Back to Journals » Breast Cancer: Targets and Therapy » Volume 8
Modulation of breast cancer cell viability by a cannabinoid receptor 2 agonist, JWH-015, is calcium dependent
Authors Hanlon K ![]() , Lozano-Ondoua A, Umaretiya P, Symons-Liguori A, Chandramouli A, Moy J, Kwass W, Mantyh P, Nelson M, Vanderah T
, Lozano-Ondoua A, Umaretiya P, Symons-Liguori A, Chandramouli A, Moy J, Kwass W, Mantyh P, Nelson M, Vanderah T
Received 12 November 2015
Accepted for publication 14 January 2016
Published 15 April 2016 Volume 2016:8 Pages 59—71
DOI https://doi.org/10.2147/BCTT.S100393
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Katherine E Hanlon,1,2 Alysia N Lozano-Ondoua,1 Puja J Umaretiya,1 Ashley M Symons-Liguori,1 Anupama Chandramouli,1 Jamie K Moy,1 William K Kwass,1 Patrick W Mantyh,1 Mark A Nelson,3 Todd W Vanderah,1
1Department of Pharmacology, University of Arizona College of Medicine, Tucson, AZ, USA; 2Department of Biomedical Sciences, University of New England College of Osteopathic Medicine, Biddeford, ME, USA; 3Department of Pathology, University of Arizona College of Medicine, Tucson, AZ, USA
Introduction: Cannabinoid compounds, both nonspecific as well as agonists selective for either cannabinoid receptor 1 (CB1) or cannabinoid receptor 2 (CB2), have been shown to modulate the tumor microenvironment by inducing apoptosis in tumor cells in several model systems. The mechanism of this modulation remains only partially delineated, and activity induced via the CB1 and CB2 receptors may be distinct despite significant sequence homology and structural similarity of ligands.
Methods: The CB2-selective agonist JWH-015 was used to investigate mechanisms downstream of CB2 activation in mouse and human breast cancer cell lines in vitro and in a murine mammary tumor model.
Results: JWH-015 treatment significantly reduced primary tumor burden and metastasis of luciferase-tagged murine mammary carcinoma 4T1 cells in immunocompetent mice in vivo. Furthermore, JWH-015 reduced the viability of murine 4T1 and human MCF7 mammary carcinoma cells in vitro by inducing apoptosis. JWH-015-mediated reduction of breast cancer cell viability was not dependent on Gαi signaling in vitro or modified by classical pharmacological blockade of CB1, GPR55, TRPV1, or TRPA1 receptors. JWH-015 effects were calcium dependent and induced changes in MAPK/ERK signaling.
Conclusion: The results of this work characterize the actions of a CB2-selective agonist on breast cancer cells in a syngeneic murine model representing how a clinical presentation of cancer progression and metastasis may be significantly modulated by a G-protein-coupled receptor.
Keywords: cannabinoid receptor-2, CB2, breast cancer, JWH-015, MAPK/ERK, apoptosis, calcium
Introduction
Breast cancer is the leading cause of cancer-related deaths among women aged 34–50 worldwide,1 and is the most commonly diagnosed metastasizing tumor in women of all ages.2 The staggering prevalence of breast cancer has made it a central focus of investigative efforts over the last 70 years. The results of such expansive research efforts are encouraging: not only have there been tremendous advances in understanding the etiology of the disease but significant improvements have also been made in detection and diagnosis of breast cancer. These advances have aided in increasing the 10-year survival rates of metastatic breast cancer from 3.3% in 1944 to 22.2% in 2004, with even greater life expectancy in patients with localized disease (76.5%).3 Despite advances in understanding breast cancer as a disease, there remains a critical need for novel disease-modifying therapeutics. The possible genetic mutations occurring in breast tumors vary widely from patient to patient,4 making protein-targeted therapies applicable to specific subsets of patients and increasing the occurrence of therapeutic resistance.5 In addition, there is an inverse relationship between disease progression and efficacy of analgesics, contributing to the 75%–95% of breast cancer patients experiencing chronic debilitating pain that is predominantly associated with metastasis.6
Nonspecific cannabinoids,7–9 cannabinoid receptor 2 (CB2)-selective,10,11 as well as cannabinoid receptor 1 (CB1)-selective compounds12,13 have yielded similar antitumor results in several tumor models. The lack of neuronal expression of CB2 receptors precludes CB2 selective compounds from inducing the psychotropic effects that typically accompany CB1 activation.14 Our group and others have shown that CB2 agonists displaying selectivity for the CB2 receptor can decrease tumor cell viability and significantly attenuate cancer-induced bone pain without displaying psychoactive or addictive properties.15,16 Additionally, CB2 receptors are markedly upregulated in many breast tumors,17 which is a curious finding given that many of these tumor cells are of epithelial origin, while in normal adults, CB2 receptor expression is primarily limited to cells of the immune system and bone maintenance cells.18
While the antitumor effects of cannabinoids have been demonstrated in a variety of tumor models, the intracellular pathway(s) by which cannabinoid antitumor activity occurs remain unclear. Several cannabinoid mechanisms have been reported, some conflicting, that identify cannabinoid activity through Gαi,19 Gαq,20 mTORC1,21 AKT/PI3K inhibition,17 AKT/PI3K stimulation,22 MAPK/ERK modulation,23 p38/MAPK modulation,24 ceramide accumulation,25 induction of reactive oxygen species,26 modification of matrix metalloproteinases,10 Id-1 inhibition,9 and the involvement of extraneous receptors including TRPV127 and GPR55.28 To further complicate matters, it has also been suggested that the action of cannabinoids on tumor cells alone is not sufficient to produce in vivo antitumor effects and also requires immune system interaction to achieve full efficacy.7 Here, we focus on the direct effects and mechanism of the CB2-selective agonist JWH-015 on breast cancer cells. We show that JWH-015 inhibits tumor cell viability and induces apoptosis of breast cancer cell lines (murine) 4T1 and (human) MCF7 in vitro and attenuates primary tumor growth and metastasis in vivo. In addition, we show that the MAPK/ERK pathway is modified by JWH-015. Finally, we demonstrate that these effects occur through a Ca2+-dependent mechanism that is unlikely to lie downstream of the traditional Gαi pathway previously associated with cannabinoid receptor activity. The antiproliferative effects of a CB2 agonist along with our previous work demonstrating significant efficacy in inhibiting bone cancer pain and slowing bone loss in a murine model of advanced breast cancer strongly suggest that CB2 agonists should be investigated in humans as adjunct therapy for advanced stages of breast cancer.
Methods
In vitro
Cell culture
4T1 and MCF7 cell lines were obtained from American Type Culture Collection Biological Resource Center (Manassas, VA, USA). All cells were cultured in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum, 100 IU mL−1 penicillin and 100 μg/mL streptomycin. Cells were plated in 10 cm tissue culture dishes and allowed to grow exponentially.
No ethical approval was sought or required for experiments involving commercially purchased cell lines, as per guidelines of the University of Arizona human subjects protection program.
Stable expression of 4T1-Luc cells
4T1 cells stably expressing the pGL4.17 (luciferase) vector (Promega, Fitchburg, WI, USA) were developed via transfection with Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) and selected with neomycin with expression being verified prior to each assay according to the manufacturer’s instructions.
CB2 small hairpin RNA knockdown
CB2 and control scramble small hairpin RNA (shRNA) DNA lentiviral particles (Santa Cruz Biotechnology, Dallas, TX, USA) were used to transiently knockdown CB2 receptor expression in 4T1 cells according to the manufacturer’s instructions.
Compounds
The National Institute on Drug Abuse provided Cannabidiol, SR144528, and SR141716. Pertussis toxin (PTX) and capsazepine were purchased from Sigma-Aldrich (St Louis, MO, USA). JWH-015, o-1602, nifedipine, ω-conotoxin MVIIC, and HC-030031 were purchased from Tocris (Ellisville, MO, USA). All compounds were dissolved in dimethyl sulfoxide (DMSO) and added to fresh culture media such that the effective DMSO concentration was 0.1% per well.
Cellular viability assay
Cellular viability was measured using the sulforhodamine B (SRB; Sigma-Aldrich) assay as previously described.29
Immunoblotting
Cells were lysed, protein extracts were separated electrophoretically, and then transferred to a polyvinylidene fluoride membrane as previously described.30 The membrane was incubated with either polyclonal anti-CB2 (Cayman Chemical, Ann Arbor, MI, USA; item number 101550) or monoclonal phospho-ERK1/2 (Cell Signaling Technology, Danvers, MA, USA) antibody followed by horseradish peroxidase-conjugated secondary IgG antibody (Cell Signaling Technology) and developed using a chemiluminescent system (Amersham Biosciences, Piscataway, NJ, USA). α-Tubulin and glyceraldehyde phosphate dehydrogenase (Cell Signaling Technology) were used as loading controls. Quantification of Western blot was performed using gel analysis in ImageJ (National Institutes of Health, Bethesda, MD, USA).
Ca2+ imaging
4T1 cells were cultured to 50% confluence in dual-chamber slides in complete media. On the day of imaging, cells were switched to serum-low, antibiotic-free Opti-minimal essential medium (Opti-MEM) media (Thermo Fisher Scientific) and equilibrated for 1 hour in a 5% CO2 incubator at 37°C. Lyophilized fura-2 acetoxymethyl (Fura-2 AM) (Thermo Fisher Scientific) dissolved at 1 mg/mL in DMSO constituted stock solution, which was then diluted to 5 mg/mL in Opti-MEM. Cells were incubated for 1 hour in 5 μg/mL Fura-2 AM for loading, followed by 30-minute incubation in Opti-MEM without Fura-2 AM to allow de-esterification of the fluorescent dye. Cells were imaged on a Nikon fluorescent microscope (Nikon Instruments, Inc., Melville, NY, USA) equipped with a charge-coupled device camera. Absorbance at 340 and 380 nm were simultaneously recorded, and the absorbance ratio (340 nm [Ca2+-bound Fura-2] over 380 nm [free Fura-2]) was plotted for a period of 60 minutes before spiking the media with potassium chloride for positive control.
Caspase 3/7 activity
Caspase-Glo 3/7 assay (Promega) was used to measure caspase 3 and 7 activities in 4T1 cells and MCF7 cells (which express caspase 7 but not caspase 3) according to the manufacturer’s instructions. Results were normalized to total protein and expressed as a percentage of control.
Immunohistochemistry
4T1 cells were allowed to grow to 50% confluence in dual-chamber slides before replacing media with serum-low Opti-MEM. Cells were treated with JWH-015 for 24 hours and fixed with 10% formalin for 20 minutes at room temperature. Cells were permeabilized with 0.25% Triton X-100 (Sigma-Aldrich) for 30 minutes and blocked with 5% bovine serum albumin for 30 minutes at room temperature. Cells were incubated in polyclonal Ki67 antibody (Abcam, Cambridge, MA, USA) diluted 1/200 in 5% bovine serum albumin overnight at 4°C. Slides were incubated for 2 hours at room temperature with Alexa Fluor 488 (Thermo Fisher Scientific) anti-rabbit IgG secondary antibody, mounted with 4′,6-diamidino-2-phenylindole (Abcam), and visualized on a fluorescent microscope (Zeiss, Oberkochen, Germany).
Cyclic adenosine monophosphate
A cyclic adenosine monophosphate (cAMP) complete enzyme immunoassay kit (Enzo Life Sciences, Farmingdale, NY, USA) was used to measure forskolin-induced cAMP formation in vitro in response to JWH-015. Assay was conducted per manufacturer instructions and normalized to total protein.
In vivo
Animals
All procedures were approved by the University of Arizona Animal Care and Use Committee and conformed to the Guidelines of the National Institutes of Health and the International Association for the Study of Pain. Female BALB/cfC3H mice (Harlan, Indianapolis, IN, USA) weighed 15–20 g prior to initiation of study. Mice were maintained in a climate-controlled room on a 12-hour light/dark cycle and allowed food and water ad libitum.
Mammary fat pad injections
Mice were injected with 4T1-Luc cells (5×105) into the c-9 mammary fat pad as previously described.31
Treatment
The CB2 receptor agonist JWH-015 with affinity at receptor (Ki) at CB2=13.8 nM and 30-fold selectivity over CB1 was dissolved in a vehicle solution of 10% DMSO, 10% Tween-80, and 80% saline. All injections were made at a volume of 10 mL/kg and consisted of once-daily intraperitoneal (ip) injections for 21 days (8–28). Randomly assigned animals were treated with JWH-015 (6 mg/kg ip) or vehicle.
Bioluminescence imaging
Animals inoculated into the mammary fat pad with 4T1-Luc cells were live-imaged on a weekly basis as previously described.31 Briefly, 4T1-Luc tumor-bearing animals were anesthetized with inhaled isoflurane and injected ip with K+ d-luciferin (Molecular Imaging Products, Bend, OR, USA) 5 minutes before imaging (5-minute exposure in a dark chamber). Dark images were assigned intensity map coloring in ImageJ and overlaid on 100 ms light exposures of the same animals.
Ex vivo
Primary tumor and metastasis analysis
Twenty-eight days after tumor inoculation, animals were sacrificed, primary tumors excised, and tumor mass recorded. Common sites of mammary tumor metastasis were harvested (brain, lung, femur, kidney, and spleen) and homogenized in the presence of protease inhibitor cocktail (Sigma-Aldrich) for ex vivo detection of metastasis using a dual-luciferase reporter assay (Promega) performed according to the manufacturer’s instructions. Data are represented as % of total samples in treatment group testing positive for luciferase activity.
Statistical analysis
SRB assays, quantified Western blots, caspase 3/7 assays, cAMP assays, and primary tumor mass were analyzed using one-way analysis of variance followed by Student–Neuman–Kuels testing for multiple comparisons in FlashCalc.32 Differences were considered to be significant if P≤0.05. When possible, potencies (or A50) were determined by regression analysis of dose–response curve (log dose [x] vs response [y]) using a 95% confidence interval (CI) according to the method of analysis of the graded dose–response.33 For the calculations of A50s, the minimal possible response was set to 0%.
Results
Breast cancer cells functionally express the CB2 receptor
Cellular expression of CB2 was positively identified in cell lysate from several immortalized mouse and human breast cancer cell lines (Figure 1A). To ascertain whether JWH-015 stimulation of the CB2 receptor engages the traditionally described Gαi pathway, cAMP was assayed in the presence of varying concentrations of JWH-015 after forskolin stimulation in 4T1 cells (Figure 1B). We found that 4T1 cells demonstrate high baseline adenylate cyclase activity that is not increased by incubation with forskolin (P>0.05) and can be blocked by low nanomolar concentrations of JWH-015. Peak inhibition of cAMP occurs at concentrations of 1 nM (P<0.05) and 10 nM (P<0.05) of JWH-015, while inhibition of cAMP accumulation is abolished at JWH-015 concentrations higher than 100 nM (P>0.05). To evaluate the functionality of CB2 expression in 4T1 mammary carcinoma cells, we performed knockdown of the CB2 receptor in 4T1 murine mammary carcinoma cells in vitro via shRNA DNA lentiviral particles (Figure 1C, D). shRNA knockdown of the CB2 receptor reduced cancer cell viability in a manner consistent with degree of knockdown (P<0.05). These data suggest the expression of the CB2 receptor as part of the vitality of the cancer cell line.
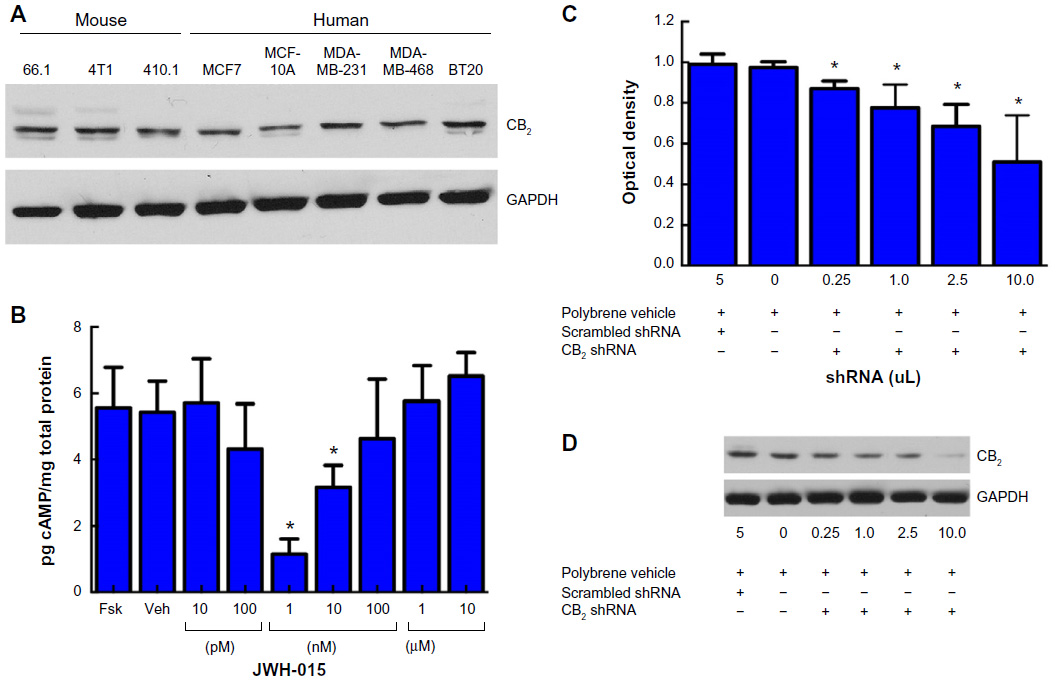 | Figure 1 Breast cancer cells functionally express the CB2 receptor. |
JWH-015 reduces primary tumor burden and decreases metastasis in vivo
4T1 cells were transfected with luciferase, and the resulting stable 4 T1-Luc cell line was injected into the c-9 mammary fat pad of immunocompetent female BALB/cfC3H mice. The reported data are combined from three separate experiments in which tumors were allowed to establish for 7 days postinoculation and mice were then treated with either CB2 agonist JWH-015 6 mg/kg ip (n=19) or vehicle (n=12) for 21 days. Mice were imaged on a weekly basis (Figure 2A) beginning on day 7 through day 28 to track primary tumor growth and sites of metastasis. On day 28, mice were sacrificed, primary tumors were immediately resected, and common metastatic site tissues were harvested. Chronic administration of JWH-015 (6 mg/kg) significantly reduced primary tumor mass (P<0.05; Figure 2B). Tissue samples from common metastatic sites were harvested for ex vivo analysis with a Promega dual-luciferase reporter assay. Detectable metastases (defined by the presence of luciferase expressing cells in the tissue sample) were reported as a percentage of the total number of the respective tissue samples per experimental group (Figure 2C). Comparison of samples isolated from either vehicle- or JWH-015 (6 mg/kg)-treated animals revealed a trend toward reduced metastasis in tissues isolated from brain, femur, kidney, spleen, lung, and liver; however, since this assay evaluated the presence or absence of luciferase-expressing cells within each tissue sample without quantification, only trends in CB2 agonist-induced reduction in metastasis can be reported along with the statistically significant reduction in primary tumor burden.
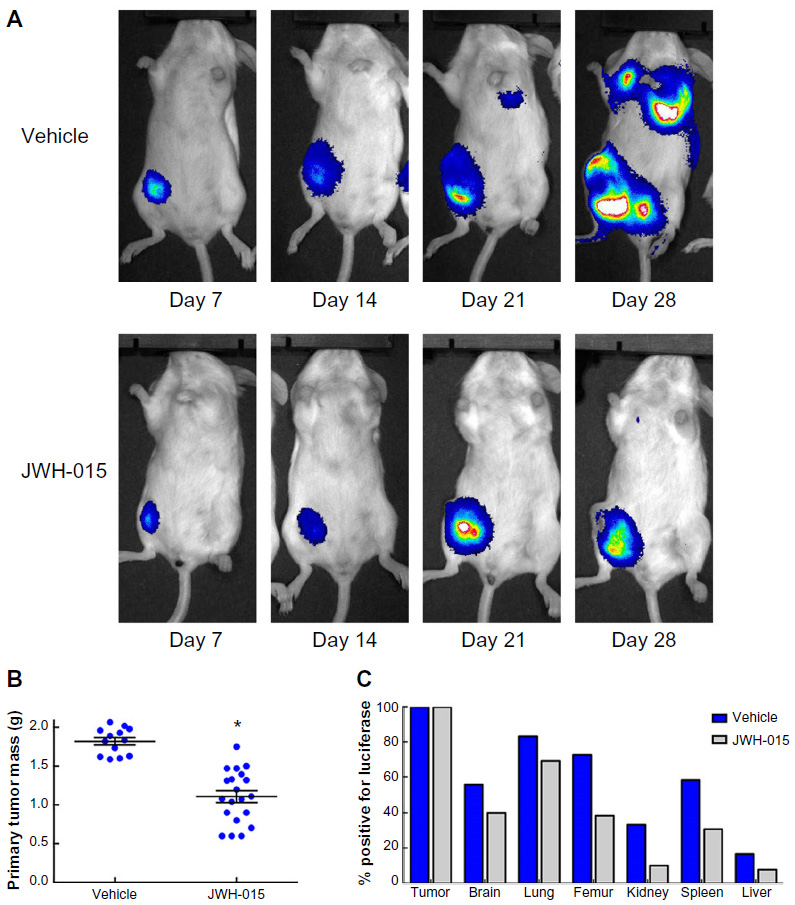 | Figure 2 In vivo administration of JWH-015 attenuates primary mammary tumor growth. |
The CB2 inverse agonist SR144528 does not block JWH-015-mediated reduction of breast cancer cell viability
The SRB assay was used to determine the viability of mouse and human (4T1 and MCF7, respectively) breast tumor cells in vitro in the presence of the CB2-specific agonist JWH-015, the CB2-specific inverse agonist SR144528, or a combination thereof (Figure 3). Treatment with JWH-015 over a 48-hour period reduced the viability of 4T1 cells (A50=2.8 μM; 95% CI =2.56–3.07) and MCF7 cells (A50=4.16 μM; 95% CI =3.24–5.34) in a dose-dependent manner. Pretreatment with the CB2 inverse agonist SR144528 1 hour prior to administration of JWH-015 did not block the viability reduction by JWH-015 in either 4T1 cells (A50=2.475 μM; 95% CI =1.77–3.46) or MCF7 cells (A50=5.78 μM; 95% CI =5.04–6.52). When SR144528 was administered alone in either 4T1 cells (A50=4.88 μM; 95% CI =4.12–5.77) or MCF7 cells (A50=7.01 μM; 95% CI =3.53–13.92), cellular viability was also reduced, suggesting that CB2-activity-modifying compounds significantly decrease breast cancer proliferation.
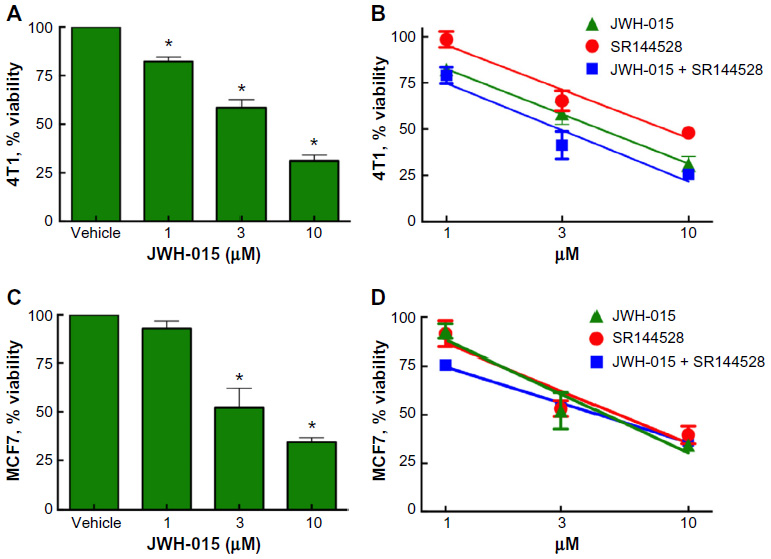 | Figure 3 CB2 inverse agonist SR144528 does not attenuate cell viability reduction induced by JWH-015 in vitro. |
JWH-015-induced apoptosis is independent of cell cycle
To determine whether loss of breast cancer cell viability is the direct result of cell killing or is an effect secondary to senescence, Ki67 imaging was done in vitro using a Ki67 primary antibody and an fluorescein isothiocyanate-conjugated secondary antibody (Figure 4A). No observable difference was seen between the number of 4T1 cell nuclei taking up 4′,6-diamidino-2-phenylindole stain and those expressing fluorescein isothiocyanate fluorescence in naïve cells, media-treated cells, or JWH-015 (10 μM)-treated cells over a 24-hour period.
 | Figure 4 In vitro administration of JWH-015 induces apoptosis of breast cancer cells independent of cell cycle. |
To verify that, consistent with other cannabinoid investigations,8,34 JWH-015 attenuates cellular viability in 4T1 and MCF7 breast cancer cells by inducing apoptosis, caspase 3/7 activity was assessed. JWH-015 produced activation of early caspases in a dose-dependent manner in vitro, 12 hours posttreatment (Figure 4B and C) in 4T1 cells and MCF7 cells (P<0.05). These data suggest a CB2 drug-induced apoptosis and not changes in the cell cycle.
JWH-015 induces calcium flux and changes in ERK phosphorylation
To determine whether JWH-015 induces calcium flux in breast cancer cells, Fura-2 AM staining was performed in vehicle- and JWH-015-treated 4T1 cells (Figure 5). Graphical representation of calcium flux was created by plotting the A340/A380 ratio of recorded cells with respect to time (Figure 5A and C), and statistical analyses were performed on individual cell recordings (Figure 5B, D; n=9 per group; P<0.05).
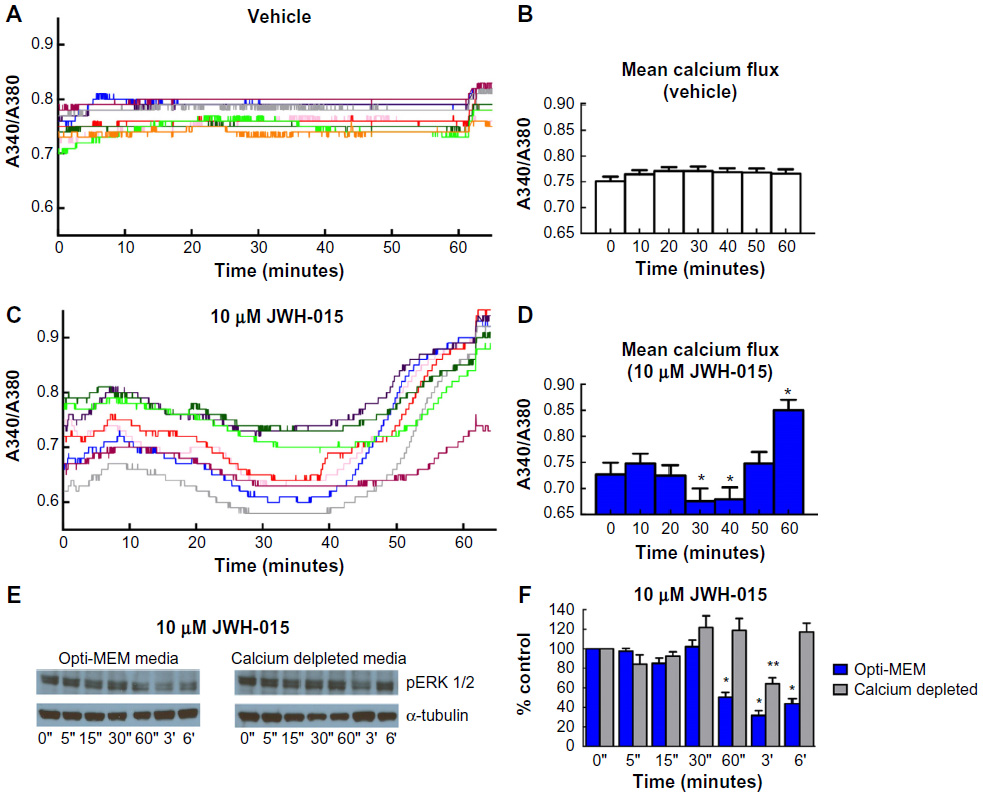 | Figure 5 JWH-015 induction of calcium flux and modulation of MAPK/ERK phosphorylation is calcium dependent. |
It has been shown previously that cannabinoids exert effects on the MAPK/ERK intracellular signaling cascade.23,35 To verify JWH-015-induced attenuation of ERK1/2 phosphorylation and to determine whether this phenomenon is downstream of JWH-015-induced calcium flux, Western blots were performed after treatment of 4T1 cells in vitro with JWH-015 for the indicated time periods in both normal or calcium-depleted media (Figure 5D). Blots were reprobed with α-tubulin as a loading control. ImageJ was used to evaluate relative density of scanned blot images and the data were normalized to relative density of control (data are averaged from three separate experiments; Figure 5F). JWH-015 (10 μM) reduced phosphorylation of ERK1/2 at 1, 3, and 6 hours postadministration (P<0.05 as compared to vehicle-treated cell lysates). JWH-015 did not reduce phosphorylation of ERK1/2 in calcium-depleted cultures (P>0.05) when compared to vehicle-treated cell lysates.
JWH-015 induction of apoptosis is dependent on cellular calcium flux
JWH-015 failed to induce caspase 3/7 activation in 4T1 cells cultured in calcium-depleted media (Figure 6A and B; P<0.05). To establish whether the loss of JWH-015 effect occurs due to intracellular or extracellular calcium channel activity, further inquiry was made by pharmacological blockade of specific classes of calcium channels during JWH-015 stimulation. SRB assays were performed on 4T1 cells in the presence of JWH-015 and either the L-type calcium-channel blocker nifedipine (Figure 6C) (A50=1.89 μM; 95% CI =0.96–3.72) or the N/P/Q-type calcium-channel inhibitor ω-conotoxin MVIIC (Figure 6C) (A50=0.41 μM; 95% CI =0.21–0.79). Neither cotreatment restored JWH-015-attenuated viability of breast cancer cells. Similarly, blockade of the transient receptor potential channels TRPV1 and TRPA1 did not alter the effects of JWH-015 on tumor cell viability. The TRPV1 antagonist capsazepine did not block JWH-015 activity (A50=2.65 μM; 95% CI =2.18–3.21), nor did pretreatment with the TRPA1 antagonist HC-030031 (A50=0.32 μM; 95% CI =0.14–0.69), suggesting the CB2-mediated effects are not from extracellular calcium sources.
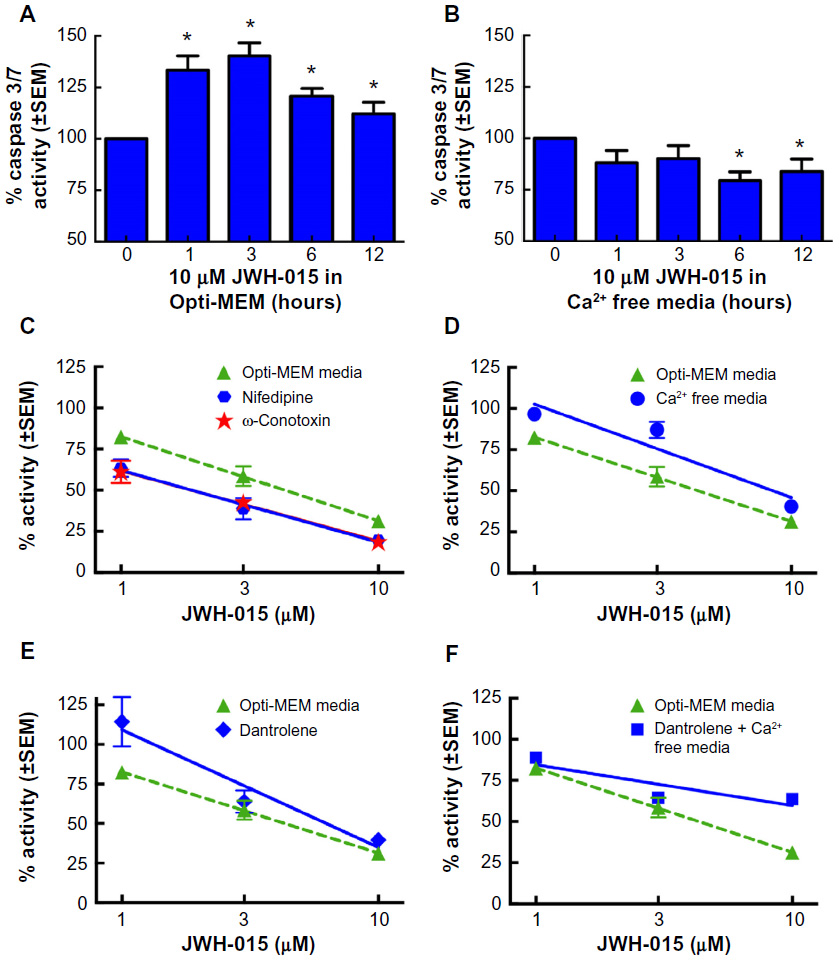 | Figure 6 Inhibition of calcium flux attenuates JWH-015-induced apoptosis in 4T1 cells. |
We next assessed JWH-015’s ability to modulate intracellular calcium flux induced by activation of the ryanodine receptor. SRB assays were carried out in the presence of JWH-015 and either the ryanodine receptor antagonist dantrolene, Ca2+-depleted media (as a mechanism to block extracellular calcium influx), or a combination thereof. Cells cultured in Ca2+-free media for 16 hours prior to treatment with JWH-015 experienced a significant dose–response curve shift (A50=9.80 μM; 95% CI =7.57–12.69) (Figure 6D). Similarly, blockade of intracellular calcium flux by pretreatment with dantrolene shifted the JWH-015 dose–response curve in 4T1 cells significantly rightward compared to JWH-015 alone (A50=18.77 μM; 95% CI =10.56–33.00) (Figure 6E), supporting an intracellular CB2-mediated calcium hypothesis. Synergistic effects were observed when SRB assays were performed in Ca2+-free media with dantrolene pretreatment (Figure 6F) as evidenced by the dose–response curve’s rightward shift of approximately 40-fold (A50=112 μM; 95% CI =60–211).
Finally, investigations were made to rule out the involvement of non-CB2 receptors or alternative G-protein coupling at relevant concentrations of JWH-015. The CB1 inverse agonist SR141716a, the GPR55 antagonist cannabidiol, or GPR55 agonist O-1602 was administered to cultures 1 hour prior to JWH-015 in SRB assays on 4T1 cells. Pretreatment with SR141716a did not alter JWH-015’s effects on cellular viability (A50=3.0 μM; 95% CI =1.82–4.94). Similarly, pretreatment with either cannabidiol or O-1602 did not block JWH-015 activity on 4T1 cells (A50=2.81 μM; 95% CI =2.21–3.43) (A50=3.21 μM; 95% CI =2.37–3.79), respectively. Treatment with either cannabidiol or O-1602 (concentrations from 0.1 to 10 μM) alone had no effect on cell viability. To determine whether the Gαi pathway is responsible for the reduction in cancer cell viability, 4T1 cells were pretreated with PTX prior to administration of JWH-015. We found that PTX inhibition of Gαi does not prevent JWH-015 reduction in 4T1 cancer cell viability (A50=3.82 μM; 95% CI =3.22–4.94).
Discussion
Despite marked advances in chemotherapeutics for early-stage breast cancer, there are few new therapies effective in slowing the progression of advanced-stage disease. Genotypic profiling of human breast cancers over the last several years has led to broad phenotypic subclassification of breast tumors, shedding light on the complexity of the disease, and demonstrating some of the genetic intratumoral differences responsible for the mixed outcomes of protein-specific therapeutics. There is a clear need for novel tumor-specific therapeutics that do not result in severe toxic side effects for the treatment of late-stage and chemotherapy-resistant breast cancer.
Cannabinoid receptors are classically defined as members of the seven transmembrane-spanning G-protein-coupled receptor family. Two distinct cannabinoid receptors have been cloned: CB1,36 which is one of the most abundantly expressed G-protein-coupled receptors on neurons, and CB2,37 found primarily in cells of the immune system and on bone maintenance cells.14 In physiologically normal systems, CB1 and CB2 were first reported to couple predominantly with Gαi,38 yet several recent studies have demonstrated CB2 receptor signaling via Gαq and other alternative coupling pathways.39,40
Several groups have shown that both nonselective cannabinoid and CB2-specific compounds decrease breast cancer viability in vitro and in vivo: Δ9-tetrahydrocannabinol and CB2-selective agonist, JWH-133, have been demonstrated to exert considerable antitumoral effects in the MMTV-neu mouse model of breast cancer, which overexpresses the EGF receptor HER2.17 Similarly, CB2-selective JWH-133 has been shown to retard growth and metastasis of aggressive spontaneous breast tumors occurring in PyMT transgenic mice, implying the cyclooxygenase-2/prostaglandin E2 signaling pathway.34 Cannabidiol inhibited human breast cancer cell line MDA-MB-231 growth in vitro by inducing apoptosis and autophagy.8 Anandamide, an endogenous cannabinoid CB1 agonist, has been shown to inhibit breast cancer cell growth and migration in multiple models,41,42 and several other studies have demonstrated CB2 agonist efficacy in breast cancer systems;43–45 however, the mechanism(s) of how cannabinoids achieve these effects is incompletely defined.
Here we demonstrate that CB2 receptors are functionally expressed on breast cancer cells and that low nanomolar concentrations of JWH-015 reduce cAMP generation, yet results in no change in cAMP levels at the higher concentrations required for in vitro cancer cell growth modification. This suggests a concentration-dependent mechanism where higher concentrations may induce activation of an alternative-coupling pathway. This effect may be due to Ca2+ modulation of adenylate cyclase.46,47 Additionally, we were unable to augment intracellular cAMP activity with forskolin stimulation, suggesting that basal cAMP formation is elevated in 4T1 murine mammary carcinoma cells. Deregulated intracellular pathway activity, particularly in growth-promoting pathways, has been repeatedly documented in cancerous cells.48 Interestingly, we also show decreased persistence of cancer cell viability following shRNA knockdown of the CB2 receptor, indicating a dependence on basal cannabinoid receptor activity, though further studies are needed to explore this phenomenon.
Several previous studies by our laboratory and others have demonstrated that CB2 receptor-mediated biological effects including analgesia,49–51 antiallodynia,52,53 and osteoclast inhibition14,54 are attenuated by the CB2 inverse agonist SR144528. However, here we present evidence that the expression level of CB2 and associated production of cAMP are substantially elevated in 4T1 mammary carcinoma cells, which may indicate a change in receptor response to pharmacological modulators including agonists and inverse agonists. Indeed, a recent study demonstrated the ability of SR144528 to act as a CB2 agonist in activated microglia.55 It is therefore likely that cell type, basal CB2 activity, and intracellular protein expression dictate whether an inverse agonist behaves like an antagonist or an agonist. We have verified here that JWH-015 reduces cellular viability by inducing cell-cycle-independent apoptosis and were unable to block the activity of JWH-015 in vitro with coadministration of the CB2 inverse agonist SR144528. Interestingly, our data also show that SR144528 attenuates proliferation of breast cancer cells, indicating that the inverse agonist may act as an agonist in this model.
We have established that the apoptosis of breast tumor cells induced by JWH-015 does not occur through a Gαi-coupled mechanism in two distinct assays: by blocking the Gαi pathway with PTX prior to treatment with JWH-015 and by direct measurement of cellular cAMP in response to treatment with JWH-015. Furthermore, we have ruled out the interaction of JWH-015 with two other Gαi-coupled receptors, CB1 and GPR55, using selective antagonists for each of the receptors. Because studies have also demonstrated that the endogenous cannabinoid anandamide can act as a low-efficacy agonist at the transient receptor potential TRPV1 calcium channels,45 we also show independence of our effect from TRPA1 and TRPV1 channels as the target for JWH-015 antitumoral effects by blocking these channels with HC-030031 and capsazepine, respectively. Finally, we have ruled out the possibility that JWH-015’s effects are mediated via L-type and N/P/Q-type calcium channels using nifedipine and ω-conotoxin MVIIC, respectively.
Although the effect of JWH-015 on breast cancer cells in vitro is not mediated by TRPA1, TRPV1, L-type, or N/P/Q-type calcium channels, we have shown that it is a calcium-dependent process. We demonstrate that by blocking either intracellular calcium flux with the ryanodine receptor antagonist dantrolene, or depleting extracellular calcium using media devoid of Ca2+, a significant rightward shift (19- and 10-fold, respectively) of the JWH-015 viability reduction dose–response curve is observed. Notably, when both intracellular and extracellular calcium fluxes are restricted simultaneously, the rightward shift (40-fold) of the JWH-015 dose–response curve observed is synergistic. Further, we show that in vitro treatment with JWH-015 induces cytoplasmic calcium flux by first depletion beginning 25 minutes postadministration and later influx beginning 40 minutes postadministration of the compound in 4T1 cells. Immediate calcium flux is not observed, as would be expected in the case that JWH-015 was primarily acting through integral membrane calcium channels. This data is supported by the work done by Felder et al56 suggesting that CB2 receptors do not modulate ion channels directly. Taken together, these data suggest that intracellular calcium flux is a secondary event necessary for induction of apoptosis that is stimulated by another primary event activated by administration of JWH-015.
Consistent with the findings on the intracellular activity of cannabinoids by other groups,7,22,57–59 we have demonstrated that CB2-selective JWH-015 reduces phosphorylation of ERK1/2 at time points relevant to peak measured caspase 3/7 activity, suggesting that the apoptosis induced by JWH-015 in breast cancer cells may be mediated by MAPK/ERK activity. Such findings are also in line with recent reports demonstrating CB2 modulation of the PI3K/AKT and MAPK/ERK pathways in hippocampal neurons.21
Our study characterizes the actions of the CB2-selective agonist JWH-015 on breast cancer cells in vitro and in vivo in a syngeneic murine model of late-stage breast cancer. Importantly, our model does not compromise immune function or produce oncogenic mutation in the host, yielding a model that bears resemblance to the clinical presentation of advanced breast cancer progression and metastasis. Additionally, because we transfected 4T1 cells with a luciferase reporter prior to host inoculation, the model utilized allows noninvasive in vivo tracking of primary tumor growth and metastasis over the entire duration of tumor establishment and treatment. In the studies presented herein, we show a reduced profile of metastasis on treatment with JWH-015 as well as reduced primary tumor burden in vivo.
Conclusion
We have demonstrated that the CB2 agonist JWH-015 induces apoptosis in a non-Gαi-mediated, calcium-dependent, and cell-cycle-independent mechanism accompanied by attenuation of activated MAPK/ERK intracellular pathway. These are findings that should be considered with our previous studies indicating that JWH-015 significantly modifies the bone microenvironment in metastatic disease54 by decreasing bone wasting in advanced stages of cancer and by significantly reducing spontaneous and evoked pain while simultaneously reducing fracture occurrence.54 Given these findings, JWH-015 and other CB2-selective agonists demand attention as adjuvant therapies, particularly in patients experiencing bone metastasis where CB2-selective compounds may not only improve disease outcome but also increase the patient’s quality of life.
Acknowledgments
This work was supported by the National Institutes of Health National Cancer Institute (R01 CA142115-01); the Arizona Cancer Center (Better than Ever grant); and the Maine Cancer Foundation (pilot research grant).
Disclosure
The authors report no conflicts of interest in this work.
References
CDC. Leading Causes of Death. 2012. Available from: http://www.cdc.gov/nchs/fastats/lcod.htm. Accessed January 19, 2016. | |
CDC. Cancer Among Women: Data and Statistics; Data by Demographic Atlanta. 2012. Available from: http://www.cdc.gov/cancer/dcpc/data/women.htm. Accessed March 30, 2012. | |
Breastcancer.org. Breast Cancer Survival Triples in Last 60 Years. 2010. Available from: http://www.breastcancer.org/symptoms/new_research/20100930.jsp. Accessed February 15, 2016. | |
Bertos NR, Park M. Breast cancer – one term, many entities? J Clin Invest. 2011;121(10):3789–3796. | |
Snijders AM, Fridlyand J, Mans DA, et al. Shaping of tumor and drug-resistant genomes by instability and selection. Oncogene. 2003;22(28):4370–4379. | |
Luger NM, Mach DB, Sevcik MA, Mantyh PW. Bone cancer pain: from model to mechanism to therapy. J Pain Symptom Manage. 2005; 29(5 Suppl):S32–S46. | |
Galve-Roperh I, Sánchez C, Cortés ML, Gómez del Pulgar T, Izquierdo M, Guzmán M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6(3):313–319. | |
Shrivastava A, Kuzontkoski PM, Groopman JE, Prasad A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol Cancer Ther. 2011;10(7):1161–1172. | |
McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther. 2007;6(11):2921–2927. | |
Blazquez C, Salazar M, Carracedo A, et al. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008;68(6):1945–1952. | |
Preet A, Qamri Z, Nasser MW, et al. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev Res (Phila). 2011;4(1):65–75. | |
Sarnataro D, Pisanti S, Santoro A, et al. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol Pharmacol. 2006;70(4):1298–1306. | |
Wang D, Wang H, Ning W, Backlund MG, Dey SK, DuBois RN. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008;68(15):6468–6476. | |
Ofek O, Karsak M, Leclerc N, et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA. 2006;103(3):696–701. | |
Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia. 2010;58(9):1017–1030. | |
Malan TP Jr, Ibrahim MM, Deng H, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93(3):239–245. | |
Caffarel MM, Andradas C, Mira E, et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol Cancer. 2010;9:196. | |
Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002;68–69:619–631. | |
Melck D, Rueda D, Galve-Roperh I, De Petrocellis L, Guzmán M, Di Marzo V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463(3):235–240. | |
Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102(52):19144–19149. | |
Palazuelos J, Ortega Z, Díaz-Alonso J, Guzmán M, Galve-Roperh I. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J Biol Chem. 2012;287(2):1198–1209. | |
Sanchez MG, Ruiz-Llorente L, Sanchez AM, Diaz-Laviada I. Activation of phosphoinositide 3-kinase/PKB pathway by CB(1) and CB(2) cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell Signal. 2003;15(9):851–859. | |
Ellert-Miklaszewska A, Kaminska B, Konarska L. Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of Bad protein. Cell Signal. 2005;17(1):25–37. | |
Herrera B, Carracedo A, Diez-Zaera M, Guzmán M, Velasco G. p38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Lett. 2005;579(22):5084–5088. | |
Velasco G, Galve-Roperh I, Sánchez C, Blázquez C, Haro A, Guzmán M. Cannabinoids and ceramide: two lipids acting hand-by-hand. Life Sci. 2005;77(14):1723–1731. | |
Massi P, Vaccani A, Ceruti S, Colombo A, Abbracchio MP, Parolaro D. Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J Pharmacol Exp Ther. 2004;308(3):838–845. | |
Patwardhan AM, Jeske NA, Price TJ, Gamper N, Akopian AN, Hargreaves KM. The cannabinoid WIN 55,212-2 inhibits transient receptor potential vanilloid 1 (TRPV1) and evokes peripheral antihyperalgesia via calcineurin. Proc Natl Acad Sci U S A. 2006;103(30):11393–11398. | |
Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther. 2010;126(3):301–313. | |
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. | |
Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112(2):195–203. | |
Tao K, Fang M, Alroy J, Sahagian GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer. 2008;8:228. | |
Vanderah TW, Gardell LR, Burgess SE, et al. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20(18):7074–7079. | |
Tallarida RJ, Murray RB. Manual of Pharmacological Calculations with Computer Programs. 2nd ed. New York, NY: Springer-Verlag Publishers; 1987. | |
Qamri Z, Preet A, Nasser MW, et al. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol Cancer Ther. 2009;8(11):3117–3129. | |
Greenhough A, Patsos HA, Williams AC, Paraskeva C. The cannabinoid delta(9)-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int J Cancer. 2007;121(10):2172–2180. | |
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–564. | |
Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. | |
Wess J. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol Ther. 1998;80(3):231–264. | |
Codd EE, Mabus JR, Murray BS, Zhang SP, Flores CM. Dynamic mass redistribution as a means to measure and differentiate signaling via opioid and cannabinoid receptors. Assay Drug Dev Technol. 2011;9(4):362–372. | |
Ho BY, Uezono Y, Takada S, Takase I, Izumi F. Coupling of the expressed cannabinoid CB1 and CB2 receptors to phospholipase C and G protein-coupled inwardly rectifying K+ channels. Receptors Channels. 1999;6(5):363–374. | |
Grimaldi C, Pisanti S, Laezza C, et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp Cell Res. 2006;312(4):363–373. | |
Ligresti A, Cascio MG, Pryce G, et al. New potent and selective inhibitors of anandamide reuptake with antispastic activity in a mouse model of multiple sclerosis. Br J Pharmacol. 2006;147(1):83–91. | |
Caffarel MM, Sarrio D, Palacios J, Guzman M, Sanchez C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006;66(13):6615–6621. | |
McAllister SD, Murase R, Christian RT, et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res Treat. 2011;129(1):37–47. | |
Melck D, Bisogno T, De Petrocellis L, et al. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem Biophys Res Commun. 1999;262(1):275–284. | |
Halls ML, Cooper DM. Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb Perspect Biol. 2011;3(1): a004143. | |
Masada N, Schaks S, Jackson SE, Sinz A, Cooper DM. Distinct mechanisms of calmodulin binding and regulation of adenylyl cyclases 1 and 8. Biochemistry. 2012;51(40):7917–7929. | |
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100(1):57–70. | |
Curto-Reyes V, Llames S, Hidalgo A, Menéndez L, Baamonde A. Spinal and peripheral analgesic effects of the CB2 cannabinoid receptor agonist AM1241 in two models of bone cancer-induced pain. Br J Pharmacol. 2010;160(3):561–573. | |
Clayton N, Marshall FH, Bountra C, O’Shaughnessy CT. CB1 and CB2 cannabinoid receptors are implicated in inflammatory pain. Pain. 2002;96(3):253–260. | |
Romero-Sandoval A, Nutile-McMenemy N, DeLeo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008;108(4):722–734. | |
Yamamoto W, Mikami T, Iwamura H. Involvement of central cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse model of neuropathic pain. Eur J Pharmacol. 2008;583(1):56–61. | |
Ibrahim MM, Deng H, Zvonok A, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci U S A. 2003;100(18):10529–10533. | |
Lozano-Ondoua AN, Hanlon KE, Largent-Milnes TM, et al. Disease modification of breast cancer-induced bone remodeling by cannabinoid 2 receptor agonists. J Bone Miner Res. 2013;28(1):92–107. | |
Ribeiro R, Wen J, Li S, Zhang Y. Involvement of ERK1/2, cPLA(2) and NF-ºB in microglia suppression by cannabinoid receptor agonists and antagonists. Prostaglandins Other Lipid Mediat. 2012;100–101:1–14. | |
Felder CC, Joyce KE, Briley EM, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48(3):443–450. | |
Bouaboula M, Poinot-Chazel C, Marchand J, et al. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem. 1996;237(3):704–711. | |
Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74(2):129–180. | |
Sanchez MG, Sanchez AM, Ruiz-Llorente L, Diaz-Laviada I. Enhancement of androgen receptor expression induced by (R)-methanandamide in prostate LNCaP cells. FEBS Lett. 2003;555(3):561–566. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.