Back to Journals » OncoTargets and Therapy » Volume 13
Mitofusin1 Is a Major Mediator in Glucose-Induced Epithelial-to-Mesenchymal Transition in Lung Adenocarcinoma Cells
Authors Liu X, Feng C, Wei G, Kong W, Meng H, Du Y, Li J
Received 15 November 2019
Accepted for publication 3 April 2020
Published 24 April 2020 Volume 2020:13 Pages 3511—3523
DOI https://doi.org/10.2147/OTT.S238714
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Xingyuan Liu,1 Chuang Feng,2 Guohua Wei,1 Wencong Kong,1 Hai Meng,3 Yaqin Du,3 Jingyuan Li4
1Pathology Department, College of Basic Medical Sciences, Jinzhou Medical University, Jinzhou, Liaoning 121001, People’s Republic of China; 2Science and Technology Department, Jinzhou Medical University, Jinzhou, Liaoning 121001, People’s Republic of China; 3Clinicopathological Center, First Affiliated Hospital of Jinzhou Medical University, Jinzhou, Liaoning 121001, People’s Republic of China; 4Faculty of Pharmaceutical Sciences, First Affiliated Hospital of Jinzhou Medical University, Jinzhou, Liaoning 121001, People’s Republic of China
Correspondence: Jingyuan Li
Faculty of Pharmaceutical Sciences, First Affiliated Hospital of Jinzhou Medical University, Jinzhou, Liaoning Province 121001, People’s Republic of China
Tel/ Fax +86-416-4145198
Email [email protected]
Background: Epithelial-to-mesenchymal transition (EMT) has been considered a latent mediator of diverse biological processes in cancer. However, the mechanisms involved in high glucose-associated EMT in lung adenocarcinoma (LAD) have not been fully clarified. In this study, we aimed to investigate whether mitofusin1 (MFN1) is involved in the EMT of LAD cells induced by glucose and to identify the molecular mechanism involved in this process.
Materials and Methods: The expression of specific proteins was analysed by Western blotting, immunohistochemistry, co-immunoprecipitation and immunofluorescence analysis. The proliferation, migration and invasion of cells were assessed by Cell Counting Kit-8, bromodeoxyuridine incorporation, wound-healing and transwell assays. Lung tissues of adjacent normal regions and lung tissues from patients with LAD and LAD combined with diabetes mellitus were collected to determine the expression and significance of MFN1.
Results: Here, we showed that the expression of MFN1 was increased in LAD tissues compared with adjacent normal tissues and expression was even higher in lung tissues from patients with LAD combined with diabetes. In the lung cancer cell line A549, increased cell proliferation, invasion and EMT induced by high glucose were inhibited by MFN1 silencing. Mechanistic studies demonstrated that inhibiting autophagy reversed the abnormal EMT triggered by high glucose conditions. In addition, our data provide novel evidence demonstrating that PTEN-induced kinase (Pink) is a potential regulator involved in MFN1-mediated cell autophagy, which eventually leads to high glucose-induced proliferation, invasion and EMT of A549 cells.
Conclusion: Taken together, our data show that MFN1 interacts with Pink to induce the autophagic process and that the abnormal occurrence of autophagy ultimately contributes to glucose-induced pathological EMT in LAD.
Keywords: lung adenocarcinoma, glucose, mitofusin1, epithelial-to-mesenchymal transition, autophagy
Introduction
Lung cancer is a heterogeneous disease clinically, biologically, histologically and molecularly with a multistep process involving genetic and epigenetic alterations.1,2 The two main types of lung cancer, non-small-cell lung cancer (NSCLC) (representing 80–85% of cases) and small cell lung cancer (SCLC) (representing 15–20% of cases), are identified based on histological, clinical and neuroendocrine characteristics.3–5 Lung adenocarcinoma (LAD), the major histological subtype of NSCLC, displays several recurrent genetic alterations including critical growth regulatory proteins (K-Ras, EGFR, FBXO17, B-RAF, MEK-1, HER2, MET, TP53, PTEN, p16, and LKB-1).6,7 Advances in the understanding of genetic alterations in patient and relevant animal models have yielded a new understanding of the characterization of LAD. However, the pathogenesis and molecular basis of LAD remain elusive.
Glucose is the primary energy source for all cells; in contrast to normal cells, tumour cells are strictly dependent on an adequate supply of glucose, which maintains a much higher rate of energy metabolism for their growth and survival.8,9 Recent studies confirmed that patients with diabetes mellitus (DM) have more risk factors for the development of cancer because increased blood glucose levels can drive malignant cell growth and mitogenesis.10,11 Coincidentally, high glucose levels were reported to induce epithelial-to-mesenchymal transition (EMT) in breast cancers via a caveolin-1-dependent mechanism.12 Evidence suggests that EMT is a pivotal event in the progression of various cancers, including the invasion and metastasis of LAD.13,14 The underlying mechanism of glucose metabolic reprogramming in EMT of LAD is not well-understood.
Mitochondria are recognized as the powerhouses of cells, which support eukaryotic life through oxidative phosphorylation.15 Due to a defect in mitochondrial oxidative phosphorylation, metabolic rearrangement occurs in most tumour cells, a phenomenon known as the Warburg effect.16 The Warburg effect was discovered by Otto Warburg in 1931 and is characterized by greatly increased glucose uptake and lactate production even under aerobic conditions.17,18 Mitofusin1 (MFN1) is a mitochondrial fusion protein that exists in the outer mitochondrial membrane. Studies in HeLa and 293T cells have demonstrated that MFN1 cooperates with mitochondrial ubiquitin ligase membrane-associated RING-CH (MARCH5) and is essential for mitochondrial homeostasis and cell survival.19 Growing evidence has shown that MFN1, as a target of microRNAs, is involved in the regulation of hypoxic pulmonary arterial hypertension and cardiomyocyte apoptosis.20,21 Nonetheless, the expression and function of MFN1 in LAD remain unclear, and the functions of MFN1 in glucose-dependent LAD EMT have not yet been reported.
In the present study, we focused on investigating the impact of MFN1 on the human LAD cell line A549 and clarifying the underlying mechanisms of glucose related EMT in LAD.
Materials and Methods
Materials
Antibodies against SQSTM1 (PB0458, 1:400) was obtained from Boster Biological Technology Co. Ltd. Antibody against MFN1 (ab107129), LC3B (ab48394), Pink (ab23707), Parkin (ab77924) and Snail (ab53519) were purchased from Abcam. Antibodies against BECN-1 (sc-48341) and Fis 1(sc-376469) were purchased from Santa Cruz Biotechnology, Inc. Antibodies against N-cadherin (#13116) and E-cadherin (#14472) were obtained from Cell Signalling Technology. The Cell Counting Kit-8 kit (C0037) was provided by Beyotime Institute of Biotechnology. Bromodeoxyuridine (BrdU) proliferation assay kit (2750) was purchased from Millipore Corporation. The immunocytochemistry detection kits (SPN-9001) obtained from ZSGB-BIO. Chloroquine diphosphate salt (C6628) was purchased Sigma. mRFP-eGFP-LC3 plasmid was obtained from Hanbio Biotechnology Co. Ltd (Shanghai, China). Enhanced chemiluminescence (ECL, RPN2236) reagents were from Amersham International. All other reagents were from common commercial sources.
Clinical Samples
Tissues were obtained from lung adenocarcinoma cancer patients, and none of them had been treated with chemotherapy or radiotherapy before surgical resection. The human specimens were separately collected within three years in the First Affiliated Hospital of Jinzhou Medical University. Written informed consent was obtained and approved by the Ethical Committee of Jinzhou Medical University (NO. JZMU20190921). The work was also conducted in accordance with the Helsinki Declaration of 1975 and its later amendments or comparable ethical standards for the use of human samples.
Immunohistochemistry (IHC)
The tissue sections were incubated in 0.1 mol/L sodium citrate buffer and heated for 20 min for antigen retrieval. After blocking endogenous peroxidase activity, the sections were incubated with anti-MFN1 antibodies (1:100). Parallel controls were run with PBS alone. After an overnight incubation, the sections were washed three times with PBS, and then the secondary antibodies were added for 30 min. Sections were visualized with 3, 3-diaminobenzidine (DAB) and counterstained using haematoxylin. Brown and yellow colours indicate positive staining. The images were recorded by digital photomicrography (Olympus, Japan) and analysed independently in a blinded manner by two experienced pathologists who selected three fields at high magnification (×400) and counted 100 cells in each field. The staining score was evaluated according to positively stained cells and staining intensity. Positive staining was scored as 0 (0%), 1 (1% ~ 25%), 2 (25% ~ 50%), 3 (50% ~ 75%), or 4 (75% ~ 100%). The staining intensity was scored as 0 (negative), 1 (weak), 2 (medium) or 3 (strong). The final histochemistry scores were calculated as a product of the positive staining score multiplied by the staining intensity score (with a score ≥ 4 as high expression, < 4 as low expression).
Cell Culture
The lung adenocarcinoma cell lines A549 and NCI-H292 were obtained from the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM containing high glucose (HG; 25 mM) or normal glucose (NG; 5 mM) with 10% foetal bovine serum, penicillin (100 U/mL) and streptomycin (100 U/mL) at 37 °C in a 5% CO2 humidified incubator (Thermo Fisher). HG treatment was used to induce the model of DM combined with A549 for 24 h.22
SiRNA (Small Interfering RNA) Transfection of A549 Cells
The specific siRNA targeted at knocking down MFN1 was synthesized by GenePharma (Shanghai, China). A non-targeted control siRNA (NC) was used as a negative control to determine and optimize the efficiency of transfection. The detailed siRNA sequences were as follows: si-MFN1:5ʹ-CUUAGAUGCUGAUGUCU-UUTT-3ʹ, si-NC sequence: 5′-UUCUCCGAACGUGUCACGUTT-3′. Transfection was implemented according to the manufacturer’s instructions of Lipofectamine 2000 Reagent. In brief, A549 cells were used at a confluence of 60%–70%; 1.5 mg of siRNA and 7.5 mL of the Lipofectamine 2000 siRNA Transfection Reagent were separately diluted in 100 μL of serum-free Opti-MEM-1 medium (31,985,070, Invitrogen) for 5 min, mixed and incubated at room temperature for 18 min. The siRNA-transfection reagent mixture was added directly to the cells. Cells were incubated for 24 h and used as required.
Cell Counting Kit-8 (CCK-8) Assay
Cells were separately cultured in 96-well plates at a density of 5000 cells per well and treated with different reagents. After 24 h of incubation at 37 °C, CCK-8 reagent (10 μL) was added to each well and incubated for 4 h in 37 °C, and the optical density (OD) was detected by a spectrophotometer at a wavelength of 450 nm.
Bromodeoxyuridine Incorporation
Cells were plated in 96-well plates at a density of 5000 cells/well, and then induced by serum deprivation for 24 h. After being treated with different agents in 5% FBS DMEM with 10 ng/mL 5-bromodeoxyuridine (BrdU) labelling solution, cells were cultured in media with different concentrations of glucose for 24 h. BrdU incorporation was measured according to the Millipore BrdU proliferation assay kit instructions. Finally, after adding 100 μL stop solution, the OD value was detected by a spectrophotometer at a wavelength of 450 nm.
Wound Healing Assay
A549 cells cultured in 6-well plates were wounded by a pipette tip (1 mm width), and cells received different concentrations of glucose. At 0, 6, 12 and 24 h after treatment, cells were photographed from the same areas as those recorded at 0 h. Photos were taken under an inverted microscope (Nikon). The experiments were performed at least three times.
Migration and Invasion Assays
The A549 cells were incubated with different groups prior to migration and invasion assays. The migration assay was performed using a modified Boyden chamber. The A549 cells were cultured in the upper chamber of the Transwell plates which were inserted into 24-well plates. The lower chamber contained the experimental reagents in 10% FBS+DMEM. Migration was measured after fixing the cells with 4% paraformaldehyde and staining them with 0.4% crystal violet. Cells on the upper part of the filter were removed, and the number of stained migrated cells was counted under an inverted microscope (Nikon). Each sample was counted randomly in nine separate locations in the centre of the membrane, and the A549 cell migration activity was reported as the number of cells migrated per field of view.
The invasion assay was also performed using a modified Boyden chamber (3422, Corning). Matrigel (35423, BD Biosciences) was diluted with cold filtered distilled water to a concentration of 25 μg/50 μL and applied to the upper chamber of 8 μm pore size polycarbonate membrane filters. The A549 cells were placed on the Matrigel in the upper chamber of the Transwell plates, and 500 uL medium containing 10% FBS was added to the lower chamber. Following 24 h incubation, the Matrigel and cells remaining on the upper membrane were wiped off using a cotton swab, and cells that invaded through the membrane were fixed with 4% paraformaldehyde and stained with 0.4% crystal violet. After imaging under an inverted microscope (Nikon), each sample was counted randomly in nine separate locations in the centre of the membrane, and the A549 cell invasion activity was reported as the number of migrated cells per field of view.
Cell Immunofluorescence
The A549 cells were grown on coverslips up to 70% confluence and treated with different agents according to the different experimental groups. After 24 h, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.01% Triton X-100 for 10 minutes, and blocked with 5% normal bovine serum for 1 h at room temperature. Cells were then incubated with primary antibodies against N-cadherin and E-cadherin overnight at 4 °C. After being washed, cells were incubated with the corresponding Cy3 (1:100) or FITC (1:100) conjugated secondary antibodies and then stained with 4ʹ,6-diamidino-2-phenylindole (DAPI; 1:100). The cells were then examined under a fluorescence microscope (Nikon).
Immunoblot Analysis
The cells were exposed to different experimental conditions. Proteins were harvested by washing cells in cold PBS three times and then scraping cells into 400 μL of lysates containing protease inhibitors on ice. The lysates were sonicated for 1 min and then centrifuged at 13,500 rpm for 15 min at 4 °C. The protein concentrations in the supernatant were determined using a Bio-Rad protein assay kit. Samples containing 20 μg of total protein were heated with 6%~12% SDS-PAGE and transferred onto nitrocellulose membranes (Millipore, USA). After blocking with 5% nonfat milk in Tris-buffered saline buffer (20 mM Tris, 150 mM NaCl, pH 7.6, Tween 20 0.1%). The membranes were incubated with specific antibodies against MFN1 (1:1000), N-cadherin (1:1000), E-cadherin (1:1000), Snail (1:500), LC3B (1:500), BECN-1 (1:500), SQSTM1 (1:500) and Pink1 (1:1000). Blots were then sequentially incubated with horseradish peroxidase-conjugated secondary antibodies and enhanced by chemiluminescence reagents.
Co-Immunoprecipitation Analysis
For the co-immunoprecipitation assays, the A549 cells were subjected to different treatments for 24 h and then lysed with RIPA buffer. Protein lysates (1 mg/mL) were pre-cleared with protein A/G beads (Santa Cruz, USA) before incubation with protein A/G beads bound to polyclonal rabbit anti-Pink 1 (10 µg) overnight at 4 °C. The antibody-protein complexes were washed four times with wash buffer (50 mM Tris, pH 7.4, 30 mM NaCl, 5 mM EDTA, 0.1% TX-100) at 4 °C. The buffer was removed, and the pellet was resuspended in Western loading buffer. The eluted samples were then assayed by Western-blot.
Evaluation of Fluorescent LC3 Puncta
LC3 puncta were indicated by the mRFP-GFP-LC3 plasmid. Briefly, A549 cells were transfected with mRFP-GFP-LC3 for 24 h before treatment. After subjecting cells to different treatments for 24 h, the A549 cells were observed under a confocal fluorescence microscope (Nikon).
Statistical Analysis
All experiments were repeated in triplicate. The data are presented as the mean ± S.E.M. Statistical analysis was performed with Student’s t-test or one-way ANOVA, followed by Dunnett’s test. Fisher’s exact test or chi square test where appropriate was utilized for calculating the potential correlations of MFN1 levels and clinicopathological features. Differences were considered to be significant at P≤0.05.
Results
MFN1 Expression Is Stimulated by High Concentrations of Glucose Both in vivo and in vitro
First, immunohistochemistry (IHC) was applied to examine the expression of MFN1 in carcinoma tissue (Figure 1A). The clinicopathologic features of the LAD patients were summarized in Table 1. The expression status of MFN1 measured was significantly associated with clinicopathologic features (combined diabetes mellitus and TNM stage). To investigate the effects of glucose on lung cancer cell lines in vitro, A549 and NCL-H292 cells were cultured and treated with glucose at different concentrations (no glucose, NO, 0 mM; low glucose, LG, 1 mM; normal glucose, NG, 5 mM; and high glucose, HG, 25 mM) to interfere with the growth of tumour cells. Figure 1B shows that the proliferation of A549 cells was increased with high glucose (HG, corresponding to patients with DM) treatment compared normal glucose treatment (NG, corresponding to physiological levels in healthy human blood). However, in NCL-H292 cells, cell viability was increased in 1 mM glucose (low glucose, LG) and remained unchanged even at high glucose concentrations (Figure 1C). To further explore whether glucose-regulated lung cancer cell dysfunction was related to mitofusin 1 (MFN1), the expression of MFN1 in A549 and NCL-H292 cells was measured by Western blotting. The results showed that high glucose caused MFN1 activation in cultured A549 cells but not in NCL-H292 cells (Figure 1D and E). Moreover, MFN1 in A549 cells was mainly found in the cytoplasm and co-localized with mitochondria as detected by immunofluorescence assays (Figure 1F). Therefore, we performed the following experiment by using A549 cells.
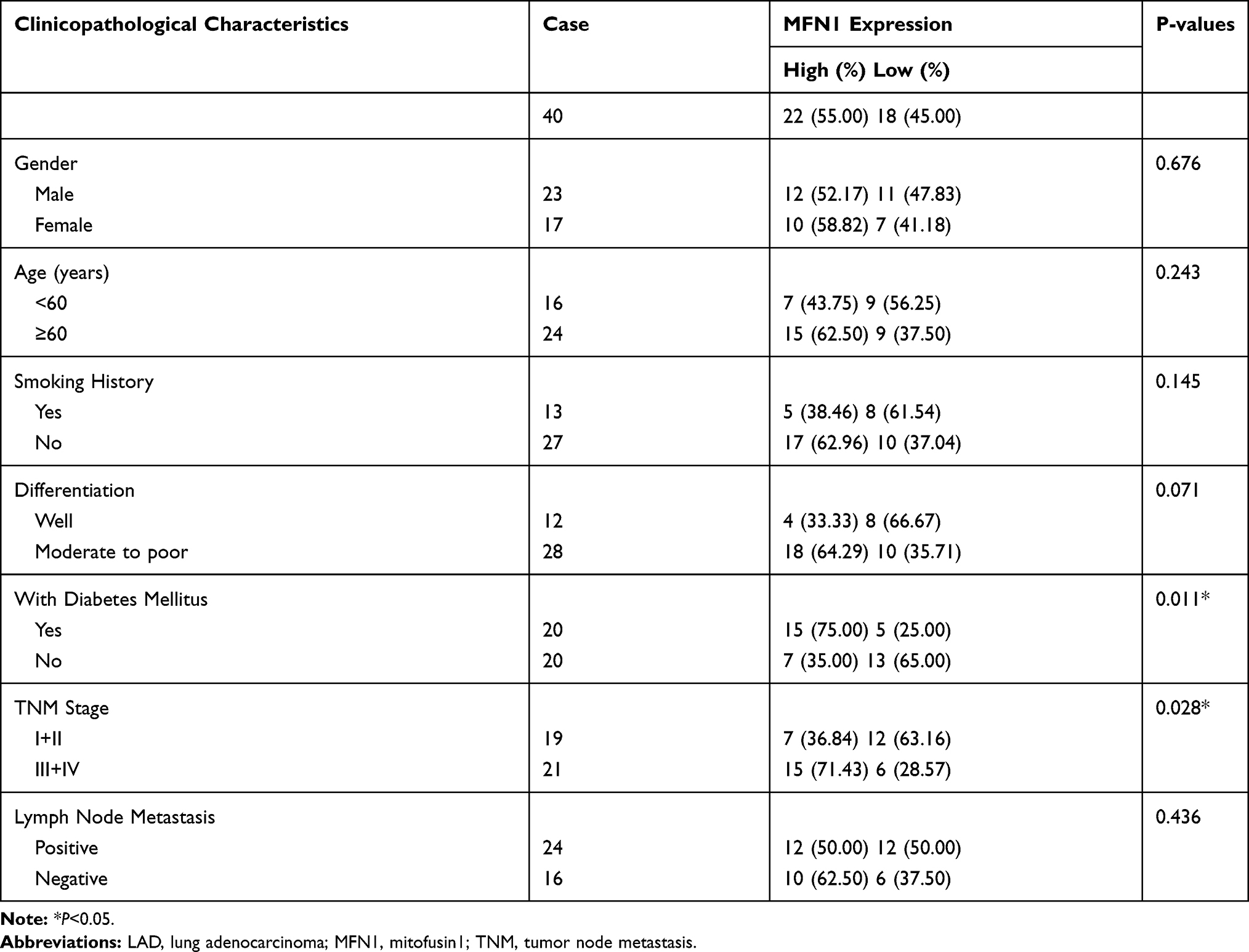 |
Table 1 Correlation Between MFN1 Expression and the Clinicopathological Characteristics of Patients with LAD |
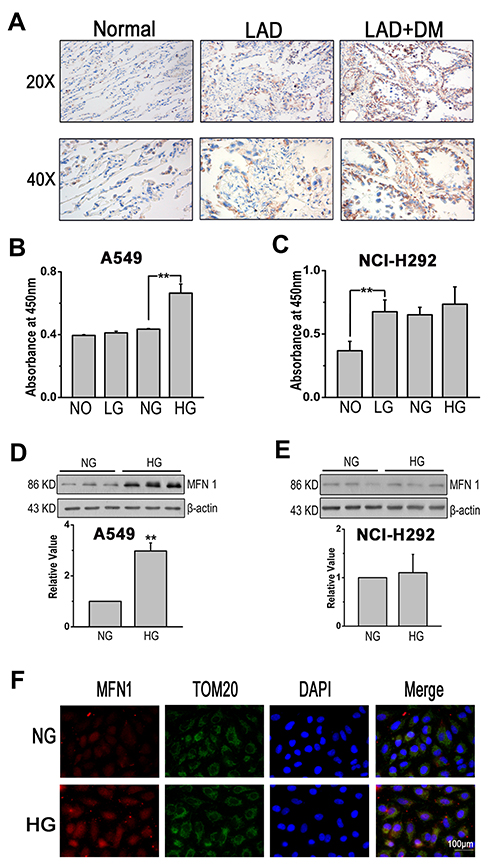 |
Figure 1 High glucose induced dysregulated cell function and aberrant expression of MFN1. (A) MFN1 in sections of lung tissues from adjacent normal, LAD and LAD+DM patients. Brown colour indicates positive staining. (B and C). Cell viability was evaluated by the CCK-8 assay in A549 and NCL-H292 cells. (D-E) The protein expression of MFN1 was determined by Western blot analysis in A549 cells (D) and in NCL-H292 cells (E). (F) A549 cells were stained with MFN1 antibody (red) and co-stained with TOM20 antibodies to visualize mitochondria (green) and DAPI staining was used to visualize cell nuclei (blue). Scale bar = 100 μm. Data shown are mean ± SEM, **P < 0.01 (n =6). Abbreviations: LAD, lung adenocarcinoma; DM, diabetes mellitus; NO, no glucose; LG, low glucose; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; TOM20, mitochondrial import receptor subunit TOM20; DAPI, 4ʹ,6-diamidino-2-phenylindole. |
MFN1 Is Involved in High Glucose-Induced EMT in A549 Cells
To investigate the effects of MFN1 on EMT. A549 cells were treated with MFN1 specific small interfering RNA (siRNA), and the expression of MFN1 protein was blocked effectively (Figure 2A). Next, the correlation of MFN1 with EMT induced by glucose was examined; as shown in Figure 2B and C, high glucose increased the expression of N-cadherin and Snail and decreased the expression of E-cadherin, and these patterns were reversed by MFN1 silencing (siMFN1). The results were further visualized by fluorescence staining in cells, which indicated that after glucose treatment, the LAD cell line exhibited increased N-cadherin and decreased E-cadherin expression, however, the effect was abolished with transient transfection of MFN1 (Figure 2D and E). Overall, these results show that MFN1 efficiently modulates the progression of high glucose-induced EMT in A549 cells.
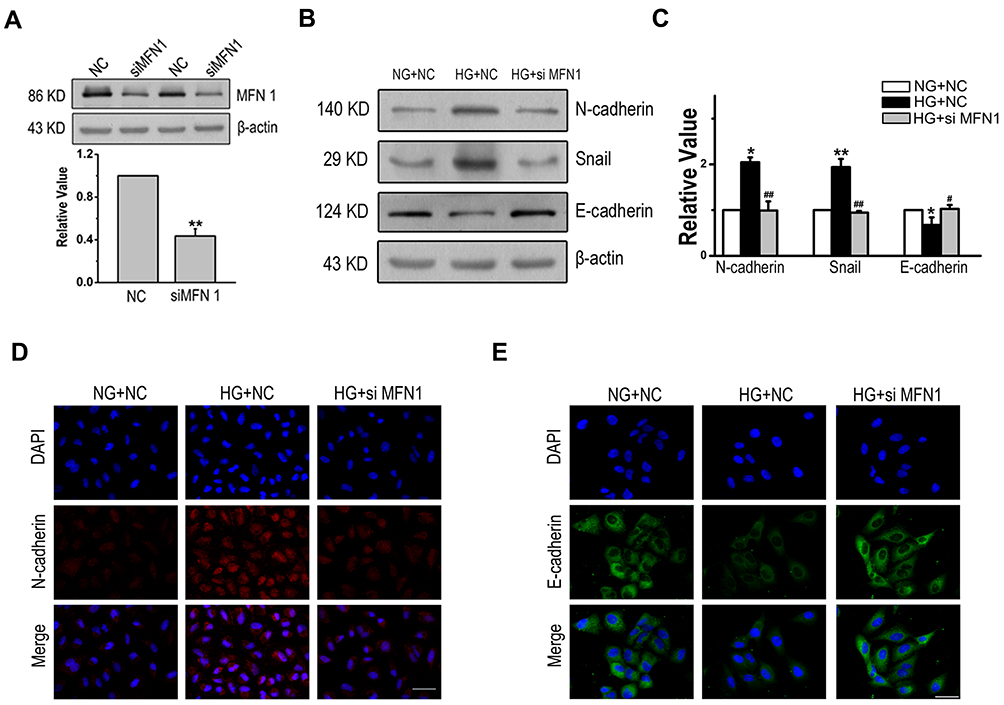 |
Figure 2 MFN1 was an important factor for EMT in human LAD. (A). The efficiency and specificity of siRNAs targeting MFN1 knockdown. The results showed that siMFN1 decreased the expression of MFN1 compared with that in cells treated with non-targeted control siRNA (NC). (B-C). The expression of mesenchymal and epithelial cell protein markers in A549 cells after high glucose treatment. (D and E). High glucose enhanced the expression of N-cadherin (red) and decreased the expression of E-cadherin (green) in the cytoplasm of A549 cells. Scale bars = 100 µm. Data shown are mean ± SEM from at least three separate experiments. *P < 0.05, **P < 0.01 vs NG+NC group. #P < 0.05, ##P < 0.01 vs HG+NC group significantly different as indicated. Abbreviations: EMT, epithelial-to-mesenchymal transition; LAD, lung adenocarcinoma; NC, non-targeted control; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; siMFN1, small interfering RNA of MFN1; DAPI, 4ʹ,6-diamidino-2-phenylindole. |
High Glucose Promotes Cell Proliferation, Migration and Invasion Through MFN1 in A549 Cells
To determine whether MFN1 influenced cell survival and growth, CCK-8 assay was performed. High glucose-enhanced cell viability was modestly inhibited after siMFN1 transfection (Figure 3A). Then cells were pulsed with 5-bromodeoxyuridine (BrdU) and stimulated with glucose; when MFN1 expression was silenced, cell BrdU incorporation was reduced accordingly (Figure 3B). To explore the role of MFN1 in high glucose-induced cellular migration, we incubated cells with 25 mM glucose and performed a scratch wound assay. There was no difference in migration within 6 and 12 hours after high glucose exposure. The effect of high glucose on cell migration was increased after 24 hours of incubation, which was blocked by siMFN1 (Figure 3C). Transwell assays indicated that glucose stimulated A549 cell migration compared with the normal control, and siMFN1 mitigated the high glucose-induced migration (Figure 3D). In addition, glucose-induced cellular invasion was also significantly inhibited in the siMFN1 group compared with the high glucose plus NC group (Figure 3E). These data demonstrate that MFN1 mediates high glucose-induced changes in proliferation, migration and invasion in the LAD cell line A549.
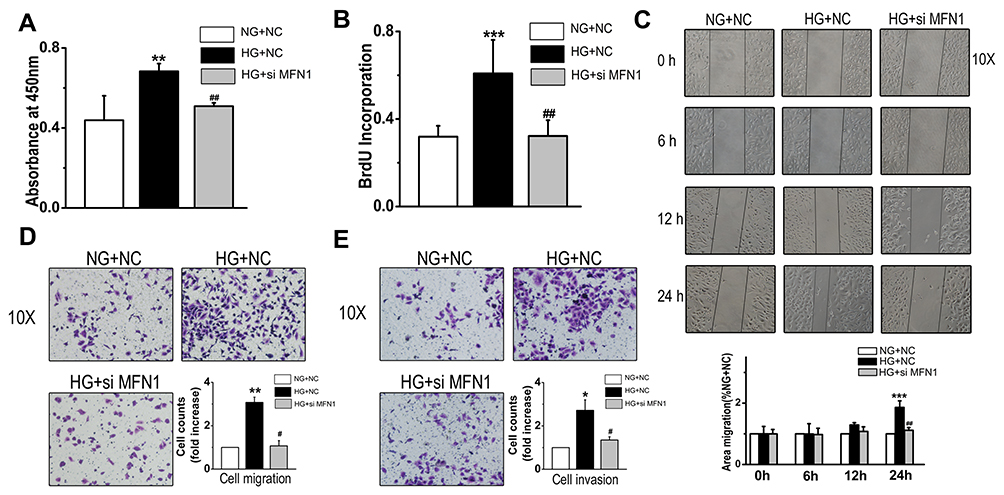 |
Figure 3 The proliferation, migration and invasion capacities of A549 cells were markedly decreased when MFN1 expression was blocked by siRNA. (A and B) High glucose increased the cell viability and BrdU incorporation of cells in a MFN1-dependent manner. (C and D) Effects of high glucose on A549 cell migration were measured by scratch wound assay (C) and Transwell assay (D). (E) SiMFN1 inhibited the high glucose-induced invasion of A549 cells. Data shown are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs NG+NC group. #P < 0.05, ##P < 0.01 vs HG+NC group (n = 6). Abbreviations: NC, non-targeted control; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; siMFN1, small interfering RNA of MFN1. |
Increased Autophagy of A549 Cells in Glucose Promotes the Progression of EMT
To clarify the role of autophagy in glucose-induced EMT, CLQ, an autophagy inhibitor by lysosomal inhibition, was administered to A549 cells in the presence of glucose. The expression of EMT markers was determined; N-cadherin and Snail were markedly increased and E-cadherin was markedly decreased in the glucose-exposed cells, and CLQ significantly attenuated the effect (Figure 4A and B). The results were confirmed by analysis of immunofluorescent images, and pretreatment with the autophagy inhibitor CLQ reversed the increase in N-cadherin and decrease in E-cadherin expression in the cytoplasm induced by high glucose (Figure 4C and D). Similar trends were found in the cell migration and invasion assays. Figure 4E and F revealed that CLQ blocked the high glucose-induced enhancement of migration and invasion in the LAD cell line A549. However, the expression of MFN1 was unaffected by CLQ, which indicates that MFN1 is the upstream mechanism of autophagy regulation (Figure 4G). These results suggest that autophagy is associated with the phenotype of EMT as well as cell migration and invasion in the LAD cell line.
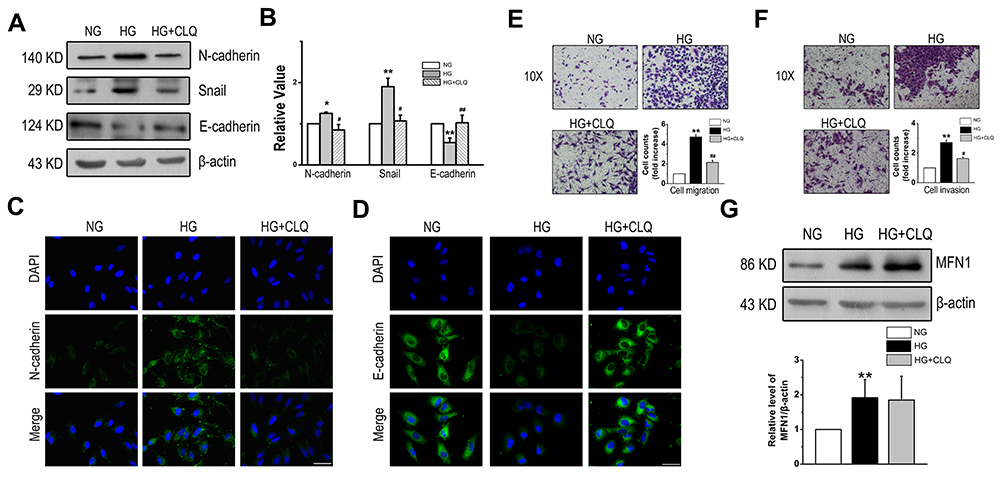 |
Figure 4 CLQ partially inhibited the progression of EMT induced by high glucose. (A and B) CLQ administration prevented high glucose-induced expression of EMT markers in A549 cells. (C and D) Immunofluorescence staining of N-cadherin and E-cadherin after CLQ treatment in high glucose conditions. (E) Boyden chamber assay results for the A549 cell line. (F) Effect of the autophagy inhibitor CLQ on A549 cell invasion. (G) The expression of MFN1 was determined after CLQ treatment. Scale bars = 100 µm. Data shown are mean ± SEM. *P < 0.05, **P < 0.01 vs NG group. #P < 0.05, ##P < 0.01 vs HG group (n = 4). Abbreviations: CLQ, chloroquine; EMT, epithelial-to-mesenchymal transition; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; DAPI, 4ʹ,6-diamidino-2-phenylindole. |
Inhibiting MFN1 Attenuates Glucose-Induced Cell Autophagy
To determine whether MFN1 promoted EMT by affecting cell autophagy, we measured the expression of LC3B, beclin (BECN)-1 and sequestosome (SQSTM) 1, which are classical autophagy-related markers. Activated expression of LC3B-II and BECN-1 and suppressed expression of SQSTM1 induced by high glucose were reversed when cells were exposed to siMFN1 (Figure 5A and B). To further detect autophagosome formation in A549 cells, we used eGFP-mRFP-LC3 fluorescence plasmid. As shown in Figure 5C, high glucose induced a massive flux of autophagy, with quenched GFP fluorescence (green) and activated autophagosomes (red) and autolysosomes (yellow) in A549 cells, and siMFN1 treatment significantly attenuated the appearance of autophagosomes.
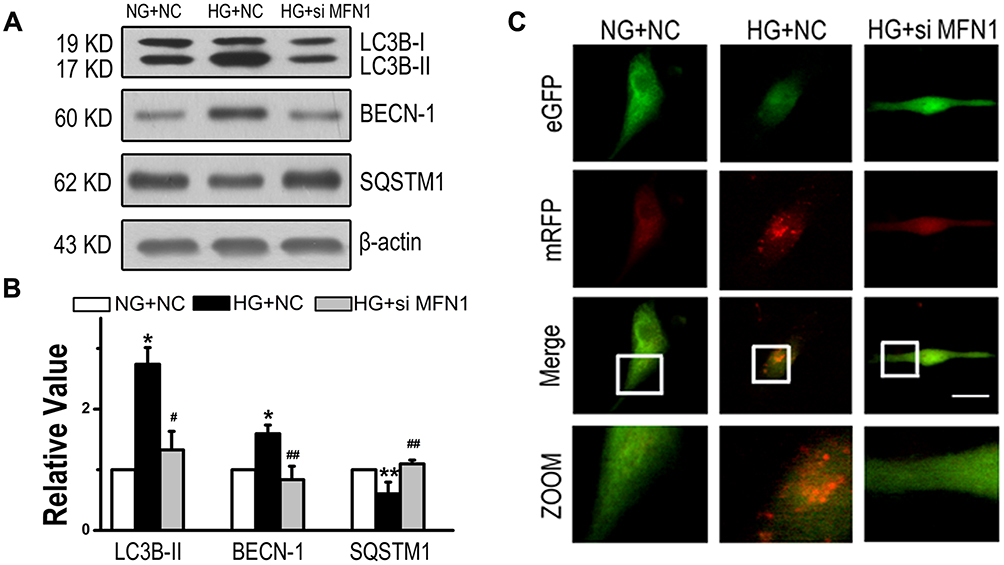 |
Figure 5 High glucose led to the induction of autophagy in the LAD cell line through MFN1. (A and B) Expression of LC3B-II, BECN-1 and SQSTM1 in A549 cells from the NG+NC, HG+NC, and HG+siMFN1 groups. (C) Cells were transfected with the eGFP-mRFP-LC3 plasmid and exposed to different concentrations of glucose for 24 h. Yellow and red dots refer to autolysosomes and autophagosomes respectively. Scale bar = 50 µm. Data shown are mean ± SEM. *P < 0.05, **P < 0.01 vs NG+NC group. #P < 0.05, ##P < 0.01 vs HG+NC group (n = 6). Abbreviations: LAD, lung adenocarcinoma; NC, non-targeted control; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; siMFN1, small interfering RNA of MFN1; LC3B, microtubule-associated proteins 1A/1B light chain 3B; BECN, beclin-1; SQSTM, sequestosome 1; eGFP, enhanced green fluorescent protein; mRFP, monomer red fluorescent protein. |
Interaction of MFN1 and PTEN Induced Kinase (Pink) Under High Glucose Conditions
Based on the observations above, it is possible that MFN1 plays an important role in glucose-induced autophagy. However, the MFN1-associated molecular chaperone in autophagy of A549 cells is still unknown. To address this issue, the STRING database was used to consolidate known and predicted protein-protein associations with MFN1 (Figure 6A). The exploration led to the identification of candidate proteins related to MFN1. The expression of Pink (PTEN induced kinase), Parkin (E3 ubiquitin ligase PARK2) and fission protein 1 (Fis 1) was determined. It appears that MFN1-associated protein expression in glucose is specific to Pink because no changes were observed in the expression of Parkin and Fis1 (Figure 6B and C). Similar results are shown in Figure 6D, in which the high glucose-enhanced expression of Pink was attenuated in the transient transfection of MFN1. Moreover, the co-immunoprecipitation assay demonstrated that there was an interaction between MFN1 and Pink (Figure 6E).
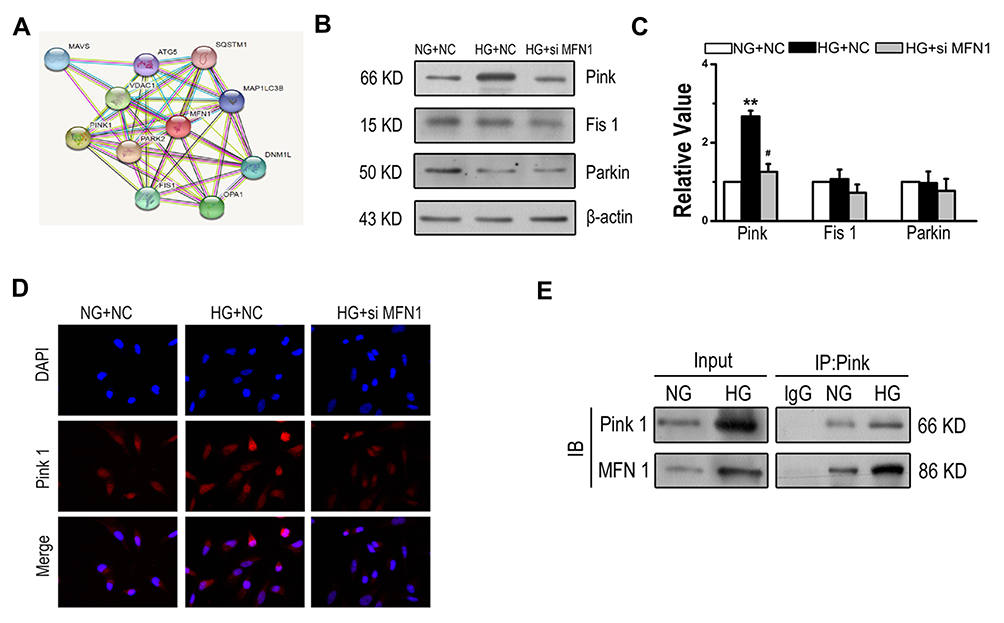 |
Figure 6 Glucose enhanced the interaction of MFN1 and Pink in A549 cells. (A) Predicted protein-protein interactions with MFN1 according to the STRING database. (B and C) The expression levels of Pink, Parkin and Fis1 were examined by Western blot in A549 cells after exposure to glucose for 24 hours. (D) Immunofluorescence of A549 cells from NG+NC, HG+NC, and HG+siMFN1. Cells were stained with DAPI (blue) and Pink (red). (E) A549 cells were exposed to 25 mM glucose, and whole cell lysates were extracted for co-immunoprecipitation with anti-Pink, and subsequent probing with anti-MFN1. Scale bar = 100 µm. Data shown are mean ± SEM. **P < 0.01 vs NG+NC group. #P < 0.05 vs HG+NC group (n = 4). Abbreviations: NC, non-targeted control; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; siMFN1, small interfering RNA of MFN1; Pink, PTEN induced kinase; DAPI, 4ʹ,6-diamidino-2-phenylindole. IB, immunoblotting; IP, immunoprecipitation; STRING, https://string-db.org/. |
MFN1 Regulates Cell Growth, Autophagy and EMT Partly Through Pink
Afterwards, experiments were carried out to determine the role of Pink in glucose-induced A549 cell dysfunction associated with MFN1. EMT markers including increased expression of N-cadherin and Snail and decreased expression of E-cadherin induced by high glucose were further exaggerated by MFN1 overexpression and suppressed by Pink knockdown (Figure 7A and B). The results from the CCK-8 assay indicated that high glucose plus MFN1 overexpression significantly promoted the growth of A549 cells compared to that with high glucose alone. Conversely, the effect was significantly reduced by co-transfection with Pink siRNA (Figure 7C). Moreover, overexpression of MFN1 caused elevation of cell migration and invasion capacities in high glucose conditions, and these effects were attenuated by siPink (Figure 7D and E). Then, A549 cells were transfected with the eGFP-mRFP-LC3 plasmid and exposed to high glucose, yellow and red dots refer to autolysosomes and autophagosomes, respectively. Enforced expression of MFN1 further promoted autophagy induced by high glucose, and this effect was blocked by siPink (Figure 7F). These data suggest that the interaction between MFN1 and Pink is important for MFN1 function and is involved in glucose-induced A549 cell dysfunction.
 |
Figure 7 MFN1 mediated the function of A549 cells mainly through Pink. (A and B) Western blot analysis was used to detect the expression of key EMT-related proteins in A549 cells after transfection with MFN1 overexpression plasmid and/or siPink. (C) The survival rate of A549 cells was studied by CCK-8 assay. (D and E) Transwell assays were used to evaluate cell infiltration capacity following overexpression of MFN1 and knockdown of Pink. (F) Representative images are presented to indicate the cellular localization patterns of the mRFP-GFP-LC3 fusion protein; scale bar = 50 μm. Data shown are mean ± SEM. **P < 0.01, ***P < 0.001 vs NG+NC group. #P < 0.05, ##P < 0.01 vs HG+NC group. $$P < 0.01, $$$P < 0.001 vs HG+MFN1 group (n = 4). Abbreviations: EMT, epithelial-to-mesenchymal transition; NC, non-targeted control; NG, normal glucose; HG, high glucose. MFN1, mitofusin1; si PINK, small interfering RNA of PTEN induced kinase; LC3B, microtubule-associated proteins 1A/1B light chain 3B; eGFP, enhanced green fluorescent protein; mRFP, monomer red fluorescent protein. |
Discussion
Tumours display reprogrammed glucose metabolism, which manifests as increased glucose uptake and glycolysis compared to that in normal tissues.23 The non-invasive assessment of glucose uptake with [18F]-fluorodeoxyglucose (FDG) positron emission tomography (PET) has been exploited as a target for anticancer diagnostics with wide clinical utility.24 Here, we demonstrate the first evidence on the uniquely specific function of MFN1 in eliciting EMT, migration and invasion in response to glucose in LAD. MFN1 interacts with Pink to induce the autophagic process, and the abnormal occurrence of autophagy finally contributes to pathological EMT in A549 cells.
The inhibition of epithelial marker (E-cadherin), increases in mesenchymal marker (N-cadherin) and transcription factor (Snail) triggers EMT, which is the propensity for metastasis and therapeutic resistance in cancer cells.25 Studies in recent years have reported that in pancreatic ductal adenocarcinoma, breast and ovarian cancers metabolic reprogramming including increased glucose uptake and lactate secretion is associated with EMT.26 In view of the important role of EMT in cancer metastasis and chemoresistance, it is important to understand the glucose metabolic regulation underlying LAD EMT. In this study, we demonstrated that the expression of MFN1 was upregulated in LAD and more seriously in LAD combined with DM patient lung tissue samples compared to normal tissues. The proliferation, invasion and migration of A549 cells were enhanced by high glucose treatment and attenuated by MFN1 siRNA. The high expression levels of N-cadherin and Snail induced by high glucose were diminished by siMFN1. These findings suggest that the effects of high glucose on A549 cells are thought to result at least in part from MFN1 in the pathogenesis of LAD, which is a novel observation worthy of further investigation.
Autophagy is a conserved biological process that maintains cellular homeostasis disruption from intracellular or environmental stresses by degrading cellular components including misfolded proteins, damaged mitochondria, and exogenous pathogens.27 Autophagy helps cancer cells overcome stressful conditions, confers resistance to cell death and provides a survival strategy for cells that are spreading outside the tumour mass by distributing substrates for metabolic balance.28,29 Accumulating evidence has demonstrated a direct connection between autophagy and EMT in solid tumours such as renal clear cell carcinoma and hepatocellular carcinoma.30 Lu et al found that the suppressed glioblastoma EMT mediated by miR-517c is associated with inhibition of autophagosomes and disrupted autophagic fux.31 Furthermore, autophagy is a strategy used by MCF7 cells to promote immune surveillance escape and acquire of the EMT phenotype in breast cancer.32 To clarify whether autophagy promoted EMT in LAD, we pretreated A549 cells with the common autophagic inhibitor CLQ and found that CLQ decreased the EMT phenotype by blocking the expression of mesenchymal markers such as N-cadherin, and Snail, which was induced by high glucose. Meanwhile, the induction of autophagy by high glucose increased cell migration and invasion in A549 cells and these effects were subsequently blocked by an inhibitor of autophagy (CLQ). Our study further indicated that inhibition of EMT by MFN1 RNA interference occurred via the inhibition of autophagy. Glucose upregulated the conversion of LC3B-II, increased the expression of BECN-1 and prevented the expression of SQSTM1 through a pathway involving MFN1. Moreover, MFN1 inhibition, can alter autophagic flux by interfering with the formation of autophagosomes demonstrating that autophagy is regulated by MFN1. This finding sheds further light on the relationship between autophagy and EMT in LAD. To the best of our knowledge, this is the first study to reveal a novel mechanism by which MFN1 in mediates the basic autophagy process that contributes to EMT in LAD at the molecular and cellular levels.
Pink protein is found in mitochondria in most cell types and spans the outer mitochondrial membrane.33 Pink is essential for maintaining mitochondrial membrane potential and facilitating mitochondrial energy metabolism.34 Pink also has tumour-promoting properties that help maintain cancer-associated phenotypes.35 More importantly, activation of Pink and the E3 ubiquitin ligase Parkin leads to recruitment of the autophagy receptors that contain the LC3-interacting region motif; these receptors bind to the LC3B-II form to initiate autophagy and mitophagy.36 Consistent with the notion, in this study, we reported that glucose stimulation promotes autophagy by increasing the crosstalk between MFN1 and Pink which are key promoters of EMT and serve as regulators in LAD. The expression of Fis1 and Parkin is unaffected by MFN1 interference. The mechanisms underlying this phenomenon need further investigation.
Conclusions
To conclude, here, we demonstrate that high glucose upregulates the expression of MFN1, which interacts with Pink and is involved in the glucose-induced dysfunction of autophagy in A549 cells. Moreover, this induction of autophagy stimulates EMT progression combined with increased migration and infiltration resulting in LAD. Future studies will also be needed to identify additional mitochondrial proteins that have potential roles in tumorigenesis and serve as molecular scaffolds in regulating LAD, especially those related to the glucose metabolism disturbance.
Abbreviations
EMT, epithelial-to-mesenchymal transition; LAD, lung adenocarcinoma; MFN1, mitofusin1; Pink, PTEN induced kinase; NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer; DM, diabetes mellitus; NC, non-targeted control siRNA; NG, normal glucose; HG, high glucose; LG, low glucose; CCK-8, Cell Counting Kit-8; BrdU, 5-bromodeoxyuridine; BECN, beclin-1; SQSTM, sequestosome 1; Parkin, E3 ubiquitin ligase PARK2; Fis 1, fission protein 1; CLQ, chloroquine.
Funding
This work was supported by the Natural Science Foundation Funding Scheme of Liaoning Province (grant number: 2019-MS-145).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Testa U, Castelli G, Pelosi E. Lung cancers: molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers (Basel). 2018;10:8. doi:10.3390/cancers10080248
2. Blandin Knight S, Crosbie PA, Balata H, Chudziak J, Hussell T, Dive C. Progress and prospects of early detection in lung cancer. Open Biol. 2017;7:9. doi:10.1098/rsob.170070
3. Larsen JE, Minna JD. Molecular biology of lung cancer: clinical implications. Clin Chest Med. 2011;32(4):703–740. doi:10.1016/j.ccm.2011.08.003
4. Ferrer I, Quintanal-Villalonga A, Molina-Pinelo S, et al. MAP17 predicts sensitivity to platinum-based therapy, EGFR inhibitors and the proteasome inhibitor bortezomib in lung adenocarcinoma. J Exp Clin Cancer Res. 2018;37(1):195. doi:10.1186/s13046-018-0871-7
5. Lou Y, Diao L, Cuentas ER, et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res. 2016;22(14):3630–3642. doi:10.1158/1078-0432.CCR-15-1434
6. Johnson JL, Pillai S, Chellappan SP. Genetic and biochemical alterations in non-small cell lung cancer. Biochem Res Int. 2012;2012:940405. doi:10.1155/2012/940405
7. Suber TL, Nikolli I, O’Brien ME, et al. FBXO17 promotes cell proliferation through activation of Akt in lung adenocarcinoma cells. Respir Res. 2018;19(1):206. doi:10.1186/s12931-018-0910-0
8. Kim NH, Cha YH, Lee J, et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat Commun. 2017;8:14374. doi:10.1038/ncomms14374
9. Koppula P, Zhang Y, Shi J, Li W, Gan B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J Biol Chem. 2017;292(34):14240–14249. doi:10.1074/jbc.M117.798405
10. Giovannucci E, Harlan DM, Archer MC, et al. Diabetes and cancer: a consensus report. CA Cancer J Clin. 2010;60(4):207–221. doi:10.3322/caac.20078
11. Han J, Zhang L, Guo H, et al. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. 2015;138(3):668–675. doi:10.1016/j.ygyno.2015.06.036
12. Zielinska HA, Holly JMP, Bahl A, Perks CM. Inhibition of FASN and ERalpha signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett. 2018;419:187–202. doi:10.1016/j.canlet.2018.01.028
13. Liu F, Song S, Yi Z, et al. HGF induces EMT in non-small-cell lung cancer through the hBVR pathway. Eur J Pharmacol. 2017;811:180–190. doi:10.1016/j.ejphar.2017.05.040
14. Zhang M, Xin W, Yi Z, et al. Human biliverdin reductase regulates the molecular mechanism underlying cancer development. J Cell Biochem. 2018;119(2):1337–1345. doi:10.1002/jcb.26285
15. van Gisbergen MW, Voets AM, Starmans MH, et al. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat Res Rev Mutat Res. 2015;764:16–30. doi:10.1016/j.mrrev.2015.01.001
16. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi:10.1016/j.ccr.2006.04.023
17. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi:10.1126/science.123.3191.309
18. Zhang C, Liu J, Liang Y, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi:10.1038/ncomms3935
19. Park YY, Nguyen OT, Kang H, Cho H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014;5:e1172. doi:10.1038/cddis.2014.142
20. Ma C, Zhang C, Ma M, et al. MiR-125a regulates mitochondrial homeostasis through targeting mitofusin 1 to control hypoxic pulmonary vascular remodeling. J Mol Med (Berl). 2017;95(9):977–993. doi:10.1007/s00109-017-1541-5
21. Li J, Li Y, Jiao J, et al. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol. 2014;34(10):1788–1799. doi:10.1128/MCB.00774-13
22. Ding CZ, Guo XF, Wang GL, et al. High glucose contributes to the proliferation and migration of non-small cell lung cancer cells via GAS5-TRIB3 axis. Biosci Rep. 2018;38. doi:10.1042/BSR20171014
23. Fong MY, Zhou W, Liu L, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17(2):183–194. doi:10.1038/ncb3094
24. Walker-Samuel S, Ramasawmy R, Torrealdea F, et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat Med. 2013;19(8):1067–1072. doi:10.1038/nm.3252
25. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16(6):488–494. doi:10.1038/ncb2976
26. Liu M, Quek LE, Sultani G, Turner N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016;4:19. doi:10.1186/s40170-016-0160-x
27. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi:10.1038/nrm1666
28. Sandilands E, Serrels B, McEwan DG, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011;14:51–60. doi:10.1038/ncb2386
29. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi:10.1038/nrc3262
30. Wang X, Gao Y, Tian N, et al. Astragaloside IV inhibits glucose-induced epithelial-mesenchymal transition of podocytes through autophagy enhancement via the SIRT-NF-kappaB p65 axis. Sci Rep. 2019;9(1):323. doi:10.1038/s41598-018-36911-1
31. Lu Y, Xiao L, Liu Y, et al. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy. 2015;11(12):2213–2232. doi:10.1080/15548627.2015.1108507
32. Gugnoni M, Sancisi V, Manzotti G, Gandolfi G and Ciarrocchi A. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016;7:e2520. doi:10.1038/cddis.2016.415
33. Salazar C, Ruiz-Hincapie P, Ruiz LM. The Interplay among PINK1/PARKIN/Dj-1 network during mitochondrial quality control in cancer biology: protein interaction analysis. Cells. 2018;7:10. doi:10.3390/cells7100154
34. Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–1166. doi:10.1038/nature04779
35. O’Flanagan CH, Morais VA, Wurst W, De Strooper B, O’Neill C. The Parkinson’s gene PINK1 regulates cell cycle progression and promotes cancer-associated phenotypes. Oncogene. 2015;34(11):1363–1374. doi:10.1038/onc.2014.81
36. Weil R, Laplantine E, Curic S, Genin P. Role of optineurin in the mitochondrial dysfunction: potential implications in neurodegenerative diseases and cancer. Front Immunol. 2018;9:1243. doi:10.3389/fimmu.2018.01243
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.