Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Modification of paclitaxel-loaded solid lipid nanoparticles with 2-hydroxypropyl-β-cyclodextrin enhances absorption and reduces nephrotoxicity associated with intravenous injection
Authors Baek J, Kim J, Park J ![]() , Cho C
, Cho C ![]()
Received 12 April 2015
Accepted for publication 22 July 2015
Published 26 August 2015 Volume 2015:10(1) Pages 5397—5405
DOI https://doi.org/10.2147/IJN.S86474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Carlos Rinaldi
Jong-Suep Baek,* Ju-Heon Kim,* Jeong-Sook Park, Cheong-Weon Cho
College of Pharmacy and Institute of Drug Research and Development, Chungnam National University, Daejeon, South Korea
*These authors contributed equally to this work
Background: Paclitaxel (PTX) solid lipid nanoparticles (SLNs) modified with 2-hydroxypropyl-β-cyclodextrin (HPCD) were evaluated for their ability to enhance PTX absorption and reduce the nephrotoxicity accompanying intravenous administration.
Methods: PTX-loaded SLNs (PS) and PTX-loaded SLNs modified using HPCD (PSC) were prepared by hot-melted sonication. The anticancer activity of PSC was evaluated in MCF-7 cells, and confocal microscopy was used to quantify the cellular uptake. The pharmacokinetic profiles of PTX released from PSC after intravenous administration were studied in rats. Furthermore, kidney toxicity was determined by measuring the kidney size and plasma creatinine level.
Results: PSC were successfully prepared by hot-melted sonication and had smaller diameters than PS. PSC exhibited improved anticancer activity and cellular uptake in MCF-7 cells. Furthermore, PSC showed higher bioavailability in rats after intravenous administration than PTX solution; however, no significant differences in kidney toxicity were observed.
Conclusion: Based on these results, PSC could be considered as a potential therapeutic PTX delivery system for breast cancer with low renal toxicity.
Keywords: paclitaxel, solid lipid nanoparticles, 2-hydroxypropyl-β-cyclodextrin, bioavailability, nephrotoxicity
Introduction
Paclitaxel (PTX), the first of a new class of microtubule-stabilizing agents, has been used as an effective chemotherapeutic agent for a wide variety of tumors such as ovarian, breast, and lung cancers.1 Available commercially as Taxol®, a concentrated solution containing 6 mg/mL PTX in cremophor EL (polyoxyl 35 castor oil) and dehydrated alcohol (1:1, v/v) because of the poor aqueous solubility of PTX (0.3–0.5 μg/mL).1–4 The use of cremophor EL in Taxol® is associated with hypersensitivity reactions such as neurotoxicity and nephrotoxicity. Hence, more effort has been put on the development of cremophor EL-free alternative carrier systems including liposomes,5 complexation with cyclodextrin,6 emulsions,7 microspheres,8 and polymeric nanoparticles.9 Abraxane® is an albumin-bound form of and the first solvent-free PTX nanoparticle to be commercialized, with a mean particle size of ~130 nm.10–12
Of the various formulations, solid lipid nanoparticles (SLNs) have been introduced as a potent carrier system for various pharmaceutical or cosmetically active compounds. SLNs have several advantages, including high biocompatibility, high bioavailability, and controlled release.13–18
We previously reported the formulation of PTX-loaded SLNs modified using 2-hydroxypropyl-β-cyclodextrin (HPCD). The resultant SLNs had improved in vitro cytotoxicity19 and oral bioavailability.20 Herein, we aimed to decrease the burst release of PTX, reduce its renal toxicity, prolong its anticancer activity, and enhance its absorption. PTX-loaded SLNs modified with HPCD (PSC) were prepared by hot-melted sonication. MCF-7 cells treated with these SLNs were assayed for cytotoxicity, cellular uptake, and apoptosis. Furthermore, tumor xenografts and in vivo bioavailability of PSC administered intravenously in rats were also investigated. Finally, renal toxicity was investigated by determining serum creatinine levels and measuring the kidney size.
Materials and methods
Materials
PTX was provided by Samyang Genex (Daejeon, South Korea). Stearic acid was purchased from Daejung Chemical Co., Ltd. (Cheongwon, South Korea). Poloxamer 188 was obtained from BASF (Ludwigshafen, Germany), and lecithin was acquired from Junsei Chemical Co., Ltd. (Tokyo, Japan). Nile red, 4′,6-diamidino-2-phenylindole (DAPI), HPCD, and mannitol were purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals and reagents used were of analytical grade.
Preparation of the PS and PSC
PTX-loaded SLNs (PS) or PSC were prepared as previously described.19 Briefly, 100 mg stearic acid was melted at 80°C in a water bath. PTX (5 mg) was dissolved in ethanol (0.25 mL) by vortexing, and the solution was warmed to 80°C in a water bath. PTX solution was injected into molten stearic acid under sonication. Lecithin (75 mg) and poloxamer 188 (75 mg) were dispersed in 3 mL of distilled water. The mixture was sonicated for 10 minutes at 80°C in a water bath using a probe-type sonicator. This mixture was added to molten stearic acid under sonication for 10 minutes at 80°C in a water bath using a probe-type sonicator. The resulting solution was dispersed in 2 mL of distilled water at 4°C and sonicated for 15 minutes. HPCD (400 mg) was added to the SLN dispersion and shaken for 40 minutes. Samples were freeze-dried until further use. Each formulation is listed in Table 1. As a control, PTX solution was prepared as the Taxol® formulation.
 | Table 1 Composition of PTX-loaded SLNs or PTX-loaded SLNs modified with HPCD |
Cell culture
MCF-7 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 100 units/mL penicillin in a humidified atmosphere of 5% CO2 at 37°C.
Proliferation assay of PS and PSC
To evaluate the proliferation of PS or PSC, the MTT assay (Sigma-Aldrich) was carried out. Briefly, MCF-7 breast cancer cells were seeded in 96-well plates at a cell density of 5×104 cells/mL (200 μL/well). After 24 hours, the cells were further incubated for 24 hours, 48 hours, or 72 hours in a fresh medium containing PS or PSC. The culture medium was replaced by MTT solution (5 mg/mL, 200 μL/well), and the cells were incubated for a further 4 hours. The supernatant was discarded, and 200 μL of dimethylsulfoxide was added to the wells. The plates were then placed in an incubator for 30 minutes. The absorbance values of each well were recorded at 570 nm using a microplate reader (Sunrise™; Tecan, Austria).
Cellular uptake using confocal microscopy
To evaluate qualitatively uptake into MCF-7 cells, Nile red-loaded SLNs (NS) or Nile red-loaded SLNs modified with HPCD (NSC) were prepared by the method described in the section “Preparation of the PS and PSC”. On glass slides (Thermo Fisher Scientific, Waltham, MA, USA), MCF-7 cells were seeded at a density of 1×106 cells per well in 1 mL growth medium and incubated for 24 hours to allow attachment. Cells were then incubated with NS or NSC in growth medium for 2 hours or 8 hours. Cells were washed twice with PBS (pH 7.4) before staining with 1 μg/mL DAPI in PBS for 3 minutes and washing twice with PBS. The cells were observed directly under a confocal microscope (CLSM, Model: LSM5LIVE; Carl Zeiss Meditec AG, Jena, Germany).
Apoptosis assay by flow cytometry
MCF-7 cells were treated with each PTX solution, PS, and PSC corresponding 10 μM PTX for 24 hours, harvested, and washed three times with PBS. The cells were first incubated with 100 μL of binding buffer and stained with 5 μL of annexin V-FITC and 5 μL of propidium iodide (PI) according to the manufacturer’s instruction (BD Biosciences, San Jose, CA, USA). The cells were then analyzed by flow cytometry (FACS Canto II, BD Biosciences). Data were analyzed and plotted for annexin V-FITC and PI in a two-way dot plot. Live, early apoptotic, late apoptotic, and necrotic cells were identified as annexin−/PI−, annexin+/PI−, annexin+/PI+ and annexin−/PI+, respectively.
Pharmacokinetic study
The pharmacokinetics of each PTX solution, PS, and PSC were investigated using male Sprague Dawley (SD) rats (NARA biotech, Seoul, Korea) weighing 200–250 g. All animal studies were conducted in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (USA), and the experimental protocols were approved by the Animal Care Committee of Chungnam National University. The rats were provided commercial rat chow diet (No 322-7-1) from Superfeed Co. (Gangwon, Korea) and tap water ad libitum. The animals were housed three per cage in laminar flow maintained at 22°C±2°C and 50%–60% relative humidity. The animals were kept in these facilities for at least 1 week before the experiment and were fasted for at least 24 hours before commencing the experiments. The different formulations of PTX were diluted with saline to 5 mg/mL PTX and administrated by intravenous injection into the tail vein at a dose of 5 mg/kg (n=3 for each group). A total of 300 μL of blood samples were collected from retro-orbital veins at different times (0.08 hour, 0.17 hour, 0.25 hour, 0.5 hour, 0.75 hour, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, and 8 hours) in polypropylene microcentrifuge tubes. The samples were immediately centrifuged at 9,425× g for 5 minutes to isolate the plasma, which was stored at -80°C and subsequently analyzed by HPLC. To determine the plasma PTX concentration, 200 μL of acetonitrile was added to 100 μL of plasma and vortexed for 5 minutes. Then, samples were centrifuged at 11,310× g for 5 minutes. The supernatant was transferred to another tube and evaporated to dryness using nitrogen gas. The residues were dissolved in 50 μL acetonitrile, and 20 μL aliquots were used for HPLC analysis. An Agilent 1,100 liquid chromatography system with an autosampler and UV detector was used with a C18 column (4.0×250 mm, 5 μm particle size, Supelco™; MetaChem, Torrance, CA USA). The flow rate was 1 mL/min, and the detection wavelength was set to 227 nm. The mobile phase was a mixture of distilled water and acetonitrile (55:45, v/v). All procedures were carried out at an ambient temperature.
Evaluation of nephrotoxicity
The severity of nephrotoxicity was assessed by both kidney weight and serum creatinine level after the pharmacokinetic study. Serum was obtained 8 hours after intravenous administration of PTX solution, PS, or PSC, and kidneys were obtained after sacrifice. Serum creatinine was measured using a creatinine assay kit from Sigma-Aldrich. The absolute and relative (organ-to-body weight ratios) kidney weights were measured for all experimental animals.
In vivo antitumor efficacy test
Briefly, a solid tumor was established by subcutaneous injection of 0.1 mL MCF-7 cell suspension (1×106 cells per mouse) in the right flank of female BALB/c nude mice (6 weeks old; Charles River Laboratories International Inc., Frederick, MD, USA). The experimental protocols were approved by the Animal Care Committee of Chungnam National University. Tumor growth was documented by measuring the length and width of the tumors using a Vernier Caliper twice weekly, and tumor volumes were calculated as length × width2/2. Intratumoral therapy was initiated when tumors reached a volume of ~100 mm3. On day 6, the mice were randomly divided into three groups, and PTX solution (100 μL 10 mg/kg PTX dispersed in saline), PSC (100 μL 10 mg/kg PTX dispersed in saline), or saline (100 μL) was intratumorally administered in three doses (on days 6, 13, and 20 after tumor inoculation). Weight changes were measured to evaluate the antitumor effects.
Statistical analysis
Student’s t-test was used to compare the groups. A P-value <0.05 was considered statistically significant. All data were expressed as the mean ± standard deviation from three independent experiments.
Results and discussion
Cell proliferation study
The antiproliferation efficiency of PS or PSC was investigated in MCF-7 cells. Incubation of MCF-7 cells for 24 hours, 48 hours, or 72 hours with PSC containing PTX at concentrations up to 10 μM efficiently inhibited proliferation. Both PS and PSC exhibited concentration- and time-dependent inhibitory effects (Figure 1). As a reference, the viability of Caco-2 cells treated with PTX solution for 72 hours was 71.45.19 The inhibition efficiency of PS and PSC containing 5 μM and 10 μM PTX increased upon increasing the incubation time from 24 hours to 48 hours. The cell viability after treatment with PSC significantly decreased upon increasing the incubation time up to 72 hours at both PTX concentrations. These results indicated that PSC exhibited a sustained and prolonged cytotoxic effect in MCF-7 cells.
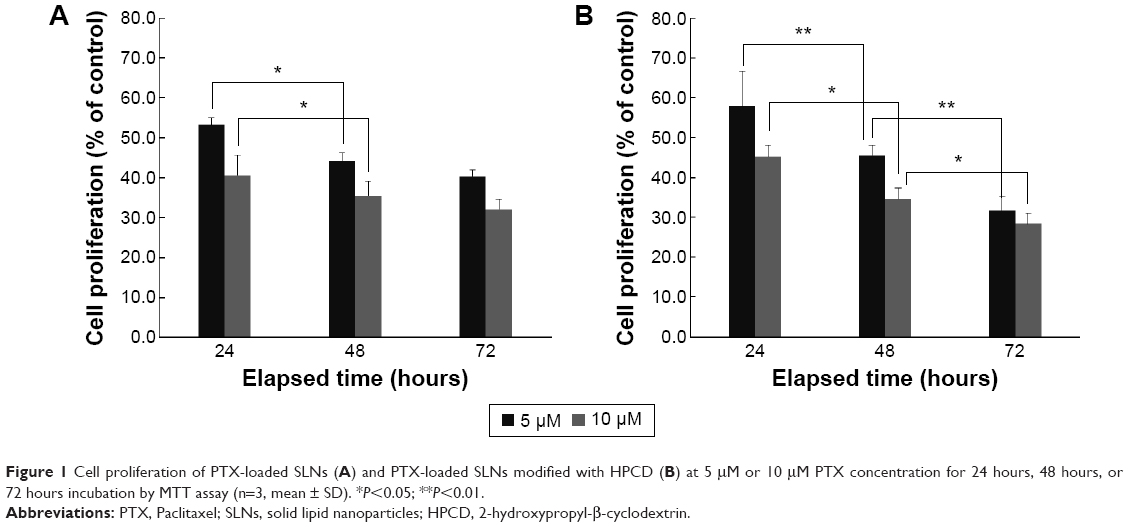 | Figure 1 Cell proliferation of PTX-loaded SLNs (A) and PTX-loaded SLNs modified with HPCD (B) at 5 μM or 10 μM PTX concentration for 24 hours, 48 hours, or 72 hours incubation by MTT assay (n=3, mean ± SD). *P<0.05; **P<0.01. |
Cellular uptake study
To observe cellular uptake by confocal microscopy, MCF-7 cells were treated with Nile red solution, NS, or NSC (Figure 2). NS and NSC exhibited more fluorescence than Nile red solution at all incubation times. Furthermore, NSC showed the highest fluorescence among the tested formulations. The enhanced uptake of Nile red from NSC might be owing to the increased solubility of Nile red by HPCD in the cell membranes. Yamazaki and Ito reported that high-solubility materials such as PEG can facilitate the cellular uptake of nanoparticles.21 Furthermore, particle size and shape were previously reported to significantly affect cellular uptake.22 Our previous study reported that the particle size of PSC was 251.4±12.0 nm. Therefore, the small PSC particle size might improve the rate and amount of cellular uptake.19 Because NSC was prepared using the same method as that of PSC, the high fluorescence observed for NSC is consistent with the results obtained previously.
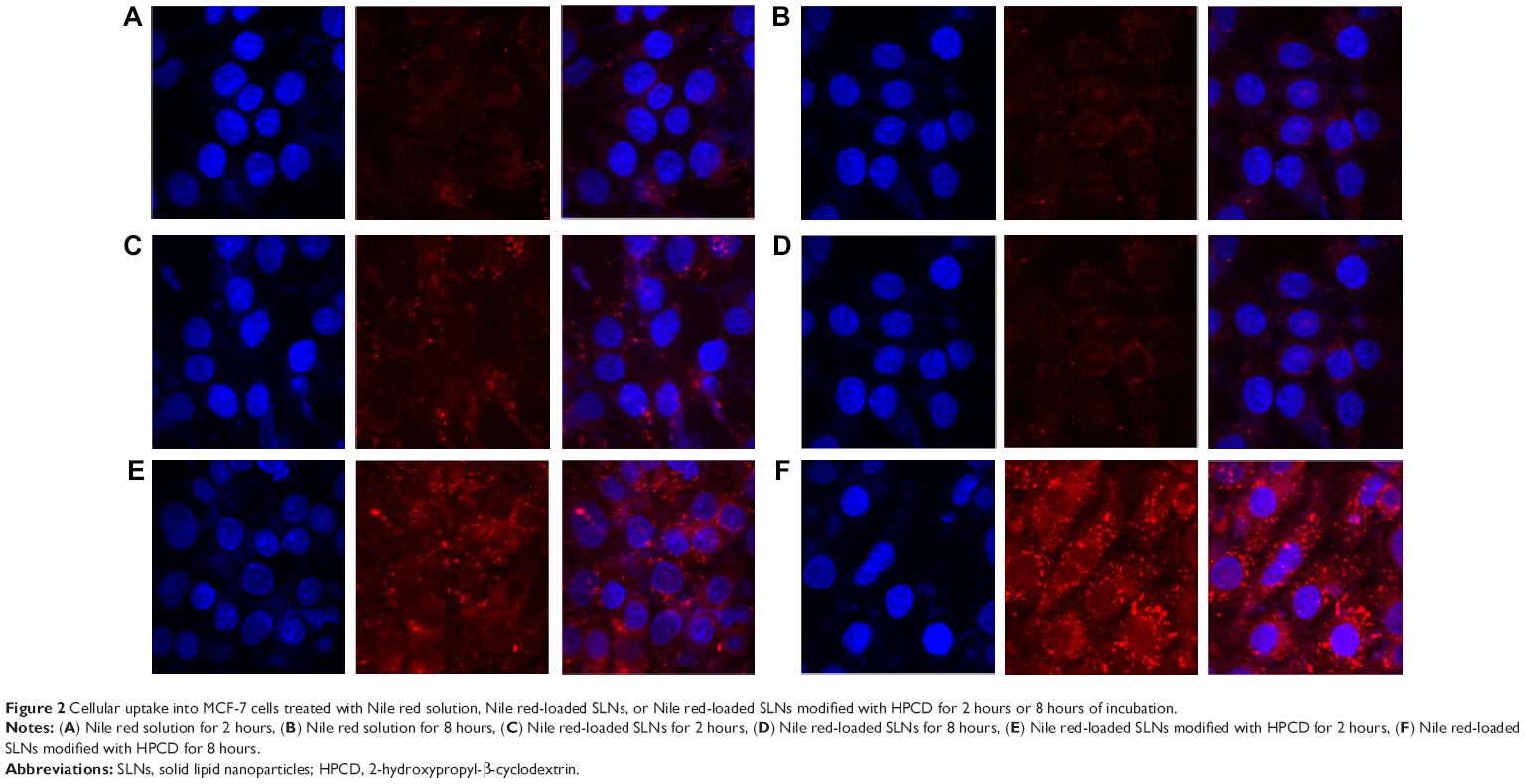 | Figure 2 Cellular uptake into MCF-7 cells treated with Nile red solution, Nile red-loaded SLNs, or Nile red-loaded SLNs modified with HPCD for 2 hours or 8 hours of incubation. |
Apoptosis assay by flow cytometry
Apoptotic effect of PTX was examined by staining PTX solution-, PS-, or PSC-treated MCF-7 cells with annexin V-FITC/PI (Figure 3). Apoptosis or programed cell death occurring after efficient cell damage plays an important role in the development and health of multicellular organisms. Dysregulation of apoptosis can disrupt the balance between cell growth and cell death and is thus an important step in cancer development.23 Evasion of apoptosis has been considered a hallmark of cancer.24 Treatment with PS or PSC for 8 hours dramatically increased the population of annexin V-positive cells to 26.0% or 44.5%, respectively, indicating the occurrence of late apoptosis in MCF-7 cells. Furthermore, PS and PSC increased the population of PI-positive cells to 17.0% and 20.9%, respectively. Notably, compared with PTX solution or PS, PSC exhibited 44.5% apoptotic cell death and 20.9% necrotic cell death because apoptosis and necrosis were more sustained at increased incubation time. Based on these results, we hypothesized that HPCD enhanced PTX-induced apoptosis in PSC, which is consistent with the cell proliferation data.
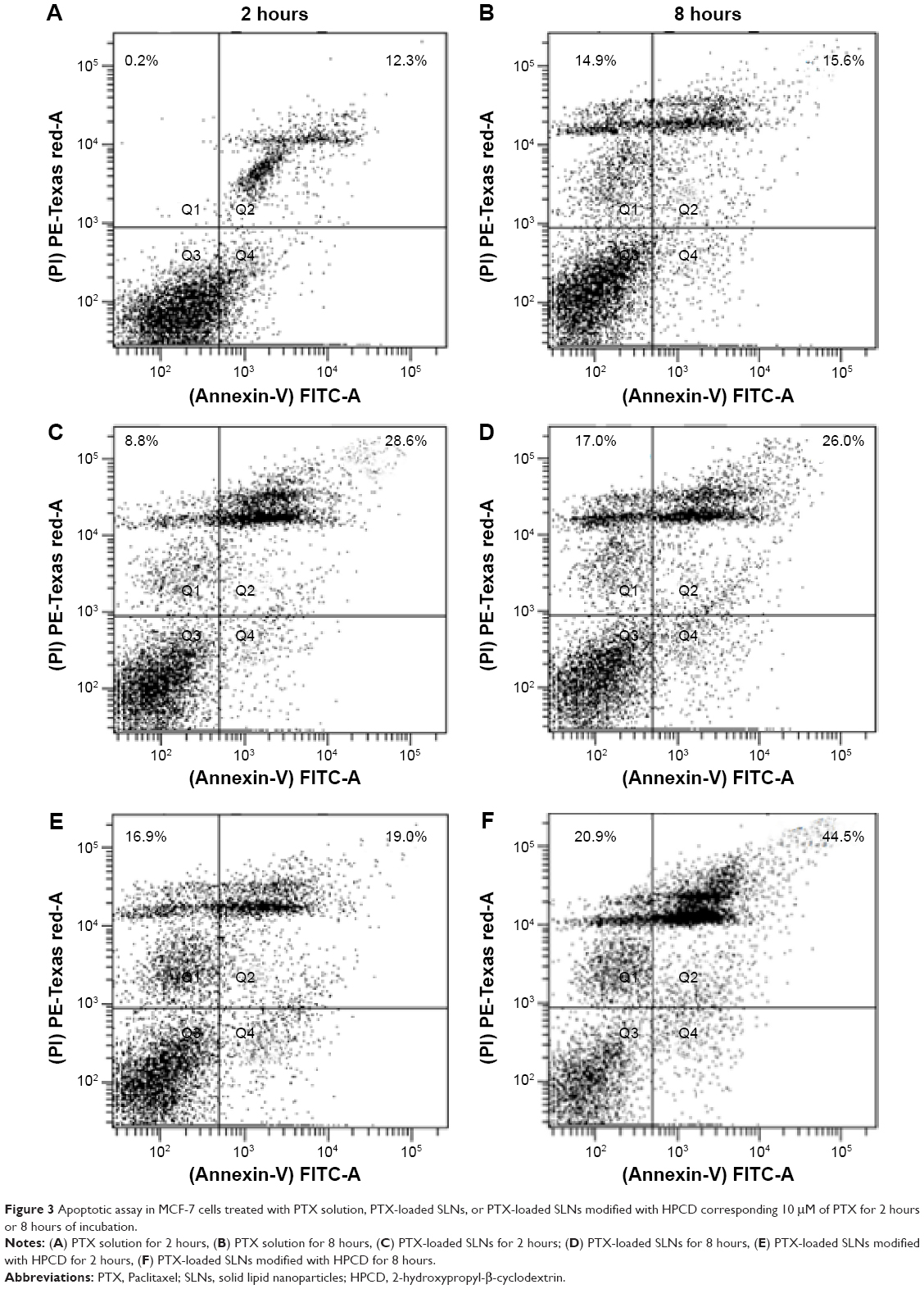 | Figure 3 Apoptotic assay in MCF-7 cells treated with PTX solution, PTX-loaded SLNs, or PTX-loaded SLNs modified with HPCD corresponding 10 μM of PTX for 2 hours or 8 hours of incubation. |
Pharmacokinetic study
The PTX concentrations in the samples were determined using a calibration curve obtained in the concentration range of 0.1–200 μg/mL, and the limit of detection was 0.05 μg/mL. The pharmacokinetic profiles of PTX after intravenous injection of PTX solution, PS, or PSC (corresponding to 5 mg/kg PTX) are shown in Figure 4. The calculated pharmacokinetic parameters such as area under the curve (AUC), plasma elimination half-life (t1/2), and total body clearance (CL) using Winnolin (Pharsight Corporation, St Louis, MO, USA) are summarized in Table 2. The plasma concentration of PTX after administration of all three formulations exhibited a biphasic decline. Although the plasma concentrations of PTX released from PS or PSC were lower than that of PTX solution at 0.08 hour and 0.17 hour after intravenous injection, both formulations significantly prolonged the retention of PTX in circulatory system. The AUC0–∞, t1/2, and CL for PTX solution were 2.57 μg·h/mL, 0.45 hour, and 1.75 L/h/kg, respectively, and those for PS were 3.31 μg·h/mL, 0.66 hour, and 1.35 L/h/kg, respectively. Interestingly, PSC showed a significant twofold increase in AUC0–∞ and 2.3-fold increase in t1/2 compared with PTX solution. Many studies have demonstrated that the intravenous nanoparticle system can significantly alter the pharmacokinetic profiles of drugs.25 Our studies also showed that PS and PSC have different pharmacokinetic properties. PSC had delayed blood clearance compared with PTX solution. It was well known that drugs can circulate for a certain period after injection before they are recognized as foreign matters and rapidly cleared by phagocytic cells of the mononuclear phagocyte system, which are abundant in special tissues and organs such as the liver and lung.26–28 The relatively higher initial plasma concentration and longer drug retention of PSC suggested that HPCD-stabilized SLNs might be a promising carrier to reduce the elimination of PTX by the mononuclear phagocyte system and extend the residence time of PTX in the circulatory system.29 Since PSC showed lower clearance and higher AUC than those of PTX solution, it was expected that PSC could prolong the exposure of cancer cells to PTX and extend its in vivo antitumor activity.
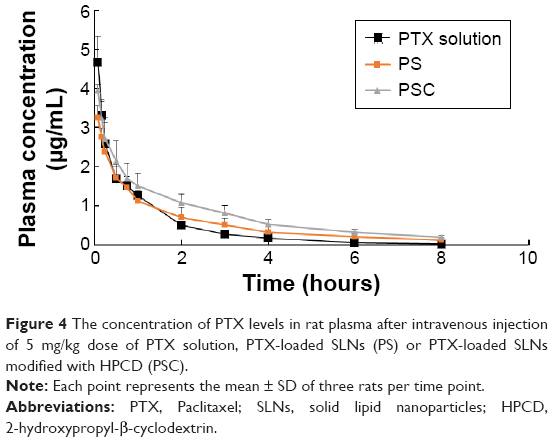 | Figure 4 The concentration of PTX levels in rat plasma after intravenous injection of 5 mg/kg dose of PTX solution, PTX-loaded SLNs (PS) or PTX-loaded SLNs modified with HPCD (PSC). |
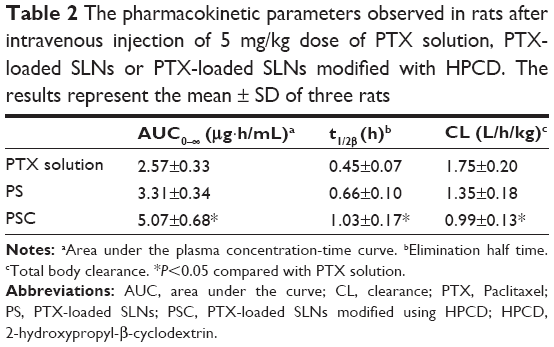 | Table 2 The pharmacokinetic parameters observed in rats after intravenous injection of 5 mg/kg dose of PTX solution, PTX-loaded SLNs or PTX-loaded SLNs modified with HPCD. The results represent the mean ± SD of three rats |
Evaluation of nephrotoxicity
The commercial formulation of PTX has resulted in cumulative sensory-dominant peripheral nephrotoxicity in humans, characterized clinically by numbness and/or paresthesia of the extremities due to the use of cremophor EL. Additionally, the damaged kidneys gain weight, and the serum creatinine level increases as the damage increases.30–34 Therefore, the renal effects of PTX solution, PS, or PSC were examined by determining the kidney weights and serum creatinine levels. The normalized kidney weight after treatment with PTX solution was 4.87±0.35 mg/g, significantly (1.35-fold) higher than that of nontreated kidney (Figure 5A). However, the normalized kidney weight after treatment with PS or PSC was 3.87±0.11 mg/g or 3.60±0.26 mg/g, respectively, and was not significantly different from that of nontreated kidney. We also measured serum creatinine levels 8 hours after intravenous administration of PTX solution, PS, or PSC (Figure 5B) as 0.52±0.06 mg/dL, 0.44±0.02 mg/dL, or 0.39±0.03 mg/dL, respectively. PS and PSC showed insignificant changes in kidney weight and plasma creatinine level after intravenous injection, suggesting no kidney toxicity compared with the commercial formulation including cremophor EL.
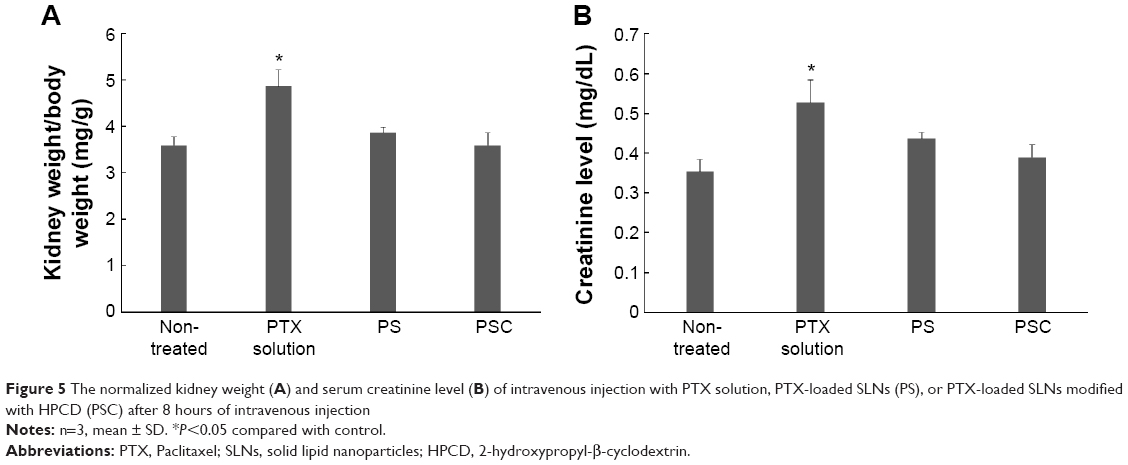 | Figure 5 The normalized kidney weight (A) and serum creatinine level (B) of intravenous injection with PTX solution, PTX-loaded SLNs (PS), or PTX-loaded SLNs modified with HPCD (PSC) after 8 hours of intravenous injection |
Antitumor efficacy
PTX solution, PSC, and a saline control were separately injected thrice (10 mg/kg each) on days 6, 13, and 20 after tumor inoculations. There were no significant differences in the sizes of tumors until day 23 between PTX solution- and PSC-treated mice (Figure 6A). However, tumor volume was remarkably small, that is, 309.6±18.7 mm3 or 264.5±23.5 mm3, respectively, when treated with PTX solution or PSC after 27 days. In particular, mice treated with PSC had smaller tumor volumes than those treated with PTX solution, indicating greater antitumor efficacy of PSC than that of PTX solution. This is most likely owing to the high cellular uptake and expected sustained release profiles of PTX from PSC within the tumor tissue based on the apoptosis results.
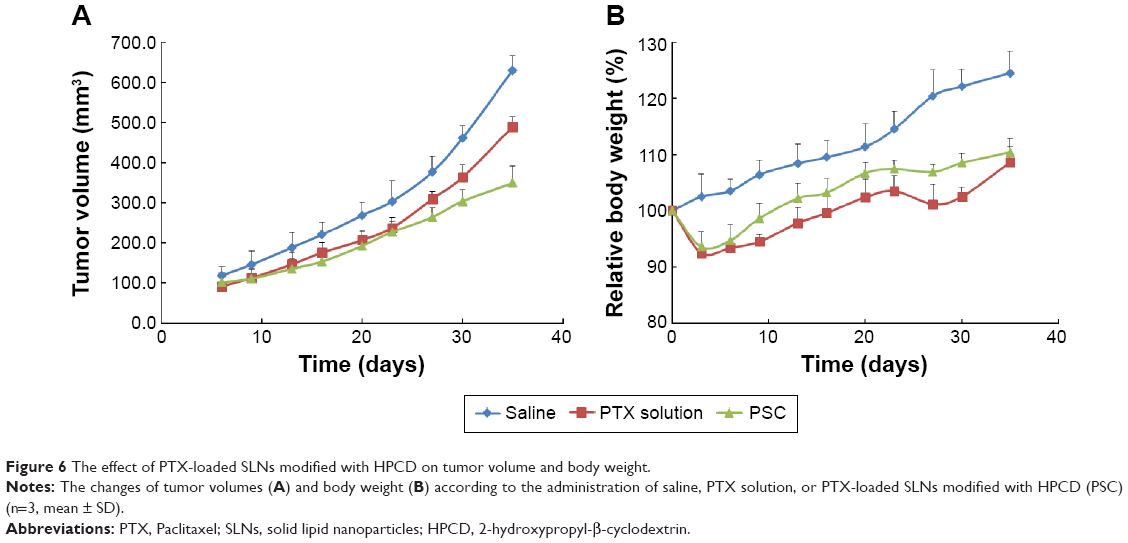 | Figure 6 The effect of PTX-loaded SLNs modified with HPCD on tumor volume and body weight. |
The mean body weight changes of mice treated with PTX solution or PSC are shown in Figure 6B. Measurement of body weight variations can be used to define the systemic toxicity of a drug. PTX solution and PSC resulted in 7.5% and 6.4%, respectively, average maximum weight loss within 9 days. Despite the similar acute reductions in body weight, weight recovery was more rapid following administration of PSC. The increased body weight of mice treated with PTX solution or PSC after 9 days may be due to normal growth coupled with growth of tumors.35 Mice treated with PSC regained their initial body weight by day 13, whereas mice treated with PTX solution regained their initial body weight by day 20. These results suggest that PSC is less toxic to normal organs compared with PTX solution.
Conclusion
PSC exhibited sustained cytotoxicity against MCF-7 cells and displayed strong antitumor activity without a corresponding change in body weight. Furthermore, PSC increased absorption of PTX and reduced nephrotoxicity when administered intravenously. Based on these results, PSC could be considered as a promising intravenous drug delivery system with enhanced absorption and reduced nephrotoxicity.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2009-0093815 and 2012R1A1B5003358). Also, we thank Ms Jisun Lee for proofreading this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235:179–192. | ||
Yoncheva K, Calleja P, Agüeros M, et al. Stabilized micelles as delivery vehicles for paclitaxel. Int J Pharm. 2012;436:258–264. | ||
Lee SC, Huh KM, Lee J, Cho YW, Galinsky RE, Park K. Hydrotropic polymeric micelles for enhanced paclitaxel solubility: in vitro and in vivo characterization. Biomacromolecules. 2007;8:202–208. | ||
Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72:191–202. | ||
Sharma A, Mayhew E, Straubinger RM. Antitumor effect of taxol-containing liposomes in a taxol-resistant murine tumor model. Cancer Res. 1993;53:5877–5881. | ||
Bilensoy E, Gürkaynak O, Doğan AL, Hincal AA. Safety and efficacy of amphiphilic beta-cyclodextrin nanoparticles for paclitaxel delivery. Int J Pharm. 2008;347:163–170. | ||
Talegaonkar S, Azeem A, Ahmad FJ, Khar RK, Pathan SA, Khan ZI. Microemulsions: a novel approach to enhanced drug delivery. Recent Pat Drug Deliv Formul. 2008;2:238–257. | ||
Dordunoo SK, Jackson JK, Arsenault LA, Oktaba AM, Hunter WL, Burt HM. Taxol encapsulation in poly(epsilon-caprolactone) microspheres. Cancer Chemother Pharmacol. 1995;36:279–282. | ||
Sharma D, Chelvi TP, Kaur J, et al. Novel taxol formulation: polyvinylpyrrolidone nanoparticle-encapsulated taxol for drug delivery in cancer therapy. Oncol Res. 1996;8:281–286. | ||
Li X, Li P, Zhang Y, et al. Novel mixed polymeric micelles for enhancing delivery of anticancer drug and overcoming multidrug resistance in tumor cell lines simultaneously. Pharm Res. 2010;27:1498–1511. | ||
Luo J, Xiao K, Li Y, et al. Well-defined, size-tunable, multifunctional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjug Chem. 2010;21:1216–1224. | ||
Xiao K, Luo J, Fowler WL, et al. A self-assembling nanoparticle for paclitaxel delivery in ovarian cancer. Biomaterials. 2009;30:6006–6016. | ||
Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci. 2008;97:3518–3590. | ||
Wissing SA, Kayser O, Muller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56:1257–1272. | ||
Souto EB, Muller RH. Cosmetic features and applications of lipid nanoparticles (SLN, NLC). Int J Cosmet Sci. 2008;30:157–165. | ||
Lippacher A, Muller RH, Mader K. Investigation on the viscoelastic properties of lipid based colloidal drug carriers. Int J Pharm. 2000;196:227–230. | ||
Marengo E, Cavalli R, Caputo O, Rodriguez L, Gasco MR. Scale-up of the preparation process of solid lipid nanospheres. Part I. Int J Pharm. 2000;205:3–13. | ||
Mehnert W, Mader K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196. | ||
Baek JS, So JW, Shin SC, Cho CW. Solid lipid nanoparticles of paclitaxel strengthened by hydroxypropyl-β-cyclodextrin as an oral delivery system. Int J Mol Med. 2012;30:953–959. | ||
Baek JS, Cho CW. 2-Hydroxypropyl-β-cyclodextrin-modified SLN of paclitaxel for overcoing p-glycoprotein function in multidrug resistant breast cancer cells. J Pharm Pharmacol. 2013;65:72–78. | ||
Yamazaki M, Ito T. Deformation and instability in membrane structure of phospholipid vesicles caused by osmophobic association: mechanical stress model for the mechanism of poly(ethylene glycol)-induced membrane fusion. Biochemistry. 1990;29:1309–1314. | ||
Gratton SE, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613–11618. | ||
Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. | ||
Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. In vivo performances. J Control Release. 2012;160:418–430. | ||
Lou H, Zhang X, Gao L, et al. In vitro and in vivo antitumor activity of oridonin nanosuspension. Int J Pharm. 2009;379:181–186. | ||
Gao L, Zhang D, Chen M, et al. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int J Pharm. 2008;355:321–327. | ||
Moqhimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanopartricles: theory to practice. Pharmacol Rev. 2001;53:283–318. | ||
Aliabadi HM, Shahin M, Brocks DR, Lavasanifar A. Disposition of drugs in block copolymer micelle delivery systems: from discovery to recovery. Clin Pharmacokinet. 2008;47:619–634. | ||
Haghighi M, Nematbakhsh M, Talebi A, et al. The role of angiotensin II receptor 1 (AT1) blockade in cisplatin-induced nephrotoxicity in rats: gender-related differences. Ren Fail. 2012;34:1046–1051. | ||
Nematbakhsh M, Ashrafi F, Safari T, et al. Administration of vitamin E and losartan as prophylaxes in cisplatin-induced nephrotoxicity model in rats. J Nephrol. 2012;25:410–417. | ||
Shimeda Y, Hirotani Y, Akimoto Y, et al. Protective effects of capsaicin against cisplatin-induced nephrotoxicity in rats. Biol Pharm Bull. 2005;28:1635–1638. | ||
Evenepoel P. Acute toxic renal failure. Best Pract Res Clin Anaesthesiol. 2004;18:37–52. | ||
Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365:417–430. | ||
Koziara JM, Whisman TR, Tseng MT, Mumper RJ. In-vivo efficacy of novel paclitaxel nanoparticles in paclitaxel-resistant human colorectal tumors. J Control Release. 2006;112:312–319. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.