Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Mitochondrial diseases caused by mtDNA mutations: a mini-review
Authors Ryzhkova AI ![]() , Sazonova MA
, Sazonova MA ![]() , Sinyov VV
, Sinyov VV ![]() , Galitsyna EV, Chicheva MM
, Galitsyna EV, Chicheva MM ![]() , Melnichenko AA
, Melnichenko AA ![]() , Grechko AV, Postnov AY, Orekhov AN
, Grechko AV, Postnov AY, Orekhov AN ![]() , Shkurat TP
, Shkurat TP
Received 23 October 2017
Accepted for publication 14 June 2018
Published 9 October 2018 Volume 2018:14 Pages 1933—1942
DOI https://doi.org/10.2147/TCRM.S154863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Anastasia I Ryzhkova,1,2 Margarita A Sazonova,1,3 Vasily V Sinyov,1 Elena V Galitsyna,4 Mariya M Chicheva,4 Alexandra A Melnichenko,4 Andrey V Grechko,5 Anton Yu Postnov,1 Alexander N Orekhov,3,6 Tatiana P Shkurat4
1Laboratory of Medical Genetics, National Medical Research Center of Cardiology, Moscow, Russian Federation; 2Department of Virology, K.I. Skryabin Moscow State Academy of Veterinary Medicine and Biotechnology-MVA, Moscow, Russian Federation; 3Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, Moscow, Russian Federation; 4Department of Genetics, Southern Federal University, Rostov-on-Don, Russian Federation; 5Federal Research and Clinical Center of Reanimatology and Rehabilitology, Moscow, Russian Federation; 6Institute for Atherosclerosis Research, Skolkovo Innovative Centre, Moscow Region, Russian Federation
Abstract: There are several types of mitochondrial cytopathies, which cause a set of disorders, arise as a result of mitochondria’s failure. Mitochondria’s functional disruption leads to development of physical, growing and cognitive disabilities and includes multiple organ pathologies, essentially disturbing the nervous and muscular systems. The origins of mitochondrial cytopathies are mutations in genes of nuclear DNA encoding mitochondrial proteins or in mitochondrial DNA. Nowadays, numerous mtDNA mutations significant to the appearance and progress of pathologies in humans are detected. In this mini-review, we accent on the mitochondrial cytopathies related to mutations of mtDNA. As well known, there are definite set of symptoms of mitochondrial cytopathies distinguishing or similar for different syndromes. The present article contains data about mutations linked with cytopathies that facilitate diagnosis of different syndromes by using genetic analysis methods. In addition, for every individual, more effective therapeutic approach could be developed after wide-range mutant background analysis of mitochondrial genome.
Keywords: mitochondrial cytopathy, mitochondrial dysfunction, mtDNA mutation, mitochondrial gene mutation
Introduction
Mitochondria are organelles, descended from ancient Alphaproteobacteria around 1.5–2 billion years ago.1,2 The presence of two membranes (the outer high-permeable membrane and the internal membrane, separating the matrix from the environment), its own ribosomes (with a smaller sedimentation coefficient than the cytoplasmic ones and divergent of its both composition and structure) and the circular double-stranded DNA in mitochondria is explained by their origin.3–7 One of the main mitochondrial functions is energy production, oxidative phosphorylation (OXPHOS) system generating ~90% of cellular energy.8 In addition, these organelles are involved in the maintaining of essential cellular processes: regulation of intracellular energy metabolism, play an important role in intracellular signaling, apoptosis, centrosome homeostasis and mitotic fidelity and chromosomal gene expression.8–11 In addition, they have a great importance in the process of development, differentiation and proliferation of cells and tissues both in normal and abnormal ways (for instance, tumorigenesis).8,10
Mitochondrial DNA is a double-stranded supercoiled ring molecule, which does not contain histones. It means that mtDNA is not packaged in the form of nucleosomes such as nuclear chromatin. However, this molecule forms a complex with >20 different proteins.12,13 Such a spherical nucleoprotein complex 100 nm in diameter is called a nucleoid and can contain one or more copy of mtDNA.13–15 MtDNA nucleoid contains two areas: core and peripheral regions that are formed by proteins such as TFAM, mtSSB, POLRMT, POLG, Twinkle helicase, TFB1M/TFB2M, TOP1M, mTERF, ATAD3, LONP1, ANT, PHB, Hsp60, M19 (MNF1) and others.12,13 Therefore, mtDNA nucleoid is anchored in the inner mitochondrial membrane by D-loop region of mtDNA, which constitutes a regulatory site for mtDNA replication and transcription.13
mtDNA is a polyploidy molecule, and each cell contains hundreds or thousands of nucleoids.16 Mammalian mitochondrial genome contains 16.5 thousand base pairs (it differs in various species), coding 37 genes, such as genes of subunits 12S and 16S of ribosomal RNA, 22 transfer RNA genes and 13 genes of protein subunits, included in the enzyme complex OXPHOS.10,17 Among 13 genes of respiratory chain proteins, seven genes encode subunits of I complex, three genes encode subunits of IV complex, two genes encode subunits of V complex and one gene encodes subunit of III complex.10 It should be emphasized that the coding region of mitochondrial genome does not contain introns. However, there is an ~1 kb noncoding region (D-loop) in this genome, which consists of the control and two hypervariable parts.10,18 Control parts of D-loop contain light strand promoter (LSP) and heavy strand promoter (HSP).13 The double strand of mitochondrial DNA subdivides to heavy and light strands (guanine and cytosine enriched, respectively).
In contrast to nuclear DNA, mitochondrial DNA is more susceptible to damages; although it has a mechanism of repair, the capacity of repair is limited.14,19 In the processes of mitogenesis, mitophagy, fusion and fission of mitochondria, the heteroplasmy level of mitochondrial genome mutations can decrease. For example, the combination of the mitochondrial genome mutation m.1494C>T with aminoglycoside stress led to mitophagy and the occurrence of oxidative stress in a cell.20 The association of mtDNA mutations with mitochondrial biogenesis was studied by Liu et al21 This research team analyzed lymphocyte cell lines obtained from patients with maternally inherited hypertension. These cells contained mitochondrial genome mutation m.4467C>A (MT-TM gene). It was found that in this cell line, containing mitochondrial genome mutation m.4467C>A (MT-TM gene), reactive oxygen species production was 114.5% higher compared with that in control cell line, not containing this mutation. However, the level of ATP in this mutant cell line was 26.4% lower compared to that in the control cell line.
However, with age, mitochondria can accumulate mutations that accelerate the aging process and degeneration.22 According to the fact that mitochondrial genome is maternally inherited, some mutations emerging in gametes can become hereditary.16,23 As noted earlier, mtDNA is a polyploid that could lead to the arising of heteroplasmy. The coexistence of more than one mtDNA variants in the same cell is a heteroplasmy. Otherwise, cell mitochondrial genome is a homoplasmic.18,24 The penetrance of mtDNA mutations in each individual is determined by many factors, including the localization of the mutation, its type and size (number of affected nucleotides) and the level of heteroplasmy.18,25–27
It is well known that a large group of human diseases is characterized by the presence of defects in the mitochondrial activity. Such diseases can be both inherited and somatic. Mitochondrial diseases may be classified into two groups:
- caused by mtDNA mutations and
- occurring because of nuclear DNA mutations.28
Cytopathies associated with mitochondrial genome mutations
The present article focuses on mitochondrial cytopathies associated with mtDNA mutations. According to the literature, mitochondrial genome mutations are associated with different mitochondrial disorders (mitochondrial cytopathies), which mainly affect nervous and muscular tissues.11 Molecular–cellular and biochemical manifestations of mitochondrial cytopathies are associated with defects of polypeptide chains belonging to the enzyme complex OXPHOS, errors in the transcription process, caused by mutations in transfer and ribosomal RNAs of mitochondrial genome.27,29 Mitochondrial dysfunction can encourage leakage of electrons from the electron transport chain (ETC), and this subsequently leads to elevation of oxidative stress in mitochondria so as in other intracellular compartment.12,13 Decrease in ATP production by OXPHOS could lead to increase in mitochondrial biogenesis (mitogenesis) or mitophagy if the mitochondrial quality control is not interrupted. Clinical manifestations of mutations in mitochondrial genome may be absent because of heteroplasmy. However, due to elevated mitogenesis for energy production or level of hypoxia reduction, the count of mutated mtDNA could be rised.30 The detection of mitochondrial genome pathologies happens at the moment when the number of copies of mutated mtDNA reaches a certain threshold at which the manifestation of the disease takes place.26,27
Symptoms associated with mitochondrial cytopathies
There is a certain range of character and symptoms, which can be used for the detection and diagnosis of mitochondrial cytopathies. It should be noted that such symptoms may be absent in healthy patients or in carriers of mtDNA mutations in the asymptomatic period of the disease.11,16
The manifestation of the character and symptoms of mitochondrial cytopathies is associated with various organs and organ systems, such as11,31
- brain and nervous system (developmental disorders and mental disorders, dementia, cramps, migraines, stroke-like episodes, atypical cerebral palsy, weakness, areflexia, gastrointestinal disorders, fainting, disturbance of thermoregulation, vision loss and blindness, hearing disorders and deafness);
- muscles (weakness, hypotonia, seizures and muscle aches);
- heart problems (cardiomyopathy, cardiac arrhythmia), hepatic problems (hypoglycemia, liver failure) and kidney problems (atrophy of proximal tubule);
- endocrine (diabetes) and exocrine (pancreatic insufficiency) disorders and
- systemic problems (weight loss, stunting, fatigue, trouble breathing).
Various symptom combinations of disease onset are possible.11,16
Syndromes of mitochondrial cytopathies
Some groups of the most frequently occurring symptom combinations of mitochondrial cytopathies are combined into syndromes. Information on mtDNA mutations associated with mitochondrial cytopathies, a review of which is given below, is presented in Table 1.
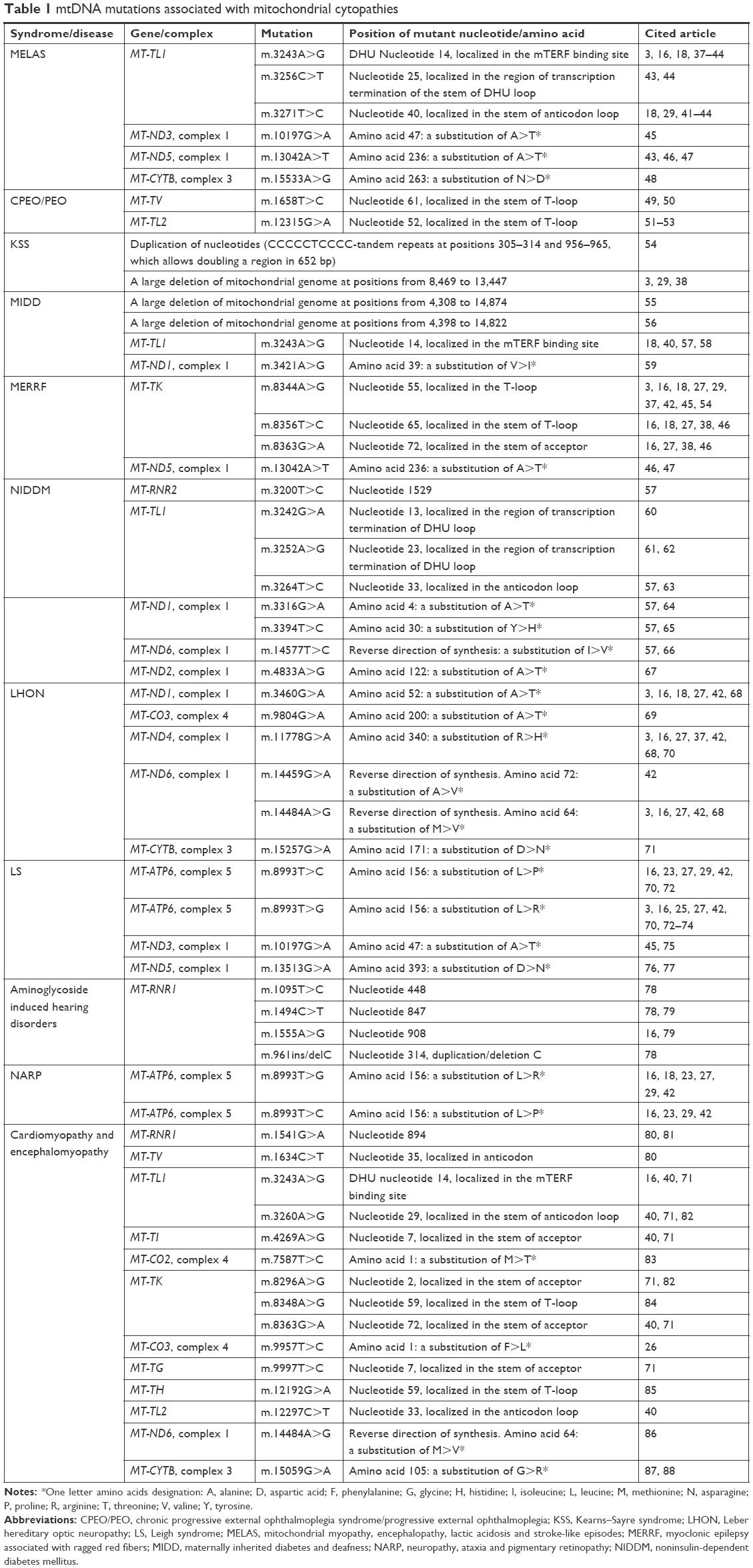 | Table 1 mtDNA mutations associated with mitochondrial cytopathies |
Cardiomyopathy and encephalomyopathy
Mitochondrial cardiomyopathy is described as a state of the myocardium, characterized by abnormal structure of the heart muscle and its functions or both of these parameters. Typical manifestations of mitochondrial disease are hypertrophic and dilated cardiomyopathy, arrhythmia and extensive myocardial infarction of left ventricle. Severe manifestations include cardiomyopathies, ventricular tachycardia and cardiac failure. The condition of patients with mitochondrial cardiomyopathy could acutely deteriorate in metabolic crisis caused by physical factors such as febrile states.32,33
Encephalomyopathies, associated with mitochondrial mutations, are characterized by lesions of gray matter of the brain and spinal cord. This pathology is caused by disturbance of the energy supply of the nervous system cells, leading to a change in membrane polarization and, as a consequence, myoclonic seizures and epilepsy. Encephalopathy is characterized by dementia, migraine-like pain, stroke episodes, sensorineural hearing loss, nerve atrophy, etc.34–36
Mitochondrial mutations, associated with cardiomyopathy and encephalopathy, can be in both the protein-coding sites and the RNA-coding portions of mtDNA. Encephalomyopathy and cardiomyopathy are some of the characteristics of certain symptom combinations of mitochondrial cytopathies, including mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy associated with ragged red fibers (MERRF), chronic progressive external ophthalmoplegia syndrome/progressive external ophthalmoplegia (CPEO/PEO) and Kearns–Sayre syndrome (KSS), as well as Alpers–Huttenlocher disease, childhood onset epilepsia partialis continua (EPC) and myoclonic epilepsy myopathy sensory ataxia (MEMSA).89,90
Aminoglycoside-induced hearing disorders
Hereditary disorders of hearing and deafness can be triggered by a conductive or sensorineural cause and also by their combination. As a rule, the prevalence of hearing disorders increases with age. However, if there is a genetic predisposition, deafness/hearing disorder can occur under the influence of a trigger factor. Such triggers can be antibiotics of aminoglycoside group, inducing ototoxicity. The use of gentamicin, tobramycin, amikacin, kanamycin, or streptomycin, even once, can lead to a bilateral hearing loss of varying severity. Aminoglycoside-induced hearing disorder is caused by damage to the auditory system, vestibular apparatus or by both of the reasons. These damages are the consequence of the cochlear hair cells’ death and the vestibular apparatus. It is well known that in individuals with aminoglycoside-induced ototoxicity, mitochondrial genome mutations, for example, m.1555A>G and m.1494C>T, are often detected.78,79,91
KSS
KSS is a special type of mitochondrial myopathy, occurring because of the large heteroplasmic deletion of mtDNA, size 1.3–10 kb. The syndrome can be both maternally inherited and somatic. It occurs in the embryonic cells at the early stages of development.38 The disease is characterized by proximal muscle weakness, retinopathy, cardiac arrhythmia and ataxia.92 The diagnostics of this disease syndrome is complicated by the similarity of several syndromes of mitochondrial cytopathies: KSS, CPEO/PEO and ophthalmoplegia-plus syndrome. If the symptoms mentioned earlier appear in an individual prior to the age of 20 years, it may be affirmed that the patient has KSS. Diagnosis of these symptoms in an individual after 20 years or the diagnosis of three and less symptoms suggests that a patient has ophthalmoplegia-plus syndrome.16,93
CPEO/PEO
CPEO/PEO is symptomatically similar to KSS. It is distinguished by the presence of visual muscles’ myopathy and ptosis, pigmentary degeneration of retina (retinitis pigmentosa) and dysfunction of central nervous system (dementia, cerebral ataxia). The manifestation of the disease occurs in childhood. In addition, this syndrome is characterized by the development of endocrine symptoms (diabetes, growth disturbance because of the growth hormone deficiency, hypoparathyroidism), dysphagia, changes in biochemical parameters and an increase in the level of lactate and protein of cerebrospinal fluid such as KSS.38,93
Leber hereditary optic neuropathy (LHON)
LHON is characterized by a sudden, complete, painless loss of central vision caused by optic nerve atrophy. Optic nerves changes in LHON develop sequentially; first, there is a loss of sight in one eye and then in the second eye. LHON symptoms may occur at any age; the average age of LHON manifestation varies from 15 to 35 years, while the proportion of men and women for this pathology is 4:1.37 Some individuals, except the core symptoms, associated with the loss of central sight, were also diagnosed with cardiac conduction disorders, sensory and motor neuropathy, tremor, ataxia and damage of basal ganglia (LHON plus).94 It is supposed that the atrophy of nerves in LHON is associated with point mtDNA mutations in genes of polypeptide chains of the first complex of OXPHOS, leading to disruption of the complex and an increased oxidative stress in the nerve endings of cells.42
MELAS
MELAS is diagnosed in early childhood or in the juvenile period. The syndrome is characterized by dilated or hypertrophic cardiomyopathy, excitation of Hiss bundles of nervous fibers (preexcitation, bundle branch block), stroke-like episodes, seizures and diabetes. It is a neurodegenerative disease. In its course, the demyelination of the nerve fibers and the gradual death of neurons happen. In addition, the symptoms of MELAS are sensorineural hearing loss, ptosis, epilepsy, muscle fatigue and pain, generalized myopathy, myalgia and severe headache.41,95,96 MELAS is diagnosed if 1–30 casual point mitochondrial genome mutations are present, meanwhile in 80% of cases, mutation m.3243A>G of gene MT-TL1 is detected.43,44 The above-noted mutation leads to destabilization of tRNA and, accordingly, to a reduction in synthesis of OXPHOS proteins and insufficiency of complexes I, III and IV.42,43
MERRF
MERRF is a chronic neurodegenerative disease that manifests in both children and adults. This syndrome is accompanied by myoclonus, seizures and cerebellar ataxia. MERRF symptoms also consist of dementia, cardiomyopathy, cardiac arrhythmia, neuropathies, pyramidal insufficiency, optic atrophy and sensorineural hearing loss.55,96 The symptoms of this cytopathy are associated with mutations in complexes of NADH-CoQ reductase and cytochrome C-oxidase (COX), some polypeptide chains of which are encoded by mitochondrial genome. It was found that MT-TK gene mutations are the cause of MERRF; in 80% of cases, mutation m.8344A>G occurs; mutations m.8356T>C and m.8363G>A are detected less frequently.27,54
Maternally inherited diabetes and deafness (MIDD)
MIDD is characterized by sensorineural hearing loss and the development of diabetes in individuals in adulthood. MIDD includes insulin-dependent diabetes mellitus (IDDM) and noninsulin-dependent diabetes mellitus (NIDDM), which are associated with diabetes mellitus type 1 and type 2, respectively.7,58 Sometimes MIDD is accompanied by other symptoms of mitochondrial cytopathies: cardiomyopathy, myopathy, retinitis pigmentosa, ptosis, disorders of the renal tubules, and psychoneurological symptoms.97 Mitochondrial cytopathy MIDD can be caused by point mutations in mtDNA or large deletions, for example, nucleotide deletion at positions 4,308–14,874 or 4,398–14,822.55,56
Leigh syndrome (LS)
LS is an infantile subacute necrotizing encephalopathy. It is a progressive neurodegenerative disease affecting children. The first signs of this cytopathy are physical and mental developmental disorders and disruption of previously acquired skills. The clinical symptoms of LS include perinatal asphyxia, respiratory dysfunction, neuropathies of cranial nerves, ataxia, dystonia and hypotension, seizures and also disturbance of the reflex activity, in particular, sucking and swallowing reflexes. The course of the disease is progressing; it is rarely undulating.98 The causes of such symptoms are mutations and functional insufficiency of NADH-CoQ reductase and cytochrome COX and also other enzymes of energy metabolism, including the ATPase, pyruvate decarboxylase and pyruvate dehydrogenase. Inheritance of LS may be recessive, linked to the X-chromosome or an autosome, and mitochondrial.70 The point nucleotide substitution of mtDNA at position 8,993 of gene of the sixth protein subunit of ATPase is linked with the development of LS. Moreover, if the level of heteroplasmy for this mutation is >90%, LS develops in the individual, and if heteroplasmy for this mutation is detected within 70%–90%, neuropathy, ataxia and pigmentary retinopathy (NARP) syndrome develops. Symptoms may not manifest in patients with the heteroplasmy level of this single nucleotide substitution <70%.16,27,42
NARP syndrome
NARP as a rule manifests in the second decade of life. In contrast to LS, the disease progresses much slower. The characteristic symptoms of NAPR are proximal neurogenic muscle weakness, sensory neuropathy, ataxia, cardiomyopathy, developmental delay and learning problems and degeneration of the retina. In addition, dementia and seizures are diagnosed.99,100
Pathogenetic mechanism of mitochondrial genome mutations
According to the literature cited in this article, the mtDNA mutations associated with mitochondrial cytopathies lead to damage in the protein subunits of mitochondrial respiratory chain enzymes or transport RNA defects (Table 1). In the first case, the synthesis of ATP decreases as a result of the dysfunction of respiratory chain complexes. This leads to an energy deficit in the mitochondria and cells of the body. In particular, the pathogenic mechanism of mitochondrial genome mutation m.8249G>A (MT-CO2 gene complex 4), leading to mitochondrial myopathy, was described in an article by Mkaouar-Rebai et al101 In the second case, tRNA dysfunction occurs, leading to reduction in the amount of protein subunits of mitochondrial respiratory chain enzymes. This also leads to a decrease of the energy level in human cells and tissues. For example, the molecular mechanism of mutation m.3243A>G (MT-TL1 gene) pathogenesis, leading to renal disease and acute kidney injury, was described in the article by Emma et al.102 Pathogenesis of m.5521G>A (MT-TW gene) associated with mitochondrial myopathy was described in the article by Mkaouar-Rebai et al.101 Unfortunately, the molecular mechanisms of mitochondrial genome mutations that lead to the occurrence and development of mitochondrial cytopathies by the world’s scientists have not been sufficiently studied. Therefore, they require further research and specification.
Relationship between nuclear DNA and mtDNA in mitochondrial disease
Mitochondrial diseases can be caused by mutations and polymorphisms in both the mitochondrial and nuclear genomes. Most of mitochondria proteins are coded by nDNA (~1,500 proteins: OXPHOS, TIM/TOM complexes, nucleoid proteins, matrix proteins, channels proteins etc.) including proteins regulated mitophagy, mitogenesis, fusion, fission, signaling proteins. Such nuclear genome mutations can cause instability in the mitochondrial genome, including the occurrence of large deletions and point mutations of mtDNA.103 For example, the combination of polymorphisms of the nuclear genomes rs6493454 and rs7182946 (locus TRPM1, chromosome 15) with mitochondrial genome mutation m.4917A>G (MT-ND2 gene) increased the risk of age-related macular degeneration (AMD). A similar effect was observed when mtDNA mutation m.12771G>A (gene MT-ND5) was combined with polymorphisms of nuclear DNA rs4932478, rs4932480, rs11459118, rs875390, rs875391, rs2351006, rs144871045 and rs2070780 (loci ABHD2/RLBP1, chromosome 15).104 In the research work by Meng et al,105 it was shown that the combination of the nuclear modifier allele A10S in the TRMU gene with mitochondrial genome mutation m.1555A>G (MT-RNR1 gene) increased the risk of deafness.
It should be noted that the number of studies investigating how a combination of mutations of the mitochondrial and nuclear genome affects the occurrence and development of diseases is now very less.
MtDNA mutations and therapy of cytopathies
Molecular–cellular mechanisms of genesis and development of mitochondrial cytopathies are still not sufficiently understood and require further investigation. Therefore, treatment of mitochondrial disorders consists of symptomatic treatment, cofactor supplementations, NO precursors and exercise.11,106
Mitochondrial genome mutations can be used for creating models to investigate the molecular–cellular mechanisms of cytopathies. Such models are already created for the study of pathologies such as MELAS, LHON, LS and MERRF.42,73,107–109
In addition, the analysis of the manifestations of mitochondrial genome mutations associated with cytopathies will allow carrying out a differentiated medical therapy for patients, choosing the very medication that would affect individuals carrying a particular mtDNA mutation.96 For example, an approach to the treatment of patients with MELAS, having mutation m.3260A>G in gene MT-TL1, is developing. Researchers believe that this mutation leads to mitochondrial dysfunction and energy deficiency in cells. For MELAS therapy, ketogenic diet and magnesium were used. It has been discovered that such treatment may lead to improvement of the function of respiratory chain complexes.110 A group of scientists from Germany made an attempt to treat a patient with mutation m.11778G>A, which was diagnosed with LHON and multiple sclerosis. The man was assigned an immunosuppressive therapy with mitoxantrone. In 12 months, the patient’s condition improved.111
Modern way for mitochondrial cytopathy therapy is gene therapy development. Several different approaches are possible in gene therapy. The first method is a heteroplasmy shifting or reduction of mutant mtDNA, the second is a transfer of normal mtDNA polypeptides into the mitochondrion and the third is direct medication of the mtDNA.106 In addition, there aroused interest in the technology of using donor mitochondria in the process of fertilization for prevention of maternally inherited mitochondrial disorders. The recent research had shown that the interaction between donor mitochondria and host cell nucleus is normal in transcriptomic and energetic profiles. Moreover, targeted treatment of mitochondrial diseases can be achieved via nanotube transmission of mtDNA from one cell to another.112,113
Conclusion
The manifestation of most mitochondrial syndromes of cytopathies has similar parameters.11,16 The use of only the biochemical and clinical research methods may not be sufficient for the appropriate diagnosis. In this case, a necessary step is the application of genetic analysis methods. If there is clear evidence of hereditary cytopathies, it is necessary to analyze mitochondrial genome mutations, for which, according to the literature, an association with the studied disease was found. It is important to note that in the development of the disease, not only a specific mutation and its heteroplasmy level are important but also a general mutant background formed by all mutant alleles of mitochondrial genome. At the same time, the symptoms of mitochondrial cytopathies in patients can occur only after reaching a certain threshold of total mutation burden of the organism.
Mitochondrial genome mutations, detected during the analysis of the literature, can be used for creating models to investigate molecular–cellular mechanisms of cytopathies. In addition, the analysis of the manifestations of mitochondrial genome mutations associated with cytopathies will allow developing cellular models for choosing drug therapy for individuals having these pathologies. These cellular models will contain mutations associated with certain types of cytopathies.
Acknowledgments
This study was supported by the Russian Science Foundation, Grant No 14-14-01038. The authors record their sincere thanks to Zukhra B Khasanova (National Medical Research Center of Cardiology, Moscow, Russian Federation) and Dr Igor A Sobenin (National Medical Research Center of Cardiology, Moscow, Russian Federation) for their facilitation and constructive comments in the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4(9):a011403. | ||
Kurland CG, Andersson SG. Origin and evolution of the mitochondrial proteome. Microbiol Mol Biol Rev. 2000;64(4):786–820. | ||
Zapico SC, Ubelaker DH. mtDNA Mutations and Their Role in Aging, Diseases and Forensic Sciences. Aging Dis. 2013;4(6):364–380. | ||
Kaushal PS, Sharma MR, Agrawal RK. The 55S mammalian mitochondrial ribosome and its tRNA-exit region. Biochimie. 2015;114:119–126. | ||
O’Brien TW. Properties of human mitochondrial ribosomes. IUBMB Life. 2003;55(9):505–513. | ||
O’Brien TW. Evolution of a protein-rich mitochondrial ribosome: implications for human genetic disease. Gene. 2002;286(1):73–79. | ||
Alcolado JC, Thomas AW. Maternally inherited diabetes mellitus: the role of mitochondrial DNA defects. Diabet Med. 1995;12(2):102–108. | ||
Druzhyna NM, Wilson GL, Ledoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008;129(7–8):383–390. | ||
Donthamsetty S, Brahmbhatt M, Pannu V, et al. Mitochondrial genome regulates mitotic fidelity by maintaining centrosomal homeostasis. Cell Cycle. 2014;13(13):2056–2063. | ||
St John JC. Mitochondrial DNA copy number and replication in reprogramming and differentiation. Semin Cell Dev Biol. 2016;52:93–101. | ||
El-Hattab AW, Scaglia F. Mitochondrial cytopathies. Cell Calcium. 2016;60(3):199–206. | ||
Lee SR, Kim N, Noh YH, et al. Mitochondrial DNA, mitochondrial dysfunction, and cardiac manifestations. Front Biosci. 2017; 22(1):1177–1194. | ||
Lee SR, Han J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid Med Cell Longev. 2017;2017:8060949. | ||
Szczepanowska K, Trifunovic A. Different faces of mitochondrial DNA mutators. Biochim Biophys Acta. 2015;1847(11):1362–1372. | ||
Bonekamp NA, Larsson NG. SnapShot: Mitochondrial Nucleoid. Cell. 2017;172(1):388.e1. | ||
Schmiedel J, Jackson S, Schafer J, Reichmann H. Mitochondrial cytopathies. J Neurol. 2003;250(3):267–277. | ||
Lee WT, St John J. The control of mitochondrial DNA replication during development and tumorigenesis. Ann N Y Acad Sci. 2015;1350:95–106. | ||
Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226(2):274–286. | ||
Fetterman JL, Holbrook M, Westbrook DG, et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc Diabetol. 2016;15(1):53. | ||
Yu J, Zheng J, Zhao X, Liu J, et al. Aminoglycoside stress together with the 12S rRNA 1494C>T mutation leads to mitophagy. PLoS One. 2014;9(12):e114650. | ||
Liu Y, Li Y, Zhu C, et al. Mitochondrial biogenesis dysfunction and metabolic dysfunction from a novel mitochondrial tRNAMet 4467 C>A mutation in a Han Chinese family with maternally inherited hypertension. Sci Rep. 2017;7(1):3034. | ||
Orogo AM, Gonzalez ER, Kubli DA, et al. Accumulation of Mitochondrial DNA Mutations Disrupts Cardiac Progenitor Cell Function and Reduces Survival. J Biol Chem. 2015;290(36):22061–22075. | ||
Nunnari J, Suomalainen A. Mitochondria: In Sickness and in Health. Cell. 2012;148(6):1145–1159. | ||
St John J. The control of mtDNA replication during differentiation and development. Biochim Biophys Acta. 1840;2014(4):1345–1354. | ||
Kauppila JHK, Stewart JB. Mitochondrial DNA: Radically free of free-radical driven mutations. Biochim Biophys Acta. 2015;1847(11):1354–1361. | ||
Arbustini E, Diegoli M, Pilotto A. Mitochondrial DNA Mutations and Cardiomyopathies. Milano: Springer Camerini F, Gavazzi A, De Maria R, editors. Cardiomyopathies: Advances in; 1998:117–127. | ||
Dimauro S. Mitochondrial diseases Biochim Biophys Acta. 2004;1658(1–2):80–88. | ||
Griffin HR, Pyle A, Blakely EL, et al. Accurate mitochondrial DNA sequencing using off-target reads provides a single test to identify pathogenic point mutations. Genet Med. 2014;16(12):962–971. | ||
Berardo A, Musumeci O, Toscano A. Cardiological manifestations of mitochondrial respiratory chain disorders. Acta Myol. 2011;30(1):9–15. | ||
Herst PM, Rowe MR, Carson GM, Berridge MV. Functional Mitochondria in Health and Disease. Front Endocrinol. 2017;8:296. | ||
Turnbull DM, Rustin P. Genetic and biochemical intricacy shapes mitochondrial cytopathies. Neurobiol Dis. 2016;92:Part A:55–63. | ||
Dominic EA, Ramezani A, Anker SD, Verma M, Mehta N, Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. 2014;100(8):611–618. | ||
Finsterer J. Stroke and Stroke-like Episodes in Muscle Disease. Open Neurol J. 2012;6:26–36. | ||
Rahman S. Mitochondrial disease and epilepsy. Dev Med Child Neurol. 2012;54(5):397–406. | ||
Khurana DS, Salganicoff L, Melvin JJ, et al. Epilepsy and Respiratory Chain Defects in Children with Mitochondrial Encephalopathies. Neuropediatrics. 2008;39(01):8–13. | ||
Parikh S. The neurologic manifestations of mitochondrial disease. Dev Disabil Res Rev. 2010;16(2):120–128. | ||
Wallace DC. A mitochondrial bioenergetic etiology of disease. J Clin Invest. 2013;123(4):1405–1412. | ||
Kabunga P, Lau AK, Phan K, et al. Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int J Cardiol. 2015;181:303–310. | ||
Zhu CC, Traboulsi EI, Parikh S. Ophthalmological findings in 74 patients with mitochondrial disease. Ophthalmic Genet. 2017;38(1):67–69. | ||
Mohammed S, Bahitham W, Chan A, Chiu B, Bamforth F, Sergi C. Mitochondrial DNA related cardiomyopathies. Front Biosci (Elite Ed). 2012;4:1706–1716. | ||
Vydt TCG, de Coo RFM, Soliman OII, et al. Cardiac involvement in adults with m.3243A>G MELAS gene mutation. Am J Cardiol. 2007;99(2):264–269. | ||
Wallace DC. Mitochondrial defects in neurodegenerative disease. Ment Retard Dev Disabil Res Rev. 2001;7(3):158–166. | ||
Lorenzoni PJ, Werneck LC, Kay CS, Silvado CE, Scola RH. When should MELAS (Mitochondrial myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like episodes) be the diagnosis? Arq Neuropsiquiatr. 2015;73(11):959–967. | ||
Wang YX, Wd L. Progress in Diagnosing Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episodes. Chin Med J. 2015;128(13):1820–1825. | ||
Leng Y, Liu Y, Fang X, et al. The mitochondrial DNA 10197 G > A mutation causes MELAS/Leigh overlap syndrome presenting with acute auditory agnosia. Mitochondrial DNA. 2015;26(2):208–212. | ||
Lorenzoni PJ, Scola RH, Csk K, Silvado CES, Werneck LC. When should MERRF (myoclonus epilepsy associated with ragged-red fibers) be the diagnosis? Arq Neuropsiquiatr. 2014;72:803–811. | ||
Schinwelski M, Kierdaszuk B, Dulski J, et al. Changing phenotypic expression in a patient with a mitochondrial encephalopathy due to 13042G>A de novo mutation – a 5 year follow up. Metab Brain Dis. 2015;30(4):1083–1085. | ||
Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, et al. Impact of the mitochondrial genetic background in complex III deficiency. PLoS One. 2010;5(9):e12801. | ||
Zhou L, Wang H, Wei J, Wang Y, Wang Y. No association between mitochondrial tRNA(Val) T1658C mutation and chronic progressive external ophthalmoplegia (CPEO). Mitochondrial DNA. 2014;25(5):385–386. | ||
Yan N, Cai S, Guo B, et al. A novel mitochondrial tRNA(Val) T1658C mutation identified in a CPEO family. Mol Vis. 2010;16:1736–1742. | ||
Elson JL, Swalwell H, Blakely EL, Mcfarland R, Taylor RW, Turnbull DM. Pathogenic mitochondrial tRNA mutations-which mutations are inherited and why? Hum Mutat. 2009;30(11):E984–E992. | ||
Karadimas CL, Salviati L, Sacconi S, et al. Mitochondrial myopathy and ophthalmoplegia in a sporadic patient with the G12315A mutation in mitochondrial DNA. Neuromuscul Disord. 2002;12(9):865–868. | ||
Cardaioli E, da Pozzo P, Malfatti E, et al. Chronic progressive external ophthalmoplegia: A new heteroplasmic tRNA(Leu(CUN)) mutation of mitochondrial DNA. J Neurol Sci. 2008;272(1):106–109. | ||
Mancuso M, Petrozzi L, Filosto M, et al. MERRF syndrome without ragged-red fibers: the need for molecular diagnosis. Biochem Biophys Res Commun. 2007;354(4):1058–1060. | ||
Ballinger SW, Shoffner JM, Hedaya EV, et al. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat Genet. 1992;1(1):11–15. | ||
Ballinger SW, Shoffner JM, Gebhart S, Koontz DA, Wallace DC. Mitochondrial diabetes revisited. Nat Genet. 1994;7(4):458–459. | ||
Zhelankin AV, Sazonova MA. Association of the mutations in the human mitochondrial genome with chronic non-inflammatory diseases: type 2 diabetes, hypertension and different types of cardiomyopathy. Patol Fiziol Eksp Ter. 2012;3:123–128. | ||
Maassen JA, van den Ouweland JM, T Hart LM, Lemkes HH. Maternally inherited diabetes and deafness: a diabetic subtype associated with a mutation in mitochondrial DNA. Horm Metab Res. 1997;29(2):50–55. | ||
Chen FL, Liu Y, Song XY, et al. A novel mitochondrial DNA missense mutation at G3421A in a family with maternally inherited diabetes and deafness. Mutat Res. 2006;602(1–2):26–33. | ||
Wortmann SB, Champion MP, van den Heuvel L, et al. Mitochondrial DNA m.3242G > A mutation. An under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction? Eur J Med Genet. 2012;55(10):552–556. | ||
Ivanova MM, Borodachev EN, Sazonova MA. Human pathologies associated with mutations of mitochondrial genome. Patol Fiziol Eksp Ter. 2012;3:115–122. | ||
Ma L, Wang H, Chen J, et al. Mitochondrial gene variation in type 2 diabetes mellitus: detection of a novel mutation associated with maternally inherited diabetes in a Chinese family. Chin Med J. 2000;113(2):111–116. | ||
Schaefer AM, Walker M, Turnbull DM, Taylor RW. Endocrine disorders in mitochondrial disease. Mol Cell Endocrinol. 2013;379(1–2):2–11. | ||
Tang J, Li J, Tian X, Kong Q, Zhang Y. Mitochondrial DNA 3243, 3316 point mutations and type 2 diabetes mellitus. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2005;22(2):198–200. | ||
Hirai M, Suzuki S, Onoda M, et al. Mitochondrial DNA 3394 mutation in the NADH dehydrogenase subunit 1 associated with non-insulin-dependent diabetes mellitus. Biochem Biophys Res Commun. 1996;219(3):951–955. | ||
Tawata M, Hayashi JI, Isobe K, et al. A new mitochondrial DNA mutation at 14577 T/C is probably a major pathogenic mutation for maternally inherited type 2 diabetes. Diabetes. 2000;49(7):1269–1272. | ||
Ohkubo E, Aida K, Chen J, et al. A patient with type 2 diabetes mellitus associated with mutations in calcium sensing receptor gene and mitochondrial DNA. Biochem Biophys Res Commun. 2000;278(3):808–813. | ||
Patsi J, Kervinen M, Finel M, Hassinen IE. Leber hereditary optic neuropathy mutations in the ND6 subunit of mitochondrial complex I affect ubiquinone reduction kinetics in a bacterial model of the enzyme. Biochem J. 2008;409(1):129–137. | ||
Tansel T, Pacal F. Ustek D. A novel ATP8 gene mutation in an infant with tetralogy of Fallot. Cardiol Young. 2014;24(3):531–533. | ||
Darin N, Oldfors A, Moslemi A-R, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: Clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001;49(3):377–383. | ||
Rossignol R, Faustin B, Rocher C, Malgat M, Mazat J-P, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370(Pt 3):751–762. | ||
Marie SKN, Oba-Shinjo SM, Marques-Dias MJ, Rosemberg S, Kok F, Reed UC. The prevalence of mitochondrial DNA mutations in Leigh syndrome in a Brazilian series. MedicalExpress. 2014;1:239–242. | ||
Trounce I, Neill S, Wallace DC. Cytoplasmic transfer of the mtDNA nt 8993 T->G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc Natl Acad Sci U S A. 1994;91(18):8334–8338. | ||
Iida Y, Fujii K, Mizuochi H, et al. Rostral brain lesions of Leigh syndrome associated with the mitochondrial DNA 8993T>G mutation. J Neurol Sci. 2016;363:77–79. | ||
Wang K, Takahashi Y, Gao ZL, et al. Mitochondrial ND3 as the novel causative gene for Leber hereditary optic neuropathy and dystonia. Neurogenetics. 2009;10(4):337–345. | ||
Wang Z, Xk Q, Yao S, et al. Phenotypic patterns of MELAS/LS overlap syndrome associated with m.13513G>A mutation, and neuropathological findings in one autopsy case. Neuropathology. 2010;30(6):606–614. | ||
Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008;65(3):368–372. | ||
Jing W, Zongjie H, Denggang F, et al. Mitochondrial mutations associated with aminoglycoside ototoxicity and hearing loss susceptibility identified by meta-analysis. J Med Genet. 2015;52(2):95–103. | ||
Huang S, Xiang G, Kang D, et al. Rapid identification of aminoglycoside-induced deafness gene mutations using multiplex real-time polymerase chain reaction. Int J Pediatr Otorhinolaryngol. 2015;79(7):1067–1072. | ||
Jaksch M, Ogilvie I, Yao J, et al. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9(5):795–801. | ||
Jaksch M, Horvath R, Horn N, et al. Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology. 2001;57(8):1440–1446. | ||
Fosslien E. Mitochondrial medicine-cardiomyopathy caused by defective oxidative phosphorylation. Ann Clin Lab Sci. 2003;33(4):371–395. | ||
Pereira L, Soares P, Radivojac P, Li B, Samuels DC. Comparing phylogeny and the predicted pathogenicity of protein variations reveals equal purifying selection across the global human mtDNA diversity. Am J Hum Genet. 2011;88(4):433–439. | ||
Ueki I, Koga Y, Povalko N, et al. Mitochondrial tRNA gene mutations in patients having mitochondrial disease with lactic acidosis. Mitochondrion. 2006;6(1):29–36. | ||
Mimaki M, Ikota A, Sato A, et al. A double mutation (G11778A and G12192A) in mitochondrial DNA associated with Leber’s hereditary optic neuropathy and cardiomyopathy. J Hum Genet. 2003;48(1):47–50. | ||
Cao Y, Ma Y, Zhang Y, et al. Detection of eight frequently encountered point mutations in mitochondria in Chinese patients suggestive of mitochondrial encephalomyopathies. Mitochondrion. 2010;10(4):330–334. | ||
Nikitin AG, Lavrikova EY, Chistiakov DA. The heteroplasmic 15059G>A mutation in the mitochondrial cytochrome b gene and essential hypertension in type 2 diabetes. Diabetes Metab Syndr. 2012;6(3):150–156. | ||
Sobenin IA, Chistiakov DA, Sazonova MA, et al. Association of the level of heteroplasmy of the 15059G>A mutation in the MT-CYB mitochondrial gene with essential hypertension. World J Cardiol. 2013;5(5):132–140. | ||
Florian A, Ludwig A, Stubbe-Dräger B, et al. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J Cardiovasc Magn Reson. 2015;17(1):40. | ||
Finsterer J, Mahjoub SZ. Presentation of adult mitochondrial epilepsy. Seizure. 2013;22(2):119–123. | ||
Leis JA, Rutka JA, Gold WL. Aminoglycoside-induced ototoxicity. CMAJ. 2015. 2015;187(1):E52. | ||
Young TJ, Shah AK, Lee MH, Hayes DL. Kearns-Sayre Syndrome: a case report and review of cardiovascular complications. Pacing Clin Electrophysiol. 2005;28(5):454–457. | ||
Schon EA, Dimauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13(12):878–890. | ||
Al-Enezi M, Al-Saleh H, Nasser M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr J Ophthalmol. 2008;15(2):81–86. | ||
Thomas T, Craigen WJ, Moore R, Czosek R, Jefferies JL. Arrhythmia as a cardiac manifestation in MELAS syndrome. Mol Genet Metab Rep. 2015;4:9–10. | ||
Finsterer J. Treatment of central nervous system manifestations in mitochondrial disorders. Eur J Neurol. 2011;18(1):28–38. | ||
Naing A, Kenchaiah M, Krishnan B, et al. Maternally inherited diabetes and deafness (MIDD): diagnosis and management. J Diabetes Complications. 2014;28(4):542–546. | ||
Martín MA, Blázquez A, Gutierrez-Solana LG, et al. Leigh syndrome associated with mitochondrial complex I deficiency due to a novel mutation in the ndufs1 gene. Arch neurol. 2005;62(4):659–661. | ||
Gelfand JM, Duncan JL, Racine CA, et al. Heterogeneous patterns of tissue injury in NARP syndrome. J Neurol. 2011;258(3):440–448. | ||
Rawle MJ, Larner AJ. NARP syndrome: a 20-year follow-up. Case Rep Neurol. 2013;5(3):204–207. | ||
Mkaouar-Rebai E, Ben Mahmoud A, Chamkha I, et al. A novel MT-CO2 m.8249G > A pathogenic variation and the MT-TW m.5521G > A mutation in patients with mitochondrial myopathy. Mitochondrial DNA. 2014;25(5):394–399. | ||
Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12(5):267–280. | ||
Rusecka J, Kaliszewska M, Bartnik E, Tońska K. Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA. J Appl Genet. 2018;59(1):43–57. | ||
Persad PJ, Heid IM, Weeks DE, et al. International Age-Related Macular Degeneration Genomics Consortium (IAMDGC). Joint Analysis of Nuclear and Mitochondrial Variants in Age-Related Macular Degeneration Identifies Novel Loci TRPM1 and ABHD2/RLBP1. Invest Ophthalmol Vis Sci. 2017;58(10):4027–4038. | ||
Meng F, Cang X, Peng Y, et al. Biochemical evidence for a nuclear modifier allele (A10S) in TRMU (methylaminomethyl-2-thiouridylate-methyltransferase) related to mitochondrial tRNA modification in the phenotypic manifestation of deafness-associated 12S rRNA mutation. J Biol Chem. 2017;292(7):2881–2892. | ||
Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. | ||
Yoshii Y, Yoneda M, Ikawa M, et al. Radiolabeled Cu-ATSM as a novel indicator of overreduced intracellular state due to mitochondrial dysfunction: studies with mitochondrial DNA-less ρ0 cells and cybrids carrying MELAS mitochondrial DNA mutation. Nucl Med Biol. 2012;39(2):177–185. | ||
Jun AS, Trounce IA, Brown MD, Shoffner JM, Wallace DC. Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol Cell Biol. 1996;16(3):771–777. | ||
Williams AJ, Murrell M, Brammah S, Minchenko J, Christodoulou J. A novel system for assigning the mode of inheritance in mitochondrial disorders using cybrids and rhodamine 6G. Hum Mol Genet. 1999;8(9):1691–1697. | ||
Steriade C, Andrade DM, Faghfoury H, Tarnopolsky MA, Tai P. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may respond to adjunctive ketogenic diet. Pediatr Neurol. 2014;50(5):498–502. | ||
Buhmann C, Gbadamosi J, Heesen C. Visual recovery in a man with the rare combination of mtDNA 11778 LHON mutation and a MS-like disease after mitoxantrone therapy. Acta Neurol Scand. 2002;106(4):236–239. | ||
Chan SSL. Inherited mitochondrial genomic instability and chemical exposures. Toxicology. 2017;391:75–83. | ||
Dahiya N, Garg S. Three-parent baby: is it ethical? Indian J Med Ethics. 2018;3(2):169. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.