Back to Archived Journals » Hypoxia » Volume 2
Modulation of hydrogen sulfide by vascular hypoxia
Received 30 April 2014
Accepted for publication 4 June 2014
Published 25 August 2014 Volume 2014:2 Pages 117—126
DOI https://doi.org/10.2147/HP.S51589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jessica M Osmond, Nancy L Kanagy
Vascular Physiology Group, Department of Cell Biology and Physiology, University of New Mexico Health Sciences Center, Albuquerque, NM, USA
Abstract: Hydrogen sulfide (H2S) has emerged as a key regulator of cardiovascular function. This gasotransmitter is produced in the vasculature and is involved in numerous processes that promote vascular homeostasis, including vasodilation and endothelial cell proliferation. Although H2S plays a role under physiological conditions, it has become clear in recent years that hypoxia modulates the production and action of H2S. Furthermore, there is growing evidence that H2S is cytoprotective in the face of hypoxic insults. This review focuses on the synthesis and signaling of H2S in hypoxic conditions in the vasculature, and highlights recent studies providing evidence that H2S is a potential therapy for preventing tissue damage in hypoxic conditions.
Keywords: H2S, cystathionine γ-lyase, vascular smooth muscle, endothelium
Introduction
It has long been known that hydrogen sulfide (H2S) is endogenously produced by multiple enzymes, including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST). This small polar molecule with a structure very similar to water was thought for many years to be a toxic metabolite with no physiological function, but recent studies have demonstrated that H2S acts in mammalian tissues as a second messenger, an antioxidant, and a sulfhydrating agent. It has important functions in regulating neurotransmission, transcription, angiogenesis, and vascular tone, and both the production and mechanism of action can be modified by exposure to hypoxia.
Within the vasculature, H2S regulates the function of smooth muscle and endothelial cells and can be produced by enzymes in the endothelium, media, and the perivascular fat.1–3 It regulates vascular smooth muscle cell (VSMC) proliferation4–8 and contraction,9–12 and endothelial cell proliferation,13–16 adhesion,17–19 and release of dilators.20 Thus, H2S causes both vasodilation and inhibition of vascular wall remodeling, similar to effects of the well-known gasotransmitter nitric oxide (NO).
It has become clear in recent years that H2S is synthesized under physiological conditions and plays an important role in vascular function (reviewed in Wang21 and Mancardi et al22). For example, global deletion of CSE, the predominant vascular H2S-synthesizing enzyme, results in hypertension and impaired endothelium-dependent vasorelaxation.23 Additionally, H2S constrains vascular tone under normoxic conditions. However, the cellular response to hypoxia induces changes in H2S synthesis and signaling. Within the vascular wall, multiple responses have been attributed to this newest member of the gasotransmitter family, and this review will focus on how H2S regulates vascular function under normoxic and hypoxic conditions.
Hypoxia and H2S synthesis
H2S is produced endogenously by desulfhydration of the amino acid cysteine by the pyridodoxyl 5′phosphate-dependent enzymes CSE and CBS or 3-MST.24 Although other H2S synthesis pathways have been described,25 the majority of endogenous H2S synthesis occurs via CSE or CBS.24 Tissue distribution of these enzymes varies, with CBS producing H2S in the central nervous system26,27 and CSE being largely responsible for H2S production in peripheral tissues, including the vasculature.2,23,28,29 Because of the tissue distribution of H2S-generating enzymes and the vascular focus of this review, more attention will be given to the effects of hypoxia on CSE generation of H2S.
Transcriptional and posttranscriptional regulation of CSE under physiological conditions is currently under investigation. One known transcriptional regulator of the CSE gene, CTH, is the ubiquitously expressed transcription factor specificity protein 1 (Sp1). Sp1 represses or activates thousands of genes involved in a variety of processes and is regulated by phosphorylation (reviewed in Tan and Khachigian30). In differentiated human aortic smooth muscle cells, Sp1 stimulates increased CTH expression.31 Sp1 levels, and in turn CSE expression, are decreased by microRNA-21 (miR-21),32 which is elevated in hypoxic conditions.33 Thus, it appears that hypoxia decreases CSE expression in the vascular wall by miR-21-induced suppression of Sp-1-dependent transcription (Figure 1).
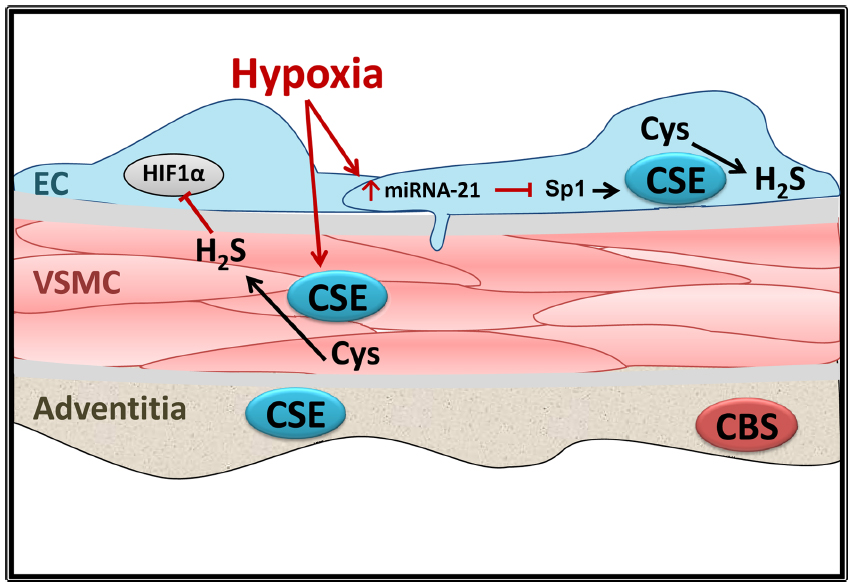 | Figure 1 Effects of hypoxia on CSE expression in the vascular wall. |
As reported by Yang et al,23 Ca2+-calmodulin may acutely activate CSE in vitro. Separately, neither Ca2+ nor calmodulin affects CSE activity but together causes a significant increase in enzyme generation of H2S. The concentration of Ca2+ used in these studies, however, was supraphysiological at 1–2 mM, and further studies are required to determine the role of Ca2+-calmodulin in CSE regulation under physiological conditions in the vasculature. Mikami et al34 assessed Ca2+ regulation of H2S production by CSE that was purified from rat liver. In solution, CSE produced H2S at Ca2+ concentrations from 0–100 nM, but when the Ca2+ concentration was increased to 300 nM, H2S production decreased. Furthermore, adding calmodulin or a calmodulin inhibitor did not affect H2S production. These results are not in alignment with the findings from Yang et al23 in intact tissues, underscoring the lack of understanding of regulation of CSE by Ca2+ or other physiological regulators.
Direct evaluation of CTH expression and CSE activity, and H2S synthesis in vivo, has also yielded conflicting results, and evaluation of these parameters in response to hypoxia is limited. Although the mechanisms of hypoxia’s regulation of H2S synthesis are not well defined, hypoxia-induced effects on H2S synthesis have been observed in many cell types,35–37 with H2S in turn promoting protective cellular responses to hypoxia.14,38,39 In a study using multiple cell culture lines, in vitro measurements of CTH promoter activity revealed a decrease in promoter activity after 2 hours of hypoxia.40 A decrease in CTH messenger RNA (mRNA) also occurred very quickly after the onset of exposure to hypoxia, but this repression was followed by a recovery of CTH mRNA and protein levels within 2 hours.40 It is not clear whether miR-21 provides a sustained inhibition of CSE expression in vivo, and additional study is needed to better understand this regulation.
As illustrated in Figure 2, H2S is involved in regulating multiple endothelial processes. It has been shown to regulate mitochondrial function under physiological conditions, acting as an electron donor at low concentrations and inhibiting mitochondrial complex IV at higher concentrations (reviewed in Szabo et al41). Studies have assessed the impact of hypoxia on H2S regulation of mitochondrial function and have shown both beneficial and deleterious effects of the gasotransmitter, depending on the concentration of H2S. Fu et al42 investigated CSE expression and activity in mitochondria of mesenteric artery VSMC under basal and hypoxic conditions. CSE expression was not detected in mitochondria under basal conditions, but exposure to the Ca2+ ionophore A23187 to mimic hypoxic cellular stress led to mitochondrial CSE expression. Translocation of CSE to mitochondria under these conditions resulted in mitochondrial H2S production and enhanced ATP production. Furthermore, the H2S donor NaHS caused a concentration-dependent decrease in ATP production in normoxia but increased ATP production during hypoxia. These results suggest that enhanced VSMC mitochondrial CSE expression and activity, triggered by cellular stressors such as hypoxia, may be one mechanism by which H2S is protective during hypoxic insults in the vasculature.
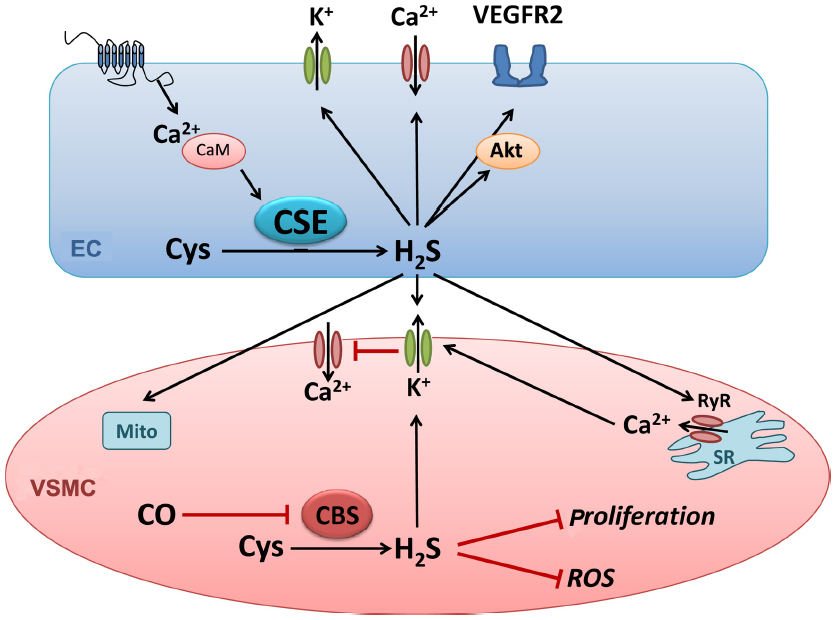 | Figure 2 Hydrogen sulfide signaling in the vascular wall. |
Evidence that CSE expression is decreased in the vasculature in conditions associated with hypoxia is growing. For example, Cindrova-Davies et al33 demonstrated that CSE mRNA and protein expression is decreased in placentas from women with intrauterine growth restriction or preeclampsia with diminished umbilical artery blood flow, two conditions associated with placental hypoxia. CSE expression is not decreased if the preeclampsia is not accompanied by diminished umbilical artery flow, suggesting that it is the functional hypoxia rather than other preeclamptic factors that decrease placental CSE expression. Expression of miR-21, which can indirectly suppress CSE expression through downregulation of Sp1 as described above,32 is also increased in intrauterine growth restriction and preeclampsia placentas. Furthermore, exposure of placental explants from healthy placentas to hypoxia for 3 hours, followed by reoxygenation for 20 hours, decreases CSE expression and increases miR-21 expression. These findings suggest that hypoxic conditions, at least during pregnancy, may decrease H2S production via a miR-21-dependent decrease in CSE expression.
Rats exposed to 3 weeks of intermittent hypoxia have decreased plasma H2S levels.43 Furthermore, CTH mRNA expression and H2S production in lung homogenates are decreased in the hypoxic group. This decrease in CSE expression and H2S availability is associated with an increase in pulmonary artery pressure. Administration of exogenous H2S prevents the hypoxia-induced increase in pressure. Taken together, these findings suggest that intermittent hypoxia exposure decreases H2S production, and the loss of H2S may be involved in the development of hypoxia-induced pulmonary hypertension. However, miR-21 and Sp1 levels have not been evaluated in this model, and it is unclear whether H2S production is decreased in a miR-21- and Sp-1-dependent manner.
Although the predominant pathway for H2S synthesis in the vasculature involves enzymatic activity of CSE, it is important to consider the indirect vascular effects of CBS deficiency. CBS activity regulates clearance of homocysteine by metabolizing it to H2S. Increased plasma homocysteine levels are a risk factor for cardiovascular disease in humans. Homozygous and heterozygous deletion of CBS has been employed to model hyperhomocysteinemia experimentally, and this deletion results in vascular complications such as endothelial dysfunction, oxidative stress, and remodeling (reviewed in Steed and Tyagi44). H2S therapy may be a beneficial treatment for the deleterious vascular effects of hyperhomocysteinemia. For example, Sen et al45 demonstrated that H2S supplementation prevents hyperhomocysteinemia-induced renal damage in mice.
Interestingly, there appears to be a link between homocysteine levels and hypoxia. Homocysteine levels were measured in patients with metabolic syndrome with and without obstructive sleep apnea. Patients with sleep apnea, who are exposed to intermittent bouts of hypoxia during sleep, have higher serum homocysteine levels than those observed in patients with metabolic syndrome alone.46 Furthermore, higher homocysteine levels correlates with more severe sleep apnea. Whether the same is true for patients without metabolic syndrome is unclear.
Hypoxia and H2S in the endothelium
The H2S-synthesizing enzymes CSE and 3-MST are expressed in endothelial cells (Figure 1),47,48 and H2S has been implicated in a number of endothelial processes, including angiogenesis, endothelium-dependent vasodilation, and regulation of inflammation (Figure 2).48–52 However, little is known about how H2S might mediate these physiological processes. In a human embryonic kidney expression system, H2S was shown to exert at least a portion of its effects through sulfhydration of cysteine residues on target proteins.53 Furthermore, H2S has been shown to act as both an autocrine and a paracrine factor, suggesting that in the endothelium, locally synthesized H2S might regulate vasodilation and angiogenesis via activation of Ca2+-activated K+ channels, vascular endothelial growth factor (VEGF) receptor 2, or other endothelial targets.50,51,54
In the endothelium, H2S promotes angiogenesis under normoxic conditions. As reported by Cai et al,49 NaHS-induced angiogenesis occurs through Akt signaling and does not involve enhanced VEGF or NO levels. There are conflicting results regarding H2S regulation of hypoxia-induced angiogenesis. A study by Wu et al55 suggested that H2S decreases the angiogenic response to hypoxia. The authors demonstrated that NaHS stimulates endothelial tube formation, as does hypoxia. During hypoxia, however, NaHS decreases tube formation. Furthermore, NaHS decreases expression of the transcription factor hypoxia inducible factor 1α (HIF1α) in endothelial cells during hypoxia has been shown to blunt hypoxia-driven increases in VEGF. This occurs through suppression of HIF1α translation and may be a mechanism by which high concentrations of H2S can limit angiogenesis.
Using a rodent model of unilateral hind limb ischemia, Wang et al56 provided evidence that H2S promotes angiogenesis in hypoxic conditions in vivo. Daily treatment with NaHS increases collateralization, capillary density, and blood flow in rat hind limbs made ischemic by femoral artery ligation. NaHS also increases VEGF protein expression in hind-limb muscles and phosphorylation of the VEGF receptor 2 in endothelial cells from ischemic hind limbs, but does not affect plasma levels of NO metabolites. Although not in agreement with the findings described above, these results highlight the potential therapeutic benefit of H2S donors for patients with peripheral artery disease.
H2S has been well described as a vasodilator in the systemic circulation, but the mechanism by which H2S elicits dilation appears to vary by vascular bed and is described in greater detail below. In at least some vascular beds, H2S-mediated vasodilation requires the endothelium,48,50,51 but there is evidence in other vascular beds and especially in larger arteries that the endothelium is not required.57,58 Additionally, endothelial CSE-derived H2S has been shown to contribute to cholinergic-mediated vasorelaxation of mesenteric arteries since loss of endothelial CTH expression blunts relaxation to methacholine.23 In addition to the evidence that H2S contributes to vasodilation under normoxic conditions, evidence is accumulating showing this gasotransmitter is involved in hypoxia-mediated vasodilation. As described in a study by Olson et al,36 hypoxia and exogenous H2S elicit similar relaxation responses in a rat’s thoracic aorta. Preventing H2S synthesis with the CSE inhibitor propargyl glycine (PAG) almost completely blocks the relaxation response to hypoxia, bolstering the conclusion that CSE-derived H2S is required for hypoxia-mediated aortic relaxation.
Where and how hypoxia regulates H2S signaling in the vasculature is not clear. In the cerebral circulation, there is evidence that hypoxia-induced dilation requires H2S but also involves carbon monoxide (CO). In cerebral arterioles, endothelial-derived CO increases vasoconstrictor tone.59 Under normoxic conditions, heme oxygenase-2 (HO-2) produces CO, which binds the heme of astrocytic CBS and inhibits CBS activity.60 As demonstrated by Morikawa et al61 in mice, hypoxia decreases cerebral CO production and elicits HO-2- and CBS-dependent vasodilation of arterioles in the cerebral cortex. These findings suggest that H2S is required for hypoxia-induced vasodilation in cerebral arterioles, but that hypoxia removes an inhibitor of H2S production rather than directly activating H2S signaling. It is unclear whether this pathway is unique to the cerebral circulation, where arterioles are in close proximity to CBS-expressing astrocytes. Indeed, the findings are somewhat confounded by evidence that hypoxia regulates expression of the enzymes that produce H2S, including an increase in both CTH mRNA and protein in hypoxia-exposed VSMC.40
Another potential but controversial role of H2S is modulation of inflammation. The cumulative data to date suggest that H2S is pro-inflammatory at high concentrations and anti-inflammatory at low concentrations.52 Zanardo et al62 demonstrated that exogenous H2S inhibits leukocyte adhesion to postcapillary mesenteric venule endothelial cells, whereas inhibiting endogenous H2S production increases leukocyte adherence to the endothelium. These compelling findings provide evidence that H2S can protect arteries from some aspects of inflammation, but little is known about the effect of hypoxia on this process in the vasculature. Fiorucci et al63 investigated the role of H2S in gastric mucosal injury caused by diminished blood flow, and thus hypoxia, after acetyl salicylic acid (ASA) treatment. NaHS reduced gastric injury and decreased inflammation. ASA treatment appeared to decrease endogenous H2S generation by reducing levels of the CTH activator Sp1. Since Sp1 is repressed by miR-21, as discussed above, and miR-21 is upregulated by hypoxia, this may be the pathway for ASA suppression of CTH expression. Studies to determine whether hypoxia does indeed decrease the anti-inflammatory effects of H2S are an intriguing area for future investigation.
In clinical studies, there is evidence that H2S signaling is impaired in atherosclerosis, a condition involving inflammation of the endothelium and potentially leading to peripheral artery disease and thus, limb ischemia and hypoxia. A recent study demonstrated that knockout of CTH in mice exacerbates the effects of an atherogenic diet, suggesting that endogenous H2S protects against atherosclerosis.64 Furthermore, administration of a slow-releasing H2S compound, GYY4137, decreases aortic plaque formation in apoE knockout mice.65 As with many of the other conditions described above, these results underscore that H2S treatment may limit the deleterious effects of atherosclerosis and other hypoxic conditions in humans.
A final consideration regarding the effects of hypoxia on H2S in the endothelium is the interaction between H2S and NO during hypoxia. The interaction between these gases has been evaluated under normoxic conditions, with conflicting results that include both H2S inhibition of NO synthase (NOS)66 and H2S activation of NOS,67 as well as formation of a nitrosothiol compound from the gases.68 Bir et al69 evaluated the interaction between NO and H2S in a rodent model of unilateral hind-limb ischemia and reported that H2S promotes ischemic vascular remodeling in an NO-dependent manner. Specifically, H2S therapy increases NO metabolites in plasma and hind-limb muscles, and scavenging NO blunts the improvement in hind-limb blood flow and prevents the enhanced angiogenesis conferred by H2S treatment. These results suggest a positive interaction between H2S and NO and provide more evidence of potential therapeutic benefit of H2S therapy in peripheral artery disease.
Hypoxia and H2S in myocytes
As illustrated in Figure 2, H2S can signal through multiple pathways. In VSMC, H2S has been shown to cause hyperpolarization and relaxation,48,57,70 and much attention has been given to the mechanism by which this occurs. It is of interest that most studies of H2S-induced hyperpolarization of VSMC have implicated at least one K+ channel, even if results do not agree on the particular type of channel involved or the concentration of H2S required for activation. In 2001, Zhao et al2 demonstrated that intravenous administration of H2S lowers blood pressure in rats and relaxes preconstricted aortic segments, and the ATP-sensitive K+ channel (KATP) blocker glibenclamide prevents the dilation, whereas inhibitors of other K+ channels do not. This was followed by studies from the same group in the rat perfused mesenteric-arterial bed, in which it was observed that the dilation to exogenous H2S was more pronounced than in the aorta, was endothelium dependent, and was only partially inhibited by glibenclamide.50 Thus, at least within the rat, H2S can cause dilation in some arteries through direct actions on the VSMC.
Several studies in rat mesenteric arteries, however, have observed dilation at much lower concentrations of H2S than those required to relax aortic segments and suggest that in this bed, vasodilation requires the endothelium and is mediated by large-conductance calcium-sensitive potassium channels (BKCa) rather than KATP.48,51,71 Thus, concentrations of H2S above 10 μM activate KATP channels to hyperpolarize and relax VSMC, but lower concentrations stimulate KATP-insensitive relaxation of VSMC, and further studies are necessary to determine whether only one or both of these pathways are physiologically relevant. In spite of the controversy over the molecular mechanism of action of H2S, there is little controversy that endogenous activity of CSE in the vasculature is affected by several hypoxia-inducing diseases and that H2S can protect VSMC from hypoxia-induced damage and death.
As described above, H2S elicits VSMC relaxation in normoxic conditions. However, hypoxia appears to diminish H2S’s ability to promote vasodilation. In a series of studies, Jackson-Weaver et al48,51 reported that isolated mesenteric arteries from rats exposed to intermittent hypoxia have decreased expression and function of CSE that results in increased myogenic tone compared with arteries from control rats. Furthermore, in rats exposed to intermittent hypoxia, an apparent decrease in CSE expression causes VSMC depolarization and diminished activity of Ca2+ sparks, a vasodilatory signaling pathway. Thus, intermittent hypoxia appears to impair H2S inhibition of myogenic tone and to promote enhanced contraction.
Evidence from Olson et al36 suggests that H2S may be an O2 sensor/transducer in hypoxic responses of vertebrate arteries. In isolated bovine pulmonary artery rings, both hypoxia and H2S (1 mM) elicit contraction when administered separately. When applied together, these agents are competitive, suggesting that they produce contraction through similar mechanisms and H2S may mediate hypoxic pulmonary vasoconstriction (HPV). Additionally, hypoxia and H2S cause VSMC membrane potential depolarization in bovine pulmonary arteries. The mechanism by which H2S mediates hypoxic vasoconstriction in pulmonary arteries is not clear, but results from a subsequent study from this group suggested H2S stimulates production of mitochondrial superoxide, which is dismutated to hydrogen peroxide to trigger downstream hypoxic vasoconstriction.72
Further support for a role for H2S in HPV is provided in a recent study by Madden et al,73 in which HPV responses were measured in intact, isolated, perfused rat lungs in conditions that promote or inhibit H2S synthesis. Providing cysteine to enhance H2S production augmented the hypoxia-induced increase in pulmonary artery pressure in isolated lungs. Conversely, inhibition of CSE with PAG diminished the pressure response to hypoxia. These findings highlight the contribution of H2S to HPV responses and suggest that this gasotransmitter is crucial for an appropriate pulmonary vascular response to hypoxia.
H2S also appears to protect VSMC from hypoxia-induced apoptosis and mitochondrial depolarization. A 2011 study by Bryan et al74 demonstrated that SMC from CSE knockout mice were more susceptible to apoptosis, mitochondrial depolarization, and cell death after exposure to 1.0% O2. There was also increased generation of mitochondrial reactive oxygen species (ROS).74 Similarly, human pulmonary VSMC exposed to CoCl2, to mimic hypoxia, had decreased H2S levels.7 However, in pulmonary VSMC, hypoxia augmented proliferation and this was ameliorated by exogenous H2S, suggesting endogenous H2S might blunt hypoxia-induced vascular remodeling and the development of pulmonary hypertension.7 In a study of VSMC from rats exposed to intermittent rather than sustained hypoxia, Hongfang et al4 reported that treatment with NaHS inhibits the hypoxia-mediated increase in pulmonary artery muscularization resulting from VSMC proliferation, as well as the increase in pulmonary artery pressure, further supporting a protective role of H2S on VSMC proliferation in conditions of pulmonary hypoxia.
The role of CBS-generated H2S under hypoxic conditions has not been extensively investigated. However, mice heterozygous for a mutated CBS gene (CBS+/−) do appear to have a defective angiogenic response to ischemia.75 The impaired formation of collateral vessels was attributed to loss of HIF-1α and VEGF signaling, suggesting that CBS generation of H2S might act permissively to promote HIF signaling in skeletal muscle arteries. In addition, hypoxia has been shown to increase the expression of CBS mRNA in the cerebral circulation in a HIF-dependent manner,76 suggesting a feedback regulation of CBS on HIF signaling. Acute regulation of CBS generation of H2S is also apparent in the cerebrovascular circulation. Studies by Morikawa et al61 reported that cerebrovascular vasodilation following hypoxia is diminished in CBS+/− compared with wild-type mice. However, it is unclear whether this is a direct vascular effect or whether it is mediated by H2S release from neurons or glial cells, since the studies were performed in mice with global knockdown of the CBS gene. Thus both acute and chronic responses to hypoxia have some dependence on CBS production of H2S.
In human keratinocytes, cell death in response to CoCl2, a surrogate for hypoxia, was ameliorated by treatment with H2S donors. Compared with cells exposed to CoCl2 alone, cells pretreated with NaHS had lower levels of ROS generation and decreased NFκB activation upstream of cyclooxygenase induction, the source of ROS.77 Thus, H2S appears to exert both direct and indirect antioxidant effects to protect VSMC and other cells from hypoxia-induced damage and cell death.
In HEP3B cells, exposure to 1.0% O2 triggered the induction of HIF1α-regulated genes, but H2S decreased the induction of these genes through destabilization of HIF1α protein.78 H2S was shown to inhibit mitochondrial oxygen consumption, leading to an increase in mitochondrial ROS generation and ubiquitination and degradation of the HIF1α protein. Wu et al55 also found that exposing cells to high levels of a H2S donor (10–100 μM NaHS) diminished hypoxia induction and stabilization of HIF1α protein. However, in the study by Wu et al, the effects were independent of ubiquitination, suggesting that H2S might regulate HIF1α protein stability by multiple mechanisms depending on the conditions and cell types in which hypoxia occurs.
H2S donors have been administered chronically in vivo to prevent hypoxia-induced morbidity. Treating rats for 3 weeks with daily injections of an H2S donor (2–200 mg/kg) protected cerebral arteries from hypoxia-induced damage.79 In a rat model of myocardial ischemia/reperfusion injury, H2S administration at the time of reperfusion decreased myocardial infarct size, inflammation, and cardiomyocyte apoptosis and preserved mitochondrial function.80 Furthermore, cardiac-specific overexpression of CSE diminished ischemia-reperfusion injury as measured by myocardial infarct size. In the kidney, ischemia markedly reduced renal production of H2S, and CSE knockout mice had increased damage and mortality after renal ischemia/reperfusion injury, suggesting that endogenous CSE activity is an important defense against hypoxic stress and preserves vascular perfusion.81 Treatment with H2S donors in this study prevented ischemia-induced renal injury in the CSE knockout mice, and overexpression of CSE reduced ROS generation in isolated tubules exposed to hypoxia. It is particularly intriguing that the level of CSE expression in kidneys from individual mice positively correlated with glomerular filtration rate and that glomeruli expressed high levels of CSE. These studies further suggest that the H2S signaling system provides important vascular protection against ischemia in many parts of the circulation.
H2S and redox regulation
Oxidative stress is increased in hypoxic environments, and many studies show that H2S is protective in conditions in which oxidative stress is elevated. Bryan et al74 demonstrated that ROS levels are higher in mesenteric artery VSMC from CSE knockout mice under normoxic conditions. Hypoxia increases ROS levels in VSMC from wildtype mice but elicits an even greater increase in cells from CSE knockout mice. These findings suggest H2S regulates redox signaling under basal and hypoxic conditions. There is some controversy over H2S’s role in regulation of redox homeostasis, with studies suggesting both direct and indirect antioxidant function for H2S. Results from a recent study by Hamar et al82 indicate that H2S is not an effective antioxidant itself. On the other hand, there is ample evidence that this gasotransmitter regulates the cellular redox environment by activating antioxidant pathways.
Calvert et al83 identified nuclear factor E2-related factor (Nrf2) signaling as one of the antioxidant pathways triggered by H2S. Pretreatment with exogenous H2S reduced cardiac damage following myocardial ischemia/reperfusion injury, and it increased expression of Nrf2, a transcription factor that regulates many antioxidant genes, and Nrf2’s downstream targets HO-1 and thioredoxin. This study, like many others, suggested a potential therapeutic benefit of exogenous H2S administration to limit oxidative damage in hypoxic conditions. How oxidative stress may affect endogenous H2S signaling, however, is less clear. Streeter et al84 reported that CTH mRNA expression is increased in cerebral arteries from diabetic rats compared with arteries from control rats, suggesting that oxidative stress may trigger upregulation of endogenous H2S production to combat redox imbalance.
H2S in the human population
In comparison with the many studies in rodents, much less is known about the distribution and function of H2S in humans. Studies of circulating levels of H2S have shown that both increased85 and decreased86 levels of H2S are associated with vascular disease.
In a study comparing tissues and plasma from 14 preeclamptic women and 14 healthy controls, Wang et al86 found lower circulating H2S and lower levels of placental CSE in women with preeclampsia compared with control patients. The decrease in CSE and H2S levels was associated with reduced fetal size. In pregnant mice, the CSE inhibitor, PAG, caused hypertension, liver dysfunction, and fetal growth restriction that was reversed by co-administration of an H2S donor. Thus, H2S production in placental vessels appears to be necessary for normal placental perfusion and fetal development, however, it is unclear whether this is mediated entirely through endothelial pathways of vessel growth or is also partially dependent on changes in VSMC that lead to enhanced constrictor sensitivity.
Jain et al87 reported that in patients with atherosclerosis, there is an inverse correlation between plasma levels of low-density lipoprotein and H2S. In this study, higher H2S was associated with less plaque development. Studies in diabetic patients also found that low levels of H2S are associated with worse outcomes in patients that have concurrent sleep apnea,88 and that renal decline progresses more rapidly in chronic kidney disease patients with low serum H2S.89 In the latter study, plasma H2S levels correlated with the rate of decline in glomerular filtration rate independently of all other predictors. Thus, declines in H2S production may contribute to multiple vascular diseases in the human population.
In contrast, a study by Peter et al85 found that plasma levels of H2S were higher in patients with peripheral artery disease than in a group of patients without detectable vascular disease. This study suggests the relationship between vascular disease and circulating H2S levels may not be as simple as that in animal models. Additional studies defining variables that affect circulating H2S levels in humans are needed, as are studies defining how circulating H2S modifies vascular responses to disease.
Summary
It has become apparent in recent years that H2S is produced endogenously and is involved in numerous physiological processes, including many that maintain vascular homeostasis. With a role in promoting endothelial proliferation, maintaining normal blood pressure, and eliciting vasodilation, this gas has emerged as a key regulator of vascular function. The protective role H2S plays in hypoxic conditions in many vascular beds is also becoming apparent. As highlighted in this review, there is exciting new evidence suggesting that this newest gasotransmitter may be a beneficial therapy for patients with a variety of conditions associated with tissue hypoxia.
Disclosure
The authors report no conflicts of interest in this work.
References
Chertok VM, Kotsyuba AE. Distribution of H2S synthesis enzymes in the walls of cerebral arteries in rats. Bull Exp Biol Med. 2012;154(1):104–107. | |
Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. Embo J. 2001;20(21):6008–6016. | |
Schleifenbaum J, Kohn C, Voblova N, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010;28(9):1875–1882. | |
Hongfang J, Cong B, Zhao B, et al. Effects of hydrogen sulfide on hypoxic pulmonary vascular structural remodeling. Life Sci. 2006;78(12):1299–1309. | |
Li H, Mani S, Cao W, et al. Interaction of hydrogen sulfide and estrogen on the proliferation of vascular smooth muscle cells. PLoS One. 2012;7(8):e41614. | |
Li L, Liu D, Bu D, et al. Brg1-dependent epigenetic control of vascular smooth muscle cell proliferation by hydrogen sulfide. Biochim Biophys Acta. 2013;1833(6):1347–1355. | |
Li Y, Liu G, Cai D, et al. H2S inhibition of chemical hypoxia-induced proliferation of HPASMCs is mediated by the upregulation of COX-2/PGI2. Int J Mol Med. 2014;33(2):359–366. | |
Yang G, Wu L, Bryan S, Khaper N, Mani S, Wang R. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovasc Res. 2010;86(3):487–495. | |
Dombkowski RA, Doellman MM, Head SK, Olson KR. Hydrogen sulfide mediates hypoxia-induced relaxation of trout urinary bladder smooth muscle. J Exp Biol. 2006;209(Pt 16):3234–3240. | |
Li Y, Zang Y, Fu S, Zhang H, Gao L, Li J. H2S relaxes vas deferens smooth muscle by modulating the large conductance Ca2+-activated K+ (BKCa) channels via a redox mechanism. J Sex Med. 2012;9(11):2806–2813. | |
Tang G, Wu L, Wang R. Interaction of hydrogen sulfide with ion channels. Clin Exp Pharmacol Physiol. 2010;37(7):753–763. | |
Tian XY, Wong WT, Sayed N, et al. NaHS relaxes rat cerebral artery in vitro via inhibition of l-type voltage-sensitive Ca2+ channel. Pharmacol Res. 2012;65(2):239–246. | |
Altaany Z, Yang G, Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. 2013;17(7):879–888. | |
Liu X, Pan L, Zhuo Y, Gong Q, Rose P, Zhu Y. Hypoxia-inducible factor-1alpha is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol Pharm Bull. 2010;33(9):1550–1554. | |
Pupo E, Pla AF, Avanzato D, et al. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic Biol Med. 2011;51(9):1765–1773. | |
Wang R. Signaling pathways for the vascular effects of hydrogen sulfide. Curr Opin Nephrol Hypertens. 2011;20(2):107–112. | |
Go YM, Lee HR, Park H. H(2)S inhibits oscillatory shear stress-induced monocyte binding to endothelial cells via nitric oxide production. Mol Cells. 2012;34(5):449–455. | |
Guan Q, Wang X, Gao L, et al. Hydrogen sulfide suppresses high glucose-induced expression of intercellular adhesion molecule-1 in endothelial cells. J Cardiovasc Pharmacol. 2013;62(3):278–284. | |
Zhang H, Zhi L, Moochhala SM, Moore PK, Bhatia M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J Leukoc Biol. 2007;82(4):894–905. | |
Kida M, Sugiyama T, Yoshimoto T, Ogawa Y. Hydrogen sulfide increases nitric oxide production with calcium-dependent activation of endothelial nitric oxide synthase in endothelial cells. Eur J Pharm Sci. 2013;48(1–2):211–215. | |
Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92(2):791–896. | |
Mancardi D, Penna C, Merlino A, Del Soldato P, Wink DA, Pagliaro P. Physiological and pharmacological features of the novel gasotransmitter: hydrogen sulfide. Biochim Biophys Acta. 2009;1787(7):864–872. | |
Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590. | |
Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206(2):267–277. | |
Hughes MN, Centelles MN, Moore KP. Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic Biol Med. 2009;47(10):1346–1353. | |
Chen X, Jhee KH, Kruger WD. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J Biol Chem. 2004;279(50):52082–52086. | |
Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16(3):1066–1071. | |
Teague B, Asiedu S, Moore PK. The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. Br J Pharmacol. 2002;137(2):139–145. | |
Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237(3):527–531. | |
Tan NY, Khachigian LM. Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol. 2009;29(10):2483–2488. | |
Yang G, Pei Y, Teng H, Cao Q, Wang R. Specificity protein-1 as a critical regulator of human cystathionine gamma-lyase in smooth muscle cells. J Biol Chem. 2011;286(30):26450–26460. | |
Yang G, Pei Y, Cao Q, Wang R. MicroRNA-21 represses human cystathionine gamma-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J Cell Physiol. 2012;227(9):3192–3200. | |
Cindrova-Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol. 2013;182(4):1448–1458. | |
Mikami Y, Shibuya N, Ogasawara Y, Kimura H. Hydrogen sulfide is produced by cystathionine gamma-lyase at the steady-state low intracellular Ca(2+) concentrations. Biochem Biophys Res Commun. 2013;431(2):131–135. | |
Beltowski J. Hypoxia in the renal medulla: implications for hydrogen sulfide signaling. J Pharmacol Exp Ther. 2010;334(2):358–363. | |
Olson KR, Dombkowski RA, Russell MJ, et al. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol. 2006;209(Pt 20):4011–4023. | |
Peng YJ, Nanduri J, Raghuraman G, et al. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107(23):10719–10724. | |
Hassan MI, Boosen M, Schaefer L, et al. Platelet-derived growth factor-BB induces cystathionine gamma-lyase expression in rat mesangial cells via a redox-dependent mechanism. Br J Pharmacol. 2012;166(8):2231–2242. | |
Tao BB, Zhang CC, Liu SY, Zhu YC. [Involvement of HIF-1 in the migration-promoting effects of hydrogen sulfide in vascular endothelial cells under normoxic conditions]. Sheng Li Xue Bao. 2012;64(2):129–134. Chinese. | |
Wang M, Guo Z, Wang S. Regulation of cystathionine gamma-lyase in mammalian cells by hypoxia. Biochem Genet. 2014;52(1–2):29–37. | |
Szabo C, Ransy C, Modis K, et al. Regulation of mitochondrial bioenergetics function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2014;171(8):2099–2122. | |
Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci U S A. 2012;109(8):2943–2948. | |
Chunyu Z, Junbao D, Dingfang B, Hui Y, Xiuying T, Chaoshu T. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun. 2003;302(4):810–816. | |
Steed MM, Tyagi SC. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal. 2011;15(7):1927–1943. | |
Sen U, Basu P, Abe OA, et al. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol. 2009;297(2):F410–F419. | |
Monneret D, Tamisier R, Ducros V, et al. The impact of obstructive sleep apnea on homocysteine and carotid remodeling in metabolic syndrome. Respir Physiol Neurobiol. 2012;180(2–3):298–304. | |
Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. 2009;146(5):623–626. | |
Jackson-Weaver O, Paredes DA, Gonzalez Bosc LV, Walker BR, Kanagy NL. Intermittent hypoxia in rats increases myogenic tone through loss of hydrogen sulfide activation of large-conductance Ca(2+)-activated potassium channels. Circ Res. 2011;108(12):1439–1447. | |
Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76(1):29–40. | |
Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287(5):H2316–H2323. | |
Jackson-Weaver O, Osmond JM, Riddle MA, et al. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca(2)(+)-activated K(+) channels and smooth muscle Ca(2)(+) sparks. Am J Physiol Heart Circ Physiol. 2013;304(11):H1446–H1454. | |
Vandiver M, Snyder SH. Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med (Berl). 2012;90(3):255–263. | |
Mustafa AK, Gadalla MM, Sen N, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2(96):ra72. | |
Tao BB, Liu SY, Zhang CC, et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid Redox Signal. 2013;19(5):448–464. | |
Wu B, Teng H, Yang G, Wu L, Wang R. Hydrogen sulfide inhibits the translational expression of hypoxia-inducible factor-1alpha. Br J Pharmacol. 2012;167(7):1492–1505. | |
Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal. 2010;12(9):1065–1077. | |
Mustafa AK, Sikka G, Gazi SK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109(11):1259–1268. | |
Streeter E, Hart J, Badoer E. An investigation of the mechanisms of hydrogen sulfide-induced vasorelaxation in rat middle cerebral arteries. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(10):991–1002. | |
Ishikawa M, Kajimura M, Adachi T, et al. Carbon monoxide from heme oxygenase-2 Is a tonic regulator against NO-dependent vasodilatation in the adult rat cerebral microcirculation. Circ Res. 2005;97(12):e104–e114. | |
Shintani T, Iwabuchi T, Soga T, et al. Cystathionine beta-synthase as a carbon monoxide-sensitive regulator of bile excretion. Hepatology. 2009;49(1):141–150. | |
Morikawa T, Kajimura M, Nakamura T, et al. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc Natl Acad Sci USA. 2012;109(4):1293–1298. | |
Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte- mediated inflammation. Faseb J. 2006;20(12):2118–2120. | |
Fiorucci S, Antonelli E, Distrutti E, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129(4):1210–1224. | |
Mani S, Li H, Untereiner A, et al. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;127(25):2523–2534. | |
Liu Z, Han Y, Li L, et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(−/−) mice. Br J Pharmacol. 2013;169(8):1795–1809. | |
Kubo S, Kurokawa Y, Doe I, Masuko T, Sekiguchi F, Kawabata A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology. 2007;241(1–2):92–97. | |
Minamishima S, Bougaki M, Sips PY, et al. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120(10):888–896. | |
Whiteman M, Li L, Kostetski I, et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun. 2006;343(1):303–310. | |
Bir SC, Kolluru GK, McCarthy P, et al. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J Am Heart Assoc. 2012;1(5):e004093. | |
Tang G, Yang G, Jiang B, Ju Y, Wu L, Wang R. H(2)S is an endothelium-derived hyperpolarizing factor. Antioxid Redox Signal. 2013;19(14):1634–1646. | |
White BJ, Smith PA, Dunn WR. Hydrogen sulphide-mediated vasodilatation involves the release of neurotransmitters from sensory nerves in pressurized mesenteric small arteries isolated from rats. Br J Pharmacol. 2013;168(4):785–793. | |
Skovgaard N, Olson KR. Hydrogen sulfide mediates hypoxic vasoconstriction through a production of mitochondrial ROS in trout gills. Am J Physiol Regul Integr Comp Physiol. 2012;303(5):R487–R494. | |
Madden JA, Ahlf SB, Dantuma MW, Olson KR, Roerig DL. Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J Appl Physiol (1985). 2012;112(3):411–418. | |
Bryan S, Yang G, Wang R, Khaper N. Cystathionine gamma-lyase-deficient smooth muscle cells exhibit redox imbalance and apoptosis under hypoxic stress conditions. Exp Clin Cardiol. 2011;16(4):e36–e41. | |
Veeranki S, Givvimani S, Pushpakumar S, Tyagi SC. Hyperhomocysteinemia attenuates angiogenesis through reduction of HIF-1alpha and PGC-1alpha levels in muscle fibers during hindlimb ischemia. Am J Physiol Heart Circ Physiol. 2014;306(8):H1116–H1127. | |
Takano N, Peng YJ, Kumar GK, et al. Hypoxia-inducible factors regulate human and rat cystathionine beta-synthase gene expression. Biochem J. 2014;458(2):203–211. | |
Yang C, Yang Z, Zhang M, et al. Hydrogen sulfide protects against chemical hypoxia-induced cytotoxicity and inflammation in HaCaT cells through inhibition of ROS/NF-kappaB/COX-2 pathway. PLoS One. 2011;6(7):e21971. | |
Kai S, Tanaka T, Daijo H, et al. Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid Redox Signal. 2012;16(3):203–216. | |
Liu L, Liu H, Sun D, et al. Effects of H(2)S on myogenic responses in rat cerebral arterioles. Circ J. 2012;76(4):1012–1019. | |
Elrod JW, Calvert JW, Morrison J, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104(39):15560–15565. | |
Bos EM, Wang R, Snijder PM, et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J Am Soc Nephrol. 2013;24(5):759–770. | |
Hamar J, Solymar M, Tanai E, et al. Bioassay-comparison of the antioxidant efficacy of hydrogen sulfide and superoxide dismutase in isolated arteries and veins. Acta Physiol Hung. 2012;99(4):411–419. | |
Calvert JW, Jha S, Gundewar S, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105(4):365–374. | |
Streeter EY, Badoer E, Woodman OL, Hart JL. Effect of type 1 diabetes on the production and vasoactivity of hydrogen sulfide in rat middle cerebral arteries. Physiol Rep. 2013;1(5):e00111. | |
Peter EA, Shen X, Shah SH, et al. Plasma free H2S levels are elevated in patients with cardiovascular disease. J Am Heart Assoc. 2013;2(5):e000387. | |
Wang K, Ahmad S, Cai M, et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation. 2013;127(25):2514–2522. | |
Jain SK, Micinski D, Lieblong BJ, Stapleton T. Relationship between hydrogen sulfide levels and HDL-cholesterol, adiponectin, and potassium levels in the blood of healthy subjects. Atherosclerosis. 2012;225(1):242–245. | |
Jain SK, Kahlon G, Morehead L, et al. The effect of sleep apnea and insomnia on blood levels of leptin, insulin resistance, IP-10, and hydrogen sulfide in type 2 diabetic patients. Metab Syndr Relat Disord. 2012;10(5):331–336. | |
Andresdottir G, Bakker SJ, Hansen HP, Parving HH, Rossing P. Urinary sulphate excretion and progression of diabetic nephropathy in Type 1 diabetes. Diabet Med. 2013;30(5):563–566. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.