Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Mitochondrial functioning abnormalities observed in blood platelets of chronic smoke-exposed guinea pigs – a pilot study
Authors Białas AJ ![]() , Siewiera K, Watała C
, Siewiera K, Watała C ![]() , Rybicka A, Grobelski B, Kośmider L
, Rybicka A, Grobelski B, Kośmider L ![]() , Kurek J, Miłkowska-Dymanowska J, Piotrowski WJ
, Kurek J, Miłkowska-Dymanowska J, Piotrowski WJ ![]() , Górski P
, Górski P ![]()
Received 26 May 2018
Accepted for publication 22 August 2018
Published 9 November 2018 Volume 2018:13 Pages 3707—3717
DOI https://doi.org/10.2147/COPD.S175444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Adam J Białas,1 Karolina Siewiera,2 Cezary Watała,2 Anna Rybicka,3 Bartłomiej Grobelski,3 Leon Kośmider,4,5 Jolanta Kurek,6 Joanna Miłkowska-Dymanowska,1 Wojciech J Piotrowski,1 Paweł Górski1
1Department of Pneumology and Allergy, Medical University of Lodz, Lodz, Poland; 2Department of Hemostasis and Hemostatic Disorders, Medical University of Lodz, Lodz, Poland; 3The animal house, Pharmaceutical Faculty, Medical University of Lodz, Lodz, Poland; 4Department of Pharmaceutics, School of Pharmacy, Virginia Commonwealth University, Richmond, VA, USA; 5Center for the Study of Tobacco Products, Virginia Commonwealth University, Richmond, VA, USA; 6Institute of Occupational Medicine and Environmental Health, Sosnowiec, Poland
Background: COPD represents a major global health issue, which is often accompanied by cardiovascular diseases. A considerable body of evidence suggests that cardiovascular risk is elevated by the activation of blood platelets, which in turn is exacerbated by inflammation. As reactive oxygen species are believed to be an important factor in platelet metabolism and functioning, the aim of our study was to perform a complex assessment of mitochondrial function in platelets in chronic smoke exposed animals with COPD-like lung lesions.
Materials and methods: Eight-week-old, male Dunkin Hartley guinea pigs (the study group) were exposed to the cigarette smoke from commercial unfiltered cigarettes (0.9 mg/cig of nicotine content) or to the air without cigarette smoke (control group), using the Candela Constructions® exposure system. The animals were exposed for 4 hours daily, 5 days a week, with 2×70 mL puff/minute, until signs of dyspnea were observed. The animals were bled, and isolated platelets were used to monitor blood platelet respiration. The mitochondrial respiratory parameters of the platelets were monitored in vitro based on continuous recording of oxygen consumption by high-resolution respirometry.
Results: An elevated respiration trend was observed in the LEAK-state (adjusted for number of platelets) in the smoke-exposed animals: 6.75 (5.09) vs 2.53 (1.28) (pmol O2/[s · 1×108 platelets]); bootstrap-boosted P1α=0.04. The study group also demonstrated lowered respiration in the ET-state (normalized for protein content): 12.31 (4.84) vs 16.48 (1.72) (pmol O2/[s · mg of protein]); bootstrap-boosted P1α=0.049.
Conclusion: Our results suggest increased proton and electron leak and decreased electron transfer system capacity in platelets from chronic smoke-exposed animals. These observations may also indicate that platelets play an important role in the pathobiology of COPD and its comorbidities and may serve as a background for possible therapeutic targeting. However, these preliminary outcomes should be further validated in studies based on larger samples.
Keywords: mitochondria, electron leak, proton leak, electron transport chain, ETC, chronic obstructive pulmonary disease, COPD, animal model
Introduction
COPD is a major global health problem.1 It is characterized by persistent respiratory symptoms and airflow limitation associated with airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases.2 Although the major risk factor is exposure to cigarette smoke,3–8 the pathobiology of this disease should be regarded as a complex interaction of several factors, including genetic vulnerability.9–14 Current treatment approaches do not satisfactorily reduce disease progression or mortality, one reason for this being the poor understanding of the cellular, molecular, and genetic mechanisms contributing to its pathobiology.15,16 Additionally, as COPD seems to be a heterogeneous disease, there is a worldwide initiative to identify relevant endotypes, and determine how understanding endotypes can improve the precision, effectiveness, and safety of the treatment provided to COPD patients.17
A significant group of COPD comorbidities is represented by cardiovascular diseases.18 A strong body of evidence suggests that inflammation may contribute to activation of blood platelets and, therefore, to increased cardiovascular risk.19,20
This important role of blood platelets in COPD may also be supported by the fact that thrombocytosis may be associated with increased short- and long-term mortality after exacerbation; and that antiplatelet therapy in this group of patients was associated with significantly lower 1-year mortality.21 Also the presence of a wider platelet distribution width seems to negatively influence the survival of patients with COPD.22
Mitochondria are multifunctional cellular organelles that play an important role in numerous aspects of cell biology, with their main roles being to act as sites of nutrient oxidation and to serve as locations for ATP synthesis; consequently, the generation of reactive oxygen and nitrogen species is the part of the normal functioning of mitochondria,23 even under normal conditions, and the electron transfer chain may “leak” 1%–2% of all processed electrons as reactive oxygen species (ROS).24,25
Nevertheless, numerous intra- and extracellular factors may increase this figure and shift the oxidative–antioxidative balance.26 Additionally, increasing evidence implicates ROS as an important factor in platelet functioning,27 thus indicating a relationship between increased ROS production and cardiovascular risk. To better clarify the nature of this relationship, the aim of the present study was to perform a complex assessment of mitochondrial function in blood platelets in COPD.
Materials and methods
Study subjects
Eight-week-old male Dunkin Hartley guinea pigs were randomly allocated to two groups: a study group (n=5) and a control group (n=5).
Cigarettes, smoke generation, and analysis of test atmospheres
The animals of the study group were exposed to smoke from unfiltered commercial cigarettes (nicotine content of 0.9 mg/cig) in the Candela exposure system (Candela Constructions®, Wola Rakowa, Poland; Figure 1) for 4 hours each day, 5 days a week, with 2×70 mL puff/minute, until signs of dyspnea were observed; these being defined as evident difficulties in breathing or tachypnea considered as ≥110 breaths per minute. Breaths were calculated manually at intervals. Five measurements per animal were performed and the mean value was calculated. The animals of the control group were exposed to air without cigarette smoke in the same exposure system. For one experimental procedure, one smoke-exposed experimental subject and one air-exposed animal were selected parallelly.
 | Figure 1 (A) Smoke-exposure system with one exposure chamber. (B) Cigarette feeder and pumping system (Candela Constructions®). |
Smoke generation quality was assessed by analyses of the test atmosphere in the breathing zone of the exposure chamber. For each series of measurements, six pumps – universal sampling pumps, three for nicotine and three for polycyclic aromatic hydrocarbons (PAHs) (SKC Ltd., Dorset, UK) – were distributed evenly in the exposure chamber. Samples for control group were taken three times (three samples for nicotine and three for PAHs) for one cycle of exposure to check if the control group is exposed to the toxins from tobacco smoke. Samples for research group were taken three times (for nicotine and PAHs) for three cycles of exposure to assure the homogeneity of exposure between cycles of exposure. Pumps for nicotine and PAH analysis were chosen randomly for each cycle. In general, 12 samples for nicotine and PAHs analysis was taken. The air was taken with 2 L/min on 42 mm Cambridge filter. Nicotine was extracted in a 50-mL flask containing 10 mL of methanol and 25 μL of internal standard (quinolone 50 mg/mL). Then, the flask was capped and shaken for 2 hours in darkness. Finally, 1 mL of extract was analyzed with gas chromatography as described previously.28 Recovery was 95.8% at the range of 0.01–20 mg/sample. Cambridge filters for PAHs analysis were extracted with 10 mL of acetonitrile (HPLC grade, LiChrosolv®, Sigma Aldrich, St Louis, MO, USA) and analyzed with AT 1200 HPLC with Zorbax Eclipse PAH 5.0 μm, 150×4.6 column, and fluorescent detector (Agilent Technologies, Santa Clara, CA, USA).
Blood and tissue collection
Under conditions of a complete anesthesia (by intramuscular administration of ketamine and xylazine into pelvic limb in doses 80 mg/kg and 10 mg/kg, respectively), blood samples (9–18 mL) were taken by cardiac puncture into tubes containing 3.2% sodium citrate (Monovette®, Sarstedt AG & Co. KG, Nümbrecht, Germany).
Total bleed was performed, leading to euthanasia of the animals. The lungs were then taken and placed in 4% formalin. All procedures were concordant with ARRIVE guidelines, Annex IV of the Directive of the European Parliament and of the European Council 2010/63/EU and were approved by the Regional Ethical Committee on Animal Research in Lodz, Poland.
Mitochondrial functioning assessment
To isolate blood platelets, blood was diluted 1:1 with PBS and centrifuged (190×g, 12 minutes, 37°C, in the presence of 62.5 ng/mL prostaglandin E1) to obtain diluted platelet-rich plasma (PRP). PRP was then centrifuged (1,000×g, 15 minutes, 37°C), the platelet pellet washed in Tyrode’s buffer (134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, pH 7.0, 5 mM glucose, 0.35% w/v BSA) with 3.2% sodium citrate (9:1, vol:vol) and centrifuged again under the same conditions.
The platelets were resuspended in Tyrode’s buffer to achieve a suspension of 1×108 platelets/mL. Mitochondrial respiratory parameters were monitored in vitro by continuous recording of oxygen consumption by high-resolution respirometry with a Clark electrode (Oxygraph-2k, Oroboros Instruments, Innsbruck, Austria). In the oxygraphic part of the study, mitochondrial respiratory parameters were monitored in intact blood platelets.29
Two milliliters of blood platelet suspension at a concentration of 1×108 platelets/mL (2×108 platelets/chamber) was added to the oxygraphic chamber and was further incubated at 37°C. Respiration was allowed to stabilize without the addition of any inhibitors or uncouplers at a ROUTINE (R) state. Afterward, oligomycin was added at a concentration of 0.2 μg/mL to evaluate the dissipative component of respiration (respiration independent of ADP phosphorylation) (LEAK-state, L). The maximum respiratory capacity was measured after titration with a chemical uncoupler, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), added in 0.25 μM aliquots until no further increase in respiration was detected; this allowed the respiratory chain condition to be assessed (ET-state, ET-capacity, E). Following this, rotenone (0.5 μM) was added to inhibit complex I and the electron transfer system (ETS) for complex II was evaluated. Finally, antimycin-A (2.5 μM), a complex III inhibitor, was added to estimate the residual, non-mitochondrial oxygen consumption (ROX). All oxygen consumption values by platelet mitochondria in the ROUTINE-state (R), LEAK-state (L) and ET-state (ET) were corrected for ROX. To determine the index of uncoupling or dyscoupling, the LEAK control ratio (L/ET) was calculated as the quotient of respiration in LEAK-state to the maximal FCCP-stimulated respiration. In addition, the ROUTINE control ratio (R/ET) was determined as the ratio expressing how close the routine respiration operates to ET-capacity. For better illustration, these procedures are summarized in Figure 2.
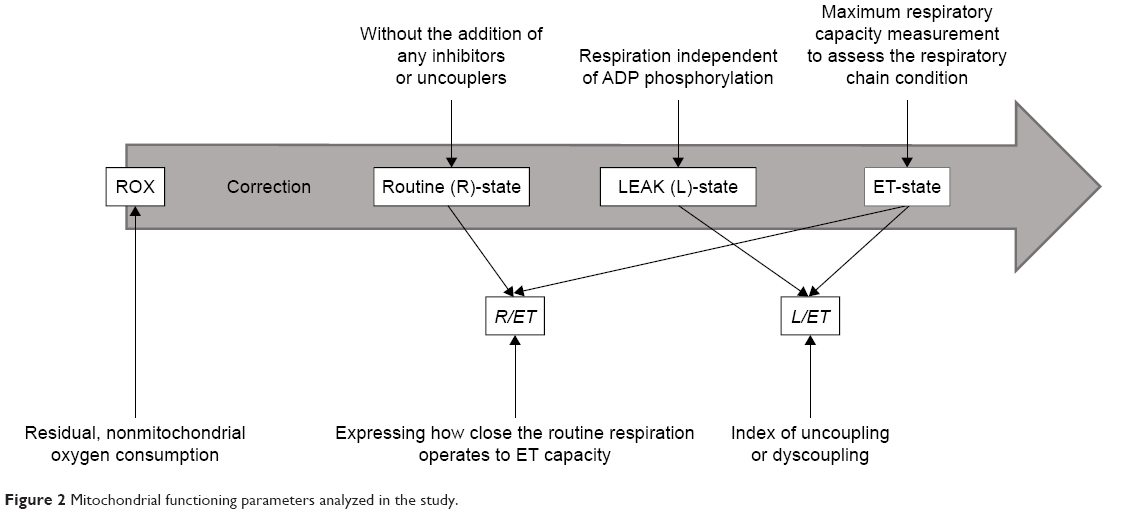 | Figure 2 Mitochondrial functioning parameters analyzed in the study. |
Since oxygen consumption should be compared between cells of identical size and in some pathological states size of cells can change, we decided to use two approaches of data normalization for a better comparison of the results. This seems to be important, due to the ongoing discussion concerning the selection of the best marker for the normalization of mitochondrial respiration data and trend to present data normalized on several different markers. Therefore, we normalized obtained data for the number of platelets in the sample and for total protein amount in the sample. Such presentation of results allows also for easier comparison of data between publications.
Protein concentration in samples was assayed spectrophotometrically with the use of Bicinchoninic Acid Protein Assay Kit (Sigma Aldrich, St Louis, MO, USA) according to the manufacturer’s protocol.
Lung histology
Lung tissue samples were stained with H&E and examined by light microscopy (Olympus, Tokyo, Japan). The fields for morphometric analysis were captured by a digital Single Lens Reflex camera – Nikon D5100 (Nikon CEE GmbH, Vienna, Austria). Images were converted into an 8-bit grayscale image by extracting the green channel and then to a binary image by thresholding. The airspaces were analyzed automatically using ImageJ software (1.51m9; Wayne Rasband, National Institutes of Health, Bethseda, MD, USA). Edge-adjacent airspaces were excluded from analysis. All images were analyzed using the same technique to minimize bias. Emphysema severity was assessed quantitatively by the calculation of D2 parameter.30,31 Inflammatory cells were identified by the morphological features.
Statistical methods
The LaMorte method was used to determine the minimal number of animals. Our leading variable used to estimate sample size was mean airspace size. Assuming the difference in means 30%–35%, effect size d=2.19 (estimated from previous study published in the study domain),32 α error probability of 0.01, and statistical power of at least 80%, we roughly estimated the approximate group sizes at five study subjects and five controls. Continuous data were presented as mean with SD (mean [SD]) or median with an interquartile range (median [IQR] from LQ [25%] to UQ [75%]), depending on the distribution of data. Variables were compared using the bootstrap-boosted inference tests: the unpaired Student’s t-test, Welch test or the Mann–Whitney U-test, depending on data normality and homogeneity of variance.
Results
The smoke was reproducibly generated and delivered to the exposure chamber. The results of analyses of test atmospheres are summarized in Table 1.
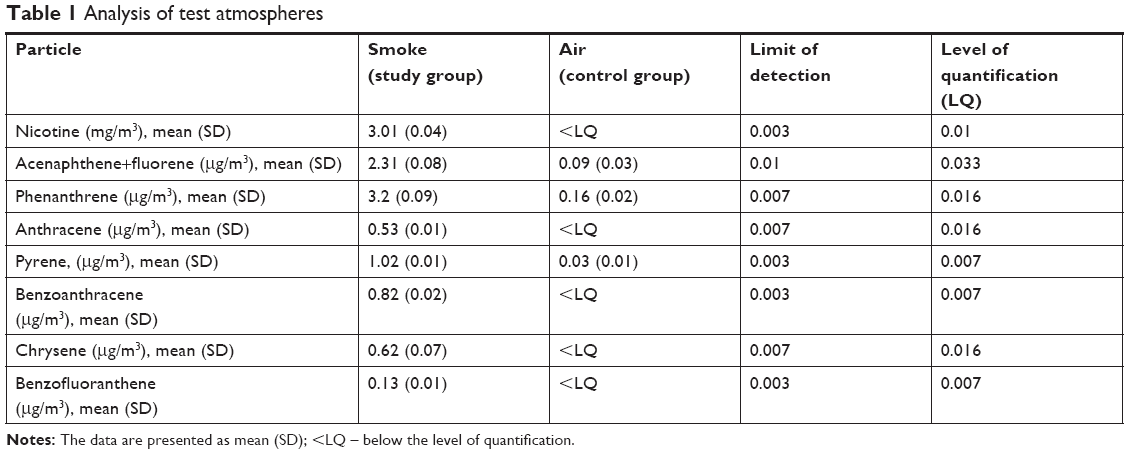 | Table 1 Analysis of test atmospheres |
The median duration of exposure of animals to cigarette smoke was 183 days (IQR: 92–183). A significant difference was observed in the number of breaths per minute (bpm) in the study group vs controls before termination: 129.92 (5.87) vs 94.85 (7.02) bpm (P=0.0002, 1–β=99.99%).
The microscopic examination of the lungs taken from the animals of the study group revealed features characteristic of emphysema (Figure 3), with significantly increased D2 index: 147.12 (131.03–162.92) vs 75.88 (74.75–86.01) μm (P=0.008, 1–β=99%).
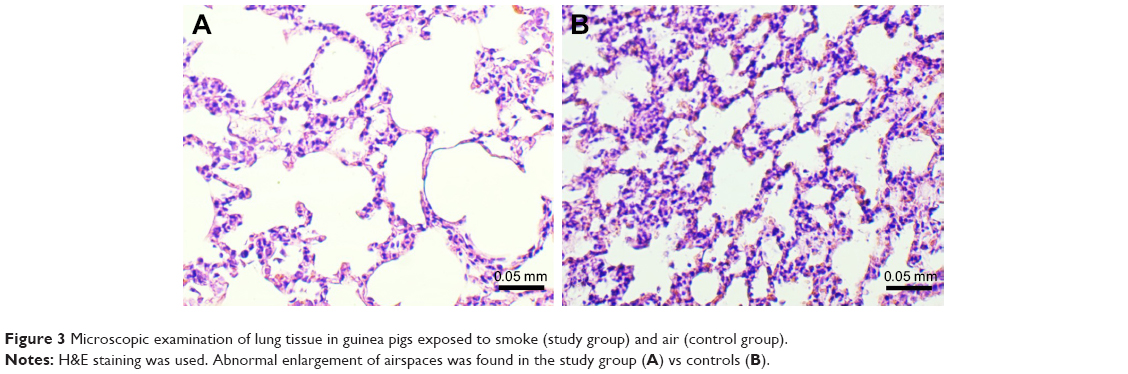 | Figure 3 Microscopic examination of lung tissue in guinea pigs exposed to smoke (study group) and air (control group). |
The inflammatory response in exposed group was constituted mainly by lymphocytes, including lymphoid follicle formation (Figure 4), macrophages, and neutrophils. Presented image could be considered as reflecting inflammatory profile observed in COPD.33–35
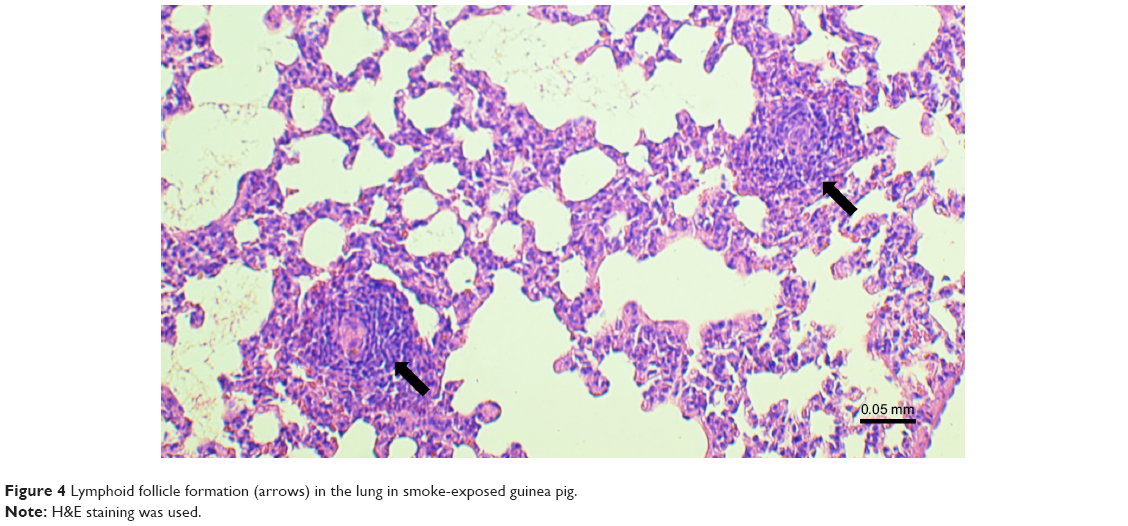 | Figure 4 Lymphoid follicle formation (arrows) in the lung in smoke-exposed guinea pig. |
The results of the analyses of mitochondrial functioning parameters (upon adjustment for the number of platelets and upon normalization for the protein content) are presented in Tables 2 and 3. In the study group, respiration was found to be elevated in the LEAK-state (upon adjustment for number of platelets): 6.75 (5.09) vs 2.53 (1.28) (pmol O2/[s · 1×108 platelets]); bootstrap-boosted P1α=0.04, and lowered in the ET-state (normalized for protein content, 12.31 [4.84] vs 16.48 [1.72] (pmol O2/[s · mg of protein]; bootstrap-boosted P1α=0.049). Also, in the chronic smoke-exposed animals, regarding samples normalized for protein content, we observed lowered R state respiration; however, the results did not achieve statistical significance (P1α=0.08).
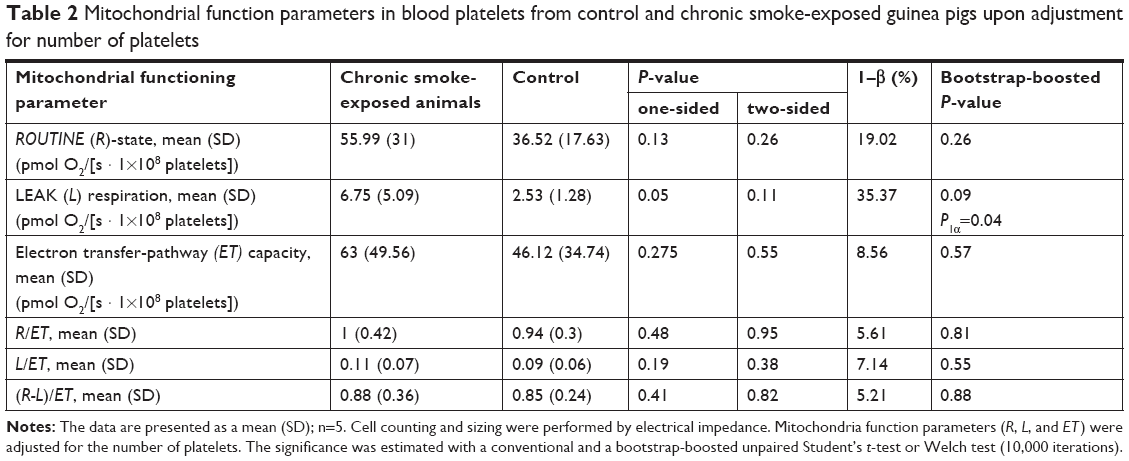 | Table 2 Mitochondrial function parameters in blood platelets from control and chronic smoke-exposed guinea pigs upon adjustment for number of platelets |
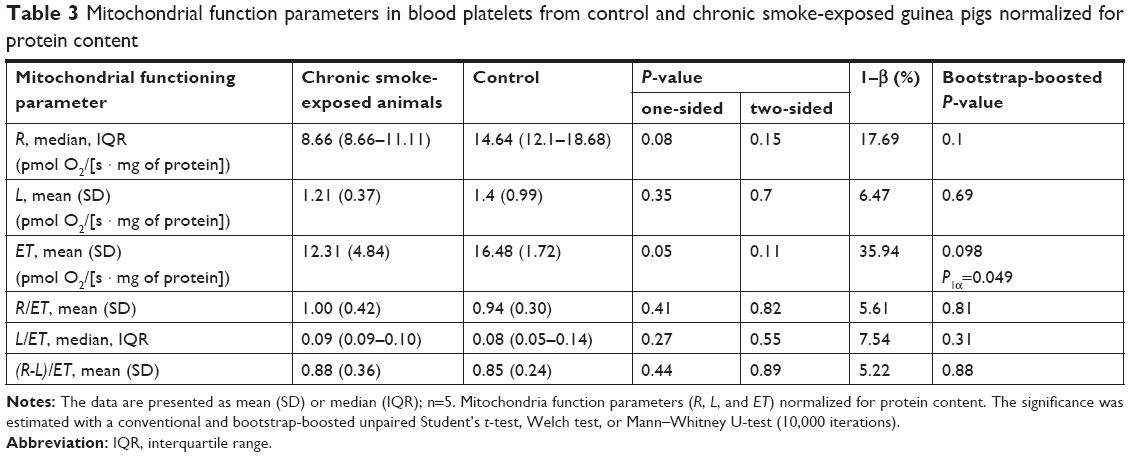 | Table 3 Mitochondrial function parameters in blood platelets from control and chronic smoke-exposed guinea pigs normalized for protein content |
For better illustration, graphical summary of the study was presented in Figure 5.
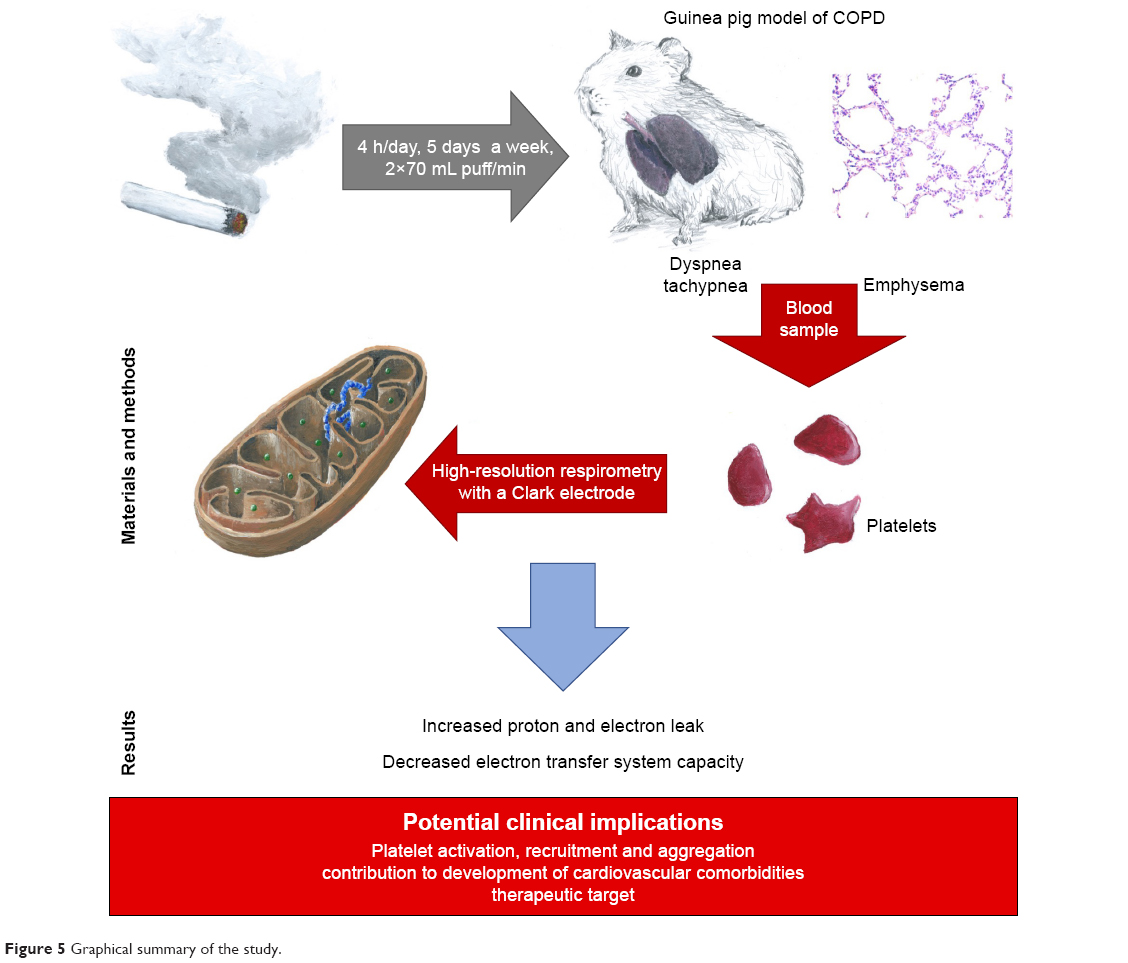 | Figure 5 Graphical summary of the study. |
Discussion
Being a complex and heterogeneous disease, both at the clinical and biological levels, COPD should be studied in homogenous populations, comparable according to disease severity and treatment approach. In addition, the results may be further influenced by the numerous comorbidities associated with COPD.18 Therefore, the assumption of studying COPD in homogenous populations would be hard to accomplish in real-life clinical studies, and it would be of clear benefit to develop a well-described animal model of COPD.
A guinea pig model was chosen for the present study because of the various anatomical and physiological similarities between their lungs and human lungs.36–40
The animals were exposed to cigarette smoke from commercial cigarettes to replicate real-life risk factors and smoke generation quality was assessed by analyses of the test atmosphere in the breathing zone of the exposure system.
Exposure was continued until signs of dyspnea were observed; hence, this approach linked the experimental model to the development of pathological lung lesions associated with cigarette smoke exposure, as confirmed by emphysema development and typical inflammatory response. In addition, this approach was also based around the development of chronic and progressive dyspnea: the most common characteristic symptom of COPD.2
The results of our mitochondrial functioning analysis suggest an increased proton and electron leak and a decreased ETS capacity in platelets in chronic smoke-exposed animals. And despite the fact that both ways of normalization indicated a different mitochondrial disorder, it does not mean that one of the findings is more correct or real.
Due to changes in both the platelet size and changes in the content of proteins between cells in different pathological states, it cannot be assumed that different ways of normalization will lead to exactly the same results. Moreover, the application of many ways of normalization may reveal differences in the case when we do not know that our original normalization method may be disturbed by some variable. In any case, both methods indicated a disturbance in the functioning of mitochondria. Increasing the number of tested animals probably will lead to disclose these changes in both cases of normalization.
Proton leak behaves in a nonohmic manner and may be divided into basal and inducible forms. The precise mechanism of basal proton leak is not fully understood.
It is unregulated and mediated mostly by the presence, but not the activity, of mitochondrial anion carrier proteins. In contrast, inducible proton leak is catalyzed by specific mitochondrial membrane proteins and can be inhibited and activated. It is catalyzed by adenine nucleotide translocase and uncoupling proteins.41 Although to our knowledge, these observations are novelties in the context of pathobiology of COPD, they seem to be logical.
Notably, the experimental evidence from previously published studies indicates that proton and electron leaks seem to be closely linked to ROS generation.42–46
The leak of electrons from donor redox centers to molecular oxygen induce ROS production.42 Also, growing body of evidence suggest the role of proton leak in this process. Talbot et al hypothesize that superoxide from complex I can act locally to stimulate proton leak.43 Additionally, Ku et al reported that state four respiration may originate from mitochondrial superoxide generation.46 However, contribution of proton leak to ROS production is more complex and seems to form a feedback loop.
Moreover, it seems that mitochondrially generated superoxide can increase proton leak, which on the other hand, could decrease superoxide generation.44 Nevertheless, it does not seem unreasonable to link proton leak with increased ROS – either as the reason or as the effect.
It is also worth to mention another evidence that suggests the correlation of proton leak and aging.44 This reflects well-established hypothesis about the role of accelerated lung aging in pathobiology of COPD.16,47
A growing body of evidence indicates that ROS play a role in regulation of the function of blood platelets; however, these relationships are not straightforward.27,48–51
ROS are involved in adhesion to and spreading on the collagen matrix if the vascular endothelial layer is injured, as well as in platelet aggregation and recruitment.27,50,52–54
Both exo- and endogenous sources of ROS may play a role in platelet activation.
Handin et al report that exogenous sources of ROS activate platelets without causing molecular oxidative damage, and that superoxide-dependent aggregation and release may be a physiologically important method to modulate hemostatic reactions; particularly in areas of inflammation or vessel injury which could have high local concentrations of superoxide anion.54 On the other hand, activation by different agonists boost endogenous ROS production.51,55–58 One important pathway for generating endogenous ROS in platelets is through ligand binding to the glycoprotein (GP)VI.51,59 Collagen is the main physiological ligand for platelet GPVI; however, fibrin and laminin have also been identified in this context.60–62 It was reported that following stimulation by collagen, platelets may produce superoxide anions, hydroxyl radicals, or hydrogen peroxide.58,63,64
Besides ROS generation, GPVI ligation initiate platelet spread, granule secretion and integrin αIIbβ3-dependent aggregation.49,57,62 Numerous signaling molecules seem to be involved in GPVI-dependent ROS generation, including tumor necrosis factor receptor-associated factor (TRAF)4, the NADPH oxidase (NOX) organizer subunit, p47phox, Hic5, proline rich tyrosine kinase 2 (Pyk2).62,65,66 There is also the evidence, which indicates that peroxynitrite (ONOO−) plays an important role in platelet stimulation. ONOO− may react with tyrosyl residues to form 3-NO2-Tyr, and with thiols to generate S-nitrosothiols, reducing the inhibitory effect of nitrogen oxide. ONOO- may also act as a substrate for cyclooxygenase peroxidase, causing prostaglandin endoperoxide H synthase activation and increased prostaglandin synthesis,67 and thus, thromboxane A2 synthesis.68
ONOO- may also induce the formation of 8-iso-prostaglandin F2α69 and, as the effect might increase calcium release from intracellular stores, induce changes in platelet morphology and amplify platelet aggregation in response to agonists.27,67 Together, these complex interactions support the possibility that oxidative stress has a proatherogenic role70 and increases cardiovascular risk. Also, we can conclude about association between ROS and the important role of cardiovascular comorbidities in COPD.18,26
Our observations also have some clinical implications. As both proton and electron leak may serve as therapeutic targets, we hypothesize that a better understanding of these mechanisms would contribute to the development of therapeutic approaches.
An interesting possibility in this regard is represented by N-acetylcysteine (NAC), a broadly used antioxidant agent. In addition to its proven direct antiaggregatory effects,71 a recent meta-analysis of 12 randomized controlled trials involving 2,691 patients with COPD indicated that NAC therapy may reduce the risk of COPD exacerbation.72
There is also some evidence that supplementing regular treatment with administration of 600 mg N-acetylcysteine once a day and a 20 minute daily walk improves the quality of life of stable COPD patients.73 This may serve as a background for further studies based on therapeutically targeting the interaction between oxidative stress, platelets, and COPD, thus contributing to a better quality of life and improved prognoses for this population of patients.
The study does have some limitations. First, the study is based on a small sample and, therefore, could only be considered as a preliminary report which should be further validated in studies with larger samples with greater statistical power. The minimal number of animals was calculated using LaMorte method; however, our leading variable used to estimate sample size was mean airspace size. Our present results will be used to determine the number of animals for our further analyses, which will aim to confirm and broaden our hypotheses. Second, although guinea pigs have many advantages as a model of COPD, any conclusions from such research should not be directly and unrestrainedly translated to human pathobiology. Therefore, the research explored herein requires further investigation in a study on humans.
Conclusions
Our results suggest increased proton and electron leak and decreased ETS capacity in platelets from chronic smoke-exposed animals. These observations may also indicate that platelets play an important role in the pathobiology of COPD and its comorbidities and may serve as a background for possible therapeutic targeting.
However, these preliminary outcomes should be further validated in studies based on larger samples.
Author contributions
Conception – PG and AJB; design – AJB, PG, CW, KS, AR, BG; drafting of the manuscript – AJB, KS, PG, CW; acquisition and analysis of data – AJB, AR, BG, KS, CW, LK, JK, JMD, WJP, PG; analysis and interpretation of data – AJB, KS, CW, PG, AR, BG, JMD, WJP; drafting the manuscript for important intellectual content – all authors; all authors also critically revised the manuscript and gave approval of the version to be published.
Disclosure
The costs of this study were defrayed from regular finances of the Department of Pneumology and Allergy, The animal house of the Pharmaceutical Faculty, Department of Hemostasis and Hemostatic Disorders, of Medical University of Lodz, Poland, and of the Department of General and Inorganic Chemistry, School of Pharmacy with the Division of Laboratory Medicine in Sosnowiec of Medical University of Silesia, and Institute of Occupational Medicine and Environmental Health in Sosnowiec.
AB is also supported by the fund for the young investigators of the Medical University of Lodz (502-03/1-151-03/502-14-364-18).
KS received the Etiuda scholarship funded by the NCN (2016/20/T/NZ3/00505).
LK is supported by the National Institute on Drug Abuse of the National Institutes of Health under Award Number P50DA036105 and the Center for Tobacco Products of the U.S. Food and Drug Administration. The content is solely the responsibility of the authors and does not necessarily represent the views of the NIH or the FDA.
LK works as an expert for the Polish National Committee for Standardization and for the European Committee for standardization of requirements and test methods for e-liquids and emissions. Additionally, JK is and LK was an employee of the Institute of Occupational Medicine and Environmental Health. One of the institute’s objectives is outsourcing for the industrial sector, including manufacturers of e-cigarettes. However, this has no influence on studies design.
The other authors report no conflicts of interest in this work.
References
Burden of COPD [in:] Chronic respiratory diseases. Available from: http://www.who.int/respiratory/copd/burden/en. Accessed 26th Dec 2015. | ||
Global Initiative for Chronic Obstructive Pulmonary Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Available from: http://goldcopd.org/gold-2017-global-strategy-diagnosis-management-prevention-copd/. Accessed 4th July 2017. | ||
Bartal M. COPD and tobacco smoke. Monaldi Arch Chest Dis. 2005;63(4):213–225. | ||
Chiba Y, Murata M, Ushikubo H, et al. Effect of cigarette smoke exposure in vivo on bronchial smooth muscle contractility in vitro in rats. Am J Respir Cell Mol Biol. 2005;33(6):574–581. | ||
Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316. | ||
Soulitzis N, Neofytou E, Psarrou M, et al. Downregulation of lung mitochondrial prohibitin in COPD. Respir Med. 2012;106(7):954–961. | ||
van Rijt SH, Keller IE, John G, et al. Acute cigarette smoke exposure impairs proteasome function in the lung. Am J Physiol Lung Cell Mol Physiol. 2012;303(9):L814–L823. | ||
Meyer A, Zoll J, Charles AL, et al. Skeletal muscle mitochondrial dysfunction during chronic obstructive pulmonary disease: central actor and therapeutic target. Exp Physiol. 2013;98(6):1063–1078. | ||
Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42(1):45–52. | ||
Pillai SG, Ge D, ICGN Investigators, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5(3):e1000421. | ||
Wilk JB, Chen TH, Gottlieb DJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5(3):e1000429. | ||
Hunninghake GM, Cho MH, Tesfaigzi Y, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361(27):2599–2608. | ||
Lambrechts D, Buysschaert I, Zanen P, et al. The 15q24/25 susceptibility variant for lung cancer and chronic obstructive pulmonary disease is associated with emphysema. Am J Respir Crit Care Med. 2010;181(5):486–493. | ||
Zöller B, Li X, Sundquist J, Sundquist K. Familial transmission of chronic obstructive pulmonary disease in adoptees: a Swedish nationwide family study. BMJ Open. 2015;5(4):e007310. | ||
Ito K, Mercado N. STOP accelerating lung aging for the treatment of COPD. Exp Gerontol. 2014;59:21–27. | ||
Miłkowska-Dymanowska J, Białas AJ, Makowska J, Wardzynska A, Górski P, Piotrowski WJ. Geroprotectors as a therapeutic strategy for COPD – where are we now? Clin Interv Aging. 2017;12:1811–1817. | ||
Agustí A, Celli B, Faner R. What does endotyping mean for treatment in chronic obstructive pulmonary disease? Lancet. 2017;390(10098):980–987. | ||
Miłkowska-Dymanowska J, Białas AJ, Zalewska-Janowska A, Górski P, Piotrowski WJ. Underrecognized comorbidities of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:1331–1341. | ||
Hansson GK, Inflammation HGK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–1695. | ||
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. | ||
Harrison MT, Short P, Williamson PA, Singanayagam A, Chalmers JD, Schembri S. Thrombocytosis is associated with increased short and long term mortality after exacerbation of chronic obstructive pulmonary disease: a role for antiplatelet therapy? Thorax. 2014;69(7):609–615. | ||
Białas AJ, Pedone C, Piotrowski WJ, Antonelli Incalzi R. Platelet distribution width as a prognostic factor in patients with COPD – pilot study. Int J Chron Obstruct Pulmon Dis. 2017;12:2261–2267. | ||
Aravamudan B, Thompson MA, Pabelick CM, Prakash YS. Mitochondria in lung diseases. Expert Rev Respir Med. 2013;7(6):631–646. | ||
Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80(5):780–787. | ||
Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134(3):707–716. | ||
Białas AJ, Sitarek P, Miłkowska-Dymanowska J, Piotrowski WJ, Górski P. The role of mitochondria and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cell Longev. 2016;2016. | ||
Chen S, Su Y, Wang J. ROS-mediated platelet generation: a microenvironment-dependent manner for megakaryocyte proliferation, differentiation, and maturation. Cell Death Dis. 2013;4(7):e722. | ||
Kosmider L, Sobczak A, Szołtysek-Bołdys I, et al. Assessment of nicotine concentration in electronic nicotine delivery system (ENDS) liquids and precision of dosing to aerosol. Przegl Lek. 2015;72(10):500–504. | ||
Sjövall F, Morota S, Hansson MJ, Friberg H, Gnaiger E, Elmér E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit Care. 2010;14(6):R214. | ||
Parameswaran H, Majumdar A, Ito S, Alencar AM, Suki B. Quantitative characterization of airspace enlargement in emphysema. J Appl Physiol. 2006;100(1):186–193. | ||
Jacob RE, Carson JP, Gideon KM, Amidan BG, Smith CL, Lee KM. Comparison of two quantitative methods of discerning airspace enlargement in smoke-exposed mice. PLoS One. 2009;4(8):e6670. | ||
Yamato H, Sun JP, Churg A, Wright JL. Cigarette smoke-induced emphysema in guinea pigs is associated with diffusely decreased capillary density and capillary narrowing. Lab Invest. 1996;75(2):211–219. | ||
Macnee W. ABC of chronic obstructive pulmonary disease. Pathology, pathogenesis, and pathophysiology. BMJ. 2006;332(7551):1202–1204. | ||
Brusselle GG, Demoor T, Bracke KR, Brandsma CA, Timens W. Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur Respir J. 2009;34(1):219–230. | ||
Kelsen SG, Aksoy MO, Georgy M, et al. Lymphoid follicle cells in chronic obstructive pulmonary disease overexpress the chemokine receptor CXCR3. Am J Respir Crit Care Med. 2009;179(9):799–805. | ||
Dalen H. An ultrastructural study of the tracheal epithelium of the guinea-pig with special reference to the ciliary structure. J Anat. 1983;136(Pt 1):47–67. | ||
Ressmeyer AR, Larsson AK, Vollmer E, Dahlèn SE, Uhlig S, Martin C. Characterisation of guinea pig precision-cut lung slices: comparison with human tissues. Eur Respir J. 2006;28(3):603–611. | ||
Muccitelli RM, Tucker SS, Hay DW, Torphy TJ, Wasserman MA. Is the guinea pig trachea a good in vitro model of human large and central airways? Comparison on leukotriene-, methacholine-, histamine- and antigen-induced contractions. J Pharmacol Exp Ther. 1987;243(2):467–473. | ||
Canning BJ, Chou Y. Using guinea pigs in studies relevant to asthma and COPD. Pulm Pharmacol Ther. 2008;21(5):702–720. | ||
Ghorani V, Boskabady MH, Khazdair MR, Kianmeher M. Experimental animal models for COPD: a methodological review. Tob Induc Dis. 2017;15:25. | ||
Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. | ||
Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. | ||
Talbot DA, Lambert AJ, Brand MD. Production of endogenous matrix superoxide from mitochondrial complex I leads to activation of uncoupling protein 3. FEBS Lett. 2004;556(1–3):111–115. | ||
Brookes PS. Mitochondrial proton leak and superoxide generation: an hypothesis. Biochem Soc Trans. 1998;26(4):S331. | ||
Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29(2):169–202. | ||
Ku HH, Sohal RS. Comparison of mitochondrial pro-oxidant generation and anti-oxidant defenses between rat and pigeon: possible basis of variation in longevity and metabolic potential. Mech Ageing Dev. 1993;72(1):67–76. | ||
Papaioannou AI, Rossios C, Kostikas K, Ito K. Can we delay the accelerated lung aging in COPD? Anti-aging molecules and interventions. Curr Drug Targets. 2013;14(2):149–157. | ||
Begonja AJ, Gambaryan S, Geiger J, et al. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106(8):2757–2760. | ||
Krötz F, Sohn HY, Pohl U. Reactive oxygen species: players in the platelet game. Arterioscler Thromb Vasc Biol. 2004;24(11):1988–1996. | ||
Jang JY, Min JH, Chae YH, et al. Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid Redox Signal. 2014;20(16):2528–2540. | ||
Qiao J, Arthur JF, Gardiner EE, Andrews RK, Zeng L, Xu K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018;14:126–130. | ||
Wang WQ, Zhang HF, Gao GX, Bai QX, Li R, Wang XM. Adiponectin inhibits hyperlipidemia-induced platelet aggregation via attenuating oxidative/nitrative stress. Physiol Res. 2011;60(2):347–354. | ||
Iuliano L, Praticò D, Ghiselli A, Bonavita MS, Violi F. Superoxide dismutase triggers activation of “primed” platelets. Arch Biochem Biophys. 1991;289(1):180–183. | ||
Handin RI, Karabin R, Boxer GJ. Enhancement of platelet function by superoxide anion. J Clin Invest. 1977;59(5):959–965. | ||
Garcia-Souza LF, Oliveira MF. Mitochondria: biological roles in platelet physiology and pathology. Int J Biochem Cell Biol. 2014;50:156–160. | ||
Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2012;32(12):2946–2955. | ||
Bakdash N, Williams MS. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic Biol Med. 2008;45(2):158–166. | ||
Pignatelli P, Pulcinelli FM, Lenti L, Gazzaniga PP, Violi F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood. 1998;91(2):484–490. | ||
Arthur JF, Qiao J, Shen Y, et al. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J Thromb Haemost. 2012;10(6):1133–1141. | ||
Inoue O, Suzuki-Inoue K, Mccarty OJ, et al. Laminin stimulates spreading of platelets through integrin alpha6beta1-dependent activation of GPVI. Blood. 2006;107(4):1405–1412. | ||
Alshehri OM, Hughes CE, Montague S, et al. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126(13):1601–1608. | ||
Carrim N, Walsh TG, Consonni A, Torti M, Berndt MC, Metharom P. Role of focal adhesion tyrosine kinases in GPVI-dependent platelet activation and reactive oxygen species formation. PLoS One. 2014;9(11):e113679. | ||
Praticó D, Pasin M, Barry OP, et al. Iron-dependent human platelet activation and hydroxyl radical formation: involvement of protein kinase C. Circulation. 1999;99(24):3118–3124. | ||
Caccese D, Praticò D, Ghiselli A, et al. Superoxide anion and hydroxyl radical release by collagen-induced platelet aggregation – role of arachidonic acid metabolism. Thromb Haemost. 2000;83(3):485–490. | ||
Arthur JF, Shen Y, Gardiner EE, et al. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J Thromb Haemost. 2011;9(1):163–172. | ||
Katsume A, Okigaki M, Matsui A, et al. Early inflammatory reactions in atherosclerosis are induced by proline-rich tyrosine kinase/reactive oxygen species-mediated release of tumor necrosis factor-alpha and subsequent activation of the p21Cip1/Ets-1/p300 system. Arterioscler Thromb Vasc Biol. 2011;31(5):1084–1092. | ||
Ferroni P, Vazzana N, Riondino S, Cuccurullo C, Guadagni F, Davì G. Platelet function in health and disease: from molecular mechanisms, redox considerations to novel therapeutic opportunities. Antioxid Redox Signal. 2012;17(10):1447–1485. | ||
Schildknecht S, van der Loo B, Weber K, Tiefenthaler K, Daiber A, Bachschmid MM. Endogenous peroxynitrite modulates PGHS-1-dependent thromboxane A2 formation and aggregation in human platelets. Free Radic Biol Med. 2008;45(4):512–520. | ||
Krötz F, Sohn HY, Gloe T, et al. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100(3):917–924. | ||
Núñez-Córdoba JM, Martínez-González MA. Antioxidant vitamins and cardiovascular disease. Curr Top Med Chem. 2011;11(14):1861–1869. | ||
Anfossi G, Russo I, Massucco P, Mattiello L, Cavalot F, Trovati M. N-acetyl-L-cysteine exerts direct anti-aggregating effect on human platelets. Eur J Clin Invest. 2001;31(5):452–461. | ||
Fowdar K, Chen H, He Z, et al. The effect of N-acetylcysteine on exacerbations of chronic obstructive pulmonary disease: a meta-analysis and systematic review. Heart Lung. 2017;46(2):120–128. | ||
Salve VT, Atram JS. N-acetylcysteine combined with home based physical activity: effect on health related quality of life in stable COPD patients: a randomised controlled trial. J Clin Diagn Res. 2016;10(12):OC16–OC19. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.