Back to Archived Journals » Pathology and Laboratory Medicine International » Volume 7
Mixed gonadal dysgenesis associated with persistent Müllerian duct syndrome – a rare anomaly
Authors Baig M ![]()
Received 15 June 2015
Accepted for publication 7 August 2015
Published 16 October 2015 Volume 2015:7 Pages 95—98
DOI https://doi.org/10.2147/PLMI.S90512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Paul Zhang
Video abstract presented by Baig
Views: 6985
Mirza Asif Baig
Shri BM Patil Medical College Hospital and Research Centre, Bijapur Liberal District Education (BLDE) University, Bijapur, Karnataka, India
Background: Persistent Müllerian duct syndrome (PMDS) is a rare autosomal recessive inherited disorder. It is a type of internal male pseudo-hermaphroditism in which Müllerian duct derivatives persist in the phenotypically and karyotypically normal male. The exact incidence is unknown, but to date, approximately 400 cases have been reported. PMDS is caused either by insufficient amount of Müllerian inhibiting factor or insensitivity of the target organs to Müllerian inhibiting factor. The case presented in this report discusses mixed gonadal dysgenesis, a condition that refers to individuals who usually have a differentiated gonad on one side and a streak gonad or testis on the other side.
Case summary: A 27-year-old male patient presented with cryptorchidism and a left inguinal hernia. On exploratory laparotomy, a uterus with bilateral adnexa was noted, and histopathology revealed the features of mixed gonadal dysgenesis associated with PMDS. The closest differential diagnosis for this condition is true hermaphroditism.
Conclusion: PMDS is a rare disorder, and is important to diagnose this condition early because 30% of the cases progress to gonadoblastomas like dysgerminoma, yolk sac tumor, and embryonal carcinoma, and hence gonadectomy is necessary. The very rare nature of this condition and grave prognosis merits its reporting.
Keywords: persistent Müllerian ducts, pseudohermaphroditism, true hermaphroditism, gonadoblastomas, MIF, cryptorchidism
Introduction
Persistent Müllerian duct syndrome (PMDS) is a rare form of internal male pseudo-hermaphroditism in which Müllerian duct derivatives (uterus, cervix, Fallopian tubes, and upper two-third of the vagina) are present in a phenotypically and karyotypically normal male.1 Its association with mixed gonadal dysgenesis (MGD) makes it a very rare condition. The exact incidence is unknown but to date approximately 400 cases of PMDS have been reported.2 The study by Farikullah et al suggests that approximately 200 cases of PMDS have been reported in the last 50 years.3
PMDS is caused either by an insufficient amount of Müllerian inhibiting factor (MIF) or due to insensitivity of the target organs to MIF. MGD refers to individuals who usually have a differentiated gonad on one side and a streak gonad or a streak testis on the other side. Few authors apply the term to the patients who show testicular differentiation on either bilateral streak testis or bilateral dysgenetic testis with 45X/46XY.1
The presence of both dysgenetic gonadal tissue and Y chromosome material is responsible for increased risk of malignancies. Thus, diagnosis of MGD with PMDS cannot be missed as it has important clinical implications for gender assignments and the decision for early gonadectomy can be taken as 30% of the cases progress to gonadoblastomas.4
The closest differential diagnosis (DD) for PMDS with asymmetric gonadal dysgenesis (AGD) is a true hermaphroditism that can only be diagnosed by the following histopathological criteria necessary for diagnosis.
Case report
Ethical approval for this case report was granted by the institutional committee. Patient consent was also obtained. A 27-year-old married male patient was referred to the general surgery clinic for painful swelling in the left inguinoscrotal region and bilateral undescended testis. The patient described the pain as dull and persistent with no clear exacerbating factors. He had a male phenotype with well-developed secondary sexual characters, and his past history included primary infertility. On examination, external genitalia were normal with empty scrotal sac and a mass in the left inguinal canal that can be reduced into the scrotum easily. The patient had bilateral undescended testes, and the right testis was palpable and left testis was impalpable. A clinical diagnosis of cryptorchidism with indirect left inguinal hernia was made.
Laboratory investigations
Complete blood count: white blood cell count =5.21×103/mm; hemoglobin =11.7 g/dL; platelet count =297×103/mm; coagulation profile: prothrombin time =12.4 seconds; partial thromboplastin time =30.5 seconds; liver function test and renal function test = normal; anti-Müllerian hormone (AMH) = undetectable; semen analysis = azoospermia; and ultrasonography revealed bilateral cryptorchidism and inguinal hernia.
Karyotyping was not performed initially due to financial reasons but later on the Government subsidized the test expenses. It was 46XY karyotype.
Exploratory laparotomy
The contents of the left inguinal canal, in particular, the left spermatic cord, were identified as emerging from the left internal inguinal ring. Adjacent to the spermatic cord, an unidentified tubulomuscular structure passing from the internal inguinal ring could be seen. A small incision was made within the structure and flexible endoscopy demonstrated a hollow tubular structure that eventually led to a blind end and had communication with the prostatic urethra, but not with the bladder. The excised structure measured 8×5×4 cm. It was seen to contain mucus only. The indirect hernia was subsequently repaired using a standard Lichtenstein repair with a plug and patch mesh.
Fowler Stephens bilateral orchidopexy was performed. The scissors were used to open the peritoneum lateral to the vessels and medial to the vas deferens. The left testis was brought down to the scrotum via the inguinal canal.
The excised tubulomuscular structure measured 8×5×4 cm with both the tubes measuring 4 cm, and two globular masses (ovary-like structures) were seen attached to each Fallopian tube.
Müllerian remnants were resected and testicular biopsy from right and left testis were sent for histopathological examination.
Gross
Uterocervix measured 8×5×4 cm with each tube measuring 4 cm. Two globular tissues (ovary-like structures) were seen attached to each Fallopian tube at the end. Each ovary-like globular tissue measured 2.5×2×1 cm, and the cut section showed grayish white solid and cystic areas (Figure 1).
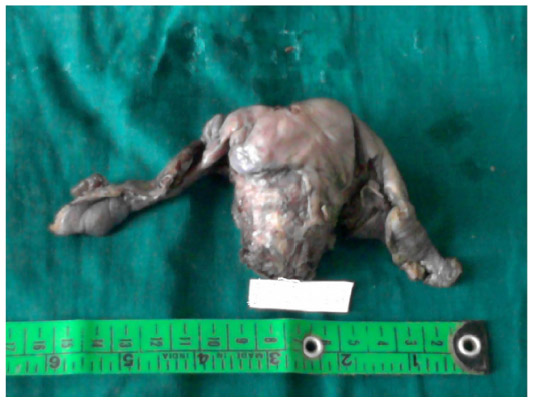 | Figure 1 Uterocervix with bilateral Fallopian tubes and ovaries. |
Microscopy
Cross section studied through the uterocervix showed endometrium in proliferative phase with small round glands and compact stroma (Figure 2) with both the Fallopian tubes showing normal histology (Figure 3). Cross section studied through globular tissue attached to Fallopian tubes showed seminiferous tubules with immature sertoli cells and few primitive germ cells (Figure 4). No ovarian stroma or primordial follicles were identified. Cross section studied through uterocervix separately sent for testicular biopsy showed seminiferous tubules showing incomplete spermatocytic maturation arrest (right testis) (Figure 5), seminiferous tubules with immature sertoli cells, and primitive spermatogonia (left testis). On the basis of the all these findings, a diagnosis of PMDS with MGD was made.
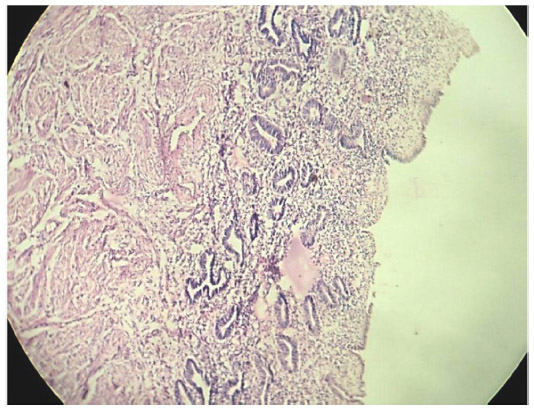 | Figure 2 H&E showing proliferative endometrium. |
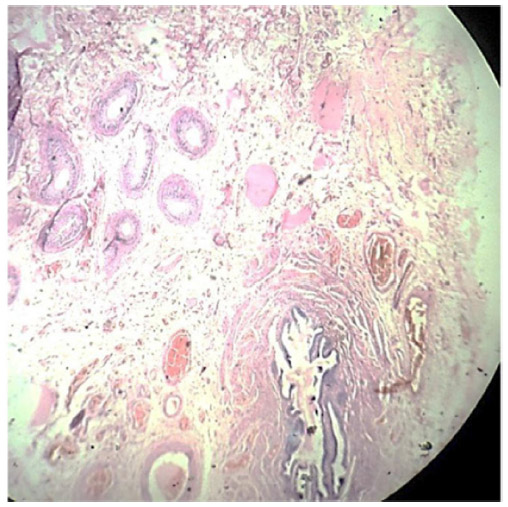 | Figure 3 LP; H&E-stained fallopian tube with fimbriae lined by simple columnar epithelium, and epididymis and vas deferens lined by columnar epithelium. |
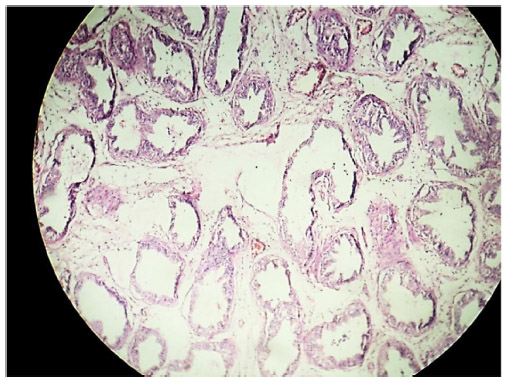 | Figure 4 LP; H&E stain showing seminiferous tubule with spermatocytic arrest and immature sertoli cells. |
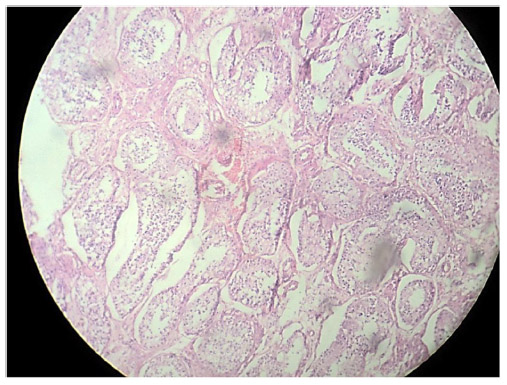 | Figure 5 H&E stain showing incomplete spermatocytic arrest with immature sertoli cells and primitive germ cells (right testis). |
Discussion
PMDS is an autosomal recessive inherited rare disorder. In the human fetus, both the Müllerian and Wolffian ducts are present at the seventh week of gestation, and in males by this time the testis differentiates. AMH is produced by testicular sertoli cells at the seventh week of gestation that causes the Müllerian ducts to disappear by epithelial–mesenchymal transformation. Subsequent to the atrophy of Müllerian ducts induced by AMH, the testes move transabdominally to the deep inguinal ring by the guidance of the gubernaculum and continue to descend into the scrotum. Defects with AMH or the receptors result in PMDS.3,5
A normal sex differentiation is controlled by testosterone, dihydrotestosterone, and MIF. Sertoli cells secrete MIF that leads to the regression of Müllerian ducts. Testosterone has a direct effect on Müllerian duct and promotes the differentiation of epididymis, vas deferens, and seminal vesicles. Dihydrotestosterone induces the development of male external genitalia.5,6
Patients with PMDS are externally normally virilized because dihydrotestosterone that induces this is not linked to AMH. PMDS is an autosomal recessive disorder, and AMH mutations occur with the increasing rate in consanguineous marriage.3
Children born with intersex disorders comprise approximately 1.7% of all live birth.7 Ovotesticular disorder of sexual differentiation or true hermaphroditism is a rare intersex disorder characterized by the presence of both ovarian tissue and testicular element regardless of their karyotype. The rarity is because most of the fetuses do not survive.8
PMDS is a rare form of internal male pseudo-hermaphroditism in which Müllerian duct derivatives (uterus, cervix, Fallopian tubes, and upper two-third of the vagina) are present in a phenotypically and karyotypically normal male patient with normal secondary sexual characters. It is due to deficiency or lack of MIF.1
MGD refers to individuals who usually have a differentiated gonad on one side and a streak gonad or a streak testis on the other side. Few authors apply the term to patients who show testicular differentiation on both sides with bilateral streak testes or bilateral dysgenetic testes with karyotype 45X/46XY.1 This condition involves a heterogeneous group of gonadal and phenotyping abnormalities with a wide clinical spectrum. The phenotype depends on the ratio of testicular tissue that induces virilization.9
PMDS = Wolffian + Müllerian ducts due to deficiency of MIFs. PMDS has two anatomical variants: Type 1(males) and Type 2 (females):
- Type 1 is more common accounting for 80%–90% of all cases. It again has two subtypes, Type 1a and Type 1b:
- Type 1a is hernia uteri inguinalis that shows descended testis and herniation of ipsilateral uterus and fallopian tube in the inguinal canal.
- Type 1b is crossed testicular ectopia that shows herniation of both the testis and the entire uterus and both the Fallopian tubes.
- Type 2 shows bilateral cryptorchidism, with the testis fixed in the round ligament in an ovarian position.1
True hermaphroditism was the closest differential diagnosis and was excluded by following the strict histological criteria necessary for diagnosis. The strict histological criterion necessary for the diagnosis of true hermaphroditism is ovotestis, that is, well-defined testicular elements like solid seminiferous tubules with immature sertoli cells, few primitive germ cells, and ovarian stroma composed of numerous primordial or mature follicles containing primary oocytes.9,10 It is important to differentiate the two conditions because it is necessary for gender assignment, and in PMDS with MGD, 30% patients develop germ cell tumors like dysgerminoma, yolk sac tumor, embryonal carcinoma, and hence gonadectomy is necessary.4
Conclusion
PMDS with MGD is a very rare disorder, and it is important to diagnose this condition because 30% of cases progress to gonadoblastomas, like dysgerminoma, yolk sac tumor, and embryonal carcinoma, and hence gonadectomy is necessary.
It should be emphasized that whenever Müllerian remnants are detected in patients, they should be resected. Immediate orchidopexy procedure and simultaneous resection of Müllerian remnants offer protection against gonadoblastomas.
The very rare nature of this condition and its dismal prognosis merit its reporting.
Disclosure
The author reports no conflicts of interest in this work.
References
Renu D, Rao BG, Rangnath K, Namitha. Persistent Mullerian duct syndrome. Indian J Radiol Imaging. 2010;20(1):72–74. | |
Yuksel B, Saygun O, Hengirmen S. Persistent Müllerian duct syndrome associated with irreducible inguinal hernia, bilateral cryptorchidism and testicular neoplasia: a case report. Acta Chir Belg. 2006;106: 119–120. | |
Farikullah J, Ehtesham, S, Nappo S, Patel L, Hennayake S. Persistent Müllerian duct syndrome: lessons learned from managing a series of eight patients over a 10-year period and review of literature regarding malignant risk from the Müllerian remnants. BJU Int. 2012;110(11 Pt C): E1084–E1089. | |
Donahoe PK, Crawford JD, Hendren WH. Mixed gonadal dysgenesis, pathogenesis, and management. J. Pediatr. Surg. 1979;14(3):287–300. | |
Moore K, Persaud T. The Developing Human: Clinically Oriented Embryology. 8th ed. Philadelphia: Elsevier Health; 2008:263–269. | |
Josso N, Picard J, Imbeaud S, di Clemente N, Rey R. Clinical aspects and molecular genetics of persistent Müllerian duct syndrome. Clin Endocrinol (Oxf). 1997;47:137–144. | |
Dekker HM, de Jong IJ, Sanders J, Wolf RF. Persistent Müllerian duct syndrome. Radiographics. 2003;23:309–313. | |
Queipo G, Nieto, K, Grether P, et al. Unusual mixed gonadal dysgenesis associated with Müllerian duct persistence, polygonadia, and a 45 X or 46 XY karyotype. Am J Med Genet A. 2005;136A(4):386–389. | |
Scully RE, Young RH, Clement PB. Atlas of Tumor Pathology: Tumors of the Ovary, Maldeveloped Gonads, Fallopian Tube, and Broad Ligament, 3rd Series. Armed Forces Institute of Pathology: Washington, DC, 1998:399–408. | |
Blackless M, Charuvastra A, Derryck A, Fausto-Sterling A, Lauzanne K, Lee E. How sexually dimorphic are we? Review and synthesis. Am J Hum Biol. 2000;12:151–166. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.