Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Mitochondrial multiorgan disorder syndrome score generated from definite mitochondrial disorders
Authors Finsterer J ![]() , Zarrouk-Mahjoub S
, Zarrouk-Mahjoub S
Received 13 August 2017
Accepted for publication 25 August 2017
Published 6 October 2017 Volume 2017:13 Pages 2569—2579
DOI https://doi.org/10.2147/NDT.S149067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Josef Finsterer,1 Sinda Zarrouk-Mahjoub2
1Municipal Hospital Rudolfstiftung, Vienna, Austria; 2Genomics Platform, Pasteur Institute of Tunis, Tunis, Tunisia
Objectives: Mitochondrial disorders (MIDs) frequently present as mitochondrial multiorgan disorder syndrome (MIMODS) at onset or evolve into MIMODS during the course. This study aimed to find which organs and/or tissues are most frequently affected by MIMODS, which are the most frequent abnormalities within an affected organ, whether there are typical MIMODS patterns, and to generate an MIMODS score to assess the diagnostic probability for an MID.
Methods: This is a retrospective evaluation of clinical, biochemical, and genetic investigations of adult patients with definite MIDs. A total of 36 definite MID patients, 19 men and 17 women, aged 29–82 years were included in this study. The diagnosis was based on genetic testing (n=21), on biochemical investigations (n=17), or on both (n=2).
Results: The number of organs most frequently affected was 4 ranging from 1 to 9. MIMODS was diagnosed in 97% of patients. The organs most frequently affected were the muscle (97%), central nervous system (CNS; 72%), endocrine glands (69%), heart (58%), intestines (55%), and peripheral nerves (50%). The most frequent CNS abnormalities were leukoencephalopathy, prolonged visually evoked potentials, and atrophy. The most frequent endocrine abnormalities included thyroid dysfunction, short stature, and diabetes. The most frequent cardiac abnormalities included arrhythmias, systolic dysfunction, and hypertrophic cardiomyopathy. The most frequent MIMODS patterns were encephalomyopathy, encephalo-myo-endocrinopathy, and encepalo-myo-endocrino-cardiopathy. The mean ± 2SD MIMODS score was 35.97±27.6 (range =11–71). An MIMODS score >10 was regarded as indicative of an MID.
Conclusion: Adult MIDs manifest as MIMODS in the vast majority of the cases. The organs most frequently affected in MIMODS are muscles, CNS, endocrine glands, and heart. An MIMODS score >10 suggests an MID.
Keywords: multisystem disease, mitochondrial cytopathy, multiorgan, mitochondriopathy, cardiomyopathy, encephalomyopathy
Introduction
Mitochondrial disorders (MIDs) frequently present as multiorgan disorder syndrome (MODS) already at the onset of the disease or evolve into a mitochondrial multiorgan disorder syndrome (MIMODS) during the disease course.1,2 The term “multi-organ disorder syndrome” should not be mixed up with multiorgan dysfunction syndrome, which is used for acute multiorgan failure during severe system disease (eg, cardiogenic shock).3 MODS may occur not only in MIDs but also in disorders other than MIDs, such as myotonic dystrophy, Fabry disease, and glycogen storage disease. In general, all tissues may be affected in an MID, but those with the highest demand of oxygen or with the highest energy requirements are most frequently involved. These organs include the skeletal muscle, the peripheral nerves, the central nervous system (CNS), endocrine organs, the eyes, the ears, the heart, the arteries, the gastrointestinal tract, the kidneys, the bone marrow, the cartilage,4,5 and the skin.1 This retrospective study was carried out to assess which organs/tissues are the most frequently affected in MIMODS, which are the most frequent manifestations within an organ, and whether there are typical patterns of organ involvement suggesting an MID and to generate a score, for estimating the probability of a phenotype to represent an MID.
Methods
All patients with a definite MID diagnosed between 1992 and 2014 were retrospectively investigated. A “definite MID” was diagnosed if biochemical investigations (the determination of the respiratory chain complex activity in the muscle homogenate) revealed a single or multiple respiratory chain complex defect(s) or if a mutation causative for the phenotype could be identified (by Southern analysis, PCR, restriction fragment length polymorphism, polyacrylamide gel electrophoresis, or long PCR in lymphocytes or muscle). From each of the included patients, all available records were assessed for diseases in any of the frequently affected organs in MIDs. As a generally accepted definition of MIMODS is not available, MIMODS was diagnosed in case more than one tissue was affected. All previous and current information about history, clinical examination, and instrumental investigations available was relevant for assessing how many organs were affected in a patient. Within an organ, it was counted how many abnormalities per organ were present. Patients who are currently alive as well as those already deceased were considered.
Patients with equivocal results on biochemical investigations or in case of uncertainty about the pathogenicity of a mutation such as mtDNA polymorphisms were excluded. Also patients with a probable and possible MID were excluded. Probable MIDs were defined as those without a biochemical or genetic abnormality but with abnormal histochemical findings on muscle biopsy (cytochrome oxidase–negative fibers, ragged red fibers, nicotinamide adenine dinucleotide–negative fibers, and succinate dehydrogenase hyperreactive fibers), increased lipid droplets or glycogen storage, abnormally shaped or structured mitochondria, paracrystalline inclusions, abnormal cycle lactate stress test, elevated resting lactate, or the presence of a lactate peak on cerebral magnetic resonance spectroscopy (MRS). Although MID patients frequently present with dyslipidemia or hyperuricemia, they were not regarded as MID manifestations because of the potential nutritional influence. Hyperhidrosis was regarded as a manifestation of the autonomic nervous system (ANS), since this etiology is the most frequent among MIDs, but hyperhidrosis may be due to other causes as well. Abnormal visually evoked potentials (VEPs) were regarded as indicative of a CNS lesion after the exclusion of ophthalmologic abnormalities. Although arterial hypertension can be frequently found in MIDs, it was not assessed as a manifestation of an MID since arterial hypertension is frequently multicausal. Atherosclerosis was regarded as MID manifestation only if classical cardiovascular risk factors (ie, diabetes, arterial hypertension, hyperlipidemia, and smoking) were absent. Dysphagia was regarded as a gastrointestinal MID manifestation only if there were no depression and no cerebral abnormality. The present study did not assess differences of organ involvement between the onset of the MID and the last follow-up, and it was conducted in accordance with the Declaration of Helsinki. Verbal informed consent was obtained from all included patients. The research was approved by the local institutional review board of the NKH Rosenhügel.
Results
In total, 36 adult, Caucasian patients with a definite MID, 19 men and 17 women, aged 29–82 years, were included (Table 1). Meanwhile, 7 patients had died. In 21 of the included patients, the diagnosis was based on genetic testing, in 17 patients on biochemical investigations of the muscle homogenate, and in 2 patients on both (Table 1). Three patients carried a single mtDNA deletion, 15 patients carried a single mtDNA point mutation, and 2 patients carried a point mutation in more than one mtDNA gene. In a single patient, a depletion syndrome was diagnosed. Among those with a biochemical defect, 9 patients had a single complex defect and 8 had combined complex defects (Table 1). The most frequent single-complex defect concerned C1 (n=6) followed by C4 (n=3). The most frequent combined complex defect concerned C1-4 (n=4). In 2 patients, biochemical investigations and genetic investigations had been carried out.
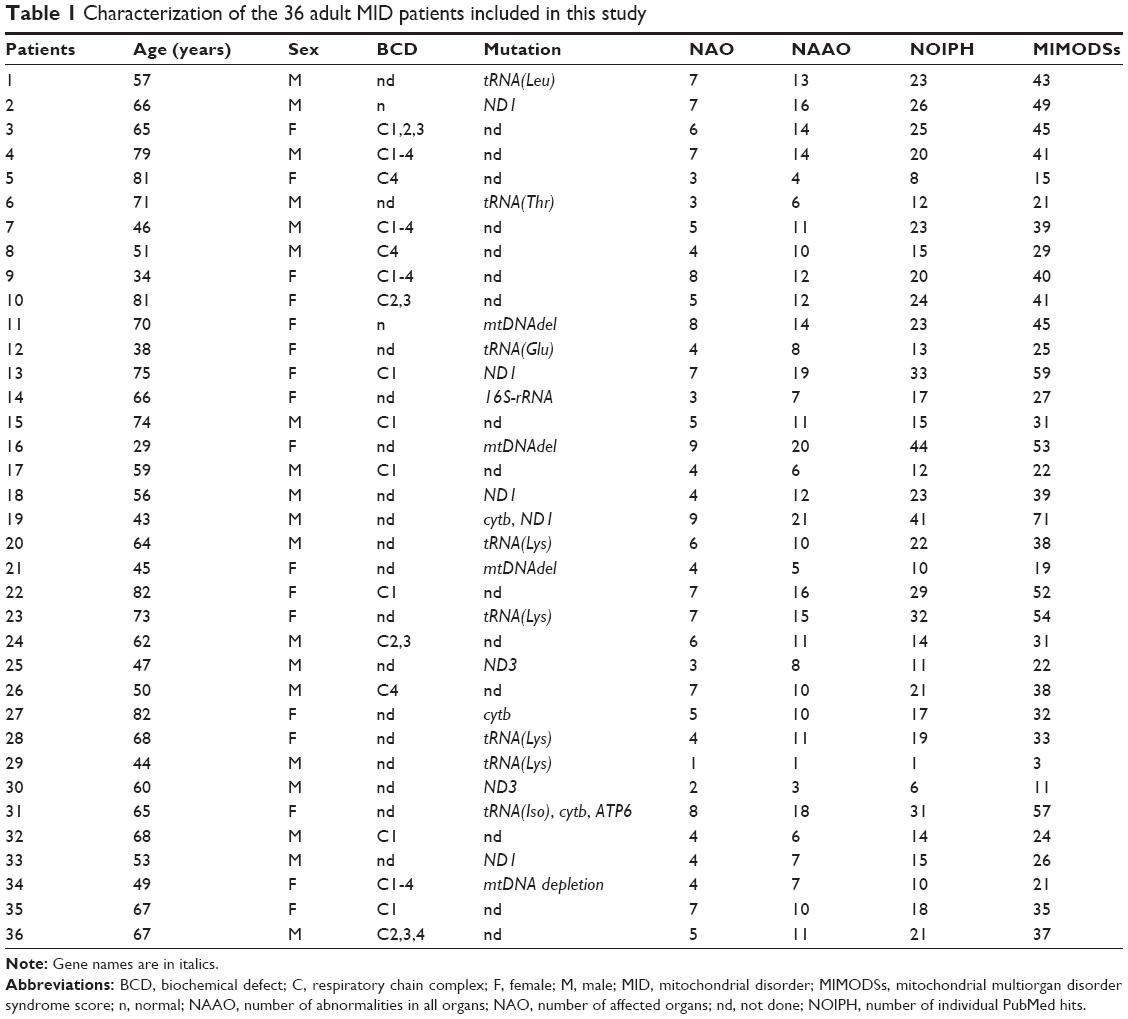 | Table 1 Characterization of the 36 adult MID patients included in this study |
In one patient, only a single organ (muscle) was affected at the time of the last available investigation. In one patient, 2 organs were affected, in 4 patients 3 organs, in 10 patients 4 organs, in 4 patients 5 organs, in 3 patients 6 organs, in 8 patients 7 organs, in 3 patients 8 organs, and in 2 patients 9 organs (Figure 1). None of the patients had involvement of >9 organs. The number of organs most frequently affected in MIMODS was 4. In total, 35 patients with MIMODS (97%) were identified. The organs most frequently affected in patients with MIMODS were the skeletal muscle (n=35), the CNS (n=27), the endocrine glands (n=26), the heart (n=21), the intestines (n=20), and the peripheral nerves (n=19; Table 2). The ears (n=11), the bone marrow (n=11), the eyes (n=8), the kidneys (n=7), the skin (n=6), and the bones (n=4) were rarely affected organs. The only organ not affected in this cohort was the vasculature and the lungs. MIMODS was as frequent in women as in men (Figure 1).
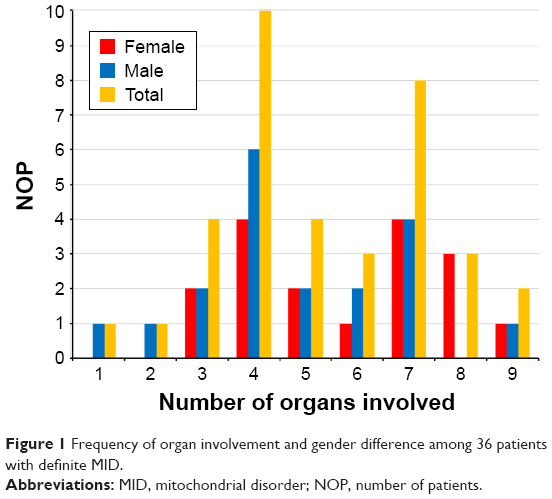 | Figure 1 Frequency of organ involvement and gender difference among 36 patients with definite MID. |
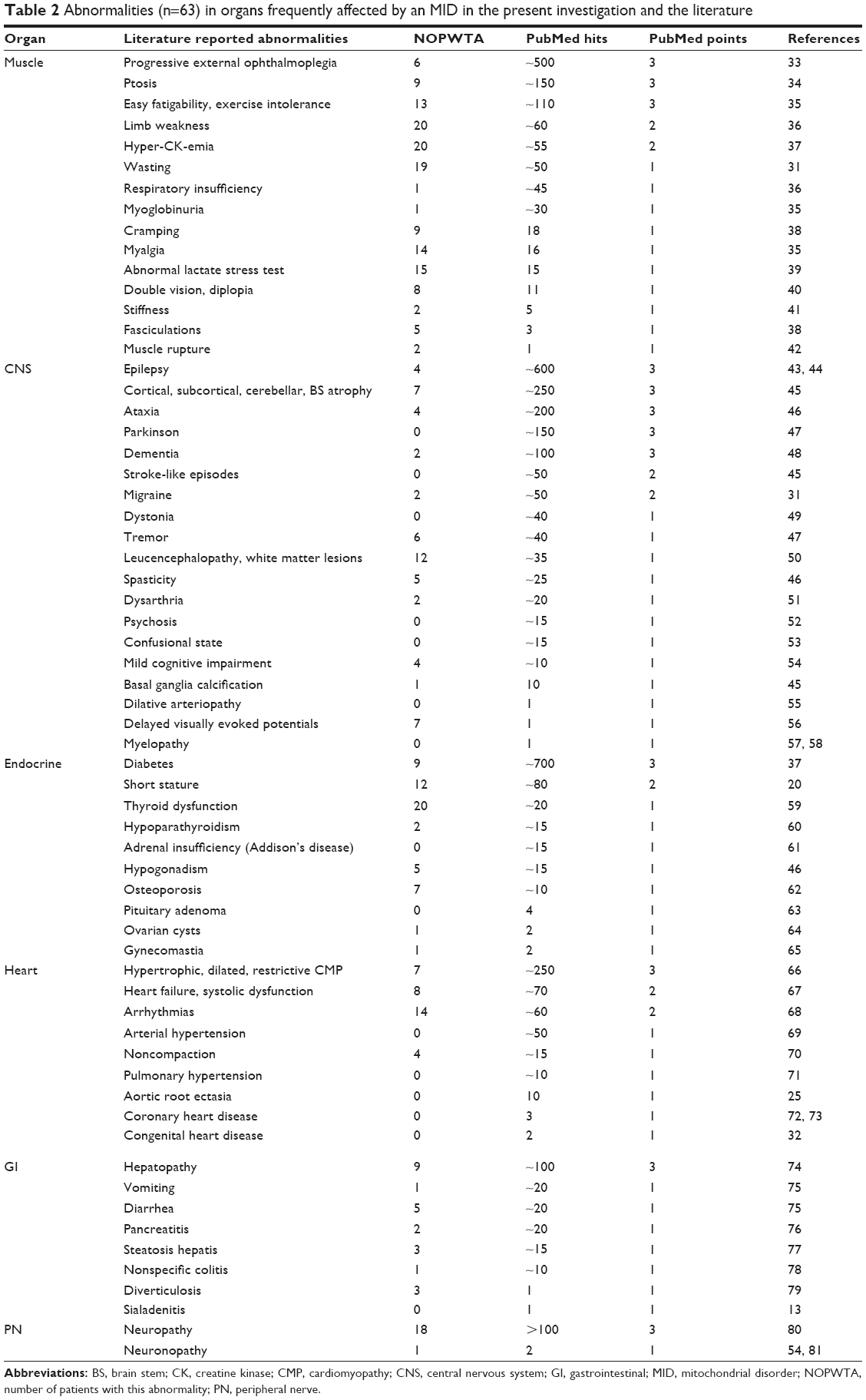 | Table 2 Abnormalities (n=63) in organs frequently affected by an MID in the present investigation and the literature |
Among the 35 patients with myopathy, 20 patients had weakness of the limb muscles, 19 had muscle wasting, 14 had myalgias, 13 had exercise intolerance, 10 had muscle cramps, 9 had ptosis, 8 had double vision, 6 had chronic progressive external ophthalmoplegia (CPEO), 6 had sore muscles, 5 had fasciculations, 2 had muscle stiffness, and 2 patients had a history of a muscle rupture. In one patient, respiratory muscles were involved. In 34 patients, creatine kinase (CK) was determined and elevated in 20 patients. In a single patient, myoglobinuria was detected. The lactate stress test on a cycle ergometer was carried out in 26 patients and was abnormal (indicative of an MID) in 15 patients. The electromyography (EMG) was carried out in 35 patients and myogenic in 13, neurogenic in 7, and normal in 15 patients. Among the 27 patients with CNS involvement, 12 had leukoencephalopathy, 7 had abnormal VEPs, 7 had focal or diffuse atrophy, 6 had tremor, 6 had cognitive impairment, 5 had spasticity, 4 had epilepsy, 4 had ataxia or dysmetria, 2 had migraine, 2 had dysarthria, 1 had basal ganglia calcification, 1 had an arachnoid cyst, and 1 had restless leg syndrome (RLS; Table 2). None of the patients underwent MRS or reported a stroke-like episode. Among the 26 patients with endocrine abnormalities, 20 had thyroid dysfunction, 12 had short stature, 9 had diabetes, 7 had osteoporosis, 5 had hypogonadism, 2 had parathyroid dysfunction, 2 had prostate hypertrophy, and 1 each had ovarian cysts, endometriosis, and gynecomastia, respectively. Among the 21 patients with cardiac involvement, 14 had arrhythmias, 8 had systolic dysfunction or heart failure, 5 had hypertrophic cardiomyopathy, 2 had dilative cardiomyopathy, 4 had valve abnormalities, 4 had noncompaction (left ventricular hypertrabeculation [LVHT]), and 1 each had pericarditis and abnormal myocardial texture, respectively (Table 2). Five were supplied with a pacemaker, but none with an implantable cardioverter defibrillator. Among the 20 patients with gastrointestinal abnormalities, 9 had hepatopathy with only elevated liver enzymes, 5 had diarrhea, 3 had diverticulosis, 3 had steatosis hepatis, 2 had pancreatitis, and 1 each had hyperemesis, hepatomegaly, nonspecific colitis, and liver cysts, respectively (Table 2). Among the 19 patients with peripheral nervous system involvement (ie, myopathy, neuropathy, or neuronopathy), 18 had neuropathy with motor, sensory, or ANS involvement and 1 had neuronopathy. The ANS was affected in 4 patients manifesting as hyperhidrosis.
Among the 11 patients with otologic involvement, 11 had hypoacusis or anacusis and 4 had tinnitus. Among the 10 patients with hematological abnormalities, 10 had anemia, 2 had thrombocytopenia, and 1 had leucopenia. Affection of the bones/cartilage resulted in polyarthralgia (n=2), arthrosis (n=2), facial dysmorphism (n=1), and hypertelorism (n=1). Among the 9 patients with ophthalmologic involvement, 2 had cataract, 3 had glaucoma, 2 had pigmentary retinopathy, and 2 had optic atrophy. Among the 7 patients with renal involvement, 5 had renal insufficiency, 2 had nephrolithiasis, and 1 had renal cysts. Seven patients had dermal involvement manifesting as lipomatosis (n=2), psoriasis (n=1), basal cell carcinoma (n=1), seborrheic eczema (n=1), and atopic dermatitis (n=1). The number of abnormalities in all the affected organs per patient ranged from 1 to 21.
Myopathy manifested with 15 of 15 different abnormalities, CNS involvement with 11 of 18 abnormalities, endocrine involvement with 7 of 10 abnormalities, cardiac involvement with 4 of 9 abnormalities, gastrointestinal involvement with 7 of 8 abnormalities, ophthalmologic involvement with 4 of 4 abnormalities, otologic involvement with 2 of 2 abnormalities, renal involvement with 3 of 4 abnormalities, hematologic involvement with 3 of 3 abnormalities, bone/cartilage involvement with 4 of 4, and dermal involvement with 4 of 10 abnormalities. In none of the patients could mitochondrial vasculopathy be unequivocally attributed to MID. Among the 10 patients with a neoplasm (28%), 6 had developed a benign neoplasm and 4 had a malignant neoplasm.
No typical pattern of organ involvement could be extracted from this study, but most of the patients presented with myopathy, CNS disease, endocrine abnormalities, and cardiac dysfunction. As 35 patients had myopathy and 27 patients had CNS involvement, encephalo-myopathy was the most frequent pattern of organ involvement. However, more frequently than 2 organs, 3 or 4 organs were simultaneously involved resulting encephalo-myo-endocrinopathy or encephalo-myo-endocrino-cardiomyopathy.
The MIMODS score was calculated as the sum of 3 figures: 1) the number of organs affected (maximal number =13); 2) the number of abnormalities within an organ (maximal number =94 [2–18 per organ]) (Table 2); and 3) the number of hits a PubMed search for each single-organ involvement by means of the search terms “specific abnormality,” “mitochondrial,” “human,” and “mutation” generated. If the number of hits exceeded 100, 3 points were additionally assigned for an abnormality, 2 points in case of 51–100 hits, and 1 point in case of 1–50 hits. The mean ± SD of the MIMODS score calculated from 35 patients (one patient did not have MIMODS) was 35.97±27.6 ranging from 11 to 71. An MIMODS score >10 was thus regarded as indicative of an MID. If MIMODS was defined as >2 organ involvement, a score >14 was indicative of MID.
Discussion
This study shows that MIMODS is frequent among nonselected patients with a definite MID. The most frequently involved organs in MIMODS are the muscle, CNS, endocrine glands, heart, and the gastrointestinal tract. Limb weakness, encephalopathy, thyroid dysfunction, and arrhythmias were the most frequent abnormalities of myopathic, CNS, endocrine, and cardiac involvement. The most frequent pattern of organ involvement in MIMODS was encephalo-myopathy, encephalo-myo-endocrinopathy, and encephalo-myo-endocrino-cardiomyopathy. An MIMODS score >10 suggests an MID.
The multiorgan nature of most MIDs is well established in the literature and supported by several studies (Tables 2 and 3).6 Whether MIMODS is more frequent in specific MIDs than in nonspecific MIDs is unknown. Among the specific MIDs, MIMODS has been particularly reported in Pearson syndrome,7–10 Kearns–Sayre syndrome (KSS), CPEO, sensory ataxic neuropathy with dysarthria and ophthalmoparesis, and mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS).11–14 However, MIMODS is also a frequent manifestation of nonspecific MIDs.15–21 The number of organs affected in MIMODS is assumed to depend on the disease duration, as MIDs are progressive disorders in the majority of cases with deterioration and affection of an increasing number of organs over time. Nearly all patients with single-organ involvement at the onset of the disease develop MIMODS over years. Often, this may occur not earlier than after several years of clinical stability.
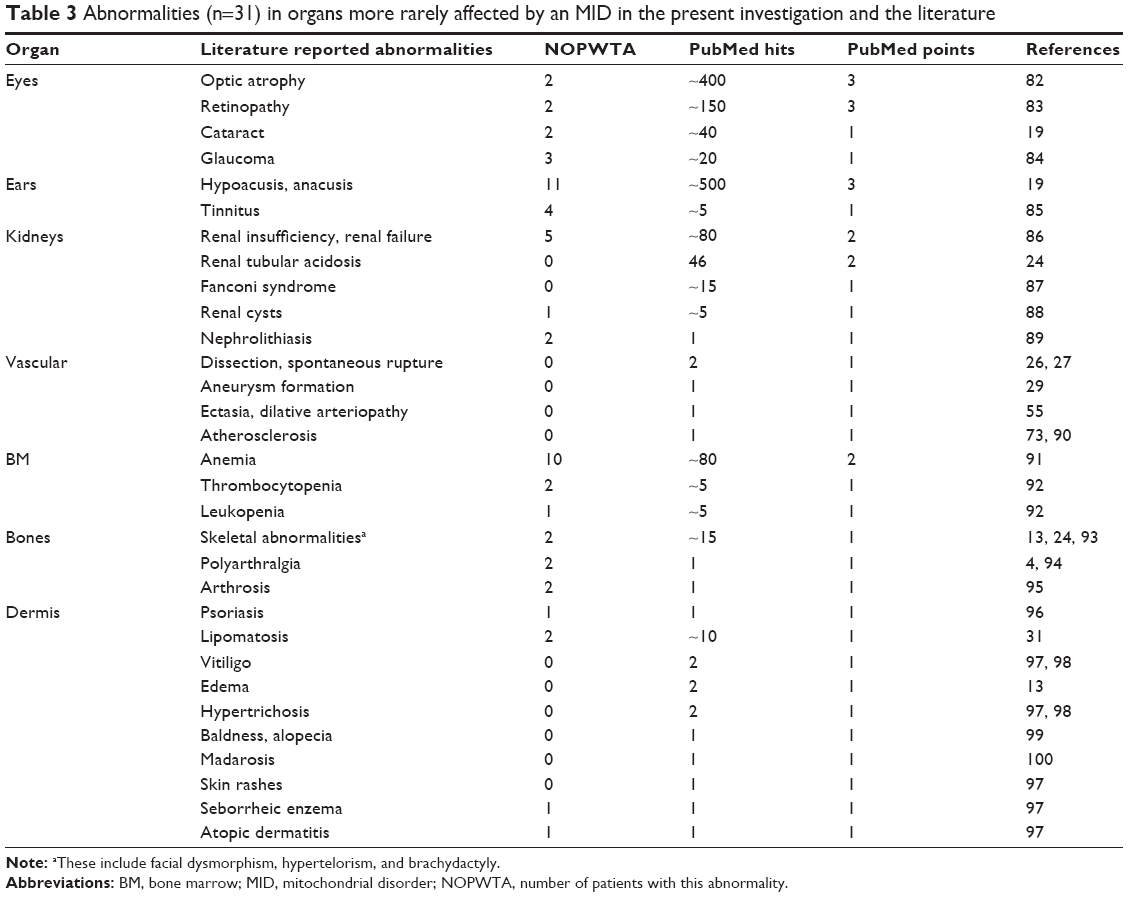 | Table 3 Abnormalities (n=31) in organs more rarely affected by an MID in the present investigation and the literature |
According to the literature, any organ may be affected in an MID.6 However, the most frequently affected organs are the muscle, the CNS, endocrine glands, and the heart, which is in line with the findings of the present study.6 Frequently affected are also the eyes and ears, which was not confirmed by the presently investigated cohort. This is most likely due to the development of ophthalmologic and otologic abnormalities only later in the disease course. More rarely involved are arteries, intestines, kidneys, the hematological system, lungs, bones, cartilage, and skin. Tables 2 and 3 list the organ abnormalities described in MIDs. Among the various abnormalities found in organs of MID patients, some are more frequent than others (Tables 2 and 3). In MID patients with myopathy, the most frequently described abnormalities include CPEO, ptosis, and exercise intolerance (>100 citations). More rarely described are limb weakness, muscle wasting, and hyper-CK-emia (51–100 citations; Table 2). Among CNS abnormalities, the most frequent are epilepsy, cerebral atrophy, ataxia, Parkinson syndrome, and dementia (>100 citations). Stroke-like episodes and migraine occur more rarely (51–100 citations). Among the abnormalities of the endocrine glands, the most frequent is by far diabetes (>100 citations). The second most frequent is short stature (51–100 citations; Table 2). Among the cardiac abnormalities, the most frequent is cardiomyopathy (>100 citations). Heart failure/systolic dysfunction, arrhythmias, and arterial hypertension occur more rarely (51–100 citations). Among the gastrointestinal abnormalities, the most frequent is hepatopathy (elevated liver enzymes; >100 citations). Affection of the peripheral nerves manifests almost exclusively as neuropathy (Table 2). The most frequent ophthalmologic abnormalities include optic atrophy and pigmentary retinopathy (Table 3). Among the otologic abnormalities, hypoacusis is by far the most frequent. Renal insufficiency and anemia are the most frequent abnormalities among the nephrological and hematological abnormalities. Thus the combination of the key features ptosis, ophthalmoparesis, easy fatigability, epilepsy, cerebral atrophy, ataxia, Parkinson syndrome, dementia, diabetes, cardiomyopathy, hepatopathy, neuropathy, optic atrophy, retinopathy, and hypoacusis (all >100 citations) is highly suggestive of an MID.
In addition to clinical features, abnormalities detectable on routine instrumental investigations were included. For example, myopathy may manifest not only with classical clinical manifestations but also with instrumental abnormalities, particularly hyper-CK-emia and abnormal cycle lactate stress test. More rarely than in muscular dystrophies, the EMG may be myogenic. Myopathy may also manifest exclusively with reduced tendon reflexes. Hyponatremia can be occasionally observed in MIDs without being attributed to drugs, diet, emesis, sweating, or renal insufficiency. In these cases, Addison’s disease or Fanconi syndrome (low molecular weight proteins, glucose, amino acid, uric acid, phosphate, and bicarbonate not reabsorbed in proximal tubules resulting in proteinuria, glucosuria, aminoaciduria, hypophosphatemia, and metabolic acidosis)22 should be considered. Renal tubular acidosis with or without Fanconi syndrome may be a rare manifestation of an MID.23,24 If the ANS is additionally affected in neuropathy, it may manifest as dry skin, hypohidrosis, or hyperhidrosis. In addition, there may be oversensitivity to light, sicca syndrome (xerostomia), decreased heart rate variability, orthostasis or syncope, gustatory sweating, obstipation, overactive bladder (detrusor overactivity), or impotence.
Diseases of the arteries such as atherosclerosis, dissection, ectasia, aneurysm formation, or spontaneous rupture are not well appreciated as a manifestation of an MID.25–29 The number of patients with such vascular abnormalities is limited to a few cases (Table 2). Nevertheless, there is an increasing evidence that these abnormalities can be clearly related to MID in the few available reports. Although hematological disease is increasingly recognized as a manifestation of MIDs, it is still underdiagnosed and underreported. Bone marrow involvement includes anemia, thrombocytopenia, and leukopenia. Among the sepcific MIDs, such hematological abnormalities occur in Pearson syndrome, KSS, Barth syndrome, mitochondrial myopathy, lactic acidosis and sideroblastic anemia, X-linked sideroblastic anemia, Leigh syndrome, Leber’s hereditary optic neuropathy, MELAS, and thiamine-responsive megaloblastic anemia.30 However, hematological abnormalities occur more frequently in nonspecific MIDs than in specific MIDs.30 Whether eosinophilia, as found in one of the patients of the present study, should be attributed to MIDs remains speculative and requires further investigations. Dermatological involvement is also hardly recognized as mitochondrial, but there is growing evidence that MIDs also manifest in the skin or its appendages.31 Dermatological involvement in MIDs includes psoriasis, vitiligo, baldness, alopecia, madarosis, hypertrichosis, skin rashes, seborrheic eczema, atopic dermatitis, or edema. Not all these aspects were found in the present study but reported in the literature (Table 2). Congenital heart disease as a manifestation of MIDs is also rare but has been described in single patients (Table 2).32 Diverticulosis is also hardly recognized as a manifestation of MIDs but is prevalent particular among nonspecific MIDs, without being frequently reported. Ischemic stroke should not be regarded as a primary manifestation of MIDs but rather as secondary in case classical risk factors for atherosclerosis, such as mitochondrial diabetes, hyperlipidemia, arterial hypertension, severe heart failure, and LVHT, are present. Altogether, only 71% of the 94 reported abnormalities described in the literature (Table 2) were also found in the present study.
Abnormalities that have not been previously described in MID patients but have been found in the present investigation include liver cysts, RLS, eosinophilia, arachnoidal cysts, prostate hypertrophy, and endometriosis. Whether these abnormalities can truly be regarded as manifestations of the mitochondrial defect and whether valve abnormalities, pericarditis, and abnormal myocardial texture should be regarded as an MID manifestation remain speculative. It is also not established that nephrolithiasis is an authentic manifestation of MIDs. According to our own experience, however, nephrolithiasis particularly occurs as an initial manifestation at the onset of adult MIDs. Syncope is multicausal but if due to a seizure or a cardiac arrhythmia an MID should be considered if no other explanations impose oneself. Syncope may be also due to arterial hypotension, which in turn could be a manifestation of adrenal insufficiency or bradyarrhythmia.
Limitations of the study were that it was retrospective, thus not all patients went through the same clinical screening, that the records could not be completely pursued in each case, that the genetic defect was not identified in each case (biochemical defects could have been secondary), that the number of included patients was low, that no control group or disease control group (eg, autoimmune disorders and myotonic dystrophy) was investigated, that the score was not validated, that the score does not predict the severity of the disease, that the number of matching PubMed citations for each MID manifestation was only approximately determined, that the number of PubMed hits may not adequately reflect the frequency of abnormalities, and that not all organs potentially affected in MIDs were also investigated in each patient. A further limitation is that biochemical and genetic studies were carried out in only 2 patients. A single biochemical defect usually reflects a mutation in one of the genes encoding the subunits of the corresponding respiratory chain complex. Multiple biochemical defects usually reflect tRNA mutations or mtDNA depletion.
Conclusion
This study shows that adult MIDs manifest as MIMODS in the vast majority of the cases; that organs most frequently affected in MIMODS are the muscle, CNS, endocrine glands, and heart; and that an MIMODS score >10 suggests an MID and thus could be a helpful parameter to assess, by nonsophisticated, clinical means, whether an MID is present or not.
Disclosure
The authors report no conflicts of interest in this work.
References
Finsterer J, Jarius C, Eichberger H. Phenotype variability in 130 adult patients with respiratory chain disorders. J Inherit Metab Dis. 2001;24:560–576. | ||
Pfeffer G, Sirrs S, Wade NK, Mezei MM. Multisystem disorder in late-onset chronic progressive external ophthalmoplegia. Can J Neurol Sci. 2011;38:119–123. | ||
Schmidt H, Lotze U, Ghanem A, et al. Relation of impaired interorgan communication and parasympathetic activity in chronic heart failure and multiple-organ dysfunction syndrome. J Crit Care. 2014;29:367–373. | ||
Lü HB, Zhou Y, Hu JZ, Lei GH, Zhu M, Li KH. Mitochondrial DNA deletion mutations in articular chondrocytes of cartilage affected by osteoarthritis. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2006;31:640–644. | ||
Chang MC, Hung SC, Chen WY, et al. Accumulation of mitochondrial DNA with 4977-bp deletion in knee cartilage – an association with idiopathic osteoarthritis. Osteoarthritis Cartilage. 2005;13:1004–1011. | ||
Schapira AH. Mitochondrial diseases. Lancet. 2012;379:1825–1834. | ||
Park J, Ryu H, Jang W, et al. Novel 5.712 kb mitochondrial DNA deletion in a patient with Pearson syndrome: a case report. Mol Med Rep. 2015;11(5):3741–3745. | ||
Williams TB, Daniels M, Puthenveetil G, Chang R, Wang RY, Abdenur JE. Pearson syndrome: unique endocrine manifestations including neonatal diabetes and adrenal insufficiency. Mol Genet Metab. 2012;106:104–107. | ||
Manea EM, Leverger G, Bellmann F, et al. Pearson syndrome in the neonatal period: two case reports and review of the literature. J Pediatr Hematol Oncol. 2009;31:947–951. | ||
Lohi O, Kuusela AL, Arola M. A novel deletion in a Pearson syndrome infant with hypospadias and cleft lip and palate. J Inherit Metab Dis. 2005;28:1165–1166. | ||
Demarest ST, Whitehead MT, Turnacioglu S, Pearl PL, Gropman AL. Phenotypic analysis of epilepsy in the mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes-associated mitochondrial DNA A3243G mutation. J Child Neurol. 2014;29:1249–1256. | ||
Gurrieri C, Kivela JE, Bojanić K, et al. Anesthetic considerations in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome: a case series. Can J Anaesth. 2011;58:751–763. | ||
Pronicki M, Sykut-Cegielska J, Mierzewska H, et al. Diversity of clinical symptoms in A3243G mitochondrial DNA mutation (MELAS syndrome mutation). Med Sci Monit. 2002;8:CR767–CR773. | ||
Bandettini di Poggio M, Nesti C, Bruno C, Meschini MC, Schenone A, Santorelli FM. Dopamine-agonist responsive Parkinsonism in a patient with the SANDO syndrome caused by POLG mutation. BMC Med Genet. 2013;14:105. | ||
Hansrote S, Croul S, Selak M, Kalman B, Schwartzman RJ. External ophthalmoplegia with severe progressive multiorgan involvement associated with the mtDNA A3243G mutation. J Neurol Sci. 2002;197:63–67. | ||
de Keyzer Y, Valayannopoulos V, Benoist JF, et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res. 2009;66:91–95. | ||
Low E, Crushell EB, Harty SB, Ryan SP, Treacy EP. Reversible multiorgan system involvement in a neonate with complex IV deficiency. Pediatr Neurol. 2008;39:368–370. | ||
Meulemans A, Seneca S, Lagae L, et al. A novel mitochondrial transfer RNA(Asn) mutation causing multiorgan failure. Arch Neurol. 2006;63:1194–1198. | ||
Lehmann D, Schubert K, Joshi PR, et al. A novel m.7539C>T point mutation in the mt-tRNA(Asp) gene associated with multisystemic mitochondrial disease. Neuromuscul Disord. 2015;25:81–84. | ||
Pitceathly RD, Taanman JW, Rahman S, et al. COX10 mutations resulting in complex multisystem mitochondrial disease that remains stable into adulthood. JAMA Neurol. 2013;70:1556–1561. | ||
Oláhová M, Haack TB, Alston CL, et al. A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency. Eur J Hum Genet. 2015;23(7):935–939. | ||
Ezgu F, Senaca S, Gunduz M, et al. Severe renal tubulopathy in a newborn due to BCS1L gene mutation: effects of different treatment modalities on the clinical course. Gene. 2013;528:364–366. | ||
Yang G, Zou L, Wang J. Mitochondrial disorders manifested as renal tubular acidosis and recurrent seizures. Chin Med J (Engl). 2014;127:1989. | ||
Gai X, Ghezzi D, Johnson MA, et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2013;93:482–495. | ||
Brunetti-Pierri N, Pignatelli R, Fouladi N, et al. Dilation of the aortic root in mitochondrial disease patients. Mol Genet Metab. 2011;103:167–170. | ||
Sakharova AV, Kalashnikova LA, Chaĭkovskaia RP, et al. [Morphological signs of mitochondrial cytopathy in skeletal muscles and micro-vessel walls in a patient with cerebral artery dissection associated with MELAS syndrome]. Arkh Patol. 2012;74:51–56. Russian. | ||
Ryther RC, Cho-Park YA, Lee JW. Carotid dissection in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes. J Neurol. 2011;258:912–914. | ||
Kalashnikova LA, Sakharova AV, Dobrynina LA, et al. [Mitochondrial arteriopathy as a cause of spontaneous dissection of cerebral arteries]. Zh Nevrol Psikhiatr Im S S Korsakova. 2010;110(Suppl 2):3–11. Russian. | ||
Tay SH, Nordli DR Jr, Bonilla E, et al. Aortic rupture in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes. Arch Neurol. 2006;63:281–283. | ||
Finsterer J, Frank M. Hematological abnormalities in mitochondrial disorders. Singapore Med J. 2015;56:412–419. | ||
Mancuso M, Orsucci D, Angelini C, et al. Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology. 2013;80:2049–2054. | ||
Kleefstra T, Wortmann SB, Rodenburg RJ, et al. Mitochondrial dysfunction and organic aciduria in five patients carrying mutations in the Ras-MAPK pathway. Eur J Hum Genet. 2011;19:138–144. | ||
Caballero PE, Candela MS, Alvarez CI, Tejerina AA. Chronic progressive external ophthalmoplegia: a report of 6 cases and a review of the literature. Neurologist. 2007;13:33–36. | ||
Spyropoulos A, Manford M, Horvath R, et al. Near-identical segregation of mtDNA heteroplasmy in blood, muscle, urinary epithelium, and hair follicles in twins with optic atrophy, ptosis, and intractable epilepsy. JAMA Neurol. 2013;70:1552–1555. | ||
Vissing CR, Duno M, Olesen JH, et al. Recurrent myoglobinuria and deranged acylcarnitines due to a mutation in the mtDNA MT-CO2 gene. Neurology. 2013;80:1908–1910. | ||
Blakely EL, Alston CL, Lecky B, et al. Distal weakness with respiratory insufficiency caused by the m.8344A>G “MERRF” mutation. Neuromuscul Disord. 2014;24:533–536. | ||
Rubegni A, Cardaioli E, Chini E, et al. A case of 3243A>G mutation in mtDNA presenting as apparently idiopathic hyperCKemia. J Neurol Sci. 2014;338:232–234. | ||
Gdynia HJ, Sperfeld AD, Knirsch U, et al. Benign symmetric lipomatosis with axonal neuropathy and abnormalities in specific mitochondrial tRNA regions. Eur J Med Res. 2006;11:545–546. | ||
Hanisch F, Müller T, Muser A, Deschauer M, Zierz S. Lactate increase and oxygen desaturation in mitochondrial disorders–evaluation of two diagnostic screening protocols. J Neurol. 2006;253:417–423. | ||
McKelvie P, Marotta R, Thorburn DR, Chin J, Punchihewa S, Collins S. A case of myelopathy, myopathy, peripheral neuropathy and subcortical grey matter degeneration associated with recessive compound heterozygous POLG1 mutations. Neuromuscul Disord. 2012;22:401–405. | ||
Deschauer M, Wieser T, Neudecker S, Lindner A, Zierz S. Mitochondrial 3243A–>G mutation (MELAS mutation) associated with painful muscle stiffness. Neuromuscul Disord. 1999;9:305–307. | ||
Barrientos A, Casademont J, Grau JM, et al. Oftalmoplejía externa progresiva y síndrome de Kearns-Sayre: estudio clínico y molecular de 6 casos [Progressive external ophthalmoplegia and the Kearns-Sayre syndrome: a clinical and molecular study of 6 cases]. Med Clin (Barc). 1995;105:180–184. Spanish. | ||
Desguerre I, Hully M, Rio M, Nabbout R. Mitochondrial disorders and epilepsy. Rev Neurol (Paris). 2014;170:375–380. | ||
Finsterer J, Zarrouk Mahjoub S. Mitochondrial epilepsy in pediatric and adult patients. Acta Neurol Scand. 2013;128:141–152. | ||
Devaux-Bricout M, Grévent D, Lebre AS, et al. Aspect en IRM cérébrale des maladies mitochondriales. Algorithme décisionnel des maladies mitochondriales les plus fréquentes [Aspect of brain MRI in mitochondrial respiratory chain deficiency. A diagnostic algorithm of the most common mitochondrial genetic mutations]. Rev Neurol (Paris). 2014;170:381–389. French. | ||
Dallabona C, Diodato D, Kevelam SH, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology. 2014;82:2063–2071. | ||
Unal Gulsuner H, Gulsuner S, Mercan FN, et al. Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proc Natl Acad Sci U S A. 2014;111:18285–18290. | ||
Inczedy-Farkas G, Trampush JW, Perczel Forintos D, et al. Mitochondrial DNA mutations and cognition: a case-series report. Arch Clin Neuropsychol. 2014;29:315–321. | ||
Doss S, Lohmann K, Seibler P, et al. Recessive dystonia-ataxia syndrome in a Turkish family caused by a COX20 (FAM36A) mutation. J Neurol. 2014;261:207–212. | ||
Zsurka G, Becker F, Heinen M, et al. Mutation in the mitochondrial tRNA(Ile) gene causes progressive myoclonus epilepsy. Seizure. 2013;22:483–486. | ||
Lovan A, ul Haq I, Balakrishnan N. Diagnostic challenges in movement disorders: Sensory Ataxia Neuropathy Dysarthria and Ophthalmoplegia (SANDO) syndrome. BMJ Case Rep. 2013;doi:10.1136/bcr-2013-010343. | ||
Anglin RE, Garside SL, Tarnopolsky MA, Mazurek MF, Rosebush PI. The psychiatric manifestations of mitochondrial disorders: a case and review of the literature. J Clin Psychiatry. 2012;73:506–512. | ||
Coomans H, Barroso B, Bertandeau E, et al. Mutisme et troubles du comportement aigus révélant un syndrome MELAS [Mutism and acute behavioral disorders revealing MELAS syndrome]. Rev Neurol (Paris). 2011;167:847–851. French. | ||
Ronchi D, Garone C, Bordoni A, et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain. 2012;135:3404–3415. | ||
Finsterer J, Bastovansky A. Dilative arteriopathy and leucencephalopathy as manifestations of a neurometabolic disease. Open Neurol J. 2015;9:28–31. | ||
Naghizadeh F, Varga ET, Molnár MJ, Holló G. [Retinal ganglion cell layer and visual function in patients with progressive external ophthalmoplegia caused by common mtDNA deletion]. Ideggyogy Sz. 2014;67:335–341. Hungarian. | ||
Haas RH, Zolkipli Z. Mitochondrial disorders affecting the nervous system. Semin Neurol. 2014;34:321–340. | ||
van Berge L, Hamilton EM, Linnankivi T, et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain. 2014;137:1019–1029. | ||
Hopkins SE, Somoza A, Gilbert DL. Rare autosomal dominant POLG1 mutation in a family with metabolic strokes, posterior column spinal degeneration, and multi-endocrine disease. J Child Neurol. 2010;25:752–756. | ||
Cassandrini D, Savasta S, Bozzola M, et al. Mitochondrial DNA deletion in a child with mitochondrial encephalomyopathy, growth hormone deficiency, and hypoparathyroidism. J Child Neurol. 2006;21:983–985. | ||
D’Aco KE, Manno M, Clarke C, Ganesh J, Meyers KE, Sondheimer N. Mitochondrial tRNA(Phe) mutation as a cause of end-stage renal disease in childhood. Pediatr Nephrol. 2013;28:515–519. | ||
Varanasi SS, Francis RM, Berger CE, Papiha SS, Datta HK. Mitochondrial DNA deletion associated oxidative stress and severe male osteoporosis. Osteoporos Int. 1999;10:143–149. | ||
Mulliez E, Blanckaert M, Blanckaert J. Acute manifestation of LHON and coincidental finding of a pituitary adenoma: a case report. Bull Soc Belge Ophtalmol. 2000;(277):35–42. | ||
Zhuo G, Feng G, Leng J, Yu L, Jiang Y. A 9-bp deletion homoplasmy in women with polycystic ovary syndrome revealed by mitochondrial genome-mutation screen. Biochem Genet. 2010;48:157–163. | ||
Finsterer J, Bittner R, Bodingbauer M, Eichberger H, Stöllberger C, Blazek G. Complex mitochondriopathy associated with 4 mtDNA transitions. Eur Neurol. 2000;44:37–41. | ||
Limongelli G, Tome-Esteban M, Dejthevaporn C, Rahman S, Hanna MG, Elliott PM. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010;12:114–121. | ||
Cardena MM, Mansur AJ, Pereira Ada C, Fridman C. A new duplication in the mitochondrially encoded tRNA proline gene in a patient with dilated cardiomyopathy. Mitochondrial DNA. 2013;24:46–49. | ||
Bates MG, Bourke JP, Giordano C, d’Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33:3023–3033. | ||
Guo H, Zhuang XY, Zhang AM, et al. Presence of mutation m.14484T>C in a Chinese family with maternally inherited essential hypertension but no expression of LHON. Biochim Biophys Acta. 2012;1822:1535–1543. | ||
Zarrouk Mahjoub S, Mehri S, Ourda F, et al. Transition m.3308T>C in the ND1 gene is associated with left ventricular hypertrabeculation/noncompaction. Cardiology. 2011;118:153–158. | ||
Hung PC, Wang HS, Chung HT, Hwang MS, Ro LS. Pulmonary hypertension in a child with mitochondrial A3243G point mutation. Brain Dev. 2012;34:866–868. | ||
Jia Z, Wang X, Qin Y, et al. Coronary heart disease is associated with a mutation in mitochondrial tRNA. Hum Mol Genet. 2013;22:4064–4073. | ||
Blanco-Grau A, Bonaventura-Ibars I, Coll-Cantí J, et al. Identification and biochemical characterization of the novel mutation m.8839G>C in the mitochondrial ATP6 gene associated with NARP syndrome. Genes Brain Behav. 2013;12:812–820. | ||
Uusimaa J, Evans J, Smith C, et al. Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur J Hum Genet. 2014;22:184–191. | ||
Cardaioli E, Da Pozzo P, Malfatti E, et al. A second MNGIE patient without typical mitochondrial skeletal muscle involvement. Neurol Sci. 2010;31:491–494. | ||
Verny C, Amati-Bonneau P, Letournel F, et al. Mitochondrial DNA A3243G mutation involved in familial diabetes, chronic intestinal pseudo-obstruction and recurrent pancreatitis. Diabetes Metab. 2008;34:620–626. | ||
Takahashi Y, Iida K, Takeno R, et al. Hepatic failure and enhanced oxidative stress in mitochondrial diabetes. Endocr J. 2008;55:509–514. | ||
Maniura-Weber K, Taylor RW, Johnson MA, et al. A novel point mutation in the mitochondrial tRNA(Trp) gene produces a neurogastrointestinal syndrome. Eur J Hum Genet. 2004;12:509–512. | ||
Perez-Atayde AR, Fox V, Teitelbaum JE, et al. Mitochondrial neurogastrointestinal encephalomyopathy: diagnosis by rectal biopsy. Am J Surg Pathol. 1998;22:1141–1147. | ||
Finsterer J. Mitochondrial neuropathy. Clin Neurol Neurosurg. 2005;107:181–186. | ||
Zhao D, Wang Z, Hong D, Zhang W, Yuan Y. Chronic progressive external ophthalmoplegia coexistent with motor neuron disease in a patient with a novel large-scale mitochondrial DNA deletion. Clin Neurol Neurosurg. 2013;115:1490–1492. | ||
La Morgia C, Caporali L, Gandini F, et al. Association of the mtDNA m.4171C>A/MT-ND1 mutation with both optic neuropathy and bilateral brainstem lesions. BMC Neurol. 2014;14:116. | ||
Duno M, Wibrand F, Baggesen K, Rosenberg T, Kjaer N, Frederiksen AL. A novel mitochondrial mutation m.8989G>C associated with neuropathy, ataxia, retinitis pigmentosa – the NARP syndrome. Gene. 2013;515:372–375. | ||
Nucci C, Martucci A, Mancino R, Cerulli L. Glaucoma progression associated with Leber’s hereditary optic neuropathy. Int Ophthalmol. 2013;33:75–77. | ||
Hiraki N, Udaka T, Yamamoto H, Kadokawa Y, Ohkubo J, Suzuki H. Mitochondrial neurogastrointestinal encephalomyopathy associated with progressive hearing loss. J Laryngol Otol. 2010;124:1007–1009. | ||
Rivera H, Martín-Hernández E, Delmiro A, et al. A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol. 2013;14:195. | ||
Liu HM, Tsai LP, Chien YH, et al. A novel 3670-base pair mitochondrial DNA deletion resulting in multi-systemic manifestations in a child. Pediatr Neonatol. 2012;53:264–268. | ||
Gürgey A, Ozalp I, Rötig A, et al. A case of Pearson syndrome associated with multiple renal cysts. Pediatr Nephrol. 1996;10:637–638. | ||
Danpure CJ, Lumb MJ, Birdsey GM, Zhang X. Alanine:glyoxylate aminotransferase peroxisome-to-mitochondrion mistargeting in human hereditary kidney stone disease. Biochim Biophys Acta. 2003;1647:70–75. | ||
Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin Dev Immunol. 2012;2012:832464. | ||
Burrage LC, Tang S, Wang J, et al. Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA) plus associated with a novel de novo mutation (m.8969G>A) in the mitochondrial encoded ATP6 gene. Mol Genet Metab. 2014;113:207–212. | ||
Muraki K, Nishimura S, Goto Y, Nonaka I, Sakura N, Ueda K. The association between haematological manifestation and mtDNA deletions in Pearson syndrome. J Inherit Metab Dis. 1997;20:697–703. | ||
Tanigawa J, Kaneko K, Honda M, et al. Two Japanese patients with Leigh syndrome caused by novel SURF1 mutations. Brain Dev. 2012;34:861–865. | ||
Harty LC, Biniecka M, O’Sullivan J, et al. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann Rheum Dis. 2012;71:582–588. | ||
Ranjbar SH, Amiri P, Amoli MM, Soltani A. A new mitochondrial mutation in a patient with diabetes mellitus, deafness, hydronephrosis and joint contractures. J Pediatr Endocrinol Metab. 2008;21:1185–1189. | ||
Finsterer J, Kovacs GG. Psoriasis, bulbar involvement, and diarrhea in late myoclonic epilepsy with ragged-red fibers-syndrome due to the m.8344A > G tRNA (Lys) mutation. Iran J Neurol. 2017;16(1):45–49. | ||
Carmi E, Defossez C, Morin G, et al. MELAS syndrome (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes. Ann Dermatol Venereol. 2001;128:1031–1035. | ||
Tulinius MH, Oldfors A, Holme E, et al. Atypical presentation of multisystem disorders in two girls with mitochondrial DNA deletions. Eur J Pediatr. 1995;154:35–42. | ||
Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. | ||
Finsterer J, Brunner S. Madarosis from mitochondriopathy. Acta Ophthalmol Scand. 2005;83:628–630. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.