")
Back to Journals » International Journal of Nanomedicine » Volume 13
Mitochondria-targeted delivery of doxorubicin to enhance antitumor activity with HER-2 peptide-mediated multifunctional pH-sensitive DQAsomes
Authors Shi M, Zhang J, Li X, Pan S, Li J, Yang C, Hu H, Qiao M, Chen D, Zhao X
Received 26 January 2018
Accepted for publication 24 April 2018
Published 18 July 2018 Volume 2018:13 Pages 4209—4226
DOI https://doi.org/10.2147/IJN.S163858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Menghao Shi,1 Jiulong Zhang,1 Xiaowei Li,1 Shuang Pan,1 Jie Li,2 Chunrong Yang,3 Haiyang Hu,1 Mingxi Qiao,1 Dawei Chen,1 Xiuli Zhao1
1School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, Liaoning 110016, People’s Republic of China; 2Department of Pharmacy, Mudanjiang Medical University, Mudanjiang, Heilongjiang 157011, People’s Republic of China; 3College Pharmacy of Jiamusi University, Jiamusi, Heilongjiang 154007, People’s Republic of China
Introduction: Multidrug resistance (MDR) of breast cancer is the major challenge to successful chemotherapy while mitochondria-targeting therapy was a promising strategy to overcome MDR.
Materials and methods: In this study, HER-2 peptide-PEG2000-Schiff base-cholesterol (HPSC) derivate was synthesized successfully and incorporated it on the surface of the doxorubicin (DOX)-loaded dequalinium (DQA) chloride vesicle (HPS-DQAsomes) to treat drug-resistant breast cancer. Evaluations were performed using human breast cancer cell and DOX-resistant breast cancer cell lines (MCF-7 and MCF-7/ADR).
Results: The particle size of HPS-DQAsomes was ~110 nm with spherical shape. In vitro cytotoxicity assay indicated that HPS-DQAsomes could increase the cytotoxicity against MCF-7/ADR cell line. Cellular uptake and mitochondria-targeting assay demonstrated that HPS-DQAsomes could target delivering therapeutical agent to mitochondria and inducing mitochondria-driven apoptosis process. In vivo antitumor assay suggested that HPS-DQAsomes could reach favorable antitumor activity due to both tumor targetability and sub-organelles’ targetability. Histological assay also indicated that HPS-DQAsomes showed a strong apoptosis-inducing effect. No obvious systematic toxicity of HPS-DQAsomes could be observed.
Conclusion: In summary, multifunctional HPS-DQAsomes provide a novel and versatile approach for overcoming MDR via mitochondrial pathway in cancer treatment.
Keywords: mitochondrial target, DQAsomes, pH responsive, HER-2 peptide
Introduction
Currently, the morbidity and mortality of malignancy increases, and it becomes one of the most menacing diseases around the world.1–3 Chemotherapy is the leading strategy for clinical treatment of cancers.4,5 However, multidrug resistance (MDR) remains a great challenge for successful chemotherapy.1,6,7 There are various mechanisms associated with MDR, often involving acquired and intrinsic resistance.8,9 Unlike acquired MDR, which mechanism was originated from the overexpression of P-glycoprotein (P-gp), an ATP-dependent efflux pump, intrinsic MDR often attributed to genetic or epigenetic changes, which perturbed the apoptosis signaling pathway.10,11 Generally, the intrinsic pathway of apoptosis is often initiated at mitochondria.
The initiation process was often induced by the following three kinds of factors: direct caspase activators, eg, cytochrome C; indirect caspase activators, eg, the second mitochondria-derived activator of caspase (Smac); and caspase-independent cell death effectors, eg, apoptosis-inducing factor (AIF).12,13 Generally, the apoptosis process was often initiated from enhancing the permeability of mitochondria membrane, followed by the release of apoptosis factors, such as cytochrome C. Then, a cascade of events activated intracellular caspase family, which resulted in a partial self-digestion, eventually.
Due to the vital role of apoptosis, mitochondria were considered to be a potential drug delivery target. Mitochondria dysfunction attributed to target drug delivery therapeutical agent to mitochondria could not only cutoff the “energy supply”, which was necessary to P-gp function, but also induce the apoptosis cascade process and sufficiently overcome MDR.14,15 To achieve mitochondria-targeted drug delivery, various strategies have been used such as protein transduction domains (PTDs), mitochondrial targeting sequences (MTSs), and lipophilic cations (eg, triphenylphosphonium and dequalinium [DQA]), which have been packed or conjugated onto small molecules or nanoparticle drug delivery systems.16–21 DQA was a cationic bola-amphiphile composed of two quinaldinium rings linked by 10 methylene groups, which was first discovered as an antimicrobial agent. However, due to its unique characteristics, DQA could self-assemble in aqueous solution into liposomes like vesicles when prepared from DQA chloride, named DQAsomes.22–24 DQAsomes displayed a positive surface charge in aqueous environment. Therefore, DQAsomes could accumulate into mitochondria in response to the electrochemical gradient across the mitochondrial membrane system. Meanwhile, DQAsomes could not only load hydrophobic drug into the DQA membrane but also load gene through electrostatic interaction between negative charge gene and positive charge DQAsomes. DQAsomes have been widely applied for the mitochondrial delivery plasmid DNA in living cells.24 While there are many advantages of DQAsomes, many obstacles do exist. Short half-life is the major drawback for its further application. Meanwhile, positive surface charge of DQAsomes may arise nonspecific toxicity. Therefore, shielding the positive charge of DQAsomes and increasing the circulation time are becoming the great challenges for designing a novel drug delivery system. It is well known that polyethylene glycol (PEG)-modified nanoparticles owe longer circulation time in blood system because PEG dilemma could avoid the reorganization of reticulo-endothelial system (RES). Furthermore, the hydrophilic PEG chain could shield surface charge of DQAsomes. However, the major obstacle of this method is that it would decrease the mitochondrial targeting capability of DQAsomes due to decreased surface potential. Therefore, it is necessary to design and construct stimuli-responsive nanoparticles, which could shield the positive charge in blood system while exposing it intracellular to target mitochondria.
It is well known that there is a significant difference between normal tissue microenvironment and tumor microenvironment, including vascular abnormalities, reactive oxygen species (ROS), and pH.25,26 All these characteristics could be utilized to design stimuli-responsive drug delivery system. For instance, various pH conditions in normal tissues (pH 7.4) and tumor acidic microenvironment (pH 6.8–5.0) could be utilized to trigger specific release of therapeutical agents. This strategy could not only increase therapeutical efficacy but also decrease nonspecific drug release and decrease systematic toxicity.
To overcome these drawbacks, we designed and constructed a multifunctional drug delivery system to efficiently deliver therapeutical agents to the mitochondria of MDR tumor cells to overcome MDR. In our previous study, we have a successful pH-responsive PEG derivate, which was relatively stable in blood system and leaked rapidly in acidic tumor microenvironment.27,28 Therefore, we used this PEG derivate (PEG2000-Schiff base bond-cholesterol [PSC]) to modify the surface of PEG2000-Schiff base bond-DQAsomes (PS-DQAsomes). Increasing evidence indicated that the improved accumulation of doxorubicin (DOX) into mitochondria in cancer cells could increase the generation of ROS and damage mitochondrial respiratory chain components, resulting in the activation of cell apoptosis effect.29–31 Furthermore, mitochondrial metabolism is critical for the survival of drug-resistant cells and mitochondrial DOX delivery may prove to be an important mechanism for bypassing and overcoming DOX resistance in cancer cells. Although PS-DQAsomes could accumulate in tumor tissue via EPR-mediated passive targetability when it was intravenously administrated into the blood system, this effect is limited. It has been reported that receptor-mediated endocytosis with ligand modification could be higher than nonmodified one.32 Therefore, we hypothesized that the efficiency of PS-DQAsomes could be significantly facilitated via receptor-mediated endocytosis. It has been reported that human epidermal growth factor receptor-2 (HER-2) is highly expressed in many tumor cell lines including breast cancer.33,34 Therefore, we hypothesized that HER-2 is a key target for intracellular delivering therapeutical agents. HER-2 peptide (YCDGFYACYMDV) is an analog of trastuzumab, which could specifically bind human epidermal growth factor receptor-2. HER-2 peptide was used to modify on the surface of PS-DQAsomes to acquire active targetability to reach higher therapeutical efficacy.
In the present study, we synthesized HER-2 peptide-modified pH-responsive PEG derivate (HER-2-PEG2000-Schiff base-cholesterol [HPSC]) and used it to modify the surface of DQAsomes (HPS-DQAsomes) (Scheme 1). We hypothesized that HPS-DQAsomes could trigger the apoptosis-resistant breast cancer cell lines by acting on the mitochondria. Accordingly, the purpose of this study was to construct HPS-DQAsomes, explore its action mechanism, and evaluate its antitumor activity both in vitro and in vivo.
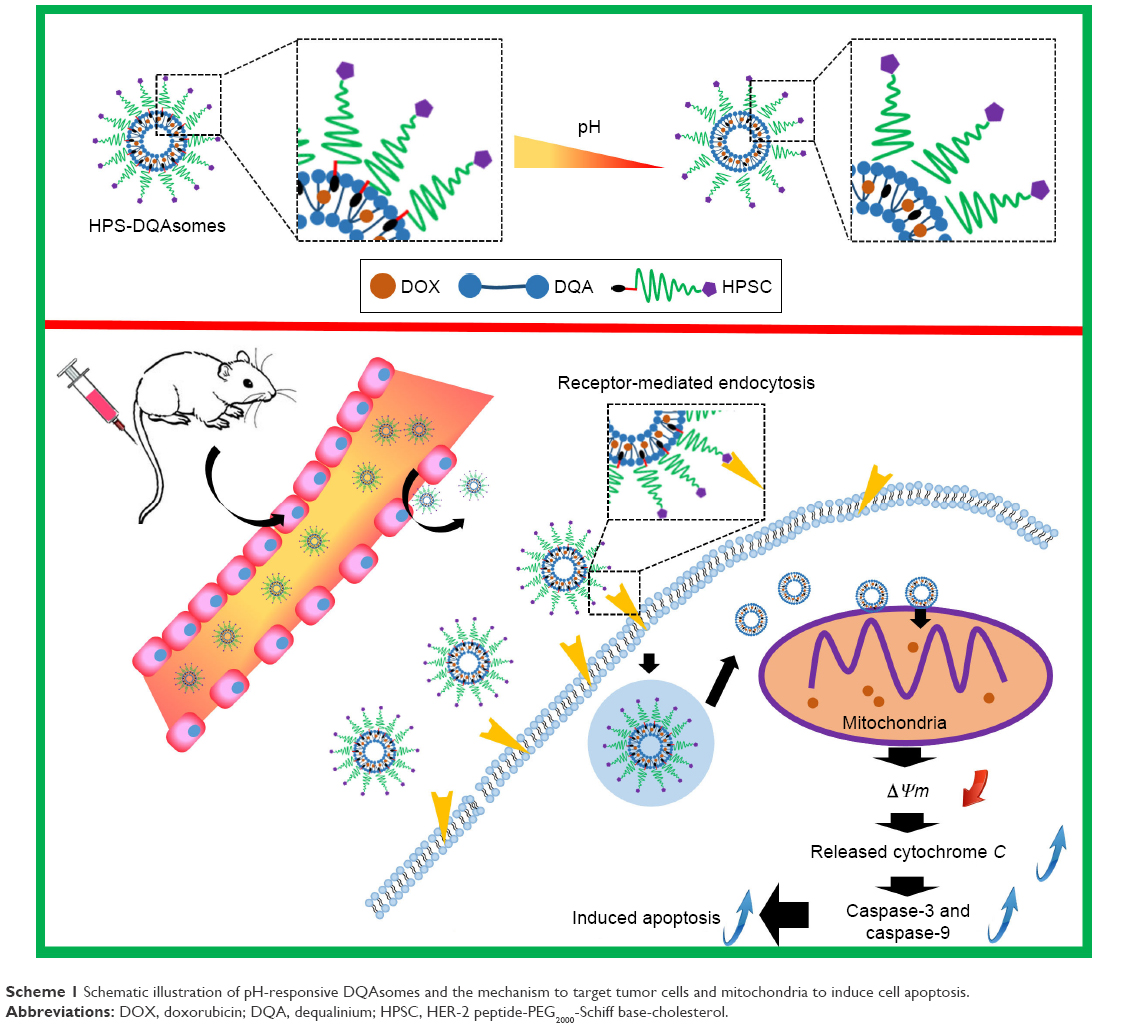 | Scheme 1 Schematic illustration of pH-responsive DQAsomes and the mechanism to target tumor cells and mitochondria to induce cell apoptosis. |
Materials and methods
Materials
DQA chloride was purchased from Sigma-Aldrich, Co. (St Louis, MO, USA). DOX·HCl was obtained from Huafeng United Technology Co., Ltd. (Beijing, People’s Republic of China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxyl-succinimide (NHS) were obtained from Shanghai Medpep Co., Ltd (Shanghai, People’s Republic of China). Cholesterol was manufactured by Shanghai Advanced Vehicle Technology (AVT) Pharmaceutical Co., Ltd. (Shanghai, People’s Republic of China) NH2-PEG2000-Schiff base-cholesterol was synthesized by our group as reported previously.27,28 HER-2 peptide (sequence: YCDGFYACYMDV) was synthesized by Qiangyao Biotechnology Co., Ltd (Shanghai, People’s Republic of China). 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was brought from Sigma-Aldrich Co. Dulbecco’s Modified Eagle’s Medium (DMEM), RPMI 1640 medium, and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). MitoTracker Green and Hoechst 33258 were purchased from Thermo Fisher Scientific. Anti-cytochrome C antibody (ab13575) and anti-Ki67 antibody (ab156956) were purchased from Abcam (Cambridge, UK). Penicillin–streptomycin kit and DAB kit were purchased from Zhongshan Goldenbridge Biotechnology, Co., Ltd (Beijing, People’s Republic of China). Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit and mitochondrial membrane potential detection kit were purchased from Beyotime Biotechnology (Shanghai, People’s Republic of China). In situ cell death detection kit-POD was manufactured by Roche (Hoffman-La Roche Ltd., Basel, Switzerland). All other chemicals and buffer solutions were of analytical grade.
Cell culture
Human breast carcinoma Michigan Cancer Foundation-7 (MCF-7) cells and DOX-resistant MCF-7 (MCF-7/ADR) cells were provided by the Chinese Academy of Sciences (Shanghai, People’s Republic of China). MCF-7 cell line was cultured in the culture medium with DMEM supplemented with 10% of fetal bovine serum (FBS), 100 μg/mL of penicillin, and 100 μg/mL of streptomycin. MCF-7/ADR cells were grown in RPMI 1640 culture medium with 10% of FBS and 1% of penicillin–streptomycin solution. To maintain the drug resistance, 200 ng/mL of DOX was added into the culture medium. Cell culture was performed in an incubator maintained at 37°C in a humidified atmosphere with 5% CO2.
Synthesis of HPSC derivate
In our previous report, we have successfully synthesized pH-sensitive PEG derivate PSC. The synthesis pathway was described as follows: cholesteryl chloroformmate was reacted with para-hydroxyl benzaldehyde with N-ethyldiisopropylamine (DIPEA) to get Chol−CHO; then, Chol−CHO and para-phenylenediamine were reacted to get NH2−SIB−Chol; and finally, COOH−PEG2000−NH2 and NH2−SIB−Chol were reacted to get PSC and the exposed amine group could be further used to react with some ligands. In brief, PSC, HER-2 peptide, EDC, and NHS were dissolved in anhydrous dimethyl sulfoxide (DMSO) with the molar ratio of 1:1:2:2. The reaction took place at room temperature for 2 days under the protection of nitrogen atmosphere. The mixture was then filtered and precipitated in cold diethyl ether overnight. The precipitate was collected and dialyzed with water for 2 days, followed by lypophilization, and HPSC was obtained.
Preparation and characterization of different DQAsomes
Preparation of DOX base
DOX base was prepared according to the previous studies.27,35 In brief, DOX·HCl was stirred with quintuple the number of mole of TEA in 12.5% methanol/chloroform mixture overnight to obtain the DOX base.
Preparation and characterization of DQAsomes
DQAsomes were prepared using the conventional thin-film hydration method as reported previously.23 In brief, 20 mg of DQA was dissolved in methanol in a round-bottom flask at 45°C and the solvent was removed using rotary evaporator. Then, the DQA thin film was hydrated using 5 mL of 4-(2-hydroxyethyl)piperazine-1-ethansulfonic acid buffer and the suspension was sonicated using probe type sonicator (200 W, 30 min in ice bath). The resulting suspension was filtered through 0.22 μm filter, and the blank DQAsomes were obtained.
For the preparation of DOX-loaded DQAsomes, DOX and DQA were dissolved in methanol with the weight ratio of 1:10 and the procedure was same as earlier. PS-DQAsomes and HPS-DQAsomes were prepared using the same method as described earlier with the weight ratio of DQA:HPSC/PSC:DOX =10:2:1.
Particle size, polydispersity index (PDI), and zeta-potential of different preparations were analyzed by Zetasizer (Nano ZS; Malvern Instruments, Malvern, UK). Morphology of DQAsomes was observed using transmission electron microscopy (TEM) (TecnaiG220; FEI, USA). In brief, different DQAsomes were diluted and placed drop wise on a TEM copper screen, damp-dried at room temperature, and stained with 2% phosphotungstic acid solution for 1 min. The drug encapsulation efficiency (EE%) was determined by the cation exchange resin-mini column centrifugation method. The EE% and loading content (LC%) were calculated as follows:
|
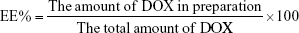
In vitro stability of different preparations
In vitro stability of different formulations was investigated in the presence of PBS and fetal bovine serum (FBS), respectively. In brief, 1 mL of preparation was mixed with 4 mL of PBS or 50% of FBS. The sample was mixed in a shaking incubator with the stirring speed of 100 rpm at 37°C. At different time points, particle size of different samples was detected using Nano ZS.
In vitro drug release of different preparations
In vitro drug release profiles were investigated using dialysis method at 37°C under pH 7.4 (normal pH) and pH 5.0 (tumor microenvironment pH). In brief, appropriate volume of different DQAsomes (containing 1.0 mg DOX) was added into the dialysis bag molecular weight cut off (MWCO 3.5 kDa). Then, the dialysis bag was put into a conical flask with 100 mL of PBS solution (containing 0.5% Tween 80) in different pH at 37°C. All flasks were placed in a shaking incubator with the stirring speed of 100 rpm. In different time points, 1 mL of sample was taken, and equal volume of release medium was added. The amount of released DOX was measured using an ultraviolent spectrophotometer at 481 nm.
In vitro cell cytotoxicity assay
Cytotoxicity of free DOX and different DQAsomes were evaluated by MTT assay. Briefly, cells were seeded in 96-well plates at a density of 5×103 cells/well and incubated for 24 h. Then, the medium was replaced with different formulations and further incubated with cells at 37°C for 24 h. At designated time intervals, 10 μL of MTT (5 mg/mL) was added into each well and further incubated for 6 h at 37°C. The medium of each well was removed, and 100 μL of DMSO was added into each well to dissolve the internalized purple formazan crystals. The absorbance of the solution at 490 nm in each well was recorded using a VersaMax Microplater Reader (Molecular Devices LLC, Sunnyvale, CA, USA). The relative cell viability (%) was calculated using the following formula:
|
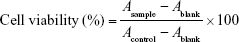
The resistant index (RI) and reversal factor (RF) were measured to evaluate the MDR effect of different formulations.
|
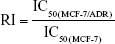
|
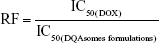
Cellular uptake study
Cellular uptake of different formulations was observed using the fluorescence microscopy and the flow cytometry, respectively. Briefly, cells were seeded in a six-well plate at a density of 5×105 cells/well and cultured for 24 h at 5% CO2 and 37°C. Following 24 h incubation, the medium was replaced with serum-free RPMI 1640 medium and different formulations were added to each well to make the final concentration of DOX 20 μg/mL and further incubated for 0.5, 2, and 4 h. Control experiment was performed by adding blank medium. After incubation, the cells were washed twice with cold PBS and fixed with 4% paraformaldehyde at room temperature for 15 min. The nuclei were stained for 15 min with Hoechst 33258. The cells were finally rinsed twice with cold PBS, and the fluorescent signal was imaged by a fluorescence microscopy (FV1000-IX81; Olympus Corporation, Tokyo, Japan).
Flow cytometry was used to quantitatively analyze the cellular uptake of various DOX-loaded formulations. Briefly, cells were seeded into six-well plates at a density of 5×105 cells/well and cultured under 5% CO2 at 37°C for 12 h. Then, different formulations were added into each well at a concentration of DOX 20 μg/mL and further incubated in different time points. The cells were washed with the PBS solution and harvested, followed by re-suspended using 0.5 mL of PBS and detected using the flow cytometry analysis (BD Biosciences, San Jose, CA, USA).
Mitochondrial targeting study
Mitochondrial colocalization assay
Confocal laser scanning microscopy (CLSM) was used to determinate the subcellular localization of different preparations against different cell lines. In brief, 5×105 cells were seeded on microscope coverslips in a six-well plate and cultured under 37°C at 5% CO2 for 24 h. The medium was replaced by serum-free RPMI 1640 medium, and cells were incubated with different DOX formulations (20 μg/mL) for 2 h. The cells were then prestained with MitoTracker Green for 2 h to visualize mitochondria. The cells were washed using cold PBS solution and fixed with 4% paraformaldehyde. Finally, the fluorescence intensity was determined by CLSM.
Annexin V-FITC apoptosis detection assay
Annexin V-FITC apoptosis detection kit was used to detect the apoptosis-inducing effect of different formulations. In brief, MCF-7/ADR cells were seeded in six-well plates and incubated overnight to allow attachment. The medium was replaced by serum-free RPMI 1640 medium, and different formulations were added into each well to make the final concentration of DOX 20 μg/mL and incubated overnight. The cells were washed with the PBS solution and stained according to the manufacturer’s instructions. Finally, the cells were analyzed using flow cytometry.
Mitochondrial membrane potential detection
JC-1 (5,5′6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl-carbocyanine iodide) was used as a fluorescence probe to detect the mitochondrial membrane potential and analyzed with flow cytometry. In brief, MCF-7/ADR cells were seeded in six-well plates at a density of 5×105 per well and incubated for 24 h and, then, different formulations (containing 20 μg/mL) were added to each well and incubated for another 24 h in an atmosphere of 5% CO2 at 37°C. The cells were washed with PBS according to the manufacturer’s instructions. The sample was finally analyzed using flow cytometry.
Released of cytochrome C
Immunocytochemistry was used to detect the release of cytochrome C when the cells were treated with different formulations. In brief, MCF-7/ADR cells were seeded into six-well plate containing coverslip with the density of 5×105 cells/well and incubated for 24 h and, then, different formulations (containing 20 μg/mL) were added to each well and incubated for another 24 h in an atmosphere of 5% CO2 at 37°C. The cells were washed using PBS and fixed using cold acetone for 20 min the cells were washed with PBS and incubated with anti-cytochrome C antibody (dilution time was 1:500) overnight. The cells were washed with PBS and further treated according to the protocol of streptavidin–peroxidase immunohistochemical kit.
Caspase-9 and caspase-3 activities
The activities of caspase-3 and caspase-9 were measured using colorimetric assay kits (KeyGen, Nanjing, People’s Republic of China). In brief, 5×105 of MCF-7/ADR cells were seeded into six-well plates and incubated for 24 h. Different formulations were added into each well (containing DOX 20 μg/mL) and incubated for 12 h in an atmosphere of 5% CO2 at 37°C. Following incubation, cells were collected and washed twice with cold PBS and, then, cells were reacted with lysis buffer (provided in the kit) for 1 h in an ice bath. The cell lysates were centrifuged at 10,000 rpm at 4°C for 1 min and treated with caspase-9 and caspase-3 substrates at 37°C for 4 h in the dark, respectively. The absorbance of the reaction was measured using the microplate reader at 405 nm. Caspase-3 activity and caspase-9 activity were expressed as a percentage relative to control according to the kit instructions.
Animal study
Animal tumor xenograft models
Female nude mice were purchased from Shenyang Changsheng Biotechnology Co., Ltd. (Liaoning province, People’s Republic of China). All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. They were maintained in a room under aseptic conditions and 12 h light/dark cycles with free access to food and sterile water and raised at 25°C±0.5°C. All the animal experiments were performed in accordance with the guidelines evaluated and approved by the Experimental Animal Administrative Committee of Shenyang Pharmaceutical University (no 13242). All the mice with subcutaneous tumors were established by injecting MCF-7/ADR cells (~1×107 cells, suspended in 200 μL of PBS) into the left backs of mice under anesthesia. When the tumor was reached ~100 mm3, the mice were used for different experiments.
In vivo antitumor activities
In vivo antitumor activities of different formulations were assessed in nude mice bearing MCF-7/ADR model. Mice were randomly separated into several groups (n=6) as follows: saline, DOX, DQAsomes, PS-DQAsomes, and HPS-DQAsomes. Different formulations were administrated through tail vein at a dose of 5 mg/kg every 3 days for four times. Mice were weighed, and tumors were measured every 2 days. The tumor size was determined with a caliper in two dimensions and calculated using the following formula:
|

The antitumor activity was evaluated in terms of tumor weight (g). The tumor growth inhibition rate (IR, %) was calculated according to following formula:
|
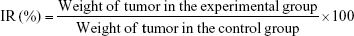
H&E staining and immunofluorescence
Tumor tissues that were harvested from pretreated mice were fixed in formalin to prepare the paraffin-embedded tumor slides (5 μm thick). The slides were dewaxed to water using xylene and different concentrations of alcohol. Then, tumor slides were treated with Tris-EDTA buffer (pH 9.0) at 95°C for 1 h and were cooled to room temperature. The slides were washed with PBS for triplicate and stained using the H&E staining kit. Terminal deoxynucleotidyl transferase (TdT) dUTP nick-Eed labeling (TUNEL) assays were used to evaluate the in situ apoptosis effect of tumor tissues using in situ cell death detection kit-POD. All the operations were done according to the kit instructions.
In vivo toxicity
Healthy female nude mice (18±2 g) were randomly assigned into five groups (n=4) and given DOX/DQA PL formulation through tail vein at a dose of 5 mg/kg every 2 days for four times, mice were weighed every 2 days, and animal behaviors were assessed. After sacrificing, liver and heart were removed and washed with PBS, fixed in 4% formaldehyde, embedded in paraffin, and cut with a microtome (5 μm thick) for H&E staining.
Statistical analysis
All the data were recorded using mean ± standard deviation (SD). Statistical significance was determined using Student’s t-test or one-way analysis of variance. P<0.05 was considered to be significant difference, and all the data were measured in triplicate.
Results and discussion
Synthesis of HPSC derivate
pH-sensitive PSC was synthesized by our laboratory, and the detailed procedure could be found in the literature.27 In order to reach active targetability, HER-2 peptide was used to link the PSC segment. HPSC was conjugated using conventional amide reaction with EDC/NHS. The typical 1H NMR spectrum is shown in Figure S1; δ3.50 ppm indicated the −OCH2CH2O− group of PEG2000, δ3.33 ppm and δ4.47 ppm indicated −CH2− and −CH− groups of HER-2 peptide, δ8.50 ppm indicated the Schiff base bond (−CH=N−), and the peak 5.4–5.5 ppm corresponded to the proton of −CONH−. All these data confirmed that HPSC was synthesized successfully.
Preparation and characterization of different DQAsomes
It has been reported that DQA could self-assemble in aqueous solution to formcationic bola liposomes like vesicle, the so-called DQAsomes.24 Due to the unique characteristics of positive surface charge, DQAsomes could be utilized as a mitochondrial targeting strategy. Meanwhile, due to the liposomes like structure, it could also load hydrophobic drug into the DQA membrane. It has been reported that improved accumulation of DOX in mitochondria of cancer cells may increase the generation of ROS and damage mitochondrial respiratory chain components, resulting in oxidative stress, lipid peroxidation, loss of cytochrome C from permeabilized mitochondria, and activation of caspase cascade.30,31 Therefore, we hypothesized that DQAsomes are the suitable carriers to carry DOX to mitochondria and increase the DOX accumulation into mitochondria region. However, significant positive charge may cause severe nonspecific toxicity and easy to be recognized by RES. Therefore, HER-2 peptide-modified pH-sensitive PEG (HPSC) was utilized to modify the surface of the DQAsomes to shield the positive charge, owing it tumor microenvironment (pH)-responsive characteristics and active targetability.
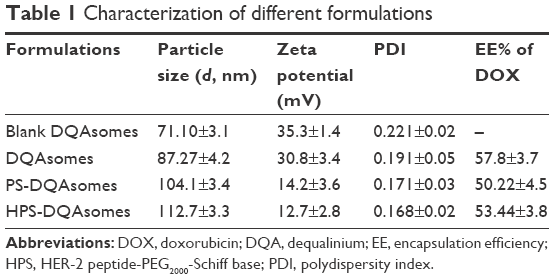
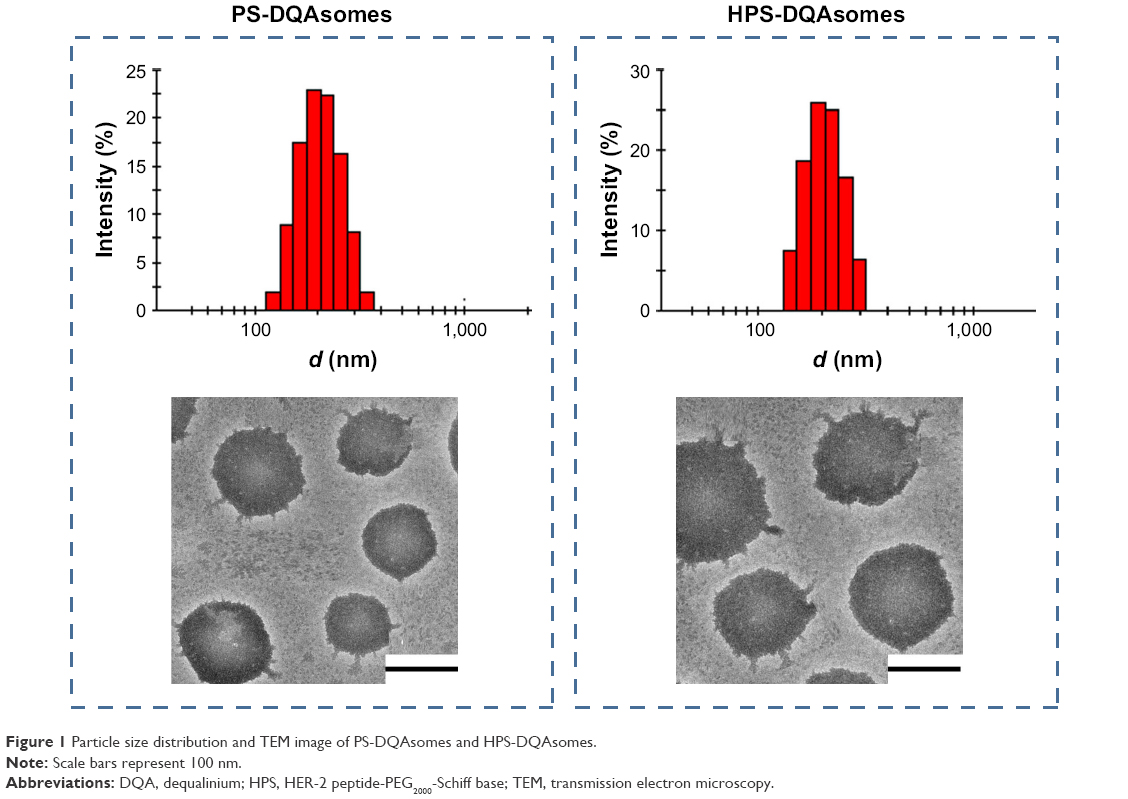
As shown in Table 1, DQAsomes were prepared using the conventional thin-film hydration method. The particle size of blank DQAsomes was ~70 nm with narrow PDI (~0.22). A slight increase in particle size could be observed for DOX-loaded DQAsomes, which was mainly attributed to the insertion of hydrophobic DOX. It is noted that both PEGylated DQAsomes showed a relative larger particle size, which was probably because the thickness of PEG segment. All preparations showed narrow particle size distribution (PDI <0.3). Meanwhile, TEM images (Figure 1) indicated that both PS-DQAsomes and HPS-DQAsomes showed a spherical shape with narrow size distribution. This result was consistent with DLS data.
 | Table 1 Characterization of different formulations |
 | Figure 1 Particle size distribution and TEM image of PS-DQAsomes and HPS-DQAsomes. |
Zeta-potential of nanoparticle is a very important factor when it was administrated into blood system. High positive charge would increase the elimination of RES and induce nonspecific toxicity.32 However, there comes a dilemma that the positive surface is the key characteristic to reach mitochondria targetability. Therefore, we utilized this pH-responsive PEG derivate to modify the surface of nanoparticles to both shield surface charge and reach long circulation time. As shown in Table 1, a significant decrease in zeta-potential could be seen for PS-DQAsomes and HPS-DQAsomes compared with DQAsomes. This result indicated that PEG shell could efficiently shield the positive surface charge of DQAsomes, which could avoid the elimination of RES and increase the circulation time. However, in our previous report, we have proved that PSC derivate owes pH-responsive characteristic, which demonstrated in mildly acidic microenvironment, and PS-DQAsomes and HPS-DQAsomes owe the mitochondrial targetability. The EE% of different DQAsomes was ~50%, which indicated that DQAsomes have a favorable capability to load DOX on the DQA membrane and the introduction of PEG derivate would influence the loading capability of DQAsomes.
It has been reported that high copolymer concentration may cause coagulation or gelation when the copolymer was administrated into blood system.32 Therefore, it is necessary to measure the in vitro stability of different formulations in the presence of FBS or PBS. As shown in Figure S2, no significant increase in particle size for both PS-DQAsomes and HPS-DQAsomes, indicating that both formulations were relatively stable when it was intravenously administrated into blood system. Neither precipitate nor visible particle was found, as well. All these results indicated that both DQAsomes were stable in blood system.
In vitro drug release from the preparations
It has been reported that there is a significant different in pH between normal tissue (pH 7.4) and tumor microenvironment (pH 6.8–5.0).36,37 Therefore, pH-responsive drug release could not only increase drug release but also decrease nonspecific toxicity. In this study, pH-independent drug release behavior was investigated at different pH values (pH 7.4 and 5.0). Significant difference of both preparations could be observed (Figure 2), indicating that both formulations owe excellent pH-responsive characteristics.
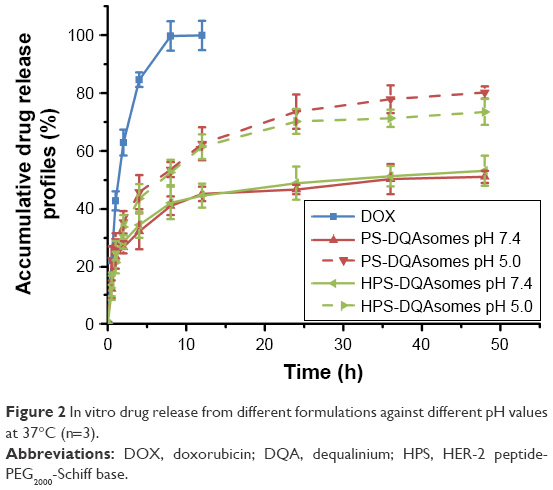 | Figure 2 In vitro drug release from different formulations against different pH values at 37°C (n=3). |
The accumulative DOX release from PS-DQAsomes and HPS-DQAsomes was 51.1% and 53.2% at pH 7.4, respectively. However, a significant increase in both preparations could be observed at pH 5.0 with the accumulative DOX release of 80.2% and 73.5%, indicating that both preparations showed a good pH-responsive feature. This result was mainly attributed to the leakage of pH-responsive Schiff base bond, which led to the detachment of PEG segment. This phenomenon would decrease the diffuse distance of DOX from the inner of the preparation to the outside environment, which resulted in a fast release profile. It is noted that there was no significant difference in release behavior between HDP-DQAsomes and DP-DQAsomes. This result suggested that the introduction of HER-2 peptide did not influence the pH responsive of HPS-DQAsomes. Taken all these together, both DQA-based drug delivery systems were expected to maintain the structure in normal pH, while rapid release therapeutical agents were expected in tumor acidic microenvironment.
In vitro cytotoxicity assay
In vitro cell cytotoxicity was studied using the standard MTT assay against sensitive MCF-7 and DOX-resistant MCF-7/ADR cell lines. Half maximal inhibitory concentration (IC50), RI, and RF of different formulations are listed in Table 2. For MCF-7, compared with DOX solution, DQAsomes group showed a higher cytotoxicity. This was probably because of the following two major reasons: 1) positive surface charge of DQAsomes increased cellular uptake and 2) DQAsomes sufficiently delivered DOX into mitochondria and induced mitochondria-oriented apoptosis process. PS-DQAsomes showed a relative lower cytotoxicity compared with DQAsomes, indicating that PEG segment formed a weak hydrophilic shell and prevented the interaction between cell membrane and preparations. HPS-DQAsomes showed the highest cytotoxicity among all groups, which implied the synergistic effect of HER-2 peptide-mediated active targetability and mitochondrial targetability of DQAsomes.
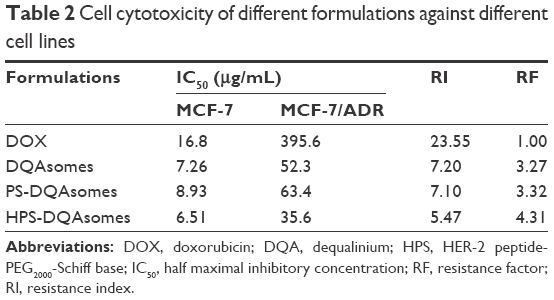 | Table 2 Cell cytotoxicity of different formulations against different cell lines |
To evaluate whether mitochondria-orientated apoptosis process could overcome MDR effect, RI and RF were calculated and the data are shown in Table 2. The RI value of DOX was 23.55, indicating that the cells owe a good resistance to DOX. RI value of DQAsomes decreased dramatically to 7.20, indicating that DQAsomes could efficiently overcome MDR effect. This phenomenon was mainly attributed to the mitochondrial targetability of DQAsomes and apoptosis-inducing effect. It was noted that HPS-DQAsomes showed the lowest RI value (5.47) and the highest RF value (4.31). This result was also attributed to the dual-targetability of HPS-DQAsomes, which could target both tumor cells and sub-cellular organelles. All these results indicated that HPS-DQAsomes were suitable carriers to overcome MDR effect of MDR tumor cell lines.
Cellular uptake assay
Cellular uptake assay was used to demonstrate the cellular uptake behavior of different formulations against different cell lines using the fluorescence microscopy and flow cytometry. As shown in Figure 3A, only weak red fluorescence could be observed for DOX group against MCF-7 and MCF-7/ADR cell lines. In comparison, stronger fluorescence could be seen for DQAsomes group for MCF-7 cell line, indicating that positive surface charge DQAsomes could increase cellular uptake. Meanwhile, for MCF-7/ADR cells, DQAsomes group also showed a stronger fluorescence. This result was mainly attributed to the mitochondrial targetability of DQAsomes and the delivery of DOX to mitochondria, which would then inhibit the energy supply and increase the drug accumulation into cytoplasm. Similarly, PS-DQAsomes showed a relatively weaker fluorescence compared with DQAsomes group against both cell lines. HPS-DQAsomes showed the strongest fluorescence intensity against both cell lines, and this result was consistent with MTT assay.
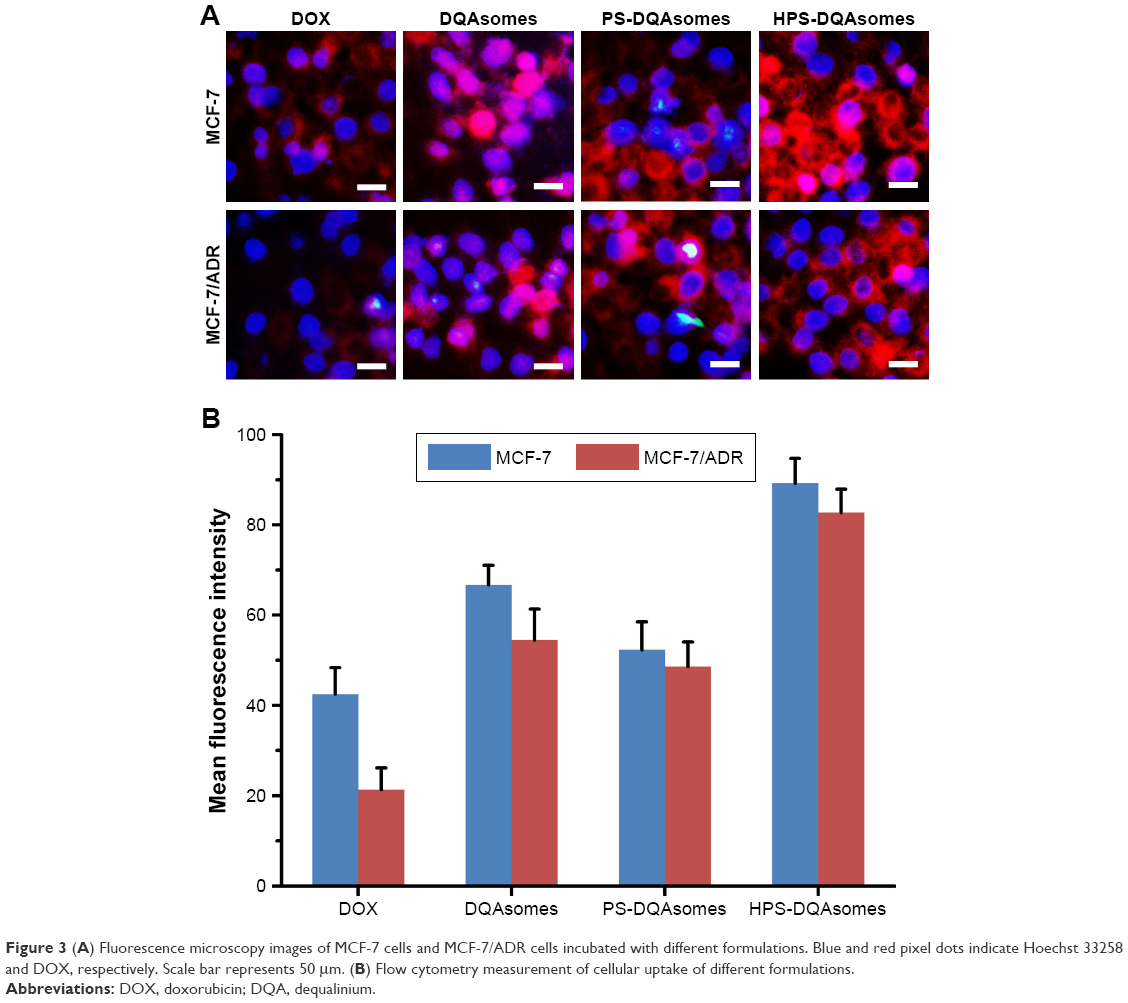 | Figure 3 (A) Fluorescence microscopy images of MCF-7 cells and MCF-7/ADR cells incubated with different formulations. Blue and red pixel dots indicate Hoechst 33258 and DOX, respectively. Scale bar represents 50 μm. (B) Flow cytometry measurement of cellular uptake of different formulations. |
Flow cytometry study was also used to quantitatively analyze the cellular uptake of different formulations against different cell lines. As illustrated in Figure 3B, the same trend could be observed compared with fluorescence microscopy. All these results indicated that HPS-DQAsomes could facilitate the drug accumulation via the synergistic effect of HER-2 peptide-mediated active targetability and DQAsomes mediated mitochondrial targetability.
Mitochondrial targeting assay
It has been reported that DQAsomes could accumulate into mitochondria of living cells in response to the mitochondrial membrane potential.38,39 However, whether these PEGylated DQAsomes could also target mitochondria has not been evaluated yet. Therefore, mitochondrial targetability of different formulations was examined using the confocal laser scanning microscopy (CLSM). As illustrated in Figure 4, red fluorescence indicated DOX while green fluorescence represented mitochondria. Yellow fluorescence indicated the co-localization of green fluorescence and red fluorescence indicated DOX, which was accumulated into mitochondria. Moderate yellow fluorescence could be observed for DOX groups against MCF-7 and MCF-7/ADR cell lines, demonstrating that DOX did not have the ability to accumulate into mitochondria. Meanwhile, the red fluorescence of MCF-7 groups was significantly higher than MCF-7/ADR groups, indicating that MCF-7/ADR cell lines owed strong resistance. In comparison, bright yellow fluorescence could be observed for both cell lines, indicating that DQAsomes could efficiently accumulate into mitochondria. It is noted that the red fluorescence increased for MCF-7/ADR groups, indicating that DOX could successfully overcome MDR effect via mitochondrial pathway. Yellow fluorescence decreased for both PS-DQAsomes against both cell lines; this phenomenon was mainly attributed to the surface-modified PEG segment, which would form a hydrophilic layer and decrease the interaction between the preparation and cell membrane. However, the yellow fluorescence was still relatively strong, indicating that this pH-responsive PEG derivate could leak in acidic microenvironment and was not influenced the mitochondrial targetability of DQAsomes. HPS-DQAsomes showed the strongest co-localization efficiency, and this result was mainly attributed to the synergistic effect of HER-2-mediated active targetability, pH-responsive PEG leakage, and mitochondrial targetability of DQAsomes.
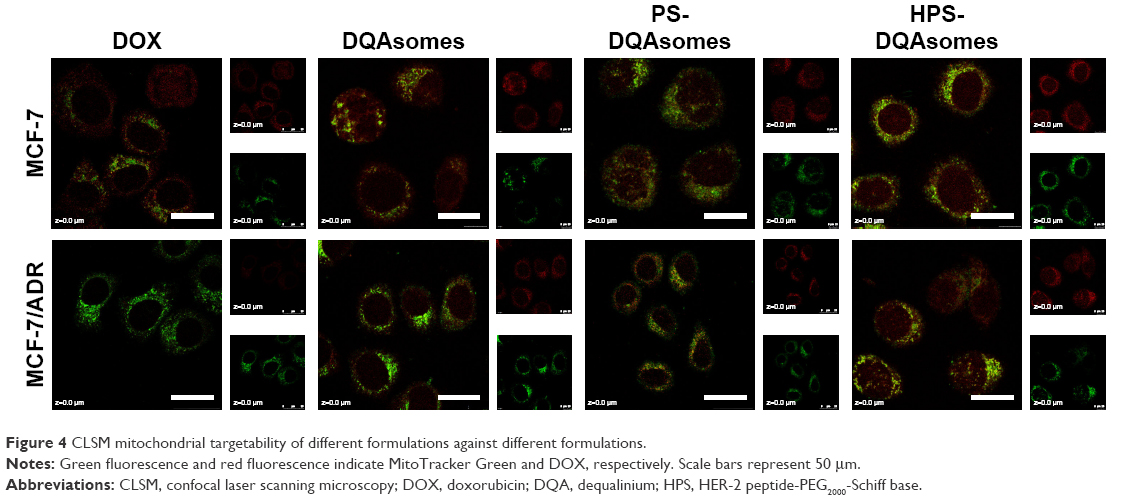 | Figure 4 CLSM mitochondrial targetability of different formulations against different formulations. |
Cell apoptosis pathway
Cell apoptosis was a programmed cell death. Reduction in mitochondrial membrane potential was considered as a sign of apoptosis. Release of cytochrome C and activation of caspase family are also needed for cell apoptosis.40,41 To find whether HPS-DQAsomes would overcome MDR effect and induce cell apoptosis via mitochondrial pathway, a series of apoptosis marker of cell apoptosis were examined in MCF-7/ADR cell lines against various DOX formulations.
Figure 5 depicts the apoptosis rate of different DOX-loaded formulations against MCF-7/ADR cell lines. The apoptosis rate of culture medium, DOX solution, DQAsomes, PS-DQAsomes, and HPS-DQAsomes was 2.51±0.31, 24.62±1.05, 83.71±0.98, 64.83±1.13, and 84.62%±1.25%, respectively. HPS-DQAsomes showed the highest apoptosis rate among all groups, indicating that HPS-DQAsomes could efficiently induce apoptosis and overcome MDR effect.
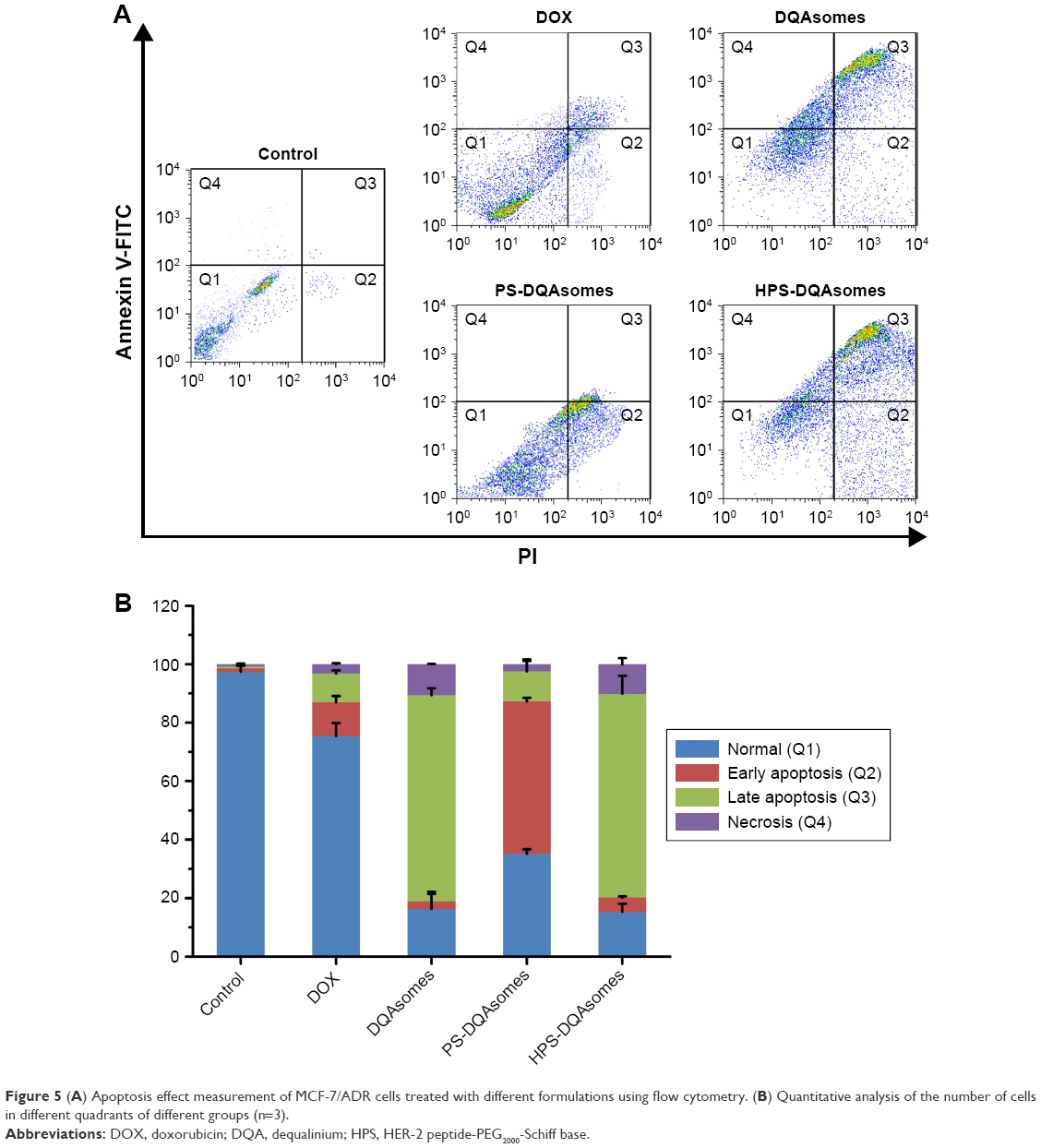 | Figure 5 (A) Apoptosis effect measurement of MCF-7/ADR cells treated with different formulations using flow cytometry. (B) Quantitative analysis of the number of cells in different quadrants of different groups (n=3). |
To demonstrate the underlying mechanism of apoptosis-inducing effect, mitochondrial membrane potential (ΔΨm) was measured using the JC-1 fluorescence dye. Figure 6 illustrates the change in ΔΨm after applying with various formulations. A decrease in ΔΨm could be observed for DOX group (46.3%±4.2%), DQAsomes (7.5%±2.1%), PS-DQAsomes (8.8%±1.3%), and HPS-DQAsomes (6.9%±0.2%) compared with negative control.
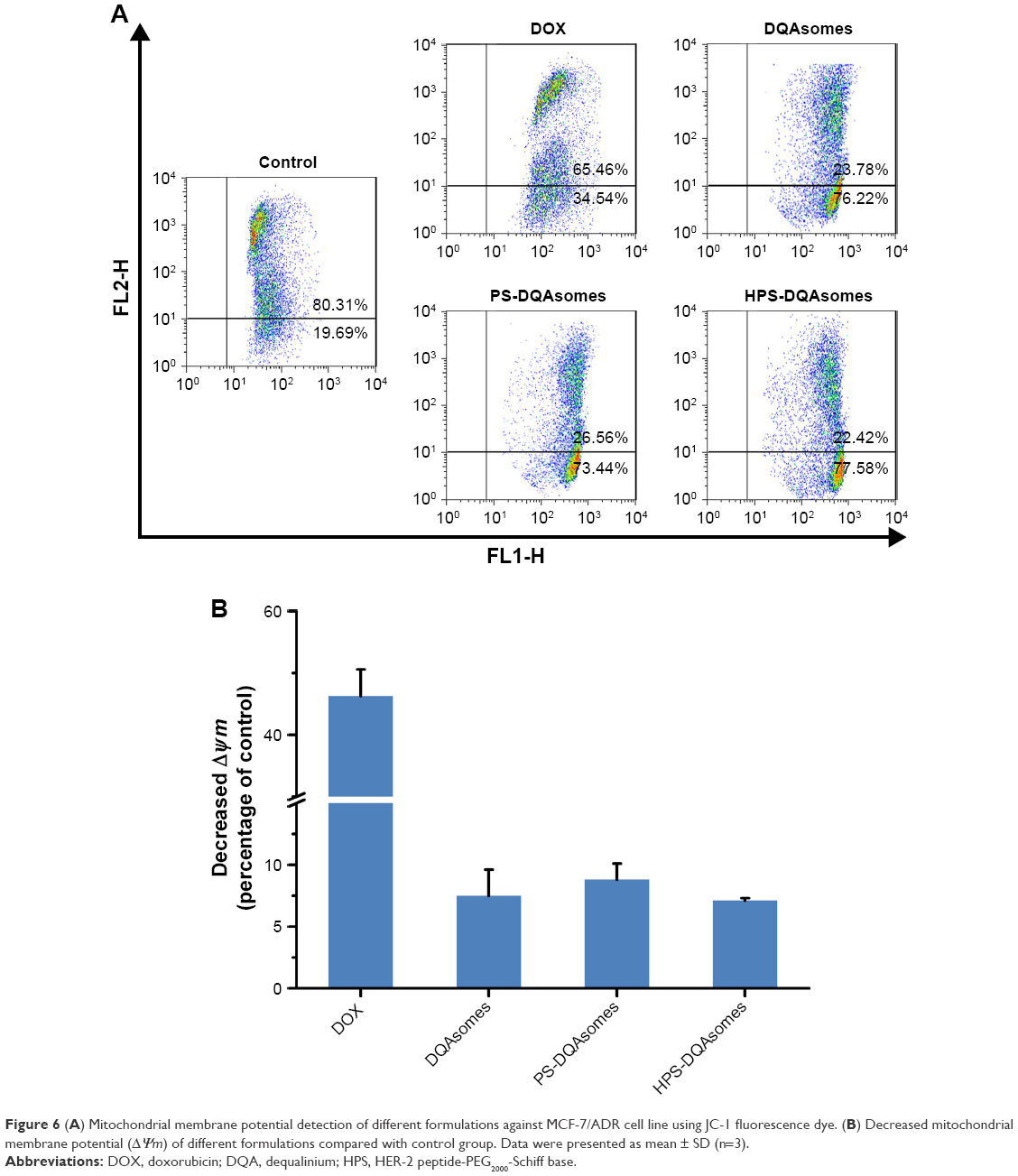 | Figure 6 (A) Mitochondrial membrane potential detection of different formulations against MCF-7/ADR cell line using JC-1 fluorescence dye. (B) Decreased mitochondrial membrane potential (ΔΨm) of different formulations compared with control group. Data were presented as mean ± SD (n=3). |
A decrease in ΔΨm would also induce the release of cytochrome C. Therefore, release of cytochrome C was also examined using immunocytochemistry assay. As depicted in Figure 7A, the blue and brown pixel dots represent the nuclei and cytochrome C, respectively. No obvious brown pixel dots could be observed for the control group, indicating that the cells were in good state. HPS-DQAsomes showed the strongest brown pixel dots. This result was consistent with previous study, further indicating that HPS-DQAsomes could efficiently induce apoptosis and overcome MDR.
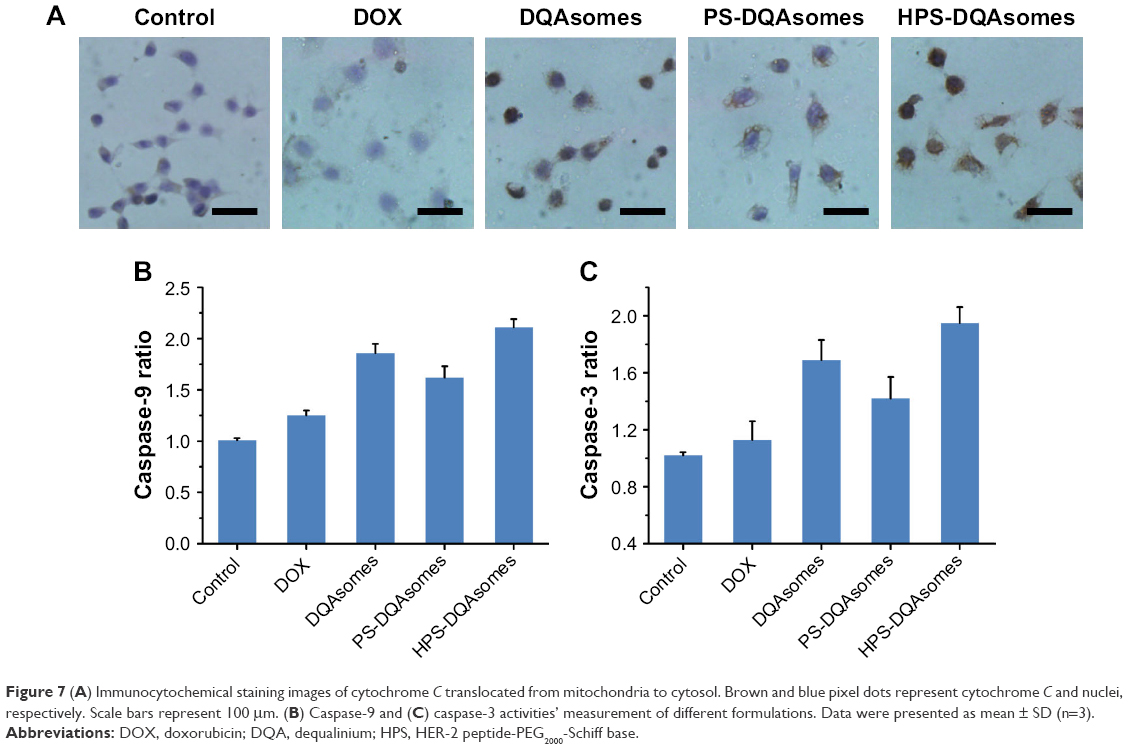 | Figure 7 (A) Immunocytochemical staining images of cytochrome C translocated from mitochondria to cytosol. Brown and blue pixel dots represent cytochrome C and nuclei, respectively. Scale bars represent 100 μm. (B) Caspase-9 and (C) caspase-3 activities’ measurement of different formulations. Data were presented as mean ± SD (n=3). |
Caspase family as a downstream biomarker of apoptosis was also detected. As shown in Figure 7B and C, caspase-9 and caspase-3 were examined in MCF-7/ADR cell line. After treatment with HPS-DQAsomes, caspase-9 and caspase-3 ratio increased dramatically compared with negative control, indicating that HPS-DQAsomes had the most evident ability for activating caspase-3 and -9 to other formulations.
From all the results described earlier, we could find that HPS-DQAsomes owe the favorable capability to induce apoptosis and overcome MDR effect against MCF-7/ADR cell line, which was mainly attributed to the HER-2 peptide-mediated endocytosis, pH-responsive of PEG segment, and mitochondrial targetability of DQAsomes.
In vivo antitumor activity
From the in vitro study, we could find that PS-DQAsomes showed unsatisfied antitumor activity. It seems that PEG is undesirable or even necessary. In fact, it is very important for the surface modification of PEG. The existence could not only shield the positive charge to reduce nonspecific toxicity but also increase the circulation time and reach the higher therapeutical efficacy in vivo. Figure 8A, C, and D illustrates the in vivo antitumor activity of different formulations. Tumor volume and IR of different DOX-loaded formulations were assessed. Control groups showed a rapid tumor increase during the experiment period, which was 900 mm3 at the 14th day. However, different DOX-treated groups showed different antitumor activities in different degrees. Compared with the DOX group (IR was 18.6%±2.5%) and the DQAsomes group (IR was 33.5%±4.2%), the PS-DQAsomes (IR was 53.6%±5.1%) and HPS-DQAsomes (IR was 72.5%±3.5%) groups showed higher antitumor activity. This result was mainly attributed to the existence of PEG, which could not only shield positive charge but also avoid the elimination of RES to reach high circulation time. Meanwhile, pH-responsive PEG leakage would not influence the mitochondrial targetability of DQAsomes and reach higher apoptosis-inducing effect. It is noted that HPS-DQAsomes showed the highest antitumor activity, which was mainly attributed to HER-2-mediated endocytosis that could significantly increase the cellular uptake and reach higher intracellular DOX accumulation.
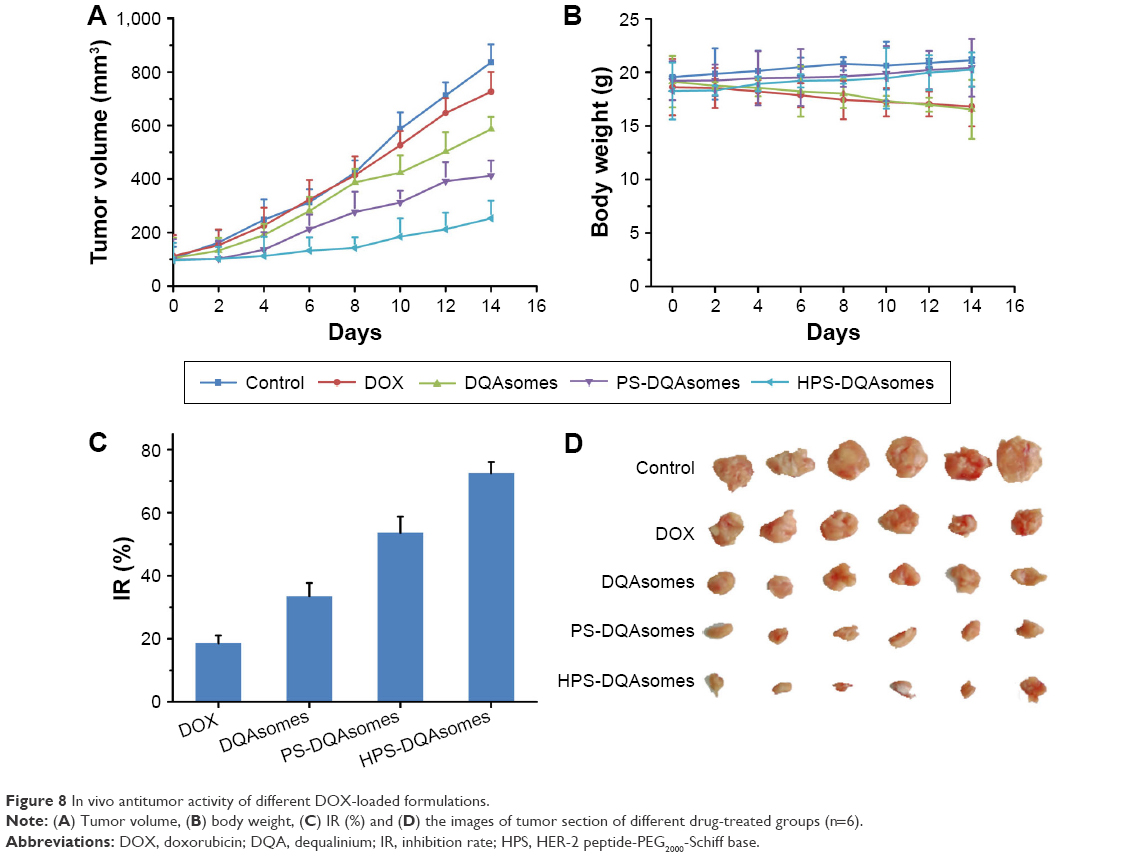 | Figure 8 In vivo antitumor activity of different DOX-loaded formulations. |
Body weight of mice was also measured during this period. As shown in Figure 8B, no obvious weight loss was observed for HPS-DQAsomes and PS-DQAsomes groups, indicating that both formulations were safety for in vivo application. However, DOX and DQAsomes groups showed a significant weight loss. It is well known that directly administrated DOX into blood system may cause severe systematic toxicity. Due to the positive surface charge of DQAsomes, it would cause nonspecific interaction in the blood system and cause systematic toxicity, as well.
All these in vivo antitumor assays demonstrated that HPS-DQAsomes owe favorable in vivo antitumor activity. The enhanced antitumor efficacy of HPS-DQAsomes could be explained by the following ways: 1) PEG segment on the surface of the formulation was shielded the positive charge of DQAsomes and reached long circulation time characteristics; 2) HER-2 peptide-mediated endocytosis was increased the cellular uptake and reached higher intracellular DOX accumulation; 3) pH-responsive PEG leakage was exposed positive charge DQAsomes; and 4) positive charge DQAsomes were targeted mitochondria to cutoff energy supply, induced apoptosis effect, and overcame MDR effect.
Histological assay
The above results indicated that HPS-DQAsomes owe sufficient antitumor activity both in vitro and in vivo. Besides these, we are also interested to investigate the ex vivo assays of tumor in order to observe the change in cancerous cells after treating with different formulations. After treating with different formulations, the tumor sections were harvested and fixed to prepare paraffin-embedded slides. H&E staining was examined, and the data are shown in Figure 9; the presence of void represents the loss of tumor cells.32 More voids could be observed for HPS-DQAsomes, implying that HPS-DQAsomes could efficiently deliver antitumor drug to tumor sites and overcome MDR effect via mitochondrial pathway.
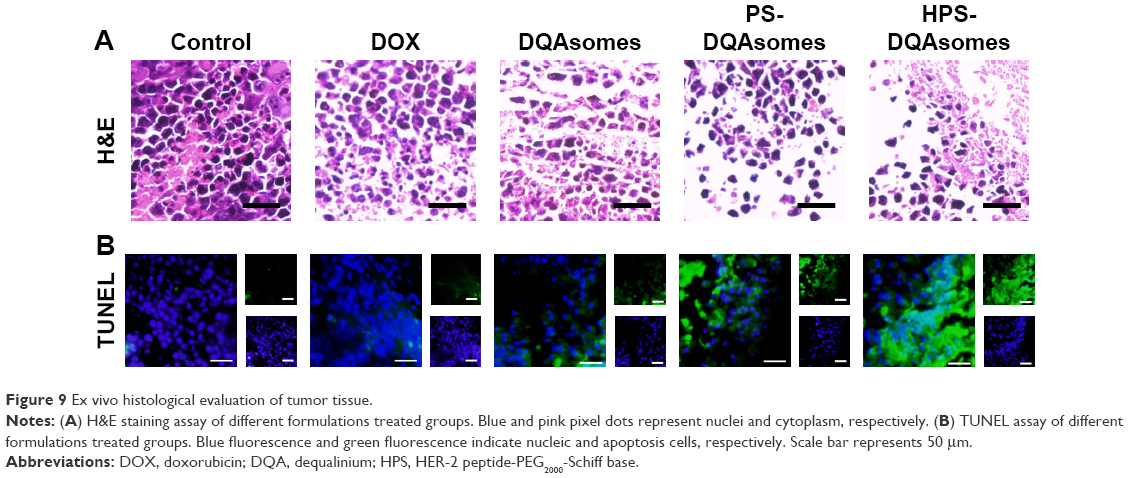 | Figure 9 Ex vivo histological evaluation of tumor tissue. |
TUNEL assay was also carried out to evaluate the apoptosis-inducing effect of different formulations in histological level. Blue fluorescence and green fluorescence indicated the nucleic and the apoptosis cells, respectively. No obvious green fluorescence could be observed for control groups, implying that the tumor was in good state. HPS-DQAsomes group showed the strongest green fluorescence among all drug-treated groups. This result was in consistent with H&E staining assay and further demonstrated the efficient antitumor activity of HPS-DQAsomes.
In vivo toxicity assay
It is well known that chemotherapy would cause severe systematic toxicity to normal tissue. Therefore, it is necessary to evaluate the security of the preparation with in vivo toxicity assay using healthy tumor-free mice. As illustrated in Figure 10, body weight of mice was measured during this period. Due to the severe cardio toxicity of DOX, heart and tissue were harvested and stained using H&E staining kit. No significant weight loss could be observed during this period. Meanwhile, compared with control group, no damage could be observed in histological level. Furthermore, no apparent signs associated with systematic toxicity such as dehydration, locomotors, impairment, and anorexia were observed during this period. All these results indicated that both PS-DQAsomes and HPS-DQAsomes were safety drug delivery systems.
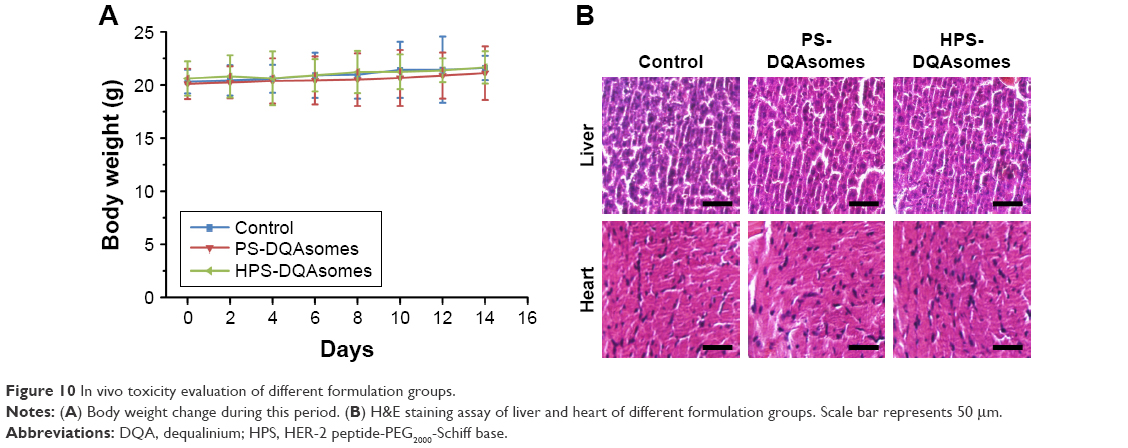 | Figure 10 In vivo toxicity evaluation of different formulation groups. |
Conclusion
In the present study, a novel multifunctional HPS-DQAsomes were constructed with HER-2 peptide-mediated active targetability, pH-responsive PEG leakage, and mitochondrial targetability to overcome MDR effect via mitochondrial pathway. HPS-DQAsomes could induce the apoptosis of MCF-7/ADR cell line by decreasing mitochondrial membrane potential, releasing cytochrome C, and activating the cascade of caspase-9 and -3. Therefore, HPS-DQAsomes were proved to be the potential nanocarriers to treat drug-resistant breast cancer.
Acknowledgment
The authors are grateful for the financial support from the National Natural Science Foundation of China (81202483, 81773668, and 81302721), Liaoning Natural Science Foundation for Excellent Talents in university (LR 2015020543), and Science and Technology project of Shenyang (F15-139-9-06 and F15-199-1-24).
Disclosure
The authors report no conflicts of interest in this work.
References
Huo X, Liu Q, Wang C, et al. Enhancement effect of P-gp inhibitors on the intestinal absorption and antiproliferative activity of bestatin. Eur J Pharm Sci. 2013;50(3):420–428. | ||
Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389(10074):1134–1150. | ||
Sun R, Liu Y, Li S-Y, et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials. 2015;37(suppl C):405–414. | ||
Wilhelmsson A, Roos M, Hagberg L, Wengström Y, Blomberg K. Motivation to uphold physical activity in women with breast cancer during adjuvant chemotherapy treatment. Eur J Oncol Nurs. 2017;29:17–22. | ||
Liu Y, Qiao L, Zhang S, et al. Dual pH-responsive multifunctional nanoparticles for targeted treatment of breast cancer by combining immunotherapy and chemotherapy. Acta Biomater. 2018;66:310–324. | ||
Joshi P, Vishwakarma RA, Bharate SB. Natural alkaloids as P-gp inhibitors for multidrug resistance reversal in cancer. Eur J Med Chem. 2017;138:273–292. | ||
Zhang Y-K, Zhang X-Y, Zhang G-N, et al. Selective reversal of BCRP-mediated MDR by VEGFR-2 inhibitor ZM323881. Biochem Pharmacol. 2017;132:29–37. | ||
Zhou J, Zhao WY, Ma X, et al. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials. 2013;34(14):3626–3638. | ||
Jiang L, Li L, He X, et al. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials. 2015;52:126–139. | ||
Singh MS, Tammam SN, Shetab Boushehri MA, Lamprecht A. MDR in cancer: addressing the underlying cellular alterations with the use of nanocarriers. Pharmacol Res. 2017;126:2–30. | ||
Alamolhodaei NS, Tsatsakis AM, Ramezani M, Hayes AW, Karimi G. Resveratrol as MDR reversion molecule in breast cancer: an overview. Food Chem Toxicol. 2017;103:223–232. | ||
Martinou J-C, Desagher S, Antonsson B. Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol. 2000;2(3):E41–E43. | ||
Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92(1):57–70. | ||
Barot M, Gokulgandhi MR, Pal D, Mitra AK. Mitochondrial localization of P-glycoprotein and peptide transporters in corneal epithelial cells – novel strategies for intracellular drug targeting. Exp Eye Res. 2013;106:47–54. | ||
Solazzo M, Fantappiè O, Lasagna N, Sassoli C, Nosi D, Mazzanti R. P-gp localization in mitochondria and its functional characterization in multiple drug-resistant cell lines. Exp Cell Res. 2006;312(20):4070–4078. | ||
Wang X-X, Li Y-B, Yao H-J, et al. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials. 2011;32(24):5673–5687. | ||
Bae Y, Jung MK, Lee S, et al. Dequalinium-based functional nanosomes show increased mitochondria targeting and anticancer effect. Eur J Pharm Biopharm. 2018;124:104–115. | ||
Paparidis G, Akrivou M, Tsachouridou V, et al. Synthesis and evaluation of 99mTc/Re-tricarbonyl complexes of the triphenylphosphonium cation for mitochondrial targeting. Nucl Med Biol. 2018;57:34–41. | ||
Sandoval-Acuña C, Fuentes-Retamal S, Guzmán-Rivera D, et al. Destabilization of mitochondrial functions as a target against breast cancer progression: role of TPP+-linked-polyhydroxybenzoates. Toxicol Appl Pharmacol. 2016;309:2–14. | ||
Yamada Y, Harashima H. Enhancement in selective mitochondrial association by direct modification of a mitochondrial targeting signal peptide on a liposomal based nanocarrier. Mitochondrion. 2013;13(5):526–532. | ||
Kawamura E, Yamada Y, Yasuzaki Y, Hyodo M, Harashima H. Intracellular observation of nanocarriers modified with a mitochondrial targeting signal peptide. J Biosci Bioeng. 2013;116(5):634–637. | ||
D’Souza GG, Boddapati SV, Weissig V. Mitochondrial leader sequence-plasmid DNA conjugates delivered into mammalian cells by DQAsomes co-localize with mitochondria. Mitochondrion. 2005;5(5):352–358. | ||
Weissig V, D’Souza GG, Torchilin VP. DQAsome/DNA complexes release DNA upon contact with isolated mouse liver mitochondria. J Control Release. 2001;75(3):401–408. | ||
D’Souza GG, Rammohan R, Cheng S-M, Torchilin VP, Weissig V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J Control Release. 2003;92(1):189–197. | ||
Yang S, Gao H. Nanoparticles for modulating tumor microenvironment to improve drug delivery and tumor therapy. Pharmacol Res. 2017;126:97–108. | ||
Overchuk M, Zheng G. Overcoming obstacles in the tumor microenvironment: recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials. 2018;156:217–237. | ||
Chen Q, Ding H, Zhou J, et al. Novel glycyrrhetinic acid conjugated pH-sensitive liposomes for the delivery of doxorubicin and its antitumor activities. RSC Adv. 2016;6(22):17782–17791. | ||
Zang X, Ding H, Zhao X, et al. Anti-EphA10 antibody-conjugated pH-sensitive liposomes for specific intracellular delivery of siRNA. Int J Nanomedicine. 2016;11:3951–3967. | ||
Theodossiou TA, Sideratou Z, Katsarou ME, Tsiourvas D. Mitochondrial delivery of doxorubicin by triphenylphosphonium-functionalized hyperbranched nanocarriers results in rapid and severe cytotoxicity. Pharm Res. 2013;30(11):2832–2842. | ||
Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol Pharm. 2014;11(8):2640–2649. | ||
Song Y-F, Liu D-Z, Cheng Y, et al. Dual subcellular compartment delivery of doxorubicin to overcome drug resistant and enhance antitumor activity. Sci Rep. 2015;5:16125. | ||
Zhang J, Zhao X, Chen Q, et al. Systematic evaluation of multifunctional paclitaxel-loaded polymeric mixed micelles as a potential anticancer remedy to overcome multidrug resistance. Acta Biomater. 2017;50:381–395. | ||
Chattopadhyay N, Cai Z, Pignol J-P, et al. Design and characterization of HER-2-targeted gold nanoparticles for enhanced X-radiation treatment of locally advanced breast cancer. Mol Pharm. 2010;7(6):2194–2206. | ||
Peng X-H, Qian X, Mao H, et al. Targeted magnetic iron oxide nanoparticles for tumor imaging and therapy. Int J Nanomedicine. 2008;3(3):311–321. | ||
Qiu L, Li Zh, Qiao M, et al. Self-assembled pH-responsive hyaluronic acid–g-poly(L-histidine) copolymer micelles for targeted intracellular delivery of doxorubicin. Acta Biomaterialia. 2014;10:2024–2035. | ||
Ko H, Son S, Jeon J, et al. Tumor microenvironment-specific nanoparticles activatable by stepwise transformation. J Control Release. 2016;234:68–78. | ||
Yhee JY, Jeon S, Yoon HY, et al. Effects of tumor microenvironments on targeted delivery of glycol chitosan nanoparticles. J Control Release. 2017;267:223–231. | ||
Weissig V, Boddapati SV, Cheng S-M, D’Souza GG. Liposomes and liposome-like vesicles for drug and DNA delivery to mitochondria. J Liposome Res. 2006;16(3):249–264. | ||
Yamada Y, Akita H, Kogure K, Kamiya H, Harashima H. Mitochondrial drug delivery and mitochondrial disease therapy – an approach to liposome-based delivery targeted to mitochondria. Mitochondrion. 2007;7(1):63–71. | ||
Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182(2):367. | ||
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. |
Supplementary materials
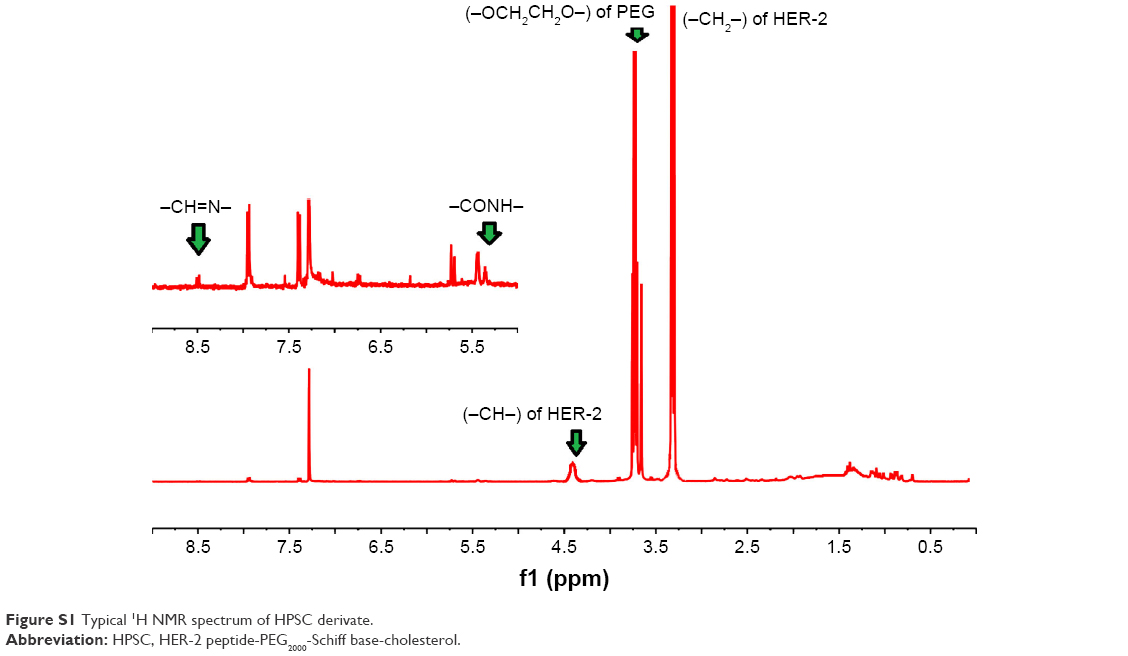 | Figure S1 Typical 1H NMR spectrum of HPSC derivate. |
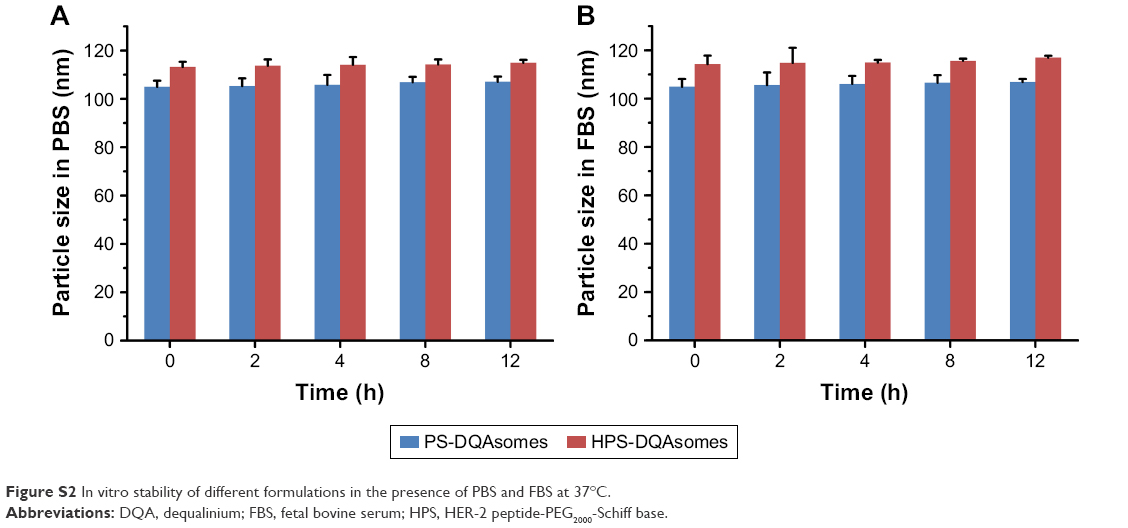 | Figure S2 In vitro stability of different formulations in the presence of PBS and FBS at 37°C. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.