conclusions
Our data show that CS considerably impacts the expression of LM-biosynthetic enzymes during macrophage polarization with implications for the subsequent LM signature profiles produced by these cells after adequate challenge. In particular, CS mainly blocked the upregulation of the inducible COX-2 and mPGES-1 in inflammatory M1-MDMs along with strong suppression of the respective pro-inflammatory LM, namely PGE2. Although CS hardly affected polarization to the M2 phenotype, it clearly impaired the capacity to produce SPMs along with reduced expression of 15-LOX-1 and -2. Therefore, despite the favorable anti-inflammatory properties of CS due to interference with pro-inflammatory LM pathways in M1-like macrophages, a detrimental impact on inflammation resolution due to the suppression of SPM generation should be considered in the overall judgment of the pharmacological profile of this natural product.
Data Sharing Statement
The datasets generated and analyzed in this study will be available by the corresponding author upon reasonable request.
Acknowledgments
The authors thank Anna König, Heidi Traber, Petra Wiecha, Katrin Fischer and Alrun Schumann for expert technical assistance.
Author Contributions
All authors significantly contributed to the reported work related to the conception, study design, execution, acquisition of data, analysis and interpretation; took part in drafting, revising, or critically reviewing the article; gave final approval of the manuscript; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Free State of Thuringia and the European Social Fund (2019 FGR 0095) and by the Deutsche Forschungsgemeinschaft (DFG), Collaborative Research Center SFB 1278 “PolyTarget” (project number 316213987, project A04) and - SFB 1127 “ChemBioSys” (project number 239748522, project A04).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi:10.1038/nature07201
2. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi:10.1038/nature13479
3. Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339(6116):166–172. doi:10.1126/science.1230720
4. Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–1832. doi:10.1038/s41591-019-0675-0
5. Mahesh G, Anil Kumar K, Reddanna P. Overview on the discovery and development of anti-inflammatory drugs: should the focus be on synthesis or degradation of PGE2? J Inflamm Res. 2021;14:253–263. doi:10.2147/JIR.S278514
6. Patil KR, Mahajan UB, Unger BS, et al. Animal models of inflammation for screening of anti-inflammatory drugs: implications for the discovery and development of phytopharmaceuticals. Int J Mol Sci. 2019;20(18):4367. doi:10.3390/ijms20184367
7. Rainsford KD. Anti-inflammatory drugs in the 21st century. Subcell Biochem. 2007;42:3–27.
8. Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24(2):121–132. doi:10.1016/j.bpg.2009.11.005
9. Peres MF, Ribeiro FV, Ruiz KG, Nociti FH
10. Kishore N, Kumar P, Shanker K, Verma AK. Human disorders associated with inflammation and the evolving role of natural products to overcome. Eur J Med Chem. 2019;179:272–309. doi:10.1016/j.ejmech.2019.06.034
11. Koeberle A, Werz O. Multi-target approach for natural products in inflammation. Drug Discov Today. 2014;19(12):1871–1882. doi:10.1016/j.drudis.2014.08.006
12. Mishra BB, Tiwari VK. Natural products: an evolving role in future drug discovery. Eur J Med Chem. 2011;46(10):4769–4807. doi:10.1016/j.ejmech.2011.07.057
13. Furst R, Zundorf I. Plant-derived anti-inflammatory compounds: hopes and disappointments regarding the translation of preclinical knowledge into clinical progress. Mediators Inflamm. 2014;2014:146832. doi:10.1155/2014/146832
14. Bennett M, Gilroy DW. Lipid mediators in inflammation. Microbiol Spectr. 2016;4(6). doi:10.1128/microbiolspec.MCHD-0035-2016
15. Chiurchiu V, Leuti A, Maccarrone M. Bioactive lipids and chronic inflammation: managing the fire within. Front Immunol. 2018;9:38. doi:10.3389/fimmu.2018.00038
16. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523. doi:10.1038/nri3859
17. Calder PC, Harwood J, Lloyd-Evans E. Eicosanoids. Essays Biochem. 2020;64(3):423–441. doi:10.1042/EBC20190083
18. Chiang N, Serhan CN. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem. 2020;64(3):443–462. doi:10.1042/EBC20200018
19. Christie WW, Harwood JL. Oxidation of polyunsaturated fatty acids to produce lipid mediators. Essays Biochem. 2020;64(3):401–421. doi:10.1042/EBC20190082
20. Leslie CC. Cytosolic phospholipase A(2): physiological function and role in disease. J Lipid Res. 2015;56(8):1386–1402. doi:10.1194/jlr.R057588
21. Sala A, Proschak E, Steinhilber D, Rovati GE. Two-pronged approach to anti-inflammatory therapy through the modulation of the arachidonic acid cascade. Biochem Pharmacol. 2018;158:161–173. doi:10.1016/j.bcp.2018.10.007
22. Dalli J, Serhan CN. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br J Pharmacol. 2019;176(8):1024–1037. doi:10.1111/bph.14336
23. Serhan CN, Gupta SK, Perretti M, et al. The Atlas of Inflammation Resolution (AIR). Mol Aspects Med. 2020;74:100894. doi:10.1016/j.mam.2020.100894
24. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128(7):2657–2669. doi:10.1172/JCI97943
25. Gilbert NC, Newcomer ME, Werz O. Untangling the web of 5-lipoxygenase-derived products from a molecular and structural perspective: the battle between pro- and anti-inflammatory lipid mediators. Biochem Pharmacol. 2021;193:114759. doi:10.1016/j.bcp.2021.114759
26. Ghiulai R, Rosca OJ, Antal DS, et al. Tetracyclic and pentacyclic triterpenes with high therapeutic efficiency in wound healing approaches. Molecules. 2020;25(23):5557. doi:10.3390/molecules25235557
27. Laszczyk MN. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med. 2009;75(15):1549–1560. doi:10.1055/s-0029-1186102
28. Safayhi H, Sailer ER. Anti-inflammatory actions of pentacyclic triterpenes. Planta Med. 1997;63(6):487–493. doi:10.1055/s-2006-957748
29. Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: molecular targets of thunder god vine. Biochem Biophys Res Commun. 2010;394(3):439–442. doi:10.1016/j.bbrc.2010.03.050
30. Cascao R, Fonseca JE, Moita LF. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front Med. 2017;4:69. doi:10.3389/fmed.2017.00069
31. Chen SR, Dai Y, Zhao J, Lin L, Wang Y, Wang Y. A mechanistic overview of triptolide and celastrol, natural products from Tripterygium wilfordii Hook F. Front Pharmacol. 2018;9:104. doi:10.3389/fphar.2018.00104
32. Ng SW, Chan Y, Chellappan DK, et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed Pharmacother. 2019;109:1785–1792. doi:10.1016/j.biopha.2018.11.051
33. Song X, Zhang Y, Dai E, Du H, Wang L. Mechanism of action of celastrol against rheumatoid arthritis: a network pharmacology analysis. Int Immunopharmacol. 2019;74:105725. doi:10.1016/j.intimp.2019.105725
34. Venkatesha SH, Dudics S, Astry B, Moudgil KD. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog Dis. 2016;74(6):ftw059. doi:10.1093/femspd/ftw059
35. Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from thunder of god vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303(1):9–20. doi:10.1016/j.canlet.2010.10.025
36. Lee JY, Lee BH, Kim ND, Lee JY. Celastrol blocks binding of lipopolysaccharides to a Toll-like receptor4/myeloid differentiation factor2 complex in a thiol-dependent manner. J Ethnopharmacol. 2015;172:254–260. doi:10.1016/j.jep.2015.06.028
37. Luo D, Guo Y, Cheng Y, Zhao J, Wang Y, Rong J. Natural product celastrol suppressed macrophage M1 polarization against inflammation in diet-induced obese mice via regulating Nrf2/HO-1, MAP kinase and NF-kappaB pathways. Aging. 2017;9(10):2069–2082. doi:10.18632/aging.101302
38. Boridy S, Le PU, Petrecca K, Maysinger D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014;5(5):e1216. doi:10.1038/cddis.2014.182
39. Joshi V, Venkatesha SH, Ramakrishnan C, et al. Celastrol modulates inflammation through inhibition of the catalytic activity of mediators of arachidonic acid pathway: secretory phospholipase A2 group IIA, 5-lipoxygenase and cyclooxygenase-2. Pharmacol Res. 2016;113(Pt A):265–275. doi:10.1016/j.phrs.2016.08.035
40. Pace S, Zhang K, Jordan PM, et al. Anti-inflammatory celastrol promotes a switch from leukotriene biosynthesis to formation of specialized pro-resolving lipid mediators. Pharmacol Res. 2021;167:105556. doi:10.1016/j.phrs.2021.105556
41. Werz O, Gerstmeier J, Libreros S, et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun. 2018;9(1):59. doi:10.1038/s41467-017-02538-5
42. Jordan PM, Gerstmeier J, Pace S, et al. Staphylococcus aureus-derived alpha-hemolysin evokes generation of specialized pro-resolving mediators promoting inflammation resolution. Cell Rep. 2020;33(2):108247. doi:10.1016/j.celrep.2020.108247
43. Werner M, Jordan PM, Romp E, et al. Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. FASEB J. 2019;33(5):6140–6153. doi:10.1096/fj.201802509R
44. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
45. Ruijter JM, Ramakers C, Hoogaars WM, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6):e45. doi:10.1093/nar/gkp045
46. Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15):e60–72. doi:10.1182/blood-2012-04-423525
47. Zhao J, Luo D, Zhang Z, et al. Celastrol-loaded PEG-PCL nanomicelles ameliorate inflammation, lipid accumulation, insulin resistance and gastrointestinal injury in diet-induced obese mice. J Control Release. 2019;310:188–197. doi:10.1016/j.jconrel.2019.08.026
48. Mosca M, Polentarutti N, Mangano G, et al. Regulation of the microsomal prostaglandin E synthase-1 in polarized mononuclear phagocytes and its constitutive expression in neutrophils. J Leukoc Biol. 2007;82(2):320–326. doi:10.1189/jlb.0906576
49. D’Acquisto F, Iuvone T, Rombola L, Sautebin L, Di Rosa M, Carnuccio R. Involvement of NF-kappaB in the regulation of cyclooxygenase-2 protein expression in LPS-stimulated J774 macrophages. FEBS Lett. 1997;418(1–2):175–178. doi:10.1016/S0014-5793(97)01377-X
50. Takada Y, Aggarwal BB. Genetic deletion of the tumor necrosis factor receptor p60 or p80 sensitizes macrophages to lipopolysaccharide-induced nuclear factor-kappa B, mitogen-activated protein kinases, and apoptosis. J Biol Chem. 2003;278(26):23390–23397. doi:10.1074/jbc.M213237200
51. Zhang K, Pace S, Jordan PM, et al. Beneficial modulation of lipid mediator biosynthesis in innate immune cells by antirheumatic Tripterygium wilfordii glycosides. Biomolecules. 2021;11(5):May. doi:10.3390/biom11050746
52. Ma X, Xu L, Alberobello AT, et al. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1alpha transcriptional axis. Cell Metab. 2015;22(4):695–708. doi:10.1016/j.cmet.2015.08.005
53. Narayan V, Ravindra KC, Chiaro C, et al. Celastrol inhibits Tat-mediated human immunodeficiency virus (HIV) transcription and replication. J Mol Biol. 2011;410(5):972–983. doi:10.1016/j.jmb.2011.04.013
54. Marshall HE, Hess DT, Stamler JS. S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl Acad Sci U S A. 2004;101(24):8841–8842. doi:10.1073/pnas.0403034101
55. Pande V, Sousa SF, Ramos MJ. Direct covalent modification as a strategy to inhibit nuclear factor-kappa B. Curr Med Chem. 2009;16(32):4261–4273. doi:10.2174/092986709789578222
56. Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79(1):541–566. doi:10.1146/annurev-physiol-022516-034339
57. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi:10.1172/JCI59643
58. Martinez FO, The GS. M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P6-13
59. Dalli J, Serhan C. Macrophage proresolving mediators-The when and where. Microbiol Spectr. 2016;4(3). doi:10.1128/microbiolspec.MCHD-0001-2014
60. Jordan PM, Werz O. Specialized pro-resolving mediators: biosynthesis and biological role in bacterial infections. FEBS J. 2021. doi:10.1111/febs.16266
61. Perry SC, Kalyanaraman C, Tourdot BE, et al. 15-Lipoxygenase-1 biosynthesis of 7S,14S-diHDHA implicates 15-lipoxygenase-2 in biosynthesis of resolvin D5. J Lipid Res. 2020;61(7):1087–1103. doi:10.1194/jlr.RA120000777
62. Jiang M, Liu X, Zhang D, et al. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an IL-33/ST2 axis-mediated microglia/macrophage M2 polarization. J Neuroinflammation. 2018;15(1):78. doi:10.1186/s12974-018-1124-6
63. Hou W, Liu B, Xu H. Celastrol: progresses in structure-modifications, structure-activity relationships, pharmacology and toxicology. Eur J Med Chem. 2020;189:112081. doi:10.1016/j.ejmech.2020.112081
64. Wagh PR, Desai P, Prabhu S, Wang J. Nanotechnology-based celastrol formulations and their therapeutic applications. Front Pharmacol. 2021;12:673209. doi:10.3389/fphar.2021.673209
65. Zomorodbakhsh S, Abbasian Y, Naghinejad M, Sheikhpour M. The effects study of isoniazid conjugated multi-wall carbon nanotubes nanofluid on Mycobacterium tuberculosis. Int J Nanomedicine. 2020;15:5901–5909. doi:10.2147/IJN.S251524
66. Kamazani FM, Sotoodehnejad Nematalahi F, Siadat SD, Pornour M, Sheikhpour M. A success targeted nano delivery to lung cancer cells with multi-walled carbon nanotubes conjugated to bromocriptine. Sci Rep. 2021;11(1):24419. doi:10.1038/s41598-021-03031-2
67. Czapka A, Grune C, Schadel P, et al. Drug delivery of 6-bromoindirubin-3’-glycerol-oxime ether employing poly(D,L-lactide-co-glycolide)-based nanoencapsulation techniques with sustainable solvents. J Nanobiotechnology. 2022;20(1):5. doi:10.1186/s12951-021-01179-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.