Back to Journals » International Journal of Nanomedicine » Volume 20
Mitochondrial Transplantation: A Paradigm Shift in Osteoporosis Therapy
Authors Shen J, Lai Y, Peng Q, Lin X, Chen S, Guo L, Xu M, Lu Y, Zhu J, Lin X, Zhang C ![]() , Liu H
, Liu H ![]()
Received 27 April 2025
Accepted for publication 22 August 2025
Published 12 September 2025 Volume 2025:20 Pages 11169—11196
DOI https://doi.org/10.2147/IJN.S537166
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Dong Wang
Jianlin Shen,1,2,* Yue Lai,3,* Qingping Peng,4,* Xuan Lin,5 Shuxuan Chen,6 Liuqian Guo,6 Miao Xu,7 Yanjin Lu,8 Jiangqi Zhu,9 Xiaoning Lin,1 Cheng Zhang,10 Huan Liu11
1Department of Orthopedics, Affiliated Hospital of Putian University, Putian, Fujian Province, 351100, People’s Republic of China; 2Central Laboratory, Affiliated Hospital of Putian University, Putian, Fujian Province, 351100, People’s Republic of China; 3The First Clinical Medical School, Guangdong Medical University, Zhanjiang, Guangdong Province, 524002, People’s Republic of China; 4College of Integrated Traditional Chinese and Western Medicine, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 5Department of Environmental and Biological Engineering, Putian University, Putian, Fujian Province, 351100, People’s Republic of China; 6Department of Clinical Medicine, Putian University, Putian, Fujian Province, People’s Republic of China; 7School of Basic Medical Sciences,Fujian Medical University, Fuzhou, Fujian Province, 350122, People’s Republic of China; 8College of Optoelectronics and Engineering, Fujian Normal University, Fuzhou, 350117, People’s Republic of China; 9Department of Laser Manufacturing, Institute of New Materials, Guangdong Academy of Sciences, Guangzhou, Guangdong, 510651, People’s Republic of China; 10Department of Trauma Center, Zhongda Hospital, Southeast University, Nanjing, 210000, People’s Republic of China; 11Department of Orthopaedics, The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huan Liu; Cheng Zhang, Email [email protected]; [email protected]
Abstract: Osteoporosis, a global health crisis marked by compromised bone mineral density and heightened fracture susceptibility, demands innovative therapeutic strategies beyond conventional anti-resorptive approaches. Mitochondrial dysfunction, characterized by impaired bioenergetics, oxidative stress overload, and calcium dysregulation, has emerged as a central driver of osteoblast-osteoclast imbalance. Recent breakthroughs in mitochondrial transplantation (MT)—a revolutionary modality involving the transfer of functional mitochondria to metabolically compromised cells—have demonstrated unprecedented efficacy in preclinical osteoporosis models, restoring bone mass, microarchitecture, and mechanical strength. This review synthesizes cutting-edge insights into mitochondrial dynamics in bone homeostasis, dissects the molecular cascades linking mitochondrial failure to osteoporotic pathogenesis, and critically evaluates MT’s potential to redefine osteoporosis management. We also discuss novel mechanisms of intercellular mitochondrial trafficking within the osteocyte dendritic network, explore bioengineered delivery platforms (eg, immunomodulatory hydrogels, nanoparticle-encapsulated mitochondria), and address emerging challenges in clinical translation, including donor source optimization, immune compatibility, and CRISPR-engineered mitochondrial genomes. By integrating single-cell omics data and AI-driven mitochondrial viability predictors, this work charts a roadmap for personalized mitochondrial medicine, positioning MT as a cornerstone of next-generation osteoporosis therapeutics.
Keywords: osteoporosis, mitochondrial transplantation, mitochondrial disease, mitochondrial transfer
Introduction
Osteoporosis, a systemic skeletal disorder characterized by diminished bone mineral density (BMD) and microarchitectural deterioration, represents a significant global health burden. This condition arises from an imbalance between bone resorption by osteoclasts and bone formation by osteoblasts, leading to increased fracture risk and associated morbidity, particularly in postmenopausal women and the elderly.1 Despite advancements in therapeutic interventions, including antiresorptive agents (eg, bisphosphonates) and anabolic drugs (eg, teriparatide), current treatments often fail to fully restore bone homeostasis and are associated with side effects, high costs, and limited long-term efficacy.2,3 Consequently, there is an urgent need for innovative therapeutic strategies that address the underlying pathophysiology of osteoporosis.
Mounting evidence establishes mitochondrial dysfunction as central to osteoporosis pathogenesis. As essential organelles, mitochondria critically regulate osteoblast and osteoclast activity by governing bone remodeling through energy production, redox homeostasis, and calcium signaling.4 Consequently, deciphering mitochondrial involvement in these processes may enable therapeutic strategies to modulate mitochondrial activity—promoting osteoblast differentiation while suppressing osteoclastogenesis to preserve bone health.Mitochondrial transfer, involving delivery of donor mitochondria to recipient cells, alters bioenergetic profiles and functional states in recipient populations.5 This capability suggests therapeutic promise for restoring homeostasis in compromised bone cells. Osteocytic dendritic networks facilitate mitochondrial transfer, supporting metabolic recovery.6 Recent work identifies mitochondrial transplantation—direct transfer of functional mitochondria to impaired cells—as a strategic approach to boost osteoblastic ATP generation, enhance respiratory capacity, and dampen osteoclastogenic cytokine expression, thereby reducing bone loss in preclinical osteoporosis models.7
This review delineates the multifaceted contributions of mitochondria to bone homeostasis and evaluates their therapeutic targeting potential via specific mitochondrial pathways to modulate osteoblast/osteoclast dynamics in osteoporosis. It synthesizes current mechanistic insights into mitochondrial dysfunction pathogenesis, advances in mitochondrial transfer techniques, and preclinical/clinical evidence for mitochondrial transplantation as a therapeutic intervention. Furthermore, we address the challenges and future directions for translating this approach into clinical practice, highlighting requirements to optimize delivery systems, ensure long-term safety, and mitigate immune responses.
Osteoporosis: A Growing Global Health Burden
Epidemiology and Current Therapies
Osteoporosis, a systemic bone disease characterized by reduced bone mineral density (BMD) and microstructural deterioration, significantly increases the risk of fractures. This condition disproportionately affects women and older adults, where osteoporotic fractures contribute substantially to disability and mortality. Osteoporosis affects over 200 million people worldwide, with osteoporotic fractures contributing significantly to disability, mortality, and healthcare costs.8–10 The growing global elderly population exacerbates the high prevalence of osteoporosis and its associated healthcare burden. Despite this pervasive impact and ongoing research into promising new areas like the gut-bone axis, osteoporosis remains an incurable condition with significant unmet clinical needs.11,12 This reality underscores the urgent need for novel therapeutic strategies, especially considering the substantial limitations of existing treatments.
Current pharmacotherapy for primary osteoporosis encompasses bone anabolic agents, anti-resorptives, dual-action compounds, and adjunctive metabolic modulators.13–15 However, these agents exhibit significant clinical limitations that constrain therapeutic efficacy and patient adherence. Regarding efficacy, many demonstrate limited capacity to increase BMD, with monotherapy typically yielding only modest gains.16,17 Furthermore, their therapeutic effects are site-specific and provide incomplete protection against certain fracture types. For example, alendronate demonstrates no significant reduction in non-vertebral fracture risk among women with osteopenia without prevalent vertebral fractures, nor significant fracture risk reduction in individuals without fracture history or for hip fractures specifically.16–19 Furthermore, long-term benefits are constrained: skeletal protection wanes with extended use, and treatment cessation may accelerate bone loss.16,20,21 Safety concerns include notable adverse events. Bisphosphonate therapy associates with gastrointestinal disturbances and 63 reported cases of osteonecrosis of the jaw (ONJ), indicating potential risk elevation.18,22,23 Prolonged bisphosphonate use increases atypical femoral fracture (AFF) risk, while agents like raloxifene elevate thrombosis risk.16,20,24 Additional constraints encompass cost and narrow therapeutic indications. These agents demonstrate suboptimal cost-effectiveness and target limited patient subsets. Critically, osteoporosis remains incurable.16,18,19,25–27 Current therapies—including calcium/vitamin D supplementation, estrogen replacement, and anti-osteoporotic agents—may attenuate disease progression but fail to restore bone remodeling homeostasis; consequently, lifelong management is requisite.28
Mitochondrial Dysfunction in Osteoporosis
Osteoporosis is characterized by an imbalance in osteoblast and osteoclast activity. Under normal physiological conditions, mitochondria play a crucial role in regulating bone metabolism and maintaining bone homeostasis. However, mitochondrial dysfunction disrupts this delicate balance, leading to intracellular abnormalities and cellular dysfunction. This disruption, in turn, affects the balance between osteoblast and osteoclast activity, contributing to the development of osteoporosis. Key features of mitochondrial dysfunction in osteoporosis include: 1. Impaired oxidative phosphorylation: Reduced ATP production compromises osteoblast differentiation and bone formation. 2. Elevated ROS production: Excessive ROS disrupts cellular signaling pathways, leading to osteocyte apoptosis and increased osteoclast activity. 3. Defective mitophagy: Impaired clearance of damaged mitochondria exacerbates cellular dysfunction and bone loss.29 Recent studies suggest that mitochondrial dysfunction specifically in osteocytes may impair their normal function and compromise their ability to maintain bone homeostasis, potentially leading to osteoporosis.30 Given the pervasive role of mitochondrial dysfunction and its associated ROS production in bone cells, it is crucial to examine how these factors specifically influence the distinct functions of osteoclasts and osteoblasts.
Impact of Mitochondrial Dysfunction on Osteoclasts: The ROS-NF-κB Axis
Mitochondrial dysfunction in osteoporosis elevates reactive oxygen species (ROS)—a pathological hallmark—driving osteoclast-mediated NF-κB activation that dysregulates bone remodeling. This mechanism integrates oxidative stress with inflammatory signaling. NF-κB transcription factors coordinate osteoclast differentiation, survival, and osteoclastogenic cytokine expression (eg, TNF-α, IL-1β).31–34 In quiescent cells, cytoplasmic NF-κB complexes with inhibitory IκB proteins. Following stimulation, IκB undergoes IKK-induced phosphorylation and ubiquitin-mediated degradation, permitting NF-κB nuclear translocation and target gene transactivation. Osteoclast ROS primarily derive from NADPH oxidases (NOX) and the mitochondrial electron transport chain. Elevated ROS activate NF-κB through covalent IKK complex modification. This multi-subunit complex comprises IKKα, IKKβ, and NEMO, undergoing direct oxidative alterations that potentiate its activity.35–37 ROS including H2O2 oxidize specific IKKβ cysteine residues (Cys179), triggering conformational activation that enables IκBα phosphorylation at Ser32/36 degradation motifs.38–41 Phosphorylated IκBα is then ubiquitinated and degraded, releasing NF-κB (eg, p65/p50 heterodimers) into the nucleus.42,43 ROS also inhibit IκB phosphatases (eg, PP2A) through oxidative modification of catalytic metal ions or active-site cysteine residues, compromising phosphatase activity and consequently potentiating IκB phosphorylation, degradation, and NF-κB activation.44–51 ROS directly activate osteoclast surface receptors (TNFR, IL-1R) and integrin signaling, initiating TAK1-dependent IKK activation. This occurs through enhanced TAK1-NEMO scaffolding within the IKK signalosome, potentiating kinase activity.Mitochondrially generated ROS modulate NF-κB signaling through dual mechanisms: impairing core mitochondrial functions (ATP synthesis, calcium handling) and inhibiting intrinsic antioxidant defenses like manganese superoxide dismutase (MnSOD). This establishes a self-amplifying oxidative cycle that sustains NF-κB activation.52–56 Physiologically, ROS function as second messengers that potentiate RANKL-induced osteoclastogenesis via NF-κB pathway activation, thereby supporting bone remodeling homeostasis.57–59 Pathological ROS elevation (eg, during inflammation or redox imbalance) induces constitutive NF-κB activation, driving osteoclast hyperactivation and prolonged lifespan that underlie osteoporosis and rheumatoid arthritis pathogenesis.60–62 In postmenopausal osteoporosis, estrogen deficiency promotes pathological ROS accumulation that potentiates osteoclastogenesis via NF-κB signaling, driving accelerated bone loss.63–66 Elevated osteoclastic ROS drive NF-κB pathway activation through direct IKK oxidation, IκB phosphatase suppression, and upstream signaling modulation. This mechanism maintains physiological bone remodeling while underpinning pathological resorption. Therapeutic targeting of the ROS-NF-κB axis through antioxidant agents or IKK inhibitors constitutes a rational approach for managing osteolytic disorders. While osteoclast activity mediates bone resorption, osteoblast function—specifically osteogenic differentiation and matrix deposition—is equally essential for osteogenesis and critically dependent on mitochondrial integrity and redox homeostasis.
Impact of Mitochondrial Dysfunction on Osteoblasts: The OXPHOS-Runx2 Axis
Mitochondrial integrity and redox homeostasis critically govern osteoblastic differentiation and bone formation—distinct from osteoclast-activating roles—primarily through master transcription factor modulation (eg, Runx2). Runx2 maintains high expression during osteoblast differentiation and is indispensable for this process.67 It orchestrates bone matrix protein synthesis at differentiation onset while sustaining preosteoblast populations, thereby inhibiting terminal maturation.68 Bioenergetic regulation involves oxidative phosphorylation (OXPHOS)—coupling electron transport chain (ETC) flux to ATP synthase activity—which powers cellular functions.69 Glutathione (GSH) scavenges reactive oxygen species (ROS) and maintains osteoblast viability.70,71 During BMSC osteogenic differentiation, GSH levels elevate preferentially in osteoblast versus adipogenic progenitors.72,73 Metabolic coupling links enhanced OXPHOS to intracellular GSH elevation, enabling mitochondrial ROS clearance and redox homeostasis preservation.74 OXPHOS dysfunction (eg, mitochondrial impairment) disrupts ROS-scavenging equilibrium, causing pathological ROS accumulation.75 Pathological consequences: ROS covalently modify Runx2 cysteines, inducing conformational disruption and 20S proteasomal degradation that compromise Runx2 stability/activity.76 OXPHOS failure reduces ATP output, elevating AMP/ATP ratios and activating AMPK.77 AMPK phosphorylates RUNX2-Ser118.78 Diabetic context: AMPK/PGC-1α downregulation correlates with diminished Runx2 expression,79 suggesting accelerated RUNX2 degradation may constitute a mitochondrial safeguard by limiting glucose utilization, suppressing OXPHOS, and attenuating ROS generation.74
Regarding Stem Cell Aging and Differentiation
COL6A3 overexpression enhances bone marrow mesenchymal stem cell (BMSC) differentiation and survival in inflammatory milieus by promoting mitochondrial autophagy (mitophagy) and preserving cristae architecture, indicating its therapeutic potential for stem cell-based osteoporosis interventions.80 Conversely, Lrrc17 knockdown activates mTOR/PI3K-mediated mitophagy, reprogramming BMSC differentiation from adipogenic to osteogenic lineages and implicating its role in aging-related differentiation shifts. In aged murine models, BMSC-specific Lrrc17 knockdown attenuates ovariectomy-induced bone loss.81 Hematopoietic stem cell transplantation (HSCT) preconditioning regimens induce MSC mitochondrial dysfunction and mitophagy impairment, consequently diminishing MSC functionality and predisposing to osteopenia/osteoporosis.82
Addressing Metabolic Regulation and Antioxidant Capacity
NAB therapy activates Nrf2 to augment mitochondrial antioxidant defenses, improve energy metabolism, and preserve trabecular architecture with calcium homeostasis. It concurrently stimulates osteogenesis via the Nrf2-GSK-3β axis, upregulating PGC-1α and TFAM. At physiological concentrations, NAB protects MC3T3-E1 osteoblasts against H2O2-induced oxidative stress, promoting differentiation and maintaining bone metabolic homeostasis.83
Focusing on Mitochondrial Quality Control and Autophagy
PINK1—a mitochondrial serine/threonine kinase regulating mitophagy—is essential for osteoblast differentiation, as evidenced by impaired differentiation in PINK1 knockout mice. This function likely involves mitochondrial homeostasis maintenance and ROS suppression.84
Collectively, these findings suggest that targeting mitochondrial dysfunction and promoting mitochondrial health may be promising therapeutic approaches for osteoporosis.
Mitochondrial Donor Sources and Compatibility Considerations
Diverse Mitochondrial Sources and Their Therapeutic Implications in Osteoporosis
Osteoporosis fundamentally stems from an imbalance between bone formation and resorption. Mitochondria, as central regulators of energy metabolism and signaling pathways, directly govern osteoblast and osteoclast activity. Mitochondria derived from distinct sources exhibit unique therapeutic profiles for osteoporosis—encompassing mechanisms, indications, advantages, and limitations—all rooted in their core metabolic functions. Skeletal muscle mitochondria function as the primary ATP source for contraction through oxidative phosphorylation (OXPHOS). Exercise induces myokine secretion (eg, irisin) from skeletal muscle, which enhances osteoblast function and bone formation via endocrine pathways.85 Their calcium-buffering capacity contributes to systemic calcium homeostasis, thereby regulating bone mineralization. Clinically, functional enhancement via exercise or pharmacotherapy promotes myokine secretion, augments osteoblast proliferation and matrix deposition, and increases mechanical loading on bone.86–91 These properties make them optimally indicated for disuse osteoporosis (prolonged immobilization or microgravity exposure) and age-related primary osteoporosis.92–94 Key advantages include non-pharmacological intervention and musculoskeletal co-benefits. Limitations involve suboptimal efficacy in advanced disease and dependence on patient-specific factors such as adherence and functional capacity.86 Impaired skeletal muscle mitochondrial function reduces mechanical loading and exacerbates bone loss, whereas enhanced activity improves bone health through myokine signaling.95–97 Hepatic mitochondria regulate systemic energy metabolism—including gluconeogenesis and fatty acid β-oxidation—and catalyze vitamin D activation, thereby establishing calcium-phosphate homeostasis to modulate bone health.98 Hepatocytes synthesize bone metabolism-related proteins to sustain nutrient supply and maintain bone tissue metabolic balance.99 Clinically, enhancing hepatic mitochondrial function improves vitamin D activation and calcium-phosphate homeostasis, indirectly promoting mineralization while mitigating lipid-induced osteotoxicity via lipid metabolism regulation.100 These agents are particularly suitable for secondary osteoporosis (eg, hepatic vitamin D deficiency or obesity-associated pathologies), as they concurrently address systemic metabolic derangements and benefit metabolic syndrome. Limitations comprise indirect mechanisms, requirement for sustained intervention, and limited direct microstructural restoration. Hepatic dysfunction impairs vitamin D activation, diminishes calcium absorption, and precipitates secondary osteoporosis; conversely, hepatic metabolites modulate bone formation by regulating osteoblastic energy metabolism.98,101–104 Mesenchymal stem cell (MSC) mitochondria drive osteogenic differentiation and critically determine pre-osteoblast fate through mitochondrial biogenesis (eg, PGC-1α pathway) and ROS signaling (low-level ROS promotes osteogenesis). MSCs additionally serve as mitochondrial donors, restoring dysfunctional bone cell mitochondria via exosomal transfer or direct transplantation, thus improving the local osteogenic microenvironment. Clinically, mitochondrial augmentation—through transplantation or pharmacological activation—enhances osteoblast differentiation and repairs compromised bone cells via exosome-mediated mitochondrial delivery.105–108 These therapies are particularly indicated for postmenopausal and glucocorticoid-induced osteoporosis, providing direct correction of impaired bone formation, efficient anabolic effects, and site-specific intervention.102,106,107 Limitations include requirements for sophisticated in vitro culture systems and transplantation protocols, immune rejection risks, and substantial costs.107,109 Crucially, MSC mitochondrial function critically regulates osteogenic differentiation: dysfunction impairs osteoblast generation and reduces bone mass, whereas augmentation substantially enhances bone-forming capacity.105
Genomic and Physiological Factors Influencing Mitochondrial Function and Compatibility
Mitochondrial DNA (mtDNA) is a covalently closed circular molecule devoid of protein-coding capacity, with a compact organization spanning 16,569 base pairs in humans. This double-stranded genome encodes 37 tightly packed genes: 22 transfer RNAs (tRNAs), two ribosomal RNA subunits (12S and 16S rRNA), and 13 polypeptides essential for the assembly and function of oxidative phosphorylation complexes.110,111 An mtDNA haplogroup denotes a phylogenetically defined cluster of mtDNA polymorphisms accumulated through maternal inheritance over evolutionary time.112 These variations modulate mitochondrial oxidative phosphorylation (OXPHOS) functional capacity, metabolic efficiency, and oxidative stress signaling.113 Mounting evidence linking mtDNA haplogroups to disease susceptibility highlights the significance of mitochondrial genetics in human health. These associations involve complex interactions among bioenergetics, redox homeostasis, immune modulation, and nuclear-mitochondrial crosstalk. As mechanistic insights advance, mtDNA haplogroups emerge as valuable precision medicine biomarkers for predicting disease risk, therapeutic responsiveness, and longevity. Realizing this potential necessitates addressing current technical and ethical constraints while adopting integrative analytical frameworks. Genomically, mitochondrial function is regulated via estrogen receptor α/β (ERα/ERβ) activation by 17β-estradiol (E2), which stimulates nuclear respiratory factor 1 (NRF1) transcription.114,115 Estrogen (E2) upregulates transcription of nuclear-encoded electron transport chain proteins via NRF1 activation, which subsequently induces Mitochondrial Transcription Factor A (TFAM) expression, thereby activating transcription of mtDNA-encoded genes.116,117 NRF-1 dimerically binds DNA response elements, recruiting coactivators PGC-1α, PGC-1β, and PRC to modulate target gene transcription in a cell-type-specific manner.118 Mitochondrially localized ERα/ERβ directly regulate mtDNA gene transcription and mitochondrial function, reflecting their tissue-specific distribution patterns.119
Aging constitutes a multifactorial process characterized by progressive functional decline, with mitochondria serving as central mediators of this deterioration.120–124 Mitochondria constitute the primary bioenergetic source essential for maintaining tissue homeostasis and supporting repair mechanisms, while their functional capacity undergoes progressive age-associated impairment.125 Aging is associated with reduced expression of mitochondrial OXPHOS genes.126,127 Building upon mitochondria’s central role in redox regulation, the Mitochondrial Free Radical Theory of Aging postulates that reactive oxygen species (ROS)-mediated oxidative damage to mtDNA initiates mitochondrial dysfunction, establishing a self-propagating cycle of escalating ROS production that ultimately disrupts cellular homeostasis.128 Among the 13 mtDNA-encoded polypeptides examined by Short et al in physiologically aging humans, nine exhibited significant reductions in elderly subjects. Critically, diminished expression of cytochrome c oxidase subunits 3 and 4 (COX3/COX4) quantitatively correlated with attenuated ATP synthesis capacity.129 ATP synthesis was assessed using Complex I-specific (glutamate/malate) and Complex II-specific (succinate + rotenone) substrates, revealing an approximately 8% decline per decade per gram tissue, or a 5% decrease normalized to mitochondrial protein content. Concomitantly, mitochondrial DNA and RNA abundance declined proportionally with ATP production in physiologically aging humans. Complementary quantitative mass spectrometry by Stauch et al of brain mitochondria from 5- (young), 12- (middle-aged), and 24-month-old (aged) mice demonstrated altered expression of catabolic pathway proteins and metabolite levels, indicating age-dependent metabolic demands influence proteomic profiles.130 Aging drives accumulation of somatic mutations and large-scale deletions in mitochondrial DNA.131,132 Concomitantly, mtDNA defects induce compensatory increases in mitochondrial DNA copy number (mtDNAcn) to mitigate functional loss from impaired mitochondria.133,134
Donor sex modulates immune cell subtype distribution, thus impacting immunological compatibility. Sex-specific disparities in immune cell proportions are evident: males demonstrate lower CD4⁺ naive T cell frequencies but elevated natural killer (NK) cell and monocyte counts compared to females, whereas females exhibit higher platelet counts.135 These disparities contribute to alterations in global immune competence, consequently affecting immunological compatibility. Immunosenescence involves age-dependent remodeling of immune cell composition and functionality, substantially impacting immunological compatibility. Notably, CD8⁺CD45RA+ naive T cell abundance declines in aged individuals, whereas CD4⁺ effector memory and CD8⁺ central memory T cell frequencies rise with aging.136–138 These alterations may affect the immune system’s capacity to recognize and respond to allogeneic tissues, thereby influencing immunological compatibility. Concomitantly, age-associated mitochondrial dysfunction may also play a role in modulating immunological compatibility. With advancing age, diminished mitochondrial function may compromise the energy supply and metabolic pathways in immune cells, consequently impairing their functional capacity and altering immunological compatibility. Importantly, certain tissues demonstrate age- and sex-specific patterns of responsiveness. Sex serves as a critical biological variable that influences multiple aspects of physiology and pathophysiology.139 Sex hormones modulate mitochondrial function.119,140,141 Studies employing young or ovariectomized animal models demonstrate that gonadal steroids regulate mitochondrial energy production via transcriptional and post-transcriptional mechanisms.140 Ovariectomy abolished the mitochondrial metabolic advantage in young female mice, whereas orchiectomy exerted no significant effect on mitochondrial function in males. These findings indicate that ovarian steroids constitute the critical factor sustaining the mitochondrial advantage observed in females.140 Sex-specific differences in mitochondrial performance are primarily mediated by endogenous estrogens, particularly estradiol (E2), acting through their cognate receptors (ERα, ERβ, GPER1) via tissue-specific and cell-type-dependent mechanisms. As transcription factors, ERα and ERβ bind estrogen response elements (EREs) in nuclear DNA, activating transcription of nuclear respiratory factor 1 (NRF-1). Evidence indicates that mammalian mtDNA inheritance is predominantly maternal, facilitated by the active degradation of paternal mitochondria post-fertilization. For instance, sperm mitochondria undergo ubiquitination and subsequent elimination. This mechanism ensures mtDNA genetic stability and minimizes pathological dysfunction arising from genetic conflicts.142 Recent murine studies have elucidated sexually dimorphic patterns in age-related mitochondrial dysfunction. In male mice, aging was associated with augmented mitochondrial activity in metabolically active tissues including cardiac muscle, skeletal myocytes, and testicular parenchyma. Conversely, age-dependent reductions in mitochondrial function were observed in female mice within thermogenic brown adipose tissue and neural components of the cerebral cortex. These findings suggest that sex-specific regulation of mitochondrial bioenergetics and reactive oxygen species homeostasis may differentially modulate immune system homeostasis through tissue-specific metabolic reprogramming.125 The observed divergence in mitochondrial adaptation to aging across sexes implies potential mechanisms underlying gender-based disparities in age-related immune dysfunction and oxidative stress susceptibility.
Challenges and Ethical Considerations in Non-Maternal Mitochondrial Transplantation
Non-maternal mitochondrial transplantation confronts multiple and complex challenges, with core conflicts centered on immunogenicity, epigenetic interference, and ethical controversies. At the immunological level, exogenous mtDNA activates the mitochondrial-specific Toll-like receptor 9 (mtDNA-TLR9), triggering an innate immune response characterized by type I interferon release and inflammasome assembly. Clinical data indicate that approximately 23% of transplant recipients develop high-titer anti-mtDNA IgG antibodies within 7 days post-transplantation, which exhibit significant positive correlation with graft dysfunction (r=0.82, p=0.003).143,144 Crucially, heterologous mitochondrial membrane proteins TOM40 and TIM23 may function as neoantigens, inducing T cell-mediated immune rejection.143 At the epigenetic level, the introduction of non-maternal mtDNA disrupts the homeostatic equilibrium established through nuclear-mitochondrial coevolution. Specifically, fluctuations in the mitochondrial metabolite α-ketoglutarate concentration inhibit TET methylcytosine dioxygenase activity, leading to abnormal hypermethylation of the IGF2-H19 imprinting control region,145,146 Concurrently, retrograde signaling dysregulation significantly reduces SIRT1 deacetylase activity, silencing the expression of nucleus-encoded mitochondrial genes such as TFAM and PGC-1α.146 The focus of ethical controversy lies in the risk that CRISPR-edited “universal donor” mitochondria pose for germline chimerism, potentially resulting in permanent alterations to the human genetic lineage.147 Furthermore, the cross-generational transmission of non-maternal mtDNA contravenes the principle of natural maternal inheritance, and modeling studies predict a potential reduction in mitochondrial haplotype diversity within the population.144,148 Addressing technical bottlenecks necessitates the development of haplotype matching systems integrating deep sequencing (coverage ≥1000×) with artificial intelligence prediction (eg, the AlphaFold-MITO model) to circumvent efficiency losses in ATP synthesis caused by defects in OXPHOS complex assembly.106
Mitochondrial Transplantation: Mechanisms and Techniques
Mitochondrial transfer refers to the endogenous intercellular movement of mitochondria through physiological conduits (eg, tunneling nanotubes [TNTs], extracellular vesicles [EVs], or cell fusion), primarily induced by cellular stress or metabolic imbalance to restore cellular function. In contrast, mitochondrial transplantation specifically denotes therapeutic procedures involving the isolation of functional mitochondria from donor cells and their exogenous administration into recipient cells. These two processes diverge substantively in methodology, biocompatibility, ethical implications, and clinical translatability. Herein, mitochondrial transfer is rigorously defined as the endogenous intercellular exchange of mitochondria mediated by TNTs, EVs, or osteocyte dendritic networks. Conversely, mitochondrial transplantation denotes the exogenous delivery of mitochondria utilizing techniques such as microinjection, nanocarrier encapsulation, or hydrogel-based loading. This conceptual distinction is mechanistically and clinically substantiated by studies contrasting natural mitochondrial trafficking6,149–152 with engineered transplantation methodologies.153–157 Having established this conceptual framework, we now examine the pivotal role of mitochondrial function in maintaining bone homeostasis and the contribution of its dysregulation to skeletal disorders.
The Critical Role of Mitochondrial Function in Bone Health
In eukaryotic cells, mitochondria orchestrate essential cellular processes, including oxidative phosphorylation, calcium homeostasis, and lipid and amino acid metabolism. They further regulate apoptotic and necrotic pathways.158 This pivotal role in cellular metabolism establishes mitochondrial dysfunction as a determinant of impaired cellular homeostasis and disease pathogenesis.159 Although mitochondrial dysfunction drives multisystem disorders, its cell-specific manifestations—such as in osteocytes and osteoblasts—increasingly contribute to distinct pathology.160
Historically, mitochondrial disease research has predominantly focused on severe multisystem disorders such as MELAS syndrome and Leigh syndrome,161,162 characterized by complex multisystem manifestations. However, subtle or localized mitochondrial dysfunction can critically impair organ-specific and cell-type-specific physiological functions. Within the bone microenvironment, mitochondrial impairment in osteoblasts, osteoclasts, and bone marrow mesenchymal stem cells (BMSCs) perturbs the tightly regulated bone remodeling equilibrium, underpinning pathologies including osteoporosis.163
Although nutritional supplements and vitamin complexes are widely utilized for general health support, their efficacy in modifying disease progression or symptom severity of mitochondrial dysfunction—particularly as therapeutics for skeletal pathologies—remains limited.30 Despite proliferating therapies targeting molecular defects in mitochondrial disorders, most confront limitations arising from etiological complexity. This inherent pathobiological intricacy frequently renders symptomatic management insufficient for arresting underlying disease progression.164 Emerging research, including mitochondrial transplantation studies, identifies novel strategies to restore mitochondrial integrity. Adaptation of these approaches to correct mitochondrial dysfunction in bone cells constitutes a viable clinical avenue for preventing and treating skeletal disorders.165 This conceptual refocusing shifts emphasis from systemic mitochondrial disorders toward mechanisms governing mitochondrial function in bone cells and skeletal integrity. Advancing mechanistic comprehension of intrinsic mitochondrial exchange processes within the bone niche is prerequisite for evaluating both endogenous reparative pathways and targeted therapeutic interventions.
Mechanisms of Intercellular Mitochondrial Transfer in Bone Homeostasis
This section examines endogenous mechanisms governing mitochondrial exchange among bone cells—a naturally occurring intercellular process. Mitochondrial transfer (MT) constitutes an evolving research domain investigating the dynamic translocation of functional mitochondria from donor to recipient cells. This translocation enables mitochondrial internalization and subsequent integration into the recipient’s metabolic architecture (Figure 1a). MT directly modifies bioenergetic parameters, metabolic plasticity, and phenotypic consequences in recipient cells.149 These mechanistically diverse processes represent specialized intercellular signaling modalities essential for maintaining tissue homeostasis and coordinating stress responses within osseous microenvironments. Current MT classification systems reflect distinct structural configurations and operational principles.
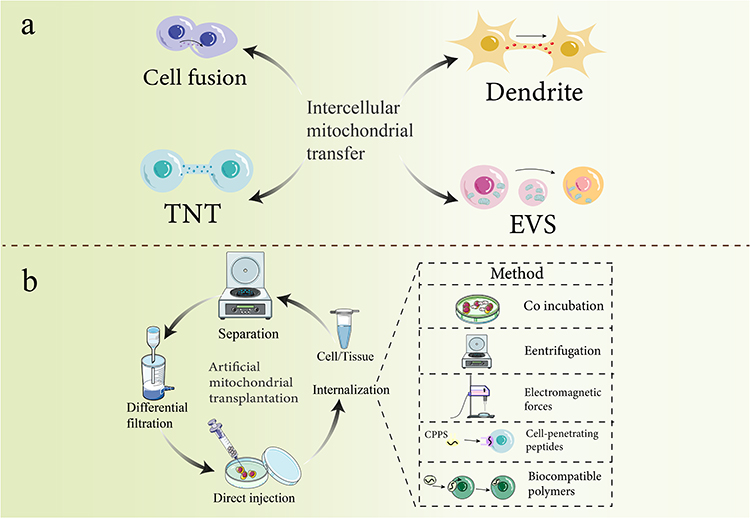 |
Figure 1 Methods of mitochondrial transfer and artificial mitochondrial transplantation. Notes: (a) The established intercellular material transport mechanisms for mitochondrial transfer encompass cell fusion, dendritic transfer, tunneling nanotubes (TNTs), and vesicle transport. (b) Artificial mitochondrial transplantation involves several steps. Cells or tissues are centrifuged, filtered, and differentiated before direct injection of mitochondria into aculture dish containing mesenchymal stem cells for subsequent internalization. Internalization methods include centrifugation, magnetic transfer, cell-penetrating peptides, and biocompatible polymers to facilitate complete mitochondrial transplantation. |
Direct Cell-to-Cell Connections for Mitochondrial Delivery
Tunneling nanotubes (TNTs): Transient, F-actin-based membrane protrusions forming direct intercellular connections. These structures mediate efficient intercellular communication and direct translocation of macromolecular cargo—including organelles such as mitochondria—between donor and recipient cells.150,151 Mitochondrial transfer via TNTs mitigates stress-induced cellular dysfunction and rescues cells during early apoptotic stages, demonstrating their functional role in cellular resilience. TNTs facilitate long-range organelle transport, enabling targeted mitochondrial delivery across cellular networks.152
Cell Fusion: Cell fusion entails permanent integration of adjacent heterotypic cells, resulting in complete sharing of cytoplasmic components and organelles, including mitochondria.166,167 Although less commonly observed than TNT-mediated transfer, cell fusion constitutes a distinct mechanism for extensive mitochondrial integration, contributing to metabolic reprogramming in recipient cells.
Vesicle-Mediated Mitochondrial Packaging and Delivery
Extracellular vesicles (EVs)—including exosomes and microvesicles—constitute a principal pathway for mitochondrial delivery. These membrane-bound structures encapsulate intact mitochondria or mitochondrial constituents prior to extracellular release. Recipient cells internalize mitochondrion-containing EVs via endocytosis or membrane fusion, facilitating integration of transferred mitochondrial cargo.168 EV-mediated mitochondrial transfer constitutes an adaptable mechanism enabling both localized and systemic mitochondrial delivery, contributing to paracrine signaling pathways and therapeutic applications.
Relevance in Bone Tissue
While mechanistic understanding of mitochondrial transfer continues to advance across biological systems, accumulating evidence establishes its essential role in bone homeostasis. Contemporary experimental approaches have enabled direct visualization and functional validation of this process within the osseous niche. Critically, mitochondrial transfer between bone cells occurs via osteocyte dendritic networks—elaborate cellular projections extensively interconnected throughout the mineralized matrix—as experimentally demonstrated.
Photoactivatable mitochondria (PhAM) studies in primary murine osteocytes and MLO-Y4 cell lines demonstrate mitochondrial transfer within dendritic networks using high-resolution confocal microscopy. Functional osteocytes transfer mitochondria to neighboring metabolically compromised osteocytes, restoring metabolic function through ER-mitochondrial axis coordination. This process specifically involves mitochondrial fusion proteins including mitofusin-2 (Mfn2), essential for osteocyte homeostasis.6
Beyond osteocyte-to-osteocyte transfer, evidence demonstrates mitochondrial transfer across additional bone cell lineages. Macrophages within the osseous niche donate mitochondria to mesenchymal stem cells (MSCs), promoting osteogenic differentiation through essential metabolic crosstalk.169 This mitochondrial transfer regulates recipient cell fate and function, critically influencing bone formation and repair processes. Under osteoporotic conditions, macrophages exhibiting phenotypic alterations release oxidatively damaged mitochondria, compromising MSC osteogenic differentiation.169
Collectively, these findings position intercellular mitochondrial transfer as a fundamental process governing homeostatic regulation in skeletal biology and pathology. Current research aims to characterize the scope and functional consequences of distinct mitochondrial transfer mechanisms in bone formation, resorption, and skeletal integrity. Given the established role of endogenous mitochondrial transfer in bone homeostasis, the following section examines emerging mitochondrial transplantation (MT) strategies for therapeutic mitochondrial restoration.
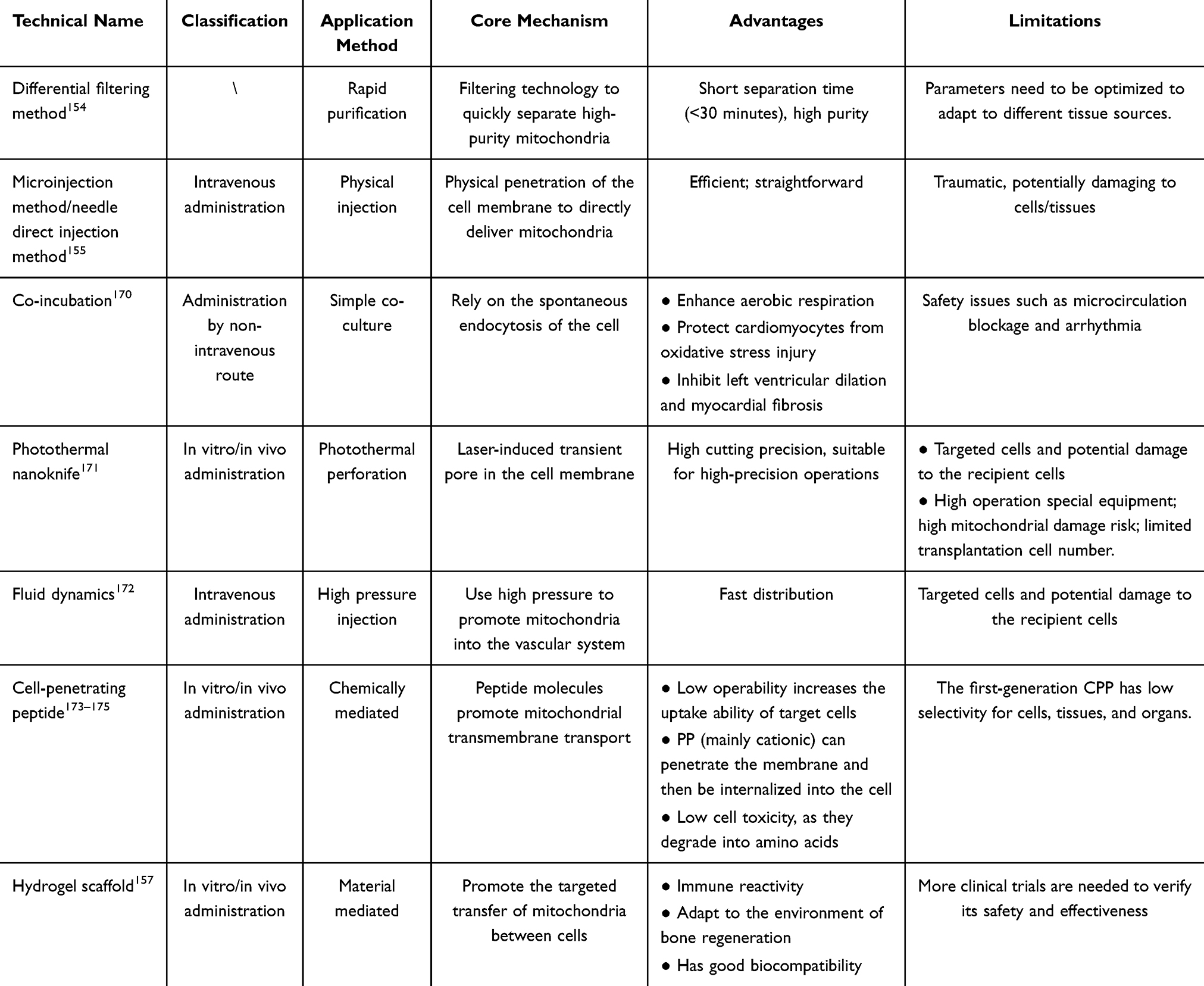 |
Table 1 Overview of Current Mitochondrial Transplantation Methods and Key Improvements |
Techniques for Mitochondrial Transfer
In light of the demonstrated benefits of mitochondrial exchange, both naturally occurring transfer and therapeutically engineered transplantation, the field has progressed to develop methodologies for the artificial delivery of mitochondria. The field of mitochondrial transfer (MT) emerged in 1982 with Clark and Shay’s pioneering technique of “mitochondrial transformation”, sparking further research despite initial uncertainties regarding the transfer mechanism.153 Artificial MT involves several steps: separation and purification of mitochondria, their transmission, and internalization by recipient cells (Figure 1b). Table 1 provides a detailed overview of the current methodologies employed in mitochondrial transplantation, outlining their key contributions and improvements in the field. Separation and purification are the most critical steps, requiring a suitable mitochondrial source. In clinical settings, autologous skeletal muscle offers advantages due to its reduced immunogenicity and ease of access.176–178 Differential and density gradient centrifugation are common separation techniques, but achieving both high yield and high purity can be challenging. Therefore, personalized centrifugation protocols may be necessary for mitochondria from different tissues. A promising approach by McCully et al utilizes differential filtration for rapid and efficient separation of highly purified, functional mitochondria, potentially meeting the time constraints of clinical applications (separation time < 30 minutes).154 Exogenous, isolated mitochondria have been successfully delivered to recipient cells in various in vitro and in vivo models. MT can be classified as in vitro or in vivo delivery. Both models often employ direct injection via microinjection or needles, a method also used in some clinical trials.155,179 For in vitro applications, researchers have explored methods beyond simple co-incubation to enhance mitochondrial internalization. These methods include centrifugation, magnetic transfer, cell-penetrating peptides, and biocompatible polymers.180 However, compared with CO incubation, invasive technologies such as photothermal nanoknife, non-injection, and hydrodynamics may be less popular due to limited targeting of cells and the potential damage to recipient cells. Studies have shown that successful mitochondrial internalization requires intact outer membranes and active mitochondria within recipient cells.181,182 Mitochondrial uptake may involve a variety of mechanisms, including actin-mediated endocytosis, macropinocytosis, and caveolin-dependent endocytosis.183,184 It is worth noting that the internalized mitochondria can avoid the lysosomal system in the cell and fuse with the host mitochondrial network.185 Further research is needed to fully elucidate the mechanisms of mitochondrial internalization, as recipient cells likely employ a combination of these pathways. To further enhance the precision, efficiency, and safety of mitochondrial delivery, advanced strategies have emerged, including exosome-mediated transfer and biomimetic coating technologies.
Exosome-Mediated Mitochondrial Transfer
Exosomes are membrane-bound extracellular vesicles with a diameter of 30–150 nm, encapsulating a diverse array of molecular cargo, including proteins, lipids, and nucleic acids. These naturally occurring nanovesicles mediate intercellular communication, modulate osteocyte activity, and exhibit favorable biocompatibility with minimal immunogenic potential.186–188 Mitochondria act as central metabolic hubs in osteocytes, orchestrating three critical physiological processes: (1) ATP generation via oxidative phosphorylation to meet cellular energy demands; (2) calcium ion buffering to maintain intracellular signaling; and (3) reactive oxygen species regulation to preserve redox equilibrium. Dysregulation of these mitochondrial functions disrupts skeletal homeostasis and represents a fundamental pathophysiological mechanism underlying osteoporotic bone loss.189–192 In response to cellular stress signals (eg, oxidative stress and bioenergetic dysfunction), donor cells (eg, mesenchymal stem cells) selectively package functional mitochondria into exosomal vesicles for intercellular transfer. Exosomes undergo cellular internalization via membrane fusion or receptor-mediated endocytosis, subsequently delivering functional mitochondrial cargo to replenish the impaired mitochondrial network in recipient cells.193–196 The exogenously delivered mitochondria functionally integrate into the metabolic networks of recipient cells, exhibiting three key biological activities: (1) restoration of ATP production through oxidative phosphorylation; (2) attenuation of reactive oxygen species (ROS) generation; and (3) modulation of calcium signaling dynamics. The exosomal payload, comprising mitochondrial DNA (mtDNA) and associated regulatory proteins (eg, mitochondrial transcription factor A, TFAM), enhances mitochondrial biogenesis and functional recovery in recipient cells, thereby reestablishing cellular bioenergetic homeostasis.197–199 Exosomes mediate the intercellular regulation of mitochondrial quality control through their molecular cargo (eg, miRNAs, cytokines), which modulates key signaling pathways (including AMPK/mTOR) in recipient cells. This coordinated action simultaneously enhances: (1) mitophagic clearance of dysfunctional mitochondria; and (2) de novo mitochondrial biogenesis, collectively optimizing cellular bioenergetic efficiency.200–203 In the therapeutic management of osteoporosis, stem cell-derived exosomes (eg, bone marrow mesenchymal stem cell exosomes) facilitate the intercellular transfer of functional mitochondria to osteoblasts, resulting in three key therapeutic effects: (1) restoration of oxidative phosphorylation capacity; (2) upregulation of osteogenic transcription factors (eg, Runx2) and bone-specific proteins (eg, osteocalcin); and (3) enhanced bone matrix deposition and mineralization.29,36 Exosome-mediated mitochondrial transfer exerts multimodal inhibitory effects on osteoclast activity through three complementary mechanisms: (1) attenuation of intracellular ROS accumulation; (2) suppression of NF-κB signaling pathways; and (3) miRNA-dependent (eg, miR-21-5p) downregulation of RANKL expression, collectively reducing osteoclastogenesis and bone resorptive capacity.204–206 Exosome-facilitated mitochondrial transfer confers cytoprotective and metabolic benefits to bone marrow stromal cells through three synergistic mechanisms: (1) augmentation of endogenous antioxidant defenses; (2) attenuation of pro-inflammatory cytokine secretion (eg, TNF-α, IL-6); and (3) metabolic reprogramming that promotes osteogenic differentiation while suppressing adipogenic commitment, thereby preserving skeletal homeostasis.102,207–209 Preclinical studies using ovariectomized rat models have demonstrated that mitochondrial-enriched mesenchymal stem cell exosomes elicit four significant therapeutic effects: (1) increased bone mineral density (p<0.01); (2) restored trabecular microarchitecture; (3) elevated osteoblast counts; and (4) reduced osteoclast numbers, confirming their osteoprotective efficacy.210,211 Exosome therapy offers advantages such as favorable biological safety (being cell-derived secretions with low immunogenicity, minimal risk of rejection, and low toxicity), strong targeting and permeability (enabling accurate delivery of mitochondria to osteocytes, penetration of biological barriers, and overcoming limitations of traditional drugs), and high modifiability (allowing modification to load specific components such as miR-146a for enhanced therapeutic specificity).212–218 Notwithstanding these advantages, critical challenges remain in clinical translation, including (1) optimization of scalable production and purification protocols; (2) incomplete elucidation of underlying molecular mechanisms; and (3) insufficient clinical validation through large-scale trials.219–221 Future clinical translation will require (1) refinement of scalable production methodologies; (2) mechanistic elucidation of mitochondrial transfer processes; (3) development of nanotechnology-enabled targeted delivery systems; and (4) rigorous multicenter clinical evaluation.
Mitochondria-Mimetic Coating-Based Immune-Evasive Enveloping Technology
Recent developments in biomimetic mitochondrial membrane engineering have yielded immunologically inert nanovehicles, marking a transformative advancement in precision therapeutics. This bioinspired approach offers two synergistic benefits: (1) superior immune evasion through molecular camouflage of surface epitopes, and (2) enhanced mitochondrial specificity via evolutionary conserved organelle targeting sequences, collectively optimizing treatment efficacy. Zheng et al established that rationally designed mitochondrial nanocarriers can reconstitute mitochondrial biogenesis pathways, effectively reversing neuronal bioenergetic deficits.222 The CSCCT nanoparticle system utilizes a hierarchical targeting approach combining blood-brain barrier (BBB) translocation with neuronal mitochondrial accumulation. Surface functionalization with DSPE-PEG2000-TPP confers: (1) mitochondriotropic specificity through triphenylphosphonium cation-mediated potential gradient recognition, (2) reduced opsonization through steric stabilization, and (3) prolonged plasma residence time by minimizing reticuloendothelial system clearance.The inherent biocompatibility of native cell membranes confers both immunotolerance and enhanced circulatory stability. The DSPE-PEG2000-TPP-modified macrophage membrane coating mediates dual functionality: (1) potentiated immune evasion through self-marker retention, and (2) selective targeting of compromised neurons via inflammation-dependent integrin-VCAM-1 molecular recognition. Focused ultrasound-mediated blood-brain barrier (BBB) disruption enables transient and reversible nanoparticle extravasation into brain parenchyma, facilitating site-specific accumulation at pathological foci. The bioengineered NC@PPN core-shell system demonstrates tumor cell-selective internalization and mitochondria-targeted delivery through coordinated surface ligand-receptor interactions and membrane fusion mechanisms. Clathrin-dependent endocytic trafficking and cholesterol-mediated cellular internalization synergistically enhance mitochondrial delivery efficiency.223 The outer shell incorporates somatostatin (SST) ligands that exhibit high-affinity binding to somatostatin receptor subtype 2 (SSTR2), which is overexpressed on MCF-7 breast cancer cell membranes, thereby mediating tumor-selective cellular recognition. The system exploits tannic acid’s (TA) polyphenolic architecture to facilitate mitochondrial localization via high-affinity interactions with voltage-dependent anion channels (VDACs) on the outer mitochondrial membrane. Quantitative colocalization analysis revealed significant spatial correlation between NC@PPNs and mitochondrial markers (Pearson’s r > 0.85), confirming targeting specificity. Peng et al developed an integrated targeting platform combining small-molecule ligands with natural biomembrane coatings. TPP-functionalized gold nanoparticles (TPP-GNPs) exploit electrostatic potential-driven interactions with mitochondrial membranes, achieving both enhanced organelle-specific accumulation and surface plasmon resonance-mediated nanoparticle assembly.224 The cancer cell membrane (CCM) encapsulation strategy forms a protective biointerface around GNA, shielding its structural integrity from biological degradation while extending its plasma half-life. Homotypic membrane fusion facilitates direct nanoparticle cytosolic entry, circumventing endolysosomal sequestration and subsequent degradation. This mechanism enables efficient TPP-mediated mitochondrial trafficking, substantially improving cellular internalization kinetics.
Programmable Nanorobots or Magnetically Guided Delivery Systems
Engineered DNA nanostructures and magnetically responsive delivery systems represent groundbreaking developments in precision mitochondrial medicine. DNA origami-based nanocarriers (eg, tetrahedral frameworks and tubular transporters) achieve cell-specific mitochondrial delivery through programmable molecular interactions and three-dimensional spatial targeting,225–228 Cell-type selective targeting is accomplished via TXNDC5 antibody-conjugated aptamer arrays immobilized on delivery substrates,229–231 A key innovation utilizes the acidic osseous milieu (pH ≤6.5) to trigger i-motif structural transitions, facilitating subsecond mitochondrial release,232–235 while ensuring nanocarrier biodegradation prevents tissue accumulation.236 Preclinical evaluation in a validated osteoporosis model revealed that a single treatment regimen increased bone mineral density by 32%, demonstrating statistically significant improvement over conventional delivery approaches (p<0.001).236 Magnetogenetic delivery systems utilizing IONP-tagged mitochondria show enhanced intercellular transfer efficiency under controlled magnetic guidance.237 The integrated platform combines magnetothermal therapy, where AMF-activated SPIONs produce localized hyperthermia, simultaneously stimulating vascularization and bone regeneration through coupled mechanisms.236 SPIONs additionally permit longitudinal, quantitative tracking of mitochondrial engraftment via MRI.238 Notwithstanding these advances, clinical implementation faces notable challenges: (1) depth-dependent magnetic field attenuation requiring high-field systems (≥3T) for deep tissue applications (>10cm), and (2) hepatotoxicity risks from persistent SPION retention.239
Application of Mitochondrial Transplantation in the Treatment of Osteoporosis
Mitochondrial Transfer in Bone Homeostasis Regulation and Osteoporosis Intervention
Accumulating evidence demonstrates that mitochondrial transfer (MT) constitutes a fundamental regulatory axis in bone homeostasis, mediating intercellular crosstalk and functional synchronization across osteogenic cell populations (Figure 2). This biological process critically regulates bone formation, remodeling kinetics, and reparative processes, thereby revealing potential therapeutic targets for osteoporosis intervention (Figure 3).
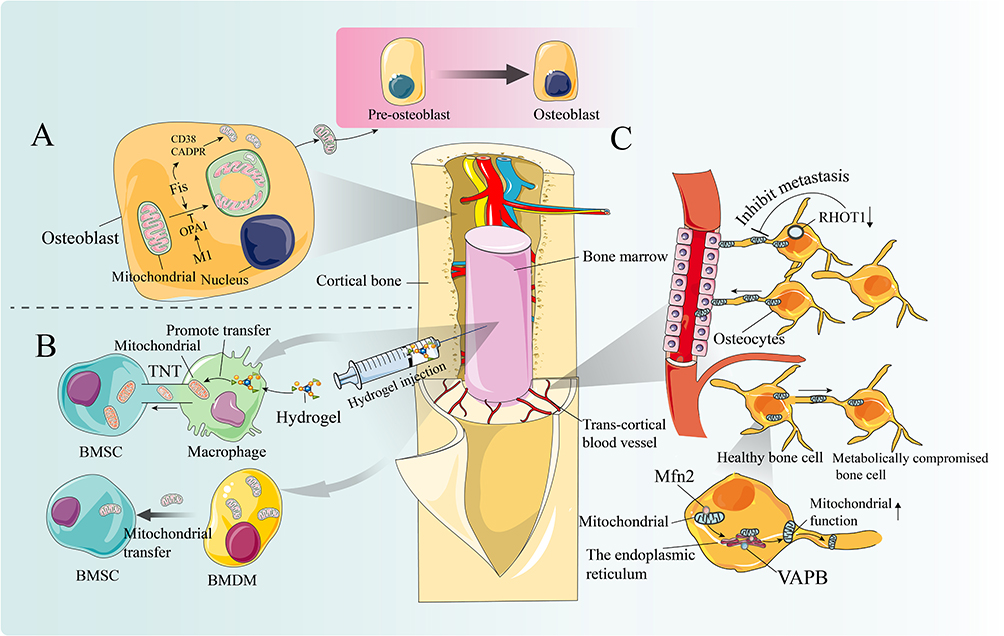 |
Figure 2 The role of mitochondrial transfer in bone cell communication and function. Notes: (A) Mature osteoblasts actively release mitochondria and mitochondrial-derived vesicles (MDVs) to facilitate the differentiation of osteoblast precursor cells. Osteogenesis induction triggers mitochondrial fragmentation, donut formation, and subsequent secretion via CD38/cADPR signaling. Disrupting mitochondrial fusion (OPA1 knockout) or enhancing fission (Fis1 overexpression) can accelerate these processes, resulting in increased mitochondrial secretion and accelerated osteogenesis. (B) The injection of an engineered hydrogel with immunoreactivity can adapt to the bone regeneration environment and promote targeted intercellular mitochondrial transfer. Macrophages can restore mitochondrial bioenergetics in bone marrow mesenchymal stem cells (BMSCs) by continuously supplying mitochondria, fundamentally addressing the issue of insufficient energy support in BMSCs caused by local inflammation during bone repair and regeneration. Another novel 3D biomimetic hydrogel scaffold with immunomodulatory properties can promote the migration of macrophage mitochondria to BMSCs, thereby stimulating osteoblastic differentiation. (C) Bone cells are interconnected through an extensive dendritic network. Mitochondrial fission protein 2 (Mfn2), aGTPase, facilitates this transfer by bridging the endoplasmic reticulum (ER) to mitochondria. Knockdown of the endoplasmic reticulum mitochondrial binding protein vesicle-associated membrane protein B(VAPB) impedes mitochondrial transfer, reducing the ability to salvage damaged cellular metabolic function (Upward black arrows indicate an increase in mitochondrial function.). The transcortical vascular system (TCV) promotes communication between the bone marrow vascular system and the external circulation. Conditional knockout of the RHOT1 gene (Downward black arrows indicate adecrease in RHOT1), encoding MIRO1, disrupts mitochondrial transfer in bone cells, leading to TCV network degradation and subsequent impairment of cortical bone healing. |
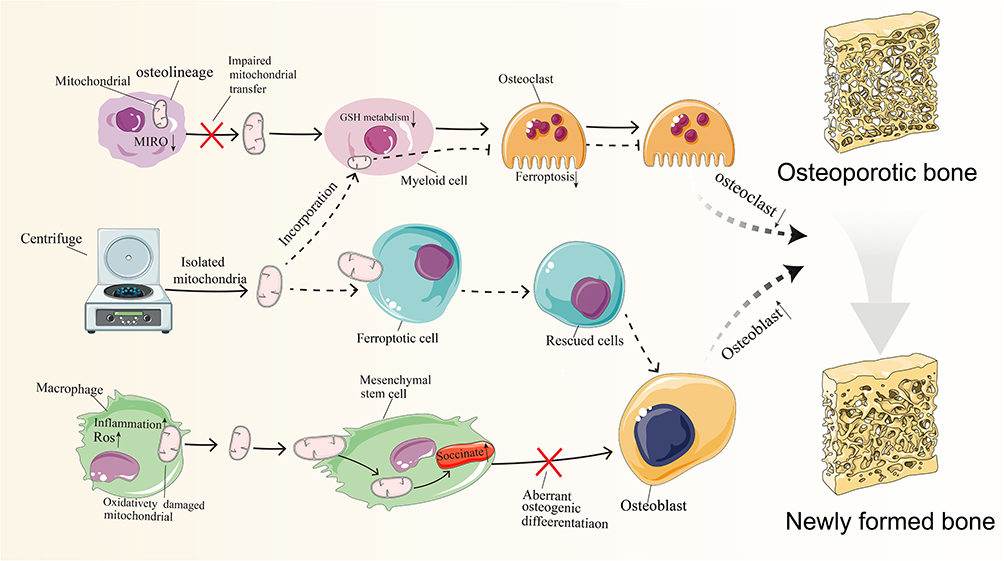 |
Figure 3 The therapeutic potential of mitochondrial transplantation in osteoporosis. Notes: Decreased MIRO1 expression in osteoblasts impairs mitochondrial transfer, leading to altered glutathione metabolism, protection of osteoclast lineage cells from iron-induced apoptosis, and promotion of osteoclast activity. In osteoporosis, macrophages with altered phenotypes and metabolic states release oxidatively damaged mitochondria, and increased mitochondrial transfer results in succinic acid accumulation, causing metabolic dysregulation in bone marrow mesenchymal stem cells. Succinic acid accumulation is apivotal factor in abnormal osteogenic differentiation. Recent research findings suggest that mitochondrial transplantation could be apromising novel approach for preventing and treating osteoporosis. Mitochondria isolated through centrifugation can be merged with cells exhibiting metabolic abnormalities, effectively protecting and repairing cellular damage, thereby restoring bone metabolic balance and promoting osteogenic differentiation. The dashed line in the figure represents the proposed hypothesis. (Black upward arrows indicate an increase in substance or expression, while black downward arrows indicate adecrease in substance or expression). |
The skeletal matrix demonstrates substantial accumulation of mitochondrial components, reflecting their active engagement in osteoblast maturation and mineral homeostasis regulation, likely facilitated by regulated extracellular vesicle trafficking.240–242 Biochemical and genetic studies confirm that fully differentiated osteoblasts actively export intact mitochondria and mitochondrial-derived vesicles (MDVs), which subsequently potentiate the osteogenic programming of mesenchymal stem cell precursors.240,243,244 Recent advances have uncovered a fundamental interdependence between mitochondrial morphological plasticity and osteogenic differentiation, whereby differentiation signals provoke mitochondrial network fragmentation and annular reorganization, ultimately leading to CD38/cADPR-dependent extracellular discharge.240,244 Experimental modulation of mitochondrial fusion-fission balance - through either fusion suppression (eg, OPA1 knockout) or fission activation (eg, Fis1 transgenic expression)–markedly amplifies mitochondrial export capacity, yielding increased extracellular mitochondrial abundance and enhanced osteogenic potential.240 These data collectively establish that the unique structural specialization of mature osteoblasts’ mitochondrial networks supports the regulated extracellular deployment of bioactive mitochondria and MDVs, which function as critical determinants of osteoblast lineage progression and bone matrix biosynthesis.240 Deciphering these regulatory circuits provides a molecular basis for therapeutic mitochondrial transfer strategies designed to stimulate bone formation and prevent osteoporosis-associated skeletal degeneration.
Osteocytes, the terminally differentiated cells embedded within the mineralized bone matrix and interconnected via an extensive canalicular network, serve as an exemplary biological system for studying mitochondrial transfer mechanisms and their role in maintaining skeletal homeostasis.6,245,246 The osteocyte lacunar-canalicular system functions as a critical mechanosensory apparatus that precisely regulates bone remodeling through paracrine communication with osteoblasts and osteoclasts.6,245,246 In this context, intercellular mitochondrial transfer represents an essential cytoprotective strategy for these long-lived cells, protecting against progressive cellular damage while maintaining their functional capacity to preserve bone structural integrity.30,247 Metabolically stressed osteocytes demonstrate the ability to internalize mitochondria from surrounding functional osteocytes, restoring bioenergetic homeostasis and maintaining their regulatory functions in bone metabolism.247 Current evidence indicates that this transfer process depends on specialized ER-mitochondria membrane contact sites, which facilitate directional mitochondrial transport through the canalicular network.6 Notably, Mitofusin 2 (Mfn2), a key mediator of mitochondrial dynamics, establishes ER-mitochondria tethering and promotes intercellular mitochondrial exchange.6,248,249 Experimental data show that VAPB knockdown significantly attenuates mitochondrial transfer, impairing metabolic recovery in dysfunctional osteocytes, suggesting that ER-mediated mitochondrial distribution involves coordinated actions of Mfn2 and VAPB-containing protein complexes.6 Age-related declines in Mfn2 expression are associated with defective mitochondrial transport and reduced transfer efficiency within the osteocyte network.6 These deficits contribute to osteocyte apoptosis, metabolic dysfunction, and ultimately promote osteoporosis development by disrupting mechanotransduction pathways and impairing the bone’s adaptive repair capacity.6
Mitochondrial transfer mechanisms involve multiple cell types beyond osteocytes, with MIRO1 (Mitochondrial Rho GTPase 1) emerging as a central regulator of microtubule-associated mitochondrial transport.250,251 The transcortical vascular system functions as a principal interface mediating molecular exchange between bone marrow vasculature and the systemic circulatory system.252 Recent investigations reveal that viable osteocytes maintain vascular integrity through mitochondrial transfer to endothelial cells, thereby preserving microvascular structure and function.252 Experimental osteocyte depletion leads to progressive deterioration of transcortical vasculature.252 Targeted deletion of Rhot1 (encoding MIRO1) in osteocytes impairs mitochondrial transport and induces vascular degeneration, establishing MIRO1-mediated transfer as essential for vascular homeostasis.252 Endothelial cells receiving osteocyte-derived mitochondria show restored functionality, increased angiogenic potential, and enhanced capacity for bone repair.252 Collectively, these findings delineate a critical osteocyte-vascular axis, revealing novel therapeutic avenues for mitochondrial transfer-based interventions in skeletal disorders associated with vascular compromise, including osteoporosis and impaired fracture healing.
Optimal bone regeneration, critical for osteoporotic fracture healing and skeletal integrity, requires tightly controlled immune regulation and efficient metabolic adaptation within the fracture callus.253 As central regulators of cellular energetics and metabolic pathways, mitochondria represent attractive therapeutic targets, with mitochondrial transfer emerging as a viable strategy for functional recovery.254,255 Recent developments in biomaterial engineering have produced immunologically active graded hydrogels specifically designed to integrate with the bone regeneration microenvironment and promote targeted mitochondrial delivery between cells.156 A prime example is the dependence of bone marrow mesenchymal stem cells (BMSCs), crucial mediators of bone repair, on mitochondrial transfer from macrophages to sustain their bioenergetic requirements. This cellular crosstalk alleviates inflammatory-mediated metabolic suppression of osteogenesis, highlighting its therapeutic potential for improving bone regeneration in osteoporosis.156 The newly engineered 3D bionic GAD/Ag PIO hydrogel platform demonstrates significant immunomodulatory capacity and promotes BMSC osteogenic differentiation through macrophage-mediated mitochondrial transfer in experimental models.157 Preclinical animal studies further validate this scaffold’s ability to enhance fracture healing.157 Together, these results position mitochondrial transfer-enabled scaffolds as a novel treatment approach for boosting bone formation and fracture repair, with important clinical implications for osteoporosis and bone regeneration therapies.157
The Therapeutic Potential of Mitochondrial Transfer From Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs) have emerged as promising therapeutic agents for tissue regeneration, owing to their triad of biological properties: immunomodulatory capacity, reactive oxygen species scavenging activity, and pro-regenerative potential.256 A fundamental mechanism mediating these therapeutic effects involves intercellular mitochondrial transfer from MSCs to recipient cells, which has been experimentally demonstrated in both animal models and cell culture systems.257 The biological significance of MSC-mediated mitochondrial transfer (MSC-MT) underscores its therapeutic potential for disorders characterized by mitochondrial impairment. Future investigations should focus on mechanistic elucidation, optimization of transfer efficacy, and development of clinically translatable therapeutic approaches.153
MSCs as Mitochondrial Donors for Tissue and Organ Repair
Bone marrow-derived mesenchymal stem cells (BM-MSCs), representing a paradigm of mesenchymal stem cell populations, exert their therapeutic effects post-transplantation via three principal mechanisms: (1) direct intercellular signaling, (2) paracrine mediator secretion, and (3) extracellular vesicle-mediated communication (encompassing both exosomes and microvesicles).258 Accumulating experimental data demonstrate that mitochondrial transfer from BM-MSCs promotes functional restoration in damaged tissues.259 This biological process assumes particular importance in disorders characterized by mitochondrial dysfunction, where BM-MSC-originated mitochondrial transfer to compromised cells can ameliorate tissue pathology. A well-documented instance involves the uptake of BM-MSC mitochondria by alveolar epithelial cells, providing cytoprotection against pulmonary injury induced by endotoxin exposure or cigarette smoke inhalation.260,261 Importantly, BM-MSC senescence, manifested as progressive functional deterioration, constitutes a major etiological factor in age-related osteoporosis. These mechanistic insights have led to the hypothesis that controlled mitochondrial transfer in vitro could enhance BM-MSC osteogenic potential, thereby offering a novel therapeutic approach for osteoporosis intervention.
Intercellular Mitochondrial Transfer Modulating MSC Function in Bone Homeostasis
In addition to serving as mitochondrial donors, MSCs exhibit functional plasticity that is modulated by mitochondrial acquisition from other cell populations, particularly macrophages. Under physiological conditions, macrophage-derived mitochondrial transfer enhances MSC osteogenic potential, contributing to skeletal homeostasis. However, in osteoporotic states, metabolically compromised macrophages predominantly release oxidatively impaired mitochondria. The subsequent uptake of these dysfunctional mitochondria, especially from pro-inflammatory M1-polarized macrophages, induces redox imbalance and succinate accumulation in recipient MSCs. This metabolic perturbation disrupts osteogenic differentiation capacity through pathological remodeling of MSC metabolism. Collectively, these observations establish bidirectional mitochondrial transfer between macrophages and MSCs as a critical metabolic checkpoint in bone homeostasis, revealing therapeutic opportunities for osteoporosis through targeted modulation of this intercellular crosstalk.262
The Role of Ferroptosis in Osteoporosis and the Potential of Mitochondrial Transplantation
Ferroptosis, an iron-dependent programmed cell death pathway driven by membrane lipid peroxidation, represents a key pathogenic mechanism in osteoporosis-related bone deterioration.263 Preserving mitochondrial integrity has emerged as a promising therapeutic strategy to counteract ferroptosis-mediated cellular damage.264 However, translating this approach into clinical applications remains challenging due to unresolved mechanistic aspects of mitochondrial dysregulation and uncertainties regarding optimal therapeutic windows.Pharmacological suppression of mitochondrial superoxide production restores bioenergetic competence and may protect against ferroptosis-induced cell death.265
Mitochondrial Transfer Regulating Ferroptosis in Bone Cells
Mitochondrial transfer (MT) serves as a critical regulatory node governing ferroptosis within the bone microenvironment.266 Substantial experimental data confirm that osteoprogenitor cells actively engage in mitochondrial donation to myeloid lineage cells. Targeted deletion of MIRO1 in osteogenic precursors impairs mitochondrial trafficking, promoting excessive osteoclast differentiation and subsequent bone loss. At the molecular level, impaired mitochondrial transfer disrupts glutathione-mediated redox balance, a key determinant of ferroptotic vulnerability, consequently inhibiting programmed cell death in osteoclast precursors while potentiating their resorptive capacity. Bidirectional mitochondrial exchange between skeletal and hematopoietic systems significantly influences glucocorticoid-induced osteoporosis pathogenesis, the most common secondary osteoporosis variant. This intercellular communication mechanism drives glutathione depletion while ameliorating disease advancement.266 Collectively, these findings establish that skeletal homeostasis depends on precise osteo-immune mitochondrial crosstalk, while identifying ferroptosis pathway modulation as a promising therapeutic strategy for glucocorticoid-associated bone loss.
Mitochondrial Transplantation as a Therapeutic Approach for Ferroptosis
Mitochondrial transplantation exhibits therapeutic efficacy in attenuating ferroptosis across diverse biological contexts, highlighting its potential application for osteoporosis management. In neurodegenerative models, transplanted mitochondria demonstrate functional engraftment within murine cortical neurons, effectively preventing ferroptosis-associated neuronal damage. Neurodegenerative disease studies further confirm that exogenously administered mitochondria integrate into neuronal networks, exerting protective effects against ferroptotic synaptic degeneration.267
Future Directions and Challenges in Mitochondrial Transplantation
Mitochondrial transplantation (MT) has emerged as a promising therapeutic strategy, demonstrating efficacy across various models, from cellular and animal studies to early human trials. These models utilize both autologous and non-autologous mitochondria. Preclinical animal studies consistently show that MT restores mitochondrial function and enhances cellular energy metabolism.146 Clinically, MT has proven effective in treating conditions like myocardial ischemia-reperfusion injury and cardiogenic shock after ischemia-reperfusion.176,268 Notably, autologous MT is a particularly attractive therapeutic approach for various diseases due to its ability to avoid significant increases in inflammatory markers like TNF-α, IL-6, and high-sensitivity C-reactive protein.179 This personalized approach, where mitochondria are processed in vitro and reintroduced into the same patient, inherently minimizes the risk of immune rejection, aligning well with principles of precision medicine. The process of mitochondrial isolation and transplantation is generally considered straightforward, time-efficient, and boasts a high success rate, offering better controllability, stability, and effectiveness compared to other enhancement strategies.106
Overcoming Translational Hurdles
Despite its considerable promise, the widespread clinical application of MT faces several uncertainties and obstacles. The primary challenge involves not only the efficient and specific delivery of functional mitochondria to target cells in vivo but also establishing the long-term stability and efficacy of transplanted mitochondria within host cells. Researchers are actively pursuing novel methods to address these issues. For instance, CRISPR gene editing technology offers the potential to correct mutations associated with mitochondrial diseases by modifying the DNA of transplanted mitochondria. Moreover, to overcome the difficulties of targeting mitochondria to specific cells in vivo, mitochondria can be encapsulated within nanoparticles for precise delivery. The development of innovative technologies such as nanoparticle-mediated delivery and CRISPR gene editing has instilled hope for the advancement of novel therapies for mitochondrial diseases.147
Mitochondrial Sourcing, Storage, and Immune Response
Although mitochondrial transplantation holds promising potential, several challenges remain for its clinical application, including mitochondrial storage, transplant rejection, and ethical considerations.145 Given the critical role of mitochondrial quality in transplantation, meticulous selection of the mitochondrial source is paramount. While autologous transplantation is generally preferred for treating mitochondrial diseases due to its ability to avoid inflammation and rejection, it is not feasible for patients with congenital mitochondrial disorders. In these cases, mitochondria derived from compatible allogeneic donors within the same species are required. To significantly expand the clinical applicability of mitochondrial transplantation, further research is essential to elucidate the immune response and associated mechanisms. The immune response remains a critical, yet understudied, aspect of mitochondrial replacement therapy.147 Mitochondria used in transplantation often originate from either autologous or allogeneic sources. While allogeneic mitochondria offer greater clinical practicality, particularly for patients with mtDNA mutations, in vitro studies have reported immune responses following their transplantation.143 Conversely, continuous injection of allogeneic mitochondria in mice has demonstrated safety.144 These conflicting findings necessitate further investigation into the underlying mechanisms.
Moreover, the limited time window for mitochondrial isolation restricts the applicability of mitochondrial transplantation. Mitochondrial activity rapidly declines following removal from the host.269 Currently, there is no effective method for long-term mitochondrial preservation, necessitating immediate use after extraction. Isolated mitochondria are susceptible to internal and external membrane damage, leading to significant decreases in function and activity.270 While refrigeration and cryopreservation are being explored as potential solutions,269 these strategies can stabilize the mitochondrial membrane but are unable to preserve normal bioenergetic functions. Consequently, the development of an effective mitochondrial storage solution is urgently needed to broaden its practical applications.269 Therefore, establishing the optimal method for separating and transplanting mitochondria is crucial for obtaining complete and feasible mitochondria for clinical treatment.271
Alternative Delivery Methods and Future Research Directions
Cell-mediated mitochondrial transfer provides a potentially safer approach compared to direct mitochondrial transplantation, eliminating the need for laborious mitochondrial isolation and the challenges associated with in vitro mitochondrial preservation. However, transfer efficiency remains a significant hurdle for cell-mediated mitochondrial transfer. Despite advancements, translating targeted mitochondrial transmission from the laboratory to clinical practice continues to face substantial challenges. The integration of functional cells can complicate the evaluation of therapeutic efficacy in cell-mediated mitochondrial transfer. Furthermore, the process of selecting donor cells and delivering mitochondria to target sites requires further standardization. Future research should prioritize the development of efficient mitochondrial storage solutions and transport methods.272
Advances in tools and technologies for measuring mitochondrial transfer in vivo, coupled with the recognition of mitochondria’s pivotal role in pathological states, have spurred rapid growth in the field of intracellular mitochondrial transfer. Despite progress, the molecular mechanisms underlying mitochondrial transfer between different cells remain largely elusive. Further research is needed to evaluate the effectiveness, stability, and safety of artificial mitochondrial transplantation technology in human trials.147 So far, only a small number of clinical trials with varying results have been conducted, and while the safety and effectiveness of mitochondrial transplantation have been validated to a certain extent, this does not necessarily indicate long-term safety and effectiveness.147 Furthermore, the effectiveness and safety of exogenous mitochondria need to be further explored in human trials.147 Future research should address several critical questions, including: the efficiency and long-term viability of mitochondria during isolation and delivery; strategies to mitigate the potential immune response to allogeneic mitochondria; the mechanisms governing fusion between transplanted mitochondria and the host cell mitochondrial network; and the preservation of unique characteristics by transplanted mitochondria within host cells. The answers to these questions, while potentially influenced by cell type, tissue, and environment, could fundamentally transform our understanding of mitochondrial biology and uncover promising avenues for therapeutic and diagnostic applications160 (Figure 4).
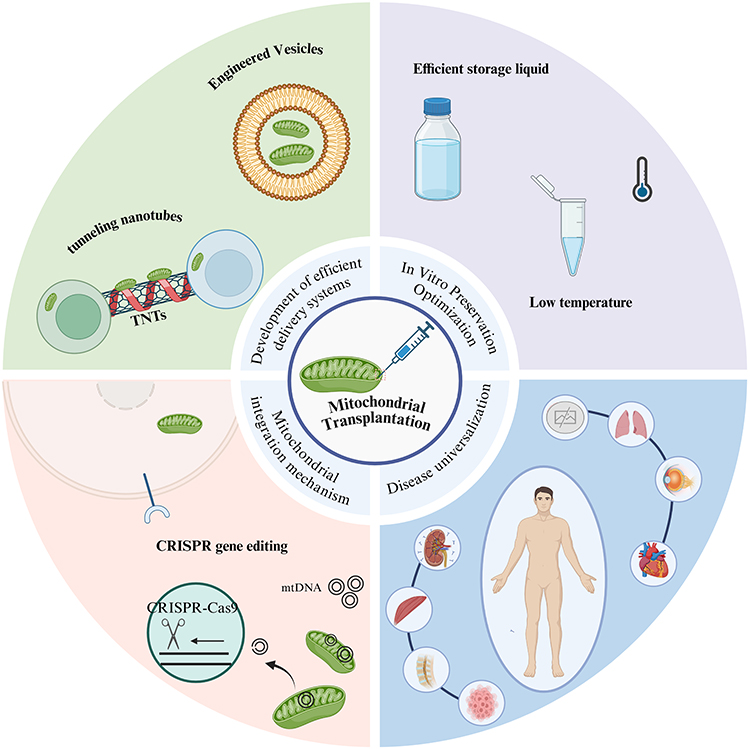 |
Figure 4 The future direction of mitochondrial transplantation. Notes: Future mitochondrial transplantation can advance through the development of efficient delivery systems, optimization of invitro preservation techniques, and innovation of mitochondrial integration mechanisms, while also being applied to various diseases. |
Conclusion
Osteoporosis, a systemic skeletal disorder characterized by progressive bone loss and microarchitectural deterioration, represents a major global health challenge necessitating therapeutic strategies beyond conventional anti-resorptive approaches. Emerging evidence implicates mitochondrial dysfunction as a central pathophysiological mechanism underlying the disrupted bone remodeling balance in osteoporosis. This review critically evaluates contemporary data demonstrating that mitochondrial quality control serves as a critical determinant of skeletal homeostasis, wherein subtle disturbances in mitochondrial function can significantly impair bone turnover processes and accelerate osteoporotic progression.
Recent progress in mitochondrial transplantation (MT)—a novel regenerative strategy involving the precise administration of functional mitochondria to cells with bioenergetic deficits—has demonstrated significant therapeutic effects in preclinical osteoporosis models, including reversal of bone mass depletion, reconstruction of trabecular networks, and enhancement of bone mechanical competence. This comprehensive review assesses various mitochondrial donor sources—particularly skeletal muscle, hepatocyte, and mesenchymal stem cell-derived mitochondria—each possessing unique therapeutic attributes for osteoporosis intervention. Of particular note, mitochondria isolated from mesenchymal stem cells exhibit pronounced osteogenic specificity, effectively restoring bone-forming capacity through both exosomal transfer pathways and direct mitochondrial incorporation.
Intercellular mitochondrial transfer occurs through phylogenetically conserved mechanisms, including tunneling nanotube-mediated transport, extracellular vesicle encapsulation, and fusogenic cellular interactions. In skeletal tissue, this process assumes particular physiological significance as osteocytes employ their extensive canalicular networks to distribute mitochondria throughout the osseous matrix, thereby maintaining metabolic homeostasis.
Despite its considerable therapeutic promise, mitochondrial transplantation faces substantial challenges in clinical implementation. Principal technical obstacles involve: (1) developing efficient methodologies for tissue-specific mitochondrial delivery, (2) ensuring sustained bioenergetic competence following cellular integration, and (3) preventing adverse immune recognition. Further limitations stem from the transient viability of isolated mitochondria and inadequate cryopreservation techniques for mitochondrial banking.
From an ethical perspective, allogeneic mitochondrial transfer necessitates careful evaluation, with particular attention to potential germline incorporation and incompatibilities between donor mitochondria and recipient nuclear genomes.Future research should prioritize the development of optimized mitochondrial preservation techniques and enhanced delivery methodologies. Emerging strategies such as CRISPR-mediated mitochondrial genome engineering and nanoparticle-facilitated mitochondrial transfer may address current technological limitations. Combining single-cell multi-omics analyses with computational models of mitochondrial function could facilitate personalized mitochondrial therapies, positioning MT as a groundbreaking approach for osteoporosis intervention. A comprehensive understanding of mitochondrial transfer mechanisms, supported by rigorous clinical evaluation, is essential for advancing mitochondrial transplantation toward clinical application in osteoporosis treatment.
Acknowledgments
We thank The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, Southwest Medical University, Affiliated Hospital of Putian University, Guangdong Medical University, Fujian Medical University and Zhongda Hospital Affiliated to Southeast University for their support of this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by National Natural Science Foundation of China (82301785); Key research and development projects of Sichuan Science and Technology Plan Project (2024YFFK0135); Fujian Provincial Natural Science Foundation of China (2024J011450); Medical Research Foundation of Putian University (2024104).
Disclosure
The authors have no relevant financial or non-financial interests to disclose.
References
1. Adejuyigbe B, Kallini J, Chiou D, et al. Osteoporosis: molecular Pathology, Diagnostics, and Therapeutics. Int J Mol Sci. 2023;24(19):14583. doi:10.3390/ijms241914583
2. Zhivodernikov IV, Kirichenko TV, Markina YV, et al. Molecular and Cellular Mechanisms of Osteoporosis. Int J Mol Sci. 2023;24(21):15772. doi:10.3390/ijms242115772
3. Foessl I, Dimai HP, Obermayer-Pietsch B. Long-term and sequential treatment for osteoporosis. Nat Rev Endocrinol. 2023;19(9):520–533. doi:10.1038/s41574-023-00866-9
4. Wang S, Deng Z, Ma Y, et al. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int J Biol Sci. 2020;16(14):2675–2691. doi:10.7150/ijbs.46627
5. Iorio R, Petricca S, Mattei V, et al. Horizontal mitochondrial transfer as a novel bioenergetic tool for mesenchymal stromal/stem cells: molecular mechanisms and therapeutic potential in a variety of diseases. J Transl Med. 2024;22(1):491. doi:10.1186/s12967-024-05047-4
6. Gao J, Qin A, Liu D, et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci Adv. 2019;5(11):eaaw7215. doi:10.1126/sciadv.aaw7215
7. Liu Z, Sun Y, Qi Z, et al. Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci. 2022;12(1):66. doi:10.1186/s13578-022-00805-7
8. Aibar-Almazán A, Voltes-Martínez A, Castellote-Caballero Y, et al. Current Status of the Diagnosis and Management of Osteoporosis. Int J Mol Sci. 2022;23(16):9465. doi:10.3390/ijms23169465
9. Harris K, Zagar CA, Lawrence KV. Osteoporosis: common Questions and Answers. Am Fam Physician. 2023;107(3):238–246.
10. Lorenc R, Głuszko P, Franek E, et al. Guidelines for the diagnosis and management of osteoporosis in Poland: update 2017. Endokrynol Pol. 2017;68(5):604–609. doi:10.5603/EP.2017.0062
11. Qi P, Chen X, Liu H, et al. Regulatory Mechanisms of Gut Homeostasis and Bone Metabolism Interplay in Osteoporosis. Phenomics. 2025. doi:10.1007/s43657-024-00207-4
12. Ji J, Cai F, Yuan C, et al. Gut Microbiota in Primary Osteoporosis: a Systematic Review. Phenomics. 2024;4(3):293–297. doi:10.1007/s43657-024-00164-y
13. Reid IR, Billingto EO. Drug therapy for osteoporosis in older adults. Lancet. 2022;399(10329):1080–1092. doi:10.1016/S0140-6736(21)02646-5
14. Cosman F, Langdahl B, Leder BZ. Treatment Sequence for Osteoporosis. Endocr Pract. 2024;30(5):490–496. doi:10.1016/j.eprac.2024.01.014
15. Langdahl BL. Overview of treatment approaches to osteoporosis. Br J Pharmacol. 2021;178(9):1891–1906. doi:10.1111/bph.15024
16. Adler RA, El-Hajj Fuleihan G, Bauer DC, et al. Managing osteoporosis in patients on long-term bisphosphonate treatment: report of a task force of the americana society for bone and mineral research. J Bone Miner Res. 2016;31(1):16–35. doi:10.1002/jbmr.2708
17. Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet. 2013;382(9886):50–56. doi:10.1016/S0140-6736(13)60856-9
18. Close P, Neuprez A, Reginster J-Y. Developments in the pharmacotherapeutic management of osteoporosis. Expert Opin Pharmacother. 2006;7(12):1603–1615. doi:10.1517/14656566.7.12.1603
19. Händel MN, Cardoso I, Von Bülow C, et al. Fracture risk reduction and safety by osteoporosis treatment compared with placebo or active comparator in postmenopausal women: systematic review, network meta-analysis, and meta-regression analysis of randomised clinical trials. BMJ. 2023;381:e068033. doi:10.1136/bmj-2021-068033
20. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296(24):2927–2938. doi:10.1001/jama.296.24.2927
21. Rizi MM, Salari A, Salesi M, et al. Comparison of bone mineral density of osteoporotic and osteopenia menopausal women treated with oral bisphosphonates before stopping the treatment and 1 year after drug holiday period. Clin Rheumatol. 2024;43(4):1375–1379. doi:10.1007/s10067-024-06906-7
22. Cranney A, Waldegger L, Zytaruk N, et al. Risedronate for the prevention and treatment of postmenopausal osteoporosis. Cochrane Database Syst Rev. 2003;4:CD004523.
23. Ruggiero SL, Mehrotra B, Rosenberg TJ, et al. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62(5):527–534. doi:10.1016/j.joms.2004.02.004
24. Vestergaard P, Schwartz K, Pinholt EM, et al. Use of bisphosphonates and raloxifene and risk of deep venous thromboembolism and pulmonary embolism. Osteoporos Int. 2010;21(9):1591–1597. doi:10.1007/s00198-009-1091-y
25. Le QA, Hay JW, Becker R, et al. Cost-effectiveness Analysis of Sequential Treatment of Abaloparatide Followed by Alendronate Versus Teriparatide Followed by Alendronate in Postmenopausal Women With Osteoporosis in the United States. Ann Pharmacother. 2019;53(2):134–143. doi:10.1177/1060028018798034
26. Hernlund E, Svedbom A, Ivergård M, et al. Osteoporosis in the European Union: medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch Osteoporos. 2013;8(1):136. doi:10.1007/s11657-013-0136-1
27. Li N, Cornelissen D, Silverman S, et al. An Updated Systematic Review of Cost-Effectiveness Analyses of Drugs for Osteoporosis. Pharmacoeconomics. 2021;39(2):181–209. doi:10.1007/s40273-020-00965-9
28. Mariniakova M, Mondockova V, Kovacova V, et al. Interrelationships among metabolic syndrome, bone-derived cytokines, and the most common metabolic syndrome-related diseases negatively affecting bone quality. Diabetol Metab Syndr. 2024;16(1):217. doi:10.1186/s13098-024-01440-7
29. Liu J, Gao Z, Liu X. Mitochondrial dysfunction and therapeutic perspectives in osteoporosis. Front Endocrinol (Lausanne). 2024;15:1325317. doi:10.3389/fendo.2024.1325317
30. Yan C, Shi Y, Yuan L, et al. Mitochondrial quality control and its role in osteoporosis. Front Endocrinol. 2023;14:1077058. doi:10.3389/fendo.2023.1077058
31. Jimi E, Takakura N, Hiura F, et al. The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: is NF-κB Inhibition “Killing Two Birds with One Stone”? Cells. 2019;8(12):1636. doi:10.3390/cells8121636
32. Noack DV. Role of NF-κB in the skeleton. Cell Res. 2011;21(1):169–182. doi:10.1038/cr.2010.159
33. Boyce BF, Xing L, Franzoso G, et al. Required and nonessential functions of nuclear factor-kappa B in bone cells. Bone. 1999;25(1):137–139. doi:10.1016/S8756-3282(99)00105-2
34. Dyson HJ, Komives EA. Role of disorder in IκB-NFκB interaction. IUBMB Life. 2012;64(6):499–505. doi:10.1002/iub.1044
35. Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2(3):a000158. doi:10.1101/cshperspect.a000158
36. Khoshnan A, Ko J, Watkin EE, et al. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci. 2004;24(37):7999–8008. doi:10.1523/JNEUROSCI.2675-04.2004
37. Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;1999(5):RE1. doi:10.1126/stke.1999.5.re1
38. Canton M, Sánchez-Rodríguez R, Spera I, et al. Reactive Oxygen Species in Macrophages: sources and Targets. Front Immunol. 2021;12:734229. doi:10.3389/fimmu.2021.734229
39. Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14(6):1065–1077. doi:10.1089/ars.2010.3376
40. Huff LA, Yan S, Clemens MG. Mechanisms of Ataxia Telangiectasia Mutated (ATM) Control in the DNA Damage Response to Oxidative Stress, Epigenetic Regulation, and Persistent Innate Immune Suppression Following Sepsis. Antioxidants. 2021;10(7). doi:10.3390/antiox10071146
41. Pagano MA, Frezzato F, Visentin A, et al. Protein Phosphorylation and Redox Status: an as Yet Elusive Dyad in Chronic Lymphocytic Leukemia. Cancers (Basel). 2022;14(19):4881. doi:10.3390/cancers14194881
42. Morgan EL, Chen Z, Van Waes C. Regulation of NFκB Signalling by Ubiquitination: a Potential Therapeutic Target in Head and Neck Squamous Cell Carcinoma? Cancers. 2020;12(10):2877. doi:10.3390/cancers12102877
43. Wang Y, Mo X, Piper MG, et al. M-CSF induces monocyte survival by activating NF-κB p65 phosphorylation at Ser276 via protein kinase C. PLoS One. 2011;6(12):e28081. doi:10.1371/journal.pone.0028081
44. Xu C, Tang J, Yang Q, et al. Histone deacetylase 5 deacetylates the phosphatase PP2A for positively regulating NF-κB signaling. J Biol Chem. 2021;297(6):101380. doi:10.1016/j.jbc.2021.101380
45. Sheu J-R, Chen Z-C, Hsu M-J, et al. CME-1, a novel polysaccharide, suppresses iNOS expression in lipopolysaccharide-stimulated macrophages through ceramide-initiated protein phosphatase 2A activation. J Cell Mol Med. 2018;22(2):999–1013. doi:10.1111/jcmm.13424
46. Barisic S, Strozyk E, Peters N, et al. Identification of PP2A as a crucial regulator of the NF-kappaB feedback loop: its inhibition by UVB turns NF-kappaB into a pro-apoptotic factor. Cell Death Differ. 2008;15(11):1681–1690. doi:10.1038/cdd.2008.98
47. Tsuchiya Y, Osaki K, Kanamoto M, et al. Distinct B subunits of PP2A regulate the NF-κB signalling pathway through dephosphorylation of IKKβ, IκBα and RelA. FEBS Lett. 2017;591(24):4083–4094. doi:10.1002/1873-3468.12912
48. Funato Y, Miki H. Reversible oxidation of PRL family protein-tyrosine phosphatases. Methods. 2014;65(2):184–189. doi:10.1016/j.ymeth.2013.06.032
49. Ostman A, Frijhoff J, Sandin A, et al. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150(4):345–356. doi:10.1093/jb/mvr104
50. Meng F-G, Zhang Z-Y. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta. 2013;1834(1):464–469. doi:10.1016/j.bbapap.2012.06.018
51. Zhang B, Pan C, Feng C, et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022;27(1):45–52. doi:10.1080/13510002.2022.2046423
52. Han W, Fessel JP, Sherrill T, et al. Enhanced Expression of Catalase in Mitochondria Modulates NF-κB-Dependent Lung Inflammation through Alteration of Metabolic Activity in Macrophages. J Immunol. 2020;205(4):1125–1134. doi:10.4049/jimmunol.1900820
53. Kastl L, Sauer SW, Ruppert T, et al. TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-κB activation in liver cells. FEBS Lett. 2014;588(1):175–183. doi:10.1016/j.febslet.2013.11.033
54. Chu T, Wang Y, Wang S, et al. Kaempferol regulating macrophage foaming and atherosclerosis through Piezo1-mediated MAPK/NF-κB and Nrf2/HO-1 signaling pathway. J Adv Res. 2024. doi:10.1016/j.jare.2024.11.016
55. Gong J, Sun P, Li L, et al. Heat stress suppresses MnSOD expression via p53-Sp1 interaction and induces oxidative stress damage in endothelial cells: protective effects of MitoQ10 and Pifithrin-α. Heliyon. 2023;9(12):e22805. doi:10.1016/j.heliyon.2023.e22805
56. Wen JJ, Garg NJ. Manganese superoxide dismutase deficiency exacerbates the mitochondrial ROS production and oxidative damage in Chagas disease. PLoS Negl Trop Dis. 2018;12(7):e0006687. doi:10.1371/journal.pntd.0006687
57. Han B, Geng H, Liu L, et al. GSH attenuates RANKL-induced osteoclast formation in vitro and LPS-induced bone loss in vivo. Biomed Pharmacother. 2020;128:110305. doi:10.1016/j.biopha.2020.110305
58. Alqranei MS, Aljohani H, Majumdar S, et al. C-phycocyanin attenuates RANKL-induced osteoclastogenesis and bone resorption in vitro through inhibiting ROS levels, NFATc1 and NF-κB activation. Sci Rep. 2020;10(1):2513. doi:10.1038/s41598-020-59363-y
59. Yang Y, Liu Z, Wu J, et al. Nrf2 Mitigates RANKL and M-CSF Induced Osteoclast Differentiation via ROS-Dependent Mechanisms. Antioxidants. 2023;12(12): 2094.
60. An Y, Zhang H, Wang C, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019;33(11):12515–12527. doi:10.1096/fj.201802805RR
61. Wang X, Chen B, Sun J, et al. Iron-induced oxidative stress stimulates osteoclast differentiation via NF-κB signaling pathway in mouse model. Metabolism. 2018;83:167–176. doi:10.1016/j.metabol.2018.01.005
62. Wang H, Cao X, Guo J, et al. BNTA alleviates inflammatory osteolysis by the SOD mediated anti-oxidation and anti-inflammation effect on inhibiting osteoclastogenesis. Front Pharmacol. 2022;13:939929. doi:10.3389/fphar.2022.939929
63. Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31(3):266–300. doi:10.1210/er.2009-0024
64. Zhou Y, Deng Y, Liu Z, et al. Cytokine-scavenging nanodecoys reconstruct osteoclast/osteoblast balance toward the treatment of postmenopausal osteoporosis. Sci Adv. 2021;7(48):eabl6432. doi:10.1126/sciadv.abl6432
65. Iantomasi T, Romagnoli C, Palmini G, et al. Oxidative Stress and Inflammation in Osteoporosis: molecular Mechanisms Involved and the Relationship with microRNAs. Int J Mol Sci. 2023;24(4):3772. doi:10.3390/ijms24043772
66. Mohamad N-V, Ima-Nirwana S, Chin K-Y. Are Oxidative Stress and Inflammation Mediators of Bone Loss Due to Estrogen Deficiency? A Review of Current Evidence. Endocr Metab Immune Disord Drug Targets. 2020;20(9):1478–1487. doi:10.2174/1871530320666200604160614
67. Goppelt-Struebe M, Hahn A, Iwanciw D, et al. Regulation of connective tissue growth factor (ccn2; ctgf) gene expression in human mesangial cells: modulation by HMG CoA reductase inhibitors (statins). Mol Pathol. 2001;54(3):176–179. doi:10.1136/mp.54.3.176
68. Pereira RC, Durant D, Canalis E. Transcriptional regulation of connective tissue growth factor by cortisol in osteoblasts. Am J Physiol Endocrinol Metab. 2000;279(3):E570–E6. doi:10.1152/ajpendo.2000.279.3.E570
69. Tufail M, Jiang C-H, Li N. Altered metabolism in cancer: insights into energy pathways and therapeutic targets. Mol Cancer. 2024;23(1):203. doi:10.1186/s12943-024-02119-3
70. Stegen S, Devignes C-S, Torrekens S, et al. Glutamine Metabolism in Osteoprogenitors Is Required for Bone Mass Accrual and PTH-Induced Bone Anabolism in Male Mice. J Bone Miner Res. 2021;36(3):604–616. doi:10.1002/jbmr.4219
71. Sharma D, Yu Y, Shen L, et al. SLC1A5 provides glutamine and asparagine necessary for bone development in mice. Elife. 2021;10.
72. Misra BB, Jayapalan S, Richards AK, et al. Untargeted metabolomics in primary murine bone marrow stromal cells reveals distinct profile throughout osteoblast differentiation. Metabolomics. 2021;17(10):86. doi:10.1007/s11306-021-01829-9
73. Tencerova M, Figeac F, Ditzel N, et al. High-Fat Diet-Induced Obesity Promotes Expansion of Bone Marrow Adipose Tissue and Impairs Skeletal Stem Cell Functions in Mice. J Bone Miner Res. 2018;33(6):1154–1165. doi:10.1002/jbmr.3408
74. Hu G, Yu Y, Sharma D, et al. Glutathione limits RUNX2 oxidation and degradation to regulate bone formation. JCI Insight. 2023;8(16). doi:10.1172/jci.insight.166888.
75. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. doi:10.1152/physrev.00018.2001
76. Sevilla F, Camejo D, Ortiz-Espín A, et al. The thioredoxin/peroxiredoxin/sulfiredoxin system: current overview on its redox function in plants and regulation by reactive oxygen and nitrogen species. J Exp Bot. 2015;66(10):2945–2955. doi:10.1093/jxb/erv146
77. Barbour JA, Turner N. Mitochondrial stress signaling promotes cellular adaptations. Int J Cell Biol. 2014;2014:156020. doi:10.1155/2014/156020
78. Chava S, Chennakesavulu S, Gayatri BM, et al. A novel phosphorylation by AMP-activated kinase regulates RUNX2 from ubiquitination in osteogenesis over adipogenesis. Cell Death Dis. 2018;9(7):754. doi:10.1038/s41419-018-0791-7
79. Zhang Y, Li M, Lou P, et al. miRNA-seq analysis of high glucose induced osteoblasts provides insight into the mechanism underlying diabetic osteoporosis. Sci Rep. 2024;14(1):13441. doi:10.1038/s41598-024-64391-z
80. Wang K, Peng X, Zhang R, et al. COL6A3 enhances the osteogenic differentiation potential of BMSCs by promoting mitophagy in the osteoporotic microenvironment. Mol Biol Rep. 2024;51(1):206. doi:10.1007/s11033-023-08918-z
81. Liu F, Yuan Y, Bai L, et al. LRRc17 controls BMSC senescence via mitophagy and inhibits the therapeutic effect of BMSCs on ovariectomy-induced bone loss. Edox Biol. 2021;43:101963.
82. Landspersky T, Stein M, Saçma M, et al. Targeting CDC42 reduces skeletal degeneration after hematopoietic stem cell transplantation. Blood Adv. 2024;8:5400–5414. doi:10.1182/bloodadvances.2024012879
83. Tang X, Ma S, Li Y, et al. Evaluating the Activity of Sodium Butyrate to Prevent Osteoporosis in Rats by Promoting Osteal GSK-3β/Nrf2 Signaling and Mitochondrial Function. J Agric Food Chem. 2020;68(24):6588–6603. doi:10.1021/acs.jafc.0c01820
84. S-Y L, H-J A, Kim JM, et al. PINK1 deficiency impairs osteoblast differentiation through aberrant mitochondrial homeostasis. Stem Cell Res Ther. 2021;12(1):589. doi:10.1186/s13287-021-02656-4
85. Liu Y, Xin L, Wang S, et al. Nanoparticles with reprogramming of mitochondrial respiratory chain complex and epigenetic modifications functions for osteoporosis treatment. Biomaterials. 2025;324:123468. doi:10.1016/j.biomaterials.2025.123468
86. Hood DA, Memme JM, Oliveira AN, et al. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu Rev Physiol. 2019;81:19–41. doi:10.1146/annurev-physiol-020518-114310
87. Smith JAB, Murach KA, Dyar KA, et al. Exercise metabolism and adaptation in skeletal muscle. Nat Rev Mol Cell Biol. 2023;24(9):607–632. doi:10.1038/s41580-023-00606-x
88. Genders AJ, Holloway GP, Bishop DJ. Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance? Int J Mol Sci. 2020;21(18):6948. doi:10.3390/ijms21186948
89. Gan Z, Fu T, Kelly DP, et al. Skeletal muscle mitochondrial remodeling in exercise and diseases. Cell Res. 2018;28(10):969–980. doi:10.1038/s41422-018-0078-7
90. Joseph A-M, Adhihetty PJ, Buford TW, et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell. 2012;11(5):801–809. doi:10.1111/j.1474-9726.2012.00844.x
91. Picca A, Faitg J, Auwerx J, et al. Mitophagy in human health, ageing and disease. Nat Metab. 2023;5(12):2047–2061. doi:10.1038/s42255-023-00930-8
92. Memme JM, Slavin M, Moradi N, et al. Mitochondrial Bioenergetics and Turnover during Chronic Muscle Disuse. Int J Mol Sci. 2021;22(10). doi:10.3390/ijms22105179.
93. Noone J, Damiot A, Kenny H, et al. The impact of 60 days of −6° head down tilt bed rest on mitochondrial content, respiration and regulators of mitochondrial dynamics. J Physiol. 2023;603:3777–3794. doi:10.1113/JP284734
94. Mackie SL, Dejaco C, Appenzeller S, et al. British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis: executive summary. Rheumatology (Oxford). 2020;59(3):487–494. doi:10.1093/rheumatology/kez664
95. Zhao C, Wu Y, Zhu S, et al. Irisin protects musculoskeletal homeostasis via a mitochondrial quality control mechanism. Int J Mol Sci. 2024;25(18):10116.
96. Engelke K, Fuerst T, Dardzinski B, et al. Odanacatib treatment affects trabecular and cortical bone in the femur of postmenopausal women: results of a two-year placebo-controlled trial. J Bone Miner Res. 2015;30(1):30–38. doi:10.1002/jbmr.2292
97. Maldonado M, Molfese DL, Viswanath H, et al. The habenula as a novel link between the homeostatic and hedonic pathways in cancer-associated weight loss: a pilot study. J Cachexia, Sarcopenia Muscle. 2018;9(3):497–504. doi:10.1002/jcsm.12286
98. A Pan, Sun X-M, Huang F-Q, et al. The mitochondrial β-oxidation enzyme HADHA restrains hepatic glucagon response by promoting β-hydroxybutyrate production. Nat Commun. 2022;13(1):386. doi:10.1038/s41467-022-28044-x
99. Thapa S, Nandy A, Rendina-Ruedy E. Endocrinal metabolic regulation on the skeletal system in post-menopausal women. Front Physiol. 2022;13:1052429. doi:10.3389/fphys.2022.1052429
100. Amar D, Gay NR, Jimenez-Morales D, et al. The mitochondrial multi-omic response to exercise training across rat tissues. Cell Metab. 2024;36(6):1411–1429.e10. doi:10.1016/j.cmet.2023.12.021
101. Handzlik-Orlik G, Holecki M, Wilczyński K, et al. Osteoporosis in liver disease: pathogenesis and management. Ther Adv Endocrinol Metab. 2016;7(3):128–135. doi:10.1177/2042018816641351
102. Lin L, Guo Z, He E, et al. SIRT2 regulates extracellular vesicle-mediated liver-bone communication. Nat Metab. 2023;5(5):821–841. doi:10.1038/s42255-023-00803-0
103. Lu D, He A, Tan M, et al. Liver ACOX1 regulates levels of circulating lipids that promote metabolic health through adipose remodeling. Nat Commun. 2024;15(1):4214. doi:10.1038/s41467-024-48471-2
104. Sohn JH, Mutlu B, Latorre-Muro P, et al. Liver mitochondrial cristae organizing protein MIC19 promotes energy expenditure and pedestrian locomotion by altering nucleotide metabolism. Cell Metab. 2023;35(8):1356–1372.e5. doi:10.1016/j.cmet.2023.06.015
105. Li M, Yu Y, Xue K, et al. Genistein mitigates senescence of bone marrow mesenchymal stem cells via ERRα-mediated mitochondrial biogenesis and mitophagy in ovariectomized rats. Redox Biol. 2023;61:102649. doi:10.1016/j.redox.2023.102649
106. Guo Y, Chi X, Wang Y, et al. Mitochondria transfer enhances proliferation, migration, and osteogenic differentiation of bone marrow mesenchymal stem cell and promotes bone defect healing. Stem Cell Res Ther. 2020;11(1):245. doi:10.1186/s13287-020-01704-9
107. Ma S, Ding R, Cao J, et al. Mitochondria transfer reverses the inhibitory effects of low stiffness on osteogenic differentiation of human mesenchymal stem cells. Er J Cell Biol. 2023;102(2):151297. doi:10.1016/j.ejcb.2023.151297
108. Wei B, Ji M, Lin Y, et al. Mitochondrial transfer from bone mesenchymal stem cells protects against tendinopathy both in vitro and in vivo. Stem Cell Res Ther. 2023;14(1):104. doi:10.1186/s13287-023-03329-0
109. X Yao, Ma Y, Zhou W, et al. In-cytoplasm mitochondrial transplantation for mesenchymal stem cells engineering and tissue regeneration. Bioeng Transl Med. 2022;7(1):e10250. doi:10.1002/btm2.10250
110. Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi:10.1038/290457a0
111. Falkenberg M. Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem. 2018;62(3):287–296. doi:10.1042/EBC20170100
112. Watson E, Forster P, Richards M, et al. Mitochondrial footprints of human expansions in Africa. Am J Hum Genet. 1997;61(3):691–704. doi:10.1086/515503
113. Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16(9):530–542. doi:10.1038/nrg3966
114. Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105(6):1342–1351. doi:10.1002/jcb.21936
115. Chen J-Q, Cammarata PR, Baines CP, et al. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;1793(10):1540–1570. doi:10.1016/j.bbamcr.2009.06.001
116. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23(9):459–466. doi:10.1016/j.tem.2012.06.006
117. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88(2):611–638. doi:10.1152/physrev.00025.2007
118. Scarpulla RC. Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim Biophys Acta. 2012;1819(9–10):1088–1097. doi:10.1016/j.bbagrm.2011.10.011
119. Klinge CM. Estrogenic control of mitochondrial function. Redox Biol. 2020;31:101435. doi:10.1016/j.redox.2020.101435
120. López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
121. Jang JY, Blum A, Liu J, et al. The role of mitochondria in aging. J Clin Invest. 2018;128(9):3662–3670. doi:10.1172/JCI120842
122. Lima T, Li TY, Mottis A, et al. Pleiotropic effects of mitochondria in aging. Nat Aging. 2022;2(3):199–213. doi:10.1038/s43587-022-00191-2
123. Lanza IR, Nair KS. Mitochondrial function as a determinant of life span. Pflugers Arch. 2010;459(2):277–289. doi:10.1007/s00424-009-0724-5
124. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016;61(5):654–666. doi:10.1016/j.molcel.2016.01.028
125. Sarver DC, Saqib M, Chen F, et al. Mitochondrial respiration atlas reveals differential changes in mitochondrial function across sex and age. Elife. 2024;13:RP96926.
126. Schaum N, Lehallier B, Hahn O, et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020;583(7817):596–602. doi:10.1038/s41586-020-2499-y
127. Zhang MJ, Pisco AO, Darmanis S, et al. Mouse aging cell atlas analysis reveals global and cell type-specific aging signatures. Elife. 2021;10:1.
128. Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20(4):145–147. doi:10.1111/j.1532-5415.1972.tb00787.x
129. Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102(15):5618–5623. doi:10.1073/pnas.0501559102
130. Stauch KL, Purnell PR, Fox HS. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging. 2014;6(4):320–334. doi:10.18632/aging.100657
131. Ye K, Lu J, Ma F, et al. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci U S A. 2014;111(29):10654–10659. doi:10.1073/pnas.1403521111
132. Zhang R, Wang Y, Ye K, et al. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genomics. 2017;18(1):890. doi:10.1186/s12864-017-4287-0
133. Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain. 2014;137(Pt 2):335–353. doi:10.1093/brain/awt343
134. Yu-wai-man P, Sitarz KS, Samuels DC, et al. OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum Mol Genet. 2010;19(15):3043–3052. doi:10.1093/hmg/ddq209
135. Rausser S, Trumpff C, Mcgill MA, et al. Mitochondrial phenotypes in purified human immune cell subtypes and cell mixtures. Elife. 2021;10:1.
136. Patin E, Hasan M, Bergstedt J, et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol. 2018;19(3):302–314. doi:10.1038/s41590-018-0049-7
137. Márquez EJ, Chung C-H, Marches R, et al. Sexual-dimorphism in human immune system aging. Nat Commun. 2020;11(1):751. doi:10.1038/s41467-020-14396-9
138. Nikolich-žugich J. Aging of the T cell compartment in mice and humans: from no naive expectations to foggy memories. J Immunol. 2014;193(6):2622–2629. doi:10.4049/jimmunol.1401174
139. Austad SN, Fischer KE. Sex Differences in Lifespan. Cell Metab. 2016;23(6):1022–1033. doi:10.1016/j.cmet.2016.05.019
140. Gaignard P, Liere P, Thérond P, et al. Role of Sex Hormones on Brain Mitochondrial Function, with Special Reference to Aging and Neurodegenerative Diseases. Front Aging Neurosci. 2017;9:406. doi:10.3389/fnagi.2017.00406
141. Sultanova RF, Schibalski R, Yankelevich IA, et al. Sex differences in renal mitochondrial function: a hormone-gous opportunity for research. Am J Physiol Renal Physiol. 2020;319(6):F1117–F24. doi:10.1152/ajprenal.00320.2020
142. Sutovsky P, Moreno RD, Ramalho-Santos J, et al. Ubiquitin tag for sperm mitochondria. Nature. 1999;402(6760):371–372. doi:10.1038/46466
143. Pollara J, Edwards RW, Lin L, et al. Circulating mitochondria in deceased organ donors are associated with immune activation and early allograft dysfunction [J]. JCI Insight. 2018;3(15). doi:10.1172/jci.insight.121622.
144. Ramirez-Barbieri G, Moskowitzova K, Shin B, et al. Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria [J]. Mitochondrion. 2019;46:103–115. doi:10.1016/j.mito.2018.03.002
145. Zhang T-G, Miao C-Y. Mitochondrial transplantation as a promising therapy for mitochondrial diseases. Acta Pharm Sin B. 2023;13(3):1028–1035. doi:10.1016/j.apsb.2022.10.008
146. Sun C, Liu X, Wang B, et al. Endocytosis-mediated mitochondrial transplantation: transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics. 2019;9(12):3595–3607. doi:10.7150/thno.33100
147. Kim JS, Lee S, Kim W-K, et al. Mitochondrial transplantation: an overview of a promising therapeutic approach. BMB Rep. 2023;56(9):488–495. doi:10.5483/BMBRep.2023-0098
148. Røyrvik EC, Burgstaller JP, Johnston IG. mtDNA diversity in human populations highlights the merit of haplotype matching in gene therapies. Mol Hum Reprod. 2016;22(11):809–817. doi:10.1093/molehr/gaw062
149. Levoux J, Prola A, Lafuste P, et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 2021;33(2):688–690. doi:10.1016/j.cmet.2021.02.003
150. Qin Y, Jiang X, Yang Q, et al. The Functions, Methods, and Mobility of Mitochondrial Transfer Between Cells. Front Oncol. 2021;11:672781. doi:10.3389/fonc.2021.672781
151. Abounit S, Zurzolo C. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J Cell Sci. 2012;125(Pt 5):1089–1098. doi:10.1242/jcs.083279
152. Wang X, Gerdes HH. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015;22(7):1181–1191. doi:10.1038/cdd.2014.211
153. Caicedo A, Aponte PM, Cabrera F, et al. Artificial Mitochondria Transfer: current Challenges, Advances, and Future Applications. Stem Cells Int. 2017;2017:7610414. doi:10.1155/2017/7610414
154. Preble JM, Pacak CA, Kondo H, et al. Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J Vis Exp. 2014;91:e51682.
155. Pinkert CA, Irwin MH, Johnson LW, et al. Mitochondria transfer into mouse ova by microinjection. Transgenic Res. 1997;6(6):379–383. doi:10.1023/A:1018431316831
156. Cai W, Mao S, Wang Y, et al. An Engineered Hierarchical Hydrogel with Immune Responsiveness and Targeted Mitochondrial Transfer to Augmented Bone Regeneration. Adv Sci. 2024;11(42):e2406287. doi:10.1002/advs.202406287
157. Qiu S, Cao L, Xiang D, et al. Enhanced osteogenic differentiation in 3D hydrogel scaffold via macrophage mitochondrial transfer. J Nanobiotechnology. 2024;22(1):540. doi:10.1186/s12951-024-02757-1
158. Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505(7483):335–343. doi:10.1038/nature12985
159. Schapira AHV. Mitochondrial diseases. Lancet. 2012;379(9828):1825–1834. doi:10.1016/S0140-6736(11)61305-6
160. Borcherding N, Brestoff JR. The power and potential of mitochondria transfer. Nature. 2023;623(7986):283–291. doi:10.1038/s41586-023-06537-z
161. Nakai R, Varnum S, Field RL, et al. Mitochondria transfer-based therapies reduce the morbidity and mortality of Leigh syndrome. Nat Metab. 2024;6(10):1886–1896. doi:10.1038/s42255-024-01125-5
162. L Liu, Yang J, Otani Y, et al. MELAS-Derived Neurons Functionally Improve by Mitochondrial Transfer from Highly Purified Mesenchymal Stem Cells (REC). Int J Mol Sci. 2023;24(24):2.
163. Li C, Ji H, Zhuang S, et al. Update on the correlation between mitochondrial function and osteonecrosis of the femoral head osteocytes. Redox Rep. 2025;30(1):2491846. doi:10.1080/13510002.2025.2491846
164. Zong Y, Li H, Liao P, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9(1):124. doi:10.1038/s41392-024-01839-8
165. Wang Y, Li W, Guo Y, et al. Mitochondria Transplantation to Bone Marrow Stromal Cells Promotes Angiogenesis During Bone Repair. Adv Sci. 2024;11(39):e2403201. doi:10.1002/advs.202403201
166. Bagheri HS, Bani F, Tasoglu S, et al. Mitochondrial donation in translational medicine; from imagination to reality. J Transl Med. 2020;18(1):367. doi:10.1186/s12967-020-02529-z
167. Aguilar PS, Baylies MK, Fleissner A, et al. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013;29(7):427–437. doi:10.1016/j.tig.2013.01.011
168. Ikeda G, Santoso MR, Tada Y, et al. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J Am Coll Cardiol. 2021;77(8):1073–1088. doi:10.1016/j.jacc.2020.12.060
169. Cai W, Zhang J, Yu Y, et al. Mitochondrial Transfer Regulates Cell Fate Through Metabolic Remodeling in Osteoporosis. Adv Sci. 2023;10(4):e2204871. doi:10.1002/advs.202204871
170. Zambrano K, Barba D, Castillo K, et al. The war against Alzheimer, the mitochondrion strikes back! Mitochondrion. 2022;64:125–135. doi:10.1016/j.mito.2022.03.003
171. Wu T-H, Sagullo E, Case D, et al. Mitochondrial Transfer by Photothermal Nanoblade Restores Metabolite Profile in Mammalian Cells. Cell Metab. 2016;23(5):921–929. doi:10.1016/j.cmet.2016.04.007
172. Gäbelein CG, Feng Q, Sarajlic E, et al. Mitochondria transplantation between living cells. PLoS Biol. 2022;20(3):e3001576. doi:10.1371/journal.pbio.3001576
173. Reissmann S. Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci. 2014;20(10):760–784. doi:10.1002/psc.2672
174. Nativo P, Prior IA, Brust M. Uptake and intracellular fate of surface-modified gold nanoparticles. ACS Nano. 2008;2(8):1639–1644. doi:10.1021/nn800330a
175. De La Fuente JM, Berry CC. Tat peptide as an efficient molecule to translocate gold nanoparticles into the cell nucleus. Bioconjug Chem. 2005;16(5):1176–1180.
176. Guariento A, Piekarski BL, Doulamis IP, et al. Autologous mitochondrial transplantation for cardiogenic shock in pediatric patients following ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2021;162(3):992–1001. doi:10.1016/j.jtcvs.2020.10.151
177. Kaza AK, Wamala I, Friehs I, et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg. 2017;153(4):934–943. doi:10.1016/j.jtcvs.2016.10.077
178. Moskowitzova K, Shin B, K Liu, et al. Mitochondrial transplantation prolongs cold ischemia time in murine heart transplantation. J Heart Lug Transplant. 2019;38(1):92–99. doi:10.1016/j.healun.2018.09.025
179. Masuzawa A, Black KM, Pacak CA, et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304(7):H966–H82. doi:10.1152/ajpheart.00883.2012
180. D’amato M, Morra F, Di Meo I, et al. Mitochondrial Transplantation in Mitochondrial Medicine: current Challenges and Future Perspectives. Int J Mol Sci. 2023;24(3):1969. doi:10.3390/ijms24031969
181. Kesner EE, Saada-Reich A, LORBERBOUM-GALSKI H. Characteristics of Mitochondrial Transformation into Human Cells. Sci Rep. 2016;6:26057. doi:10.1038/srep26057
182. Mccully JD, Cowan DB, Pacak CA, et al. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 2009;296(1):H94–H105. doi:10.1152/ajpheart.00567.2008
183. Pacak CA, Preble JM, Kondo H, et al. Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol Open. 2015;4(5):622–626. doi:10.1242/bio.201511478
184. Pfeiffer ER, Wright AT, Edwards AG, et al. Caveolae in ventricular myocytes are required for stretch-dependent conduction slowing. J Mol Cell Cardiol. 2014;76:265–274. doi:10.1016/j.yjmcc.2014.09.014
185. Cowan DB, Yao R, Thedsanamoorthy JK, et al. Transit and integration of extracellular mitochondria in human heart cells. Sci Rep. 2017;7(1):17450. doi:10.1038/s41598-017-17813-0
186. Boriachek K, Islam MN, Möller A, et al. Biological Functions and Current Advances in Isolation and Detection Strategies for Exosome Nanovesicles. Small. 2018;14(6). doi:10.1002/smll.201702153.
187. Ibrahim A, Marbán E. Exosomes: fundamental Biology and Roles in Cardiovascular Physiology. Annu Rev Physiol. 2016;78:67–83. doi:10.1146/annurev-physiol-021115-104929
188. Chen H, Wang L, Zeng X, et al. Exosomes, a New Star for Targeted Delivery. Front Cell Dev Biol. 2021;9:751079. doi:10.3389/fcell.2021.751079
189. Wang F-S, Wu R-W, Chen Y-S, et al. Biophysical Modulation of the Mitochondrial Metabolism and Redox in Bone Homeostasis and Osteoporosis: how Biophysics Converts into Bioenergetics. Antioxidants. 2021;10(9):3.
190. Rizzuto R, De Stefani D, Raffaello A, et al. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13(9):566–578. doi:10.1038/nrm3412
191. Walkon LL, Strubbe-Rivera JO, Bazil JN. Calcium Overload and Mitochondrial Metabolism. Biomolecules. 2022;12(12):1891. doi:10.3390/biom12121891
192. Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol. 1999;516(Pt 1):1–17. doi:10.1111/j.1469-7793.1999.001aa.x
193. Madison MN, Okeoma CM. Exosomes: implications in HIV-1 Pathogenesis. Viruses. 2015;7(7):4093–4118. doi:10.3390/v7072810
194. Fahey M, Bennett M, Thomas M, et al. Mesenchymal stromal cells donate mitochondria to articular chondrocytes exposed to mitochondrial, environmental, and mechanical stress. Sci Rep. 2022;12(1):21525. doi:10.1038/s41598-022-25844-5
195. Thomas MA, Fahey MJ, Pugliese BR, et al. Human mesenchymal stromal cells release functional mitochondria in extracellular vesicles. Front Bioeng Biotechnol. 2022;10:870193. doi:10.3389/fbioe.2022.870193
196. L Xia, Zhang C, Lv N, et al. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics. 2022;12(6):2928–2947. doi:10.7150/thno.69533
197. Wang X, Berkowicz A, King K, et al. Pharmacologic enrichment of exosome yields and mitochondrial cargo. Mitochondrion. 2022;64:136–144. doi:10.1016/j.mito.2022.04.001
198. Tashiro R, Bautista-Garrido J, Ozaki D, et al. Transplantation of Astrocytic Mitochondria Modulates Neuronal Antioxidant Defense and Neuroplasticity and Promotes Functional Recovery after Intracerebral Hemorrhage. J Neurosci. 2022;42(36):7001–7014. doi:10.1523/JNEUROSCI.2222-21.2022
199. Kim MJ, Hwang JW, Yun C-K, et al. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci Rep. 2018;8(1):3330. doi:10.1038/s41598-018-21539-y
200. Li N, Zhao L, Geng X, et al. Stimulation by exosomes from hypoxia-preconditioned hair follicle mesenchymal stem cells facilitates mitophagy by inhibiting the PI3K/AKT/mTOR signaling pathway to alleviate ulcerative colitis. Theranostics. 2024;14(11):4278–4296. doi:10.7150/thno.96038
201. Chen Z, Zhang M, Xu Q, et al. Huangqi-Danshen decoction improves heart failure by regulating pericardial adipose tissue derived extracellular vesicular miR-27a-3p to activate AMPKα2 mediated mitophagy. Phytomedicine. 2024;135:156187. doi:10.1016/j.phymed.2024.156187
202. Tomasetti M, Lee W, Santarelli L, et al. Exosome-derived microRNAs in cancer metabolism: possible implications in cancer diagnostics and therapy. Exp Mol Med. 2017;49(1):e285. doi:10.1038/emm.2016.153
203. Li N, Shu J, Yang X, et al. Exosomes Derived From M2 Microglia Cells Attenuates Neuronal Impairment and Mitochondrial Dysfunction in Alzheimer’s Disease Through the PINK1/Parkin Pathway. Front Cell Neurosci. 2022;16:874102. doi:10.3389/fncel.2022.874102
204. Lee KS, Lee J, Kim HK, et al. Extracellular vesicles from adipose tissue-derived stem cells alleviate osteoporosis through osteoprotegerin and miR-21-5p. J Extracell Vesicles. 2021;10(12):e12152. doi:10.1002/jev2.12152
205. Minamizaki T, Nakao Y, Irie Y, et al. The matrix vesicle cargo miR-125b accumulates in the bone matrix, inhibiting bone resorption in mice. Commun Biol. 2020;3(1):30. doi:10.1038/s42003-020-0754-2
206. Wang T, Mo L, Ou J, et al. Proteus mirabilis Vesicles Induce Mitochondrial Apoptosis by Regulating miR96-5p/Abca1 to Inhibit Osteoclastogenesis and Bone Loss. Front Immunol. 2022;13:833040. doi:10.3389/fimmu.2022.833040
207. Wang X, Yang Y, Li W, et al. Umbilical mesenchymal stem cell-derived exosomes promote spinal cord functional recovery through the miR-146b/TLR4 -mediated NF-κB p65 signaling pathway in rats. Biochem Biophys Rep. 2023;35:101497. doi:10.1016/j.bbrep.2023.101497
208. Ma S, Xing X, Huang H, et al. Skeletal muscle-derived extracellular vesicles transport glycolytic enzymes to mediate muscle-to-bone crosstalk. Cell Metab. 2023;35(11):2028–2043.e7. doi:10.1016/j.cmet.2023.10.013
209. Tan SHS, Wong JRY, Sim SJY, et al. Mesenchymal stem cell exosomes in bone regenerative strategies-a systematic review of preclinical studies. Mater Today Bio. 2020;7:100067. doi:10.1016/j.mtbio.2020.100067
210. He X, Wang Y, Z Liu, et al. Osteoporosis treatment using stem cell-derived exosomes: a systematic review and meta-analysis of preclinical studies. Stem Cell Res Ther. 2023;14(1):72. doi:10.1186/s13287-023-03317-4
211. Song H, Li X, Zhao Z, et al. Reversal of Osteoporotic Activity by Endothelial Cell-Secreted Bone Targeting and Biocompatible Exosomes. Nano Lett. 2019;19(5):3040–3048. doi:10.1021/acs.nanolett.9b00287
212. Koh HB, Kim HJ, Kang S-W, et al. Exosome-Based Drug Delivery: translation from Bench to Clinic. Pharmaceutics. 2023;15(8):2042. doi:10.3390/pharmaceutics15082042
213. Zhang B, Gong J, He L, et al. Exosomes based advancements for application in medical aesthetics. Front Bioeng Biotechnol. 2022;10:1083640. doi:10.3389/fbioe.2022.1083640
214. Rao D, Huang D, Sang C, et al. Advances in Mesenchymal Stem Cell-Derived Exosomes as Drug Delivery Vehicles. Front Bioeng Biotechnol. 2021;9:797359. doi:10.3389/fbioe.2021.797359
215. Wang J, Li X, Wang S, et al. Bone-Targeted Exosomes: strategies and Applications. Adv Healthc Mater. 2023;12(18):e2203361. doi:10.1002/adhm.202203361
216. Zhao M, Li Q, Chai Y, et al. An anti-CD19-exosome delivery system navigates the blood-brain barrier for targeting of central nervous system lymphoma. J Nanobiotechnology. 2025;23(1):173. doi:10.1186/s12951-025-03238-9
217. Powell SK. Celebrations of Life—Why Wait? Professional Case management. 2021;26(6):273–274. doi:10.1097/NCM.0000000000000532
218. Akbari A, Nazari-Khanamiri F, Ahmadi M, et al. Engineered Exosomes for Tumor-Targeted Drug Delivery: a Focus on Genetic and Chemical Functionalization. Pharmaceutics. 2022;15(1):66. doi:10.3390/pharmaceutics15010066
219. Zheng X, Ai H, Qian K, et al. Small extracellular vesicles purification and scale-up. Front Immunol. 2024;15:1344681. doi:10.3389/fimmu.2024.1344681
220. Ludwig N, Whiteside TL, Reichert TE. Challenges in Exosome Isolation and Analysis in Health and Disease. Int J Mol Sci. 2019;20(19):4684. doi:10.3390/ijms20194684
221. Lai JJ, Chau ZL, Chen S-Y, et al. Exosome Processing and Characterization Approaches for Research and Technology Development. Adv Sci. 2022;9(15):e2103222. doi:10.1002/advs.202103222
222. Zheng Q, Liu H, Zhang H, et al. Ameliorating Mitochondrial Dysfunction of Neurons by Biomimetic Targeting Nanoparticles Mediated Mitochondrial Biogenesis to Boost the Therapy of Parkinson’Parkinson’s Disease. Adv Sci. 2023;10(22):e2300758. doi:10.1002/advs.202300758
223. Sun W, Liu L, Liu Y, et al. Biomimetic Coacervates Protocells for Dual Delivery Pathways and Apoptosis/Pyroptosis Based Drug-Free Therapeutics. Small. 2025;21(29):e2503027. doi:10.1002/smll.202503027
224. Ning P, Huang L, Bao Y, et al. Portfolio Targeting Strategy To Realize the Assembly and Membrane Fusion-Mediated Delivery of Gold Nanoparticles to Mitochondria for Enhanced NIR Photothermal Therapies. Bioconjug Chem. 2020;31(12):2719–2725. doi:10.1021/acs.bioconjchem.0c00518
225. Zhang Y, Pan V, Li X, et al. Dynamic DNA Structures. Small. 2019;15(26):e1900228. doi:10.1002/smll.201900228
226. Kumar V, Palazzolo S, Bayda S, et al. DNA Nanotechnology for Cancer Therapeutics. Theranostics. 2016;6(5):710–725. doi:10.7150/thno.14203
227. Kearney CJ, Lucas CR, O’Brien FJ, et al. DNA Origami: folded DNA-Nanodevices That Can Direct and Interpret Cell Behavior. Adv Mater. 2016;28(27):5509–5524. doi:10.1002/adma.201504733
228. Liu F, Liu X, Gao W, et al. Transmembrane capability of DNA origami sheet enhanced by 3D configurational changes. iScience. 2023;26(3):1.
229. Liang C, Guo B, Wu H, et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference–based bone anabolic strategy. Nat Med. 2015;21(3):288–294. doi:10.1038/nm.3791
230. Liu X, Hu J, Ning Y, et al. Aptamer Technology and Its Applications in Bone Diseases. Cell transplantation. 2023;32:9636897221144949. doi:10.1177/09636897221144949
231. Mao M-Z, Zheng M-H, Guo B, et al. FNDC5/irisin-enriched sEVs conjugated with bone-targeting aptamer alleviate osteoporosis: a potential alternative to exercise. J Nanobiotechnology. 2025;23(1):504. doi:10.1186/s12951-025-03587-5
232. Yoneda T, Hiasa M, Nagata Y, et al. Acidic microenvironment and bone pain in cancer-colonized bone. BoneKEy reports. 2015;4:690. doi:10.1038/bonekey.2015.58
233. Peppicelli S, Calorini L, Bianchini F, et al. Acidity and hypoxia of tumor microenvironment, a positive interplay in extracellular vesicle release by tumor cells. Cellular Oncology. 2025;48(1):27–41. doi:10.1007/s13402-024-00969-z
234. Wu M, Liu Y, Zhu X, et al. Advances in i-motif structures: stability, gene expression, and therapeutic applications. Int J Biol Macromol. 2025;311(Pt 4):143555. doi:10.1016/j.ijbiomac.2025.143555
235. Huang P, Tang X, Zhao S, et al. DNA nanorobot for mitochondria-targeted microRNAs detection and tailored regulation. Theranostics. 2025;15(10):4638–4653. doi:10.7150/thno.105762
236. Wang N, Xie Y, Xi Z, et al. Hope for bone regeneration: the versatility of iron oxide nanoparticles. Front Bioeng Biotechnol. 2022;10:–2022.
237. Huang T, Zhang T, Jiang X, et al. Iron oxide nanoparticles augment the intercellular mitochondrial transfer–mediated therapy. Sci Adv. 2021;7(40):eabj0534. doi:10.1126/sciadv.abj0534
238. Pant A, Singh G, Barnwal RP, et al. QbD-driven development and characterization of superparamagnetic iron oxide nanoparticles (SPIONS) of a bone-targeting peptide for early detection of osteoporosis. Int J Pharm. 2024;654:123936. doi:10.1016/j.ijpharm.2024.123936
239. Chen S, Zhuang D, Jia Q, et al. Advances in Noninvasive Molecular Imaging Probes for Liver Fibrosis Diagnosis. Biomater Res. 2024;28:0042. doi:10.34133/bmr.0042
240. Suh J, Kim N-K, Shim W, et al. Mitochondrial fragmentation and donut formation enhance mitochondrial secretion to promote osteogenesis. Cell Metab. 2023;35(2):345–360.e7. doi:10.1016/j.cmet.2023.01.003
241. Iwayama T, Okada T, Ueda T, et al. Osteoblastic lysosome plays a central role in mineralization. Sci Adv. 2019;5(7):eaax0672. doi:10.1126/sciadv.aax0672
242. Pei D-D, Sun J-L, Zhu C-H, et al. Contribution of Mitophagy to Cell-Mediated Mineralization: revisiting a 50-Year-Old Conundrum. Adv Sci. 2018;5(10):1800873. doi:10.1002/advs.201800873
243. Kim H-R, Woo S, Cho HB, et al. Osteoblast-Derived Mitochondria Formulated with Cationic Liposome Guide Mesenchymal Stem Cells into Osteogenic Differentiation. Adv Sci. 2025;12(12):e2412621. doi:10.1002/advs.202412621
244. Suh J, Lee Y-S. The multifaceted roles of mitochondria in osteoblasts: from energy production to mitochondrial-derived vesicle secretion. J Bone Miner Res. 2024;39(9):1205–1214. doi:10.1093/jbmr/zjae088
245. Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–1234. doi:10.1038/nm.2452
246. Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5(6):464–475. doi:10.1016/j.cmet.2007.05.001
247. Jin J, Nolte PA. Mitochondrial Distribution and Osteocyte Mechanosensitivity. Curr Osteoporos Rep. 2025;23(1):22. doi:10.1007/s11914-025-00918-1
248. Naon D, Zaninello M, Giacomello M, et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc Natl Acad Sci U S A. 2016;113(40):11249–11254. doi:10.1073/pnas.1606786113
249. Han S, Zhao F, Hsia J, et al. The role of Mfn2 in the structure and function of endoplasmic reticulum-mitochondrial tethering in vivo. J Cell Sci. 2021;134(13). doi:10.1242/jcs.253443.
250. López‐Doménech G, Covill‐Cooke C, Ivankovic D, et al. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018;37(3):321–336. doi:10.15252/embj.201696380
251. Tang BL. MIRO GTPases in Mitochondrial Transport, Homeostasis and Pathology. Cells. 2015;5(1):1. doi:10.3390/cells5010001
252. Liao P, Chen L, Zhou H, et al. Osteocyte mitochondria regulate angiogenesis of transcortical vessels. Nat Commun. 2024;15(1):2529. doi:10.1038/s41467-024-46095-0
253. Yang N, Liu Y. The Role of the Immune Microenvironment in Bone Regeneration. Int J Med Sci. 2021;18(16):3697–3707. doi:10.7150/ijms.61080
254. Clemente-Suárez VJ, Martín-Rodríguez A, Yáñez-Sepúlveda R, et al. Mitochondrial Transfer as a Novel Therapeutic Approach in Disease Diagnosis and Treatment. Int J Mol Sci. 2023;24(10):8848. doi:10.3390/ijms24108848
255. Liu Y, Dissanayaka WL, Yiu C. Therapeutic implications of mitochondrial transfer on stem cell fate in regenerative medicine. J Transl Med. 2025;23(1):568. doi:10.1186/s12967-025-06472-9
256. Planat-Benard V, Varin A, Casteilla L. MSCs and Inflammatory Cells Crosstalk in Regenerative Medicine: concerted Actions for Optimized Resolution Driven by Energy Metabolism. Front Immunol. 2021;12:626755. doi:10.3389/fimmu.2021.626755
257. Hsu Y-C, Wu Y-T, Yu T-H, et al. Mitochondria in mesenchymal stem cell biology and cell therapy: from cellular differentiation to mitochondrial transfer. Semin Cell Dev Biol. 2016;52:119–131. doi:10.1016/j.semcdb.2016.02.011
258. Zhu Y, Luo M, Bai X, et al. Administration of mesenchymal stem cells in diabetic kidney disease: mechanisms, signaling pathways, and preclinical evidence. Mol Cell Biochem. 2022;477(8):2073–2092. doi:10.1007/s11010-022-04421-4
259. Rodriguez A-M, Nakhle J, Griessinger E, et al. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle. 2018;17(6):712–721. doi:10.1080/15384101.2018.1445906
260. Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18(5):759–765. doi:10.1038/nm.2736
261. Li X, Zhang Y, Yeung SC, et al. Mitochondrial Transfer of Induced Pluripotent Stem Cell–Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke–Induced Damage. Am J Respir Cell Mol Biol. 2014;51(3):455–465. doi:10.1165/rcmb.2013-0529OC
262. Liu H, Li X, Li M, et al. POSTN-Mediated Interplay of M1 Polarized Macrophage with Tendon-Derived Stem Cells to Drive Traumatic Heterotopic Ossification Formation through PTK7/ATK Signaling? Adv Sci;2025:e07951. doi:10.1002/advs.202507951
263. Xu P, Lin B, Deng X, et al. VDR activation attenuates osteoblastic ferroptosis and senescence by stimulating the Nrf2/GPX4 pathway in age-related osteoporosis. Free Radic Biol Med. 2022;193(Pt 2):720–735. doi:10.1016/j.freeradbiomed.2022.11.013
264. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
265. Shadel G, Horvath T. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569. doi:10.1016/j.cell.2015.10.001
266. Ding P, Gao C, Zhou J, et al. Mitochondria from osteolineage cells regulate myeloid cell-mediated bone resorption. Nat Commun. 2024;15(1):5094. doi:10.1038/s41467-024-49159-3
267. Chen T, Majerníková NA, Marmolejo-Garza A, et al. Mitochondrial transplantation rescues neuronal cells from ferroptosis. Free Radic Biol Med. 2023;208:62–72. doi:10.1016/j.freeradbiomed.2023.07.034
268. Wu Z, Chen L, Guo W, et al. Oral mitochondrial transplantation using nanomotors to treat ischaemic heart disease. Nat Nanotechnol. 2024;19(9):1375–1385. doi:10.1038/s41565-024-01681-7
269. Nukala VN, Singh IN, Davis LM, et al. Cryopreservation of brain mitochondria: a novel methodology for functional studies. J Neurosci Methods. 2006;152(1–2):48–54. doi:10.1016/j.jneumeth.2005.08.017
270. Yamada Y, Ito M, Arai M, et al. Challenges in Promoting Mitochondrial Transplantation Therapy. Int J Mol Sci. 2020;21(17):6365. doi:10.3390/ijms21176365
271. Hu C, Shi Z, Liu X, et al. The Research Progress of Mitochondrial Transplantation in the Treatment of Mitochondrial Defective Diseases. Int J Mol Sci. 2024;25(2):1.
272. Huang T, Zhang T, Gao J. Targeted mitochondrial delivery: a therapeutic new era for disease treatment. J Control Release. 2022;343:89–106.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.