Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Missense Genetic Polymorphisms of Microsomal (EPHX1) and Soluble Epoxide Hydrolase (EPHX2) and Their Relation to the Risk of Large Artery Atherosclerotic Ischemic Stroke in a Turkish Population
Authors Can Demirdöğen B, Miçooğulları Y, Türkanoğlu Özçelik A, Adalı O ![]()
Received 9 October 2019
Accepted for publication 8 January 2020
Published 7 May 2021 Volume 2020:16 Pages 3251—3265
DOI https://doi.org/10.2147/NDT.S233992
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Birsen Can Demirdöğen,1 Yağmur Miçooğulları,2 Aysun Türkanoğlu Özçelik,3 Orhan Adalı4
1Department of Biomedical Engineering, TOBB University of Economics and Technology, Ankara, Turkey; 2Institute of Natural and Applied Sciences, Department of Biology, Middle East Technical University, Ankara, Turkey; 3Food Safety and Agricultural Research Center, Akdeniz University, Antalya, Turkey; 4Department of Biological Sciences, Joint Graduate Program in Biochemistry, Middle East Technical University, Ankara, Turkey
Correspondence: Birsen Can Demirdöğen
Department of Biomedical Engineering, TOBB University of Economics and Technology, Ankara, Turkey
Tel +90 312 292 42 79
Email [email protected]
Purpose: Soluble epoxide hydrolase (sEH) and microsomal epoxide hydrolase (mEH) both catalyze the metabolism of epoxyeicosatrienoic acids (EETs), lipid signaling molecules that are protective against ischemic brain injury owing to their participation in the regulation of vascular tone and cerebral blood flow. In addition, mEH metabolizes polycyclic aromatic hydrocarbons, one of the causative factors of atherosclerotic lesion development. In this study, we aimed to investigate the association of enzyme activity-modifying missense single nucleotide polymorphisms (SNPs) of the sEH gene (EPHX2) and mEH gene (EPHX1) and ischemic stroke risk in a Turkish population.
Patients and Methods: Genomic DNA of patients with large artery atherosclerotic ischemic stroke (n=237) and controls (n=120) was isolated from blood samples, and genotypes for Tyr113His (rs1051740) and His139Arg (rs2234922) SNPs of EPHX1 and Arg287Gln (rs751141) SNP of EPHX2 were attained by the PCR/RFLP method.
Results: Minor allele frequency and genotype distributions for Arg287Gln, Tyr113His and His139Arg SNPs did not differ significantly between stroke patients and controls. However, hypertension- and diabetes-associated ischemic stroke risk was decreased by EPHX1 and increased by EPHX2 variants in stratification analyses.
Conclusion: This study has shown for the first time that the polymorphic alleles of EPHX1 were unlikely to be associated with large artery atherosclerotic ischemic stroke susceptibility; however, protective effects were evident within subgroups of hypertension and diabetes. In addition, EPHX2 Arg287Gln polymorphism, which has been studied for the first time in a Turkish population, was not significantly related to ischemic stroke, but increased the stroke risk in subgroup analysis.
Keywords: Arg287Gln, epoxyeicosatrienoic acids, EETs, His139Arg, SNP, Tyr113His
Introduction
Stroke, the second leading cause of death after ischemic heart disease worldwide, is characterized by the loss of one or more functions of the body owing to inadequate blood supply to the brain. Ischemic stroke is the most common type, accounting for more than 80% of all cases, and results from the obstruction of a blood vessel that supplies blood to the brain.1 If the obstruction is due to a blood clot that forms on an atherosclerotic plaque within a blood vessel in the brain, this subtype is called an atherothrombotic ischemic stroke. The mechanisms underlying ischemic stroke have not been completely explained.2,3
Epoxyeicosatrienoic acids (EETs) are signaling molecules derived from the 20-carbon polyunsaturated fatty acid arachidonic acid. EETs exhibit a wide array of potentially beneficial actions in stroke, including vasodilation, neuroprotection and promotion of angiogenesis, and suppression of platelet aggregation, oxidative stress and post-ischemic inflammation. Like other epoxides, EETs are metabolized to their supposedly less active form, dihydroxyeicosatrienoic acids (DHETs), by epoxide hydrolase enzymes.4
There are two main epoxide hydrolases: microsomal and soluble. Microsomal epoxide hydrolase (mEH, EPHX1, EC 3.3.2.9) is a critical biotransformation enzyme that catalyzes the metabolism of various xenobiotic epoxide substrates and polycyclic aromatic hydrocarbons (PAHs), including carcinogenic and mutagenic compounds in tobacco smoke, to more polar diol metabolites. These mutagens stimulate the formation of DNA adducts, which, apart from carcinogenesis, lead to genetic alterations in blood vessels.5 Animal studies have shown that tobacco smoke mutagens such as PAHs and heterocyclic amines directly increase the development of atherosclerotic lesions.6 Thus, activity of mEH may alter the atherogenic PAH levels and the risk of ischemic stroke. Moreover, mEH also plays a role in EET metabolism. Although sEH mediates the bulk of EET metabolism, a significant contribution of mEH was observed in mouse brain, pointing to a novel role of this enzyme in the regulation of physiological processes.7 Two of the missense single-nucleotide polymorphisms (SNPs) in the EPHX1 gene result in critical amino acid substitutions. rs1051740T>C SNP (Tyr113His) is located in exon 3 of the gene and the polymorphic allele changes the codon from that of tyrosine to histidine at the 113th position. The presence of a histidine residue instead of a tyrosine leads to a 39% reduction in enzyme activity.8 The other missense SNP (rs2234922A>G) is located in exon 4 and causes an amino acid change from histidine to arginine at the 139th position (His139Arg). It has been shown that His139Arg results in a 25% increase in enzyme activity.8 Association studies have demonstrated that polymorphisms in the EPHX1 gene may be important risk factors for susceptibility to various cancers;9–12 however, EPHX1 genetic polymorphisms have not been investigated in relation to ischemic stroke before.
Soluble epoxide hydrolase (sEH, EC 3.3.2.10) is the major enzyme that plays a role in the conversion of EETs to diol metabolites.13,14 sEH inhibitors demonstrated protection against cardiovascular and ischemic brain damage in animal models.15 A missense SNP in exon 8 of the gene coding for sEH (EPHX2) (rs751141G>A) causes an amino acid substitution from Arg to Gln at position 287 of the enzyme, which leads to a significant reduction in sEH activity (25–58% of that of the wild-type enzyme) in vitro.16 sEH Arg287Gln variant was observed to have a protective effect against ischemic neuronal injury.17 This finding overlaps with the proposed effect of sEH activity on EET bioavailability.18,19 There are several previous studies in other populations on the relationship between EPHX2 and cardiovascular disease, including ischemic stroke risk;20–25 however, this relationship has not been investigated before in a Turkish population.
It is well established that the frequencies of polymorphic alleles vary among populations with different ethnicities. Therefore, to the best of our knowledge, this study for the first time analyzed the association between Tyr113His and His139Arg polymorphisms of EPHX1 on ischemic stroke risk. Moreover, the association of Arg287Gln polymorphism of EPHX2 with ischemic stroke risk was sought for the first time in a Turkish population.
Materials and Methods
Subjects
Blood samples of 237 large artery atherosclerotic ischemic stroke patients (138 males and 99 females) and 120 symptom-free controls (56 males and 64 females), who were unrelated, Caucasian people, were collected from Central Anatolia, Turkey, with the collaboration of Gülhane Medical Academy, Department of Neurology, Ankara, Turkey. All participating individuals were informed about the study and signed informed consent forms were obtained. This study was approved by the Ethical Committee of Gülhane Medical Academy and carried out according to the principles of the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.26 All laboratory studies were performed blinded to the medical conditions of the subjects.
The details of inclusion and exclusion criteria were as described before.27,28 In brief, atherothrombotic ischemic stroke patients were recruited to this study. The initial diagnosis of cerebral infarction was made on the basis of a neurological examination and brain computed tomography (CT) scan, and then a transthoracic echocardiographic examination, Holter study and transcranial Doppler emboli detection procedure were carried out to rule out embolic sources. The inclusion criterion for patients was having had an anterior circulation stroke. The “large-vessel disease” group described by the Trial of ORG 10172 in Acute Stroke Treatment (TOAST), in addition to the “total anterior” and “partial anterior circulation infarcts” groups described by the Oxfordshire Community Stroke Project (OCSP), were selected as cases.
The exclusion criteria included having other major illnesses, including autoimmune diseases, coagulopathies, neoplasms, renal or hepatic failure, any known embolic source (aortic arch, cardiac or carotid), a family history of hematological, autoimmune or chronic inflammatory diseases, and a history of myocardial infarction within 3 weeks, or of transient ischemic attack or stroke at any time.
The control subjects were selected randomly from the neurology outpatient clinics from individuals who had not had a stroke or transient ischemic attack at any time, or carotid stenosis (lumen narrowing) >50% or ulcerated carotid plaque. All exclusion criteria were exactly applied to the controls.
Both cases and controls gave a detailed history of conventional vascular risk factors and conditions. Moreover, they underwent routine laboratory tests, including complete blood count, leukocyte differential count and erythrocyte sedimentation rate; routine biochemistry tests, including fasting glucose, lipid profile (triglycerides, total cholesterol, low-density lipoprotein [LDL]-cholesterol and high-density lipoprotein [HDL]-cholesterol), creatinine, sodium, potassium, bilirubin, liver function tests, routine urine tests and rheumatological screening tests; and electrocardiogram and chest X-ray.
Genotype Determination
A manual salting-out DNA isolation protocol, with minor modifications, was used to isolate DNA from whole blood samples of the subjects.29 In brief, 750 µL of TKM buffer (10 mM Trizma buffer pH 7.6, containing 10 mM KCl, 2 mM EDTA and 4 mM MgCl2) was added to 750 µL of total blood. Then, 20 µL of Triton X-100 was added to the tube and the solution was mixed with several inversions to lyse the cells. Centrifugation was performed at 1000× g at room temperature (25°C) using a Sigma 1–15 benchtop microcentrifuge (Sigma, Osterode am Harz, Germany) for 10 min. Two phases were observed after the centrifugation; the pellet containing the DNA was saved and two more washing steps with TKM buffer were performed. The final pellet was resuspended in 200 µL of TKM buffer and then 10 µL of 10% sodium dodecyl sulfate (SDS) was added. The suspension was thoroughly mixed and incubated at 58°C for 10 min. Next, 75 µL of cold saturated NaCl was added, which resulted in the precipitation of proteins. Then, centrifugation was performed at 14,000×g for 10 min at 4°C. The DNA-containing supernatant was transferred into a clean tube and 2× volume ice-cold absolute ethanol was added to precipitate the DNA. The tube was inverted several times, kept at −20°C for at least 30 min, and then centrifuged at 10,000×g for 10 min at 4°C. The supernatant was discarded, and the pellet containing DNA was dried until the ethanol was completely removed, and dissolved in 100 µL of TE buffer, pH 8.0. The tube was then incubated at 37°C for at least 2 h.
Genotyping for all polymorphisms was carried out using the polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) method. Determination of genotypes for Tyr113His (rs1051740T>C) polymorphism of EPHX1 included initial amplification of a 200-bp region in exon 3 of the gene. The PCR mixture contained 1× amplification buffer, 1.25 mM MgCl2, 200 μM dNTP mixture, 40 pmol of each primer, 2.5 U Taq DNA polymerase and approximately 200 ng DNA template. The sequences of the forward and reverse primers were 5ʹ- GGGGTCCTGAATTTTGCTCC-3ʹ and 5ʹ-CAATCTTAGTCTTGAAGTGACGGT-3ʹ, respectively.30 The primers were checked to ensure binding to the EPHX1 gene exon 3 region using the Primer-BLAST web tool.31 PCR was carried out using a Techne TC-4000 thermal cycler (Techne, Duxford, Cambridge, UK) with the following thermal cycling conditions: initial denaturation at 95°C for 5 min, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1 min. Three units of PsyI (Tth111) (MBI Fermentas, USA) endonuclease restriction enzyme was used to digest the 200-bp PCR products and results were analyzed on 2.5% agarose gel (Figure 1). A single band of 200 bp represents the banding pattern of the wild-type homozygous genotype (TT or 113Tyr/113Tyr), while two bands of 177 and 23 bp represent the homozygous polymorphic genotype (CC or 113His/113His). The banding pattern of the heterozygous genotype (TC or 113Tyr/113His) is three bands, at 200, 177 and 23 bp.
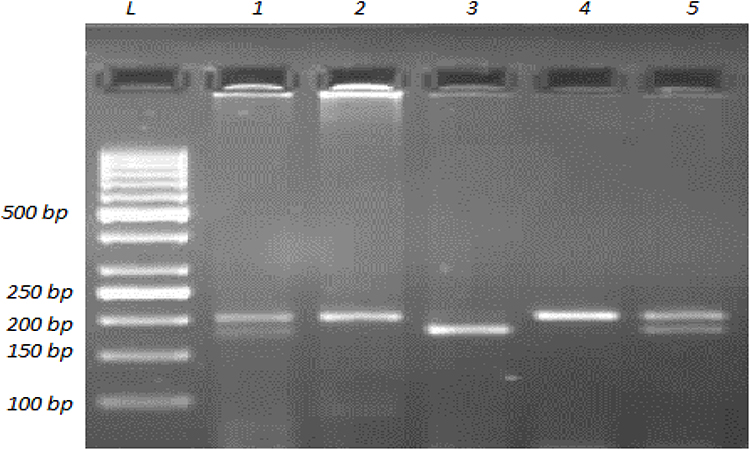 |
Figure 1 Agarose gel image showing the representative banding patterns for determining genotypes for EPHX1 Tyr113His (rs1051740T>C) polymorphism. A 200-bp fragment in EPHX1 gene exon 3 was amplified with PCR and then the PCR product was subjected to restriction endonuclease digestion with PsyI (Tth111). Digestion products were resolved by 2.5% agarose gel electrophoresis with ethidium bromide staining and the DNA bands were visualized by UV illumination. The lane labeled with L was loaded with a DNA ladder (1000 to 50 bp). Lanes 2 and 4 contain a band at 200 bp, which represents the banding pattern of the wild-type homozygous genotype (TT or 113Tyr/113Tyr). Lane 3 contains two bands at 177 and 23 bp, showing the banding pattern of the homozygous polymorphic genotype (CC or 113His/113His). Lanes 1 and 5 show the banding pattern of the heterozygous genotype (TC or 113Tyr/113His), which is three bands, at 200, 177, and 23 bp, as expected. |
The PCR fragments for His139Arg (rs2234922A>G) polymorphism on EPHX1 were obtained using the following primer pair: forward 5ʹ-ACATCCACTTCATCCACGT-3ʹ and reverse 5ʹ- ATGCCTCTGAGAAGCCAT-3ʹ.32 The primers were checked to ensure binding to the EPHX1 gene exon 4 region using the Primer-BLAST web tool.31 PCR was performed as described for Tyr113His polymorphism of the same gene. The expected PCR product length was 210 bp. Restriction endonuclease digestion with 10-unit RsaI (MBI Fermentas, USA) was performed for each PCR product with the correct band length and the results were analyzed on 2.5% agarose gel (Figure 2). Digestion resulted in a single undigested 210-bp fragment for the homozygous wild-type genotype (AA or 139His/139His), and 163 and 47-bp fragments for the homozygous polymorphic genotype (GG or 139Arg/139Arg). Three bands indicated that the person was heterozygous for the His139Arg polymorphism (AG or 139His/139Arg).
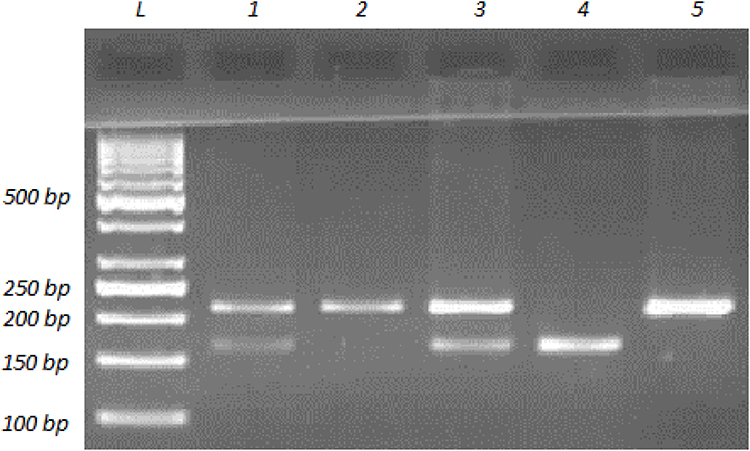 |
Figure 2 Agarose gel image showing the representative banding patterns for determining genotypes for EPHX1 His139Arg (rs2234922A>G) polymorphism. A 210-bp fragment in EPHX1 gene exon 4 was amplified with PCR and then the PCR product was subjected to restriction endonuclease digestion with RsaI. Digestion products were resolved by 2.5% agarose gel electrophoresis with ethidium bromide staining and the DNA bands were visualized by UV illumination. The lane labeled with L was loaded with a DNA ladder (1000 to 50 bp). Lanes 2 and 5 contain a band at 210 bp, which represents the banding pattern of the wild-type homozygous genotype (AA or 139His/139His). Lane 4 contains two bands, at 164 and 46 bp (the 46-bp band was at the very end of gel, which could not be photographed), showing the banding pattern of the homozygous polymorphic genotype (GG or 139Arg/139Arg). Lanes 1 and 3 show the banding pattern of the heterozygous genotype (AG or 139His/139Arg), which is three bands, at 210, 164 and 46 bp, as expected. |
To determine the genotypes for the EPHX2 Arg287Gln (rs751141G>A) polymorphism, PCR was carried out using 20 pmol of each of the following primers: forward 5ʹ-AGGAGGGTGACTCCAGACCT-3ʹ and reverse 5ʹ-CCTTGGAGCATGAGCCTTAG-3ʹ 1× PCR buffer, 2 mM MgCl2, 200 µM dNTPs, 1.5 U of Taq DNA polymerase (MBI Fermentas, USA) and about 200 ng of genomic DNA in 50 µL total reaction volume. The primers were designed using the Primer-BLAST web tool.31 The PCR conditions were: initial denaturation at 95°C for 2 min, followed by 32 cycles of denaturation at 95°C for 15 s, annealing at 55°C for 30 s and extension at 72°C for 30 s, and a final extension step at 72°C for 3 min. The expected length of PCR products was 683 bp, containing three or two recognition sites for the MspI (MBI Fermentas, USA) restriction enzyme, depending on the presence of G or A at the SNP site, respectively. Thus, incubation of the 683-bp PCR product with 4-unit MspI resulted in four fragments of 244, 237, 152 and 50 bp for the G allele, which looked like three bands on 2.5% agarose gel electrophoresis because the 244-bp and 237-bp bands could not be distinguished (Figure 3). On the other hand, three fragments of 244, 237 and 202-bp length were obtained for the A allele, which gave two bands on agarose gel electrophoresis. Digestion products obtained from heterozygotes (GA or 287Arg/287Gln) contained 244, 237, 202, 152 and 50-bp fragments; however. the 244-bp and 237-bp bands were not distinguishable when separated on 2.5% agarose gel and, thus, looked like a darker single band.
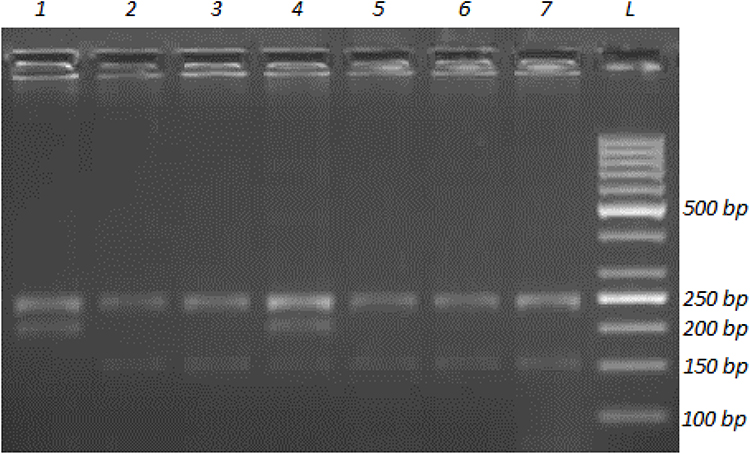 |
Figure 3 Agarose gel image showing the representative banding patterns for determining genotypes for EPHX2 Arg287Gln (rs751141G>A) polymorphism. A 683-bp fragment in EPHX2 gene exon 8 was amplified with PCR and then the PCR product was subjected to restriction endonuclease digestion with MspI. Digestion products were resolved by 2.5% agarose gel electrophoresis with ethidium bromide staining and the DNA bands were visualized by UV illumination. The lane labeled with L was loaded with a DNA ladder (1000 to 50 bp). Lanes 2, 3, 5, 6 and 7 show the banding pattern of the homozygous wild-type genotype (GG or 287Arg/287Arg), which is three bands at 240–236, 150 and 50 bp. Lane 1 contains two bands, at 240–236 and 200 bp, representing the banding pattern of the homozygous polymorphic genotype (AA or 287Gln/287Gln). Lane 4 shows the banding pattern of the heterozygous genotype (GA or 287Arg/287Gln), which is four bands, at 240–236, 200, 150 and 50 bp, as expected. |
Statistical Analysis
The Kolmogorov–Smirnov test was used to test normality of the distribution of continuous variables (age, total cholesterol, triglycerides, LDL-cholesterol, HDL-cholesterol). Differences for continuous data which followed a normal distribution (total cholesterol, triglycerides, LDL-cholesterol, HDL-cholesterol) between patients and controls were analyzed using an independent samples t-test. Continuous data which did not follow a normal distribution (age) were compared using the Mann–Whitney U-test. Categorical variables such as sex, hypertension, smoking status, diabetes and obesity were expressed as proportions and compared using the chi-squared test. Comparisons of genotype distribution and allele frequencies were assessed by Pearson’s chi-square statistics with 2 and 1 df, respectively. Fisher’s exact test was used to calculate the P values when the expected values in any of the cells of a contingency table were ≤5. Departure from the Hardy–Weinberg equilibrium was evaluated by the chi-squared test. Logistic regression analysis with backward selection was employed to investigate the effects of the vascular risk factors, age, sex, lipid parameters and genotypes on ischemic stroke in this Turkish population. Two-tailed probability values with 95% confidence intervals (CIs) were used in the estimation of odds ratios (ORs). Calibration of the logistic regression analysis was carried out with the Hosmer–Lemeshow goodness of fit test. A P value of less than 0.05 was evaluated as statistically significant unless otherwise indicated. The cut-off level for significance was adjusted at α/n using Bonferroni correction for multiple testing. These statistical analyses were performed using the SPSS 18.0 software program package (SPSS, Chicago, IL, USA). A power calculation to test the sufficiency of the sample size was carried out using G Power 3.1 software. A power value >50% was considered adequate.
Results
Demographic features, prevalences of conventional risk factors and serum lipid levels of the study population are given in Table 1. The age of the study population varied from 20 to 81 years in the stroke group, and from 39 to 90 years in the control group. There was no significant differences between patient and control groups in terms of age. We also evaluated the age range separately in males and females. In the male population, the age ranged from 20 to 81 years for stroke patients and from 42 to 87 years in the control group, and there was no significant difference in the mean age of male stroke patients and male controls. On the other hand, in the female population, the age ranged from 31 to 81 years for stroke patients and from 41 to 90 years in the control group; the mean age was significantly higher in female stroke patients compared with female controls (P=0.014). The conventional risk factors, namely male gender, hypertension, diabetes, smoking status and obesity, were found to be significantly more prevalent in the ischemic stroke group. In addition, total cholesterol and LDL-cholesterol levels were markedly higher, while the HDL-cholesterol level was significantly lower in ischemic stroke patients compared with the control group.
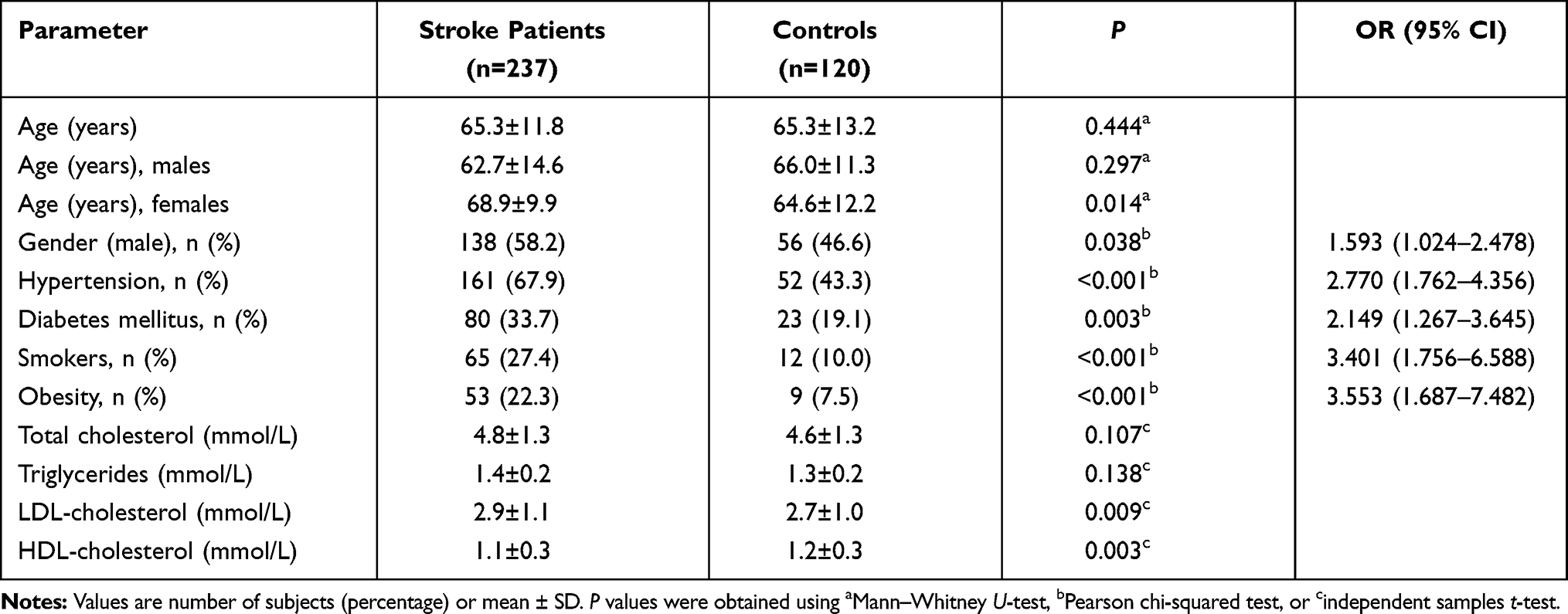 |
Table 1 Effects of Demographic Features, Conventional Risk Factors and Clinical Characteristics on Ischemic Stroke Risk |
The frequency distributions of Tyr113His (EPHX1), His139Arg (EPHX1) and Arg287Gln (EPHX2) genotypes and their corresponding alleles in ischemic stroke patients and controls are shown in Tables 2, 3 and 4. There was no deviation of genotype frequencies from Hardy–Weinberg equilibrium. We observed that there were no significant differences between ischemic stroke patients and controls in terms of genotype distributions and minor allele frequencies for any of the three analyzed genetic polymorphisms. The genotype and allele frequencies were also calculated separately for male and female subpopulations (Tables 2–4). The distribution of genotypes and the alleles for EPHX2 Arg287Gln polymorphism were significantly different in the female subpopulation (Table 4).
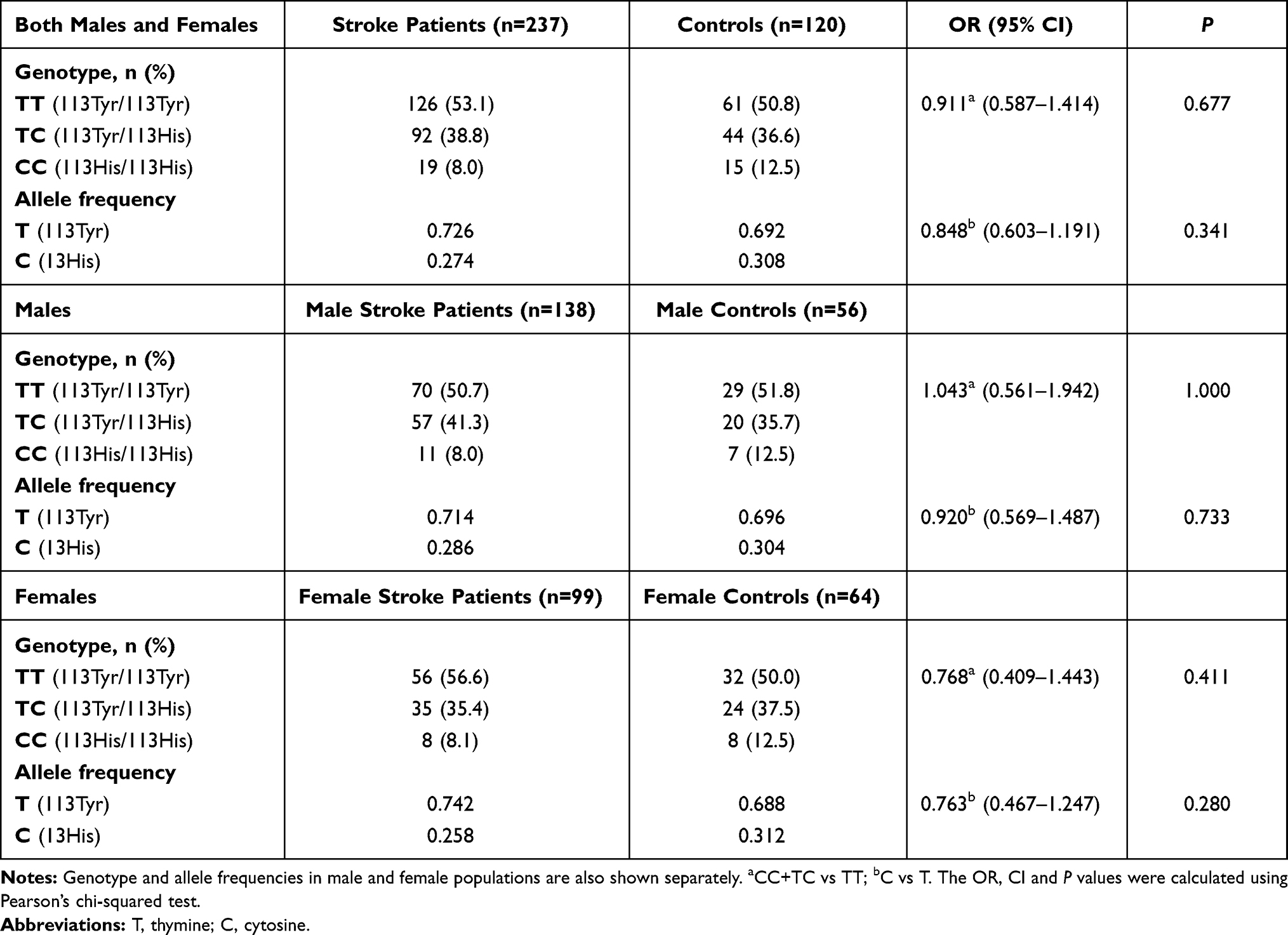 |
Table 2 Distribution of Genotypes and Allele Frequencies for EPHX1 Tyr113His (rs1051740 T>C) Polymorphism in Ischemic Stroke Patients and Controls |
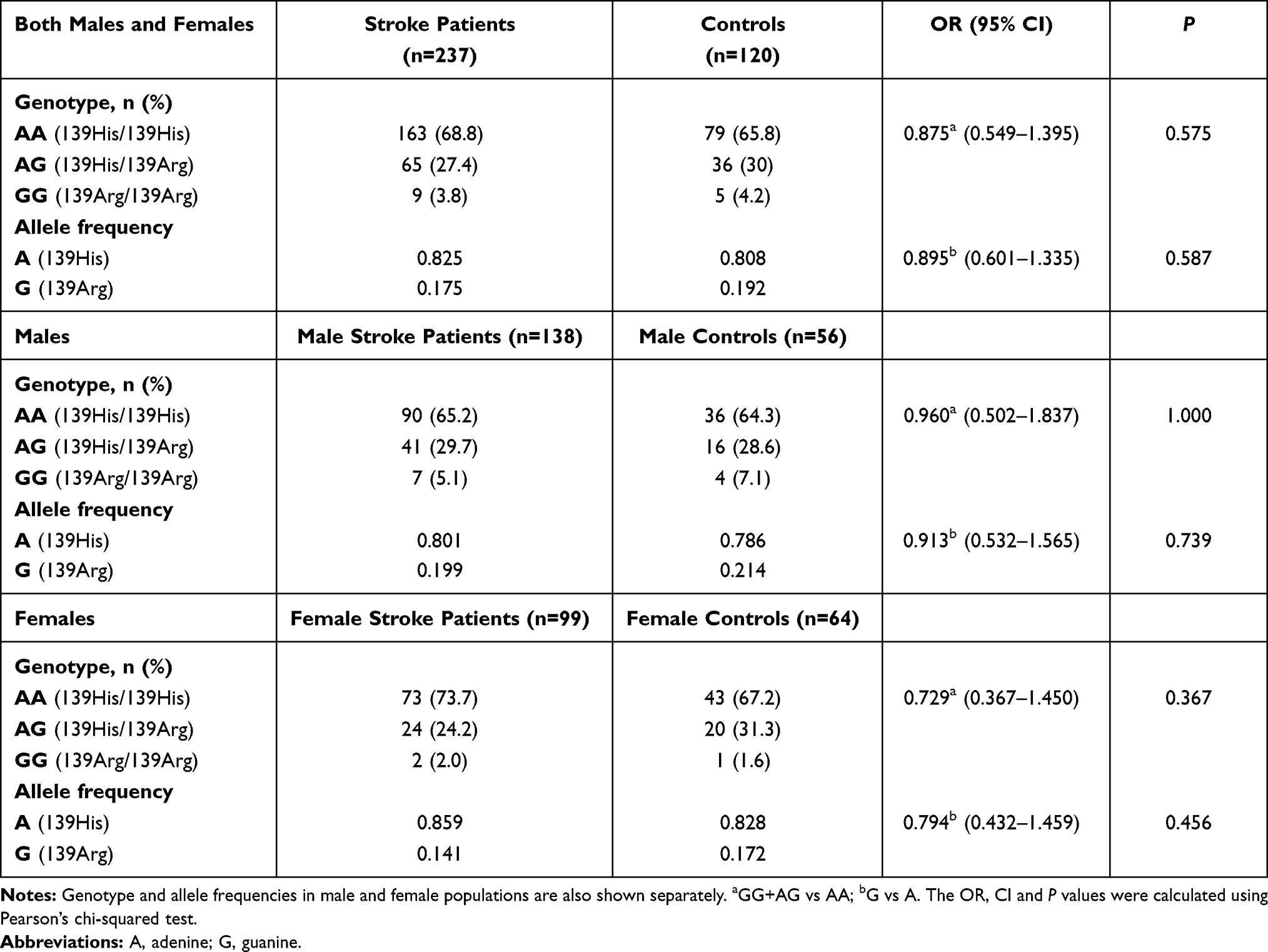 |
Table 3 Distribution of Genotypes and Allele Frequencies for EPHX1 His139Arg (rs2234922A>G) Polymorphism in Ischemic Stroke Patients and Controls |
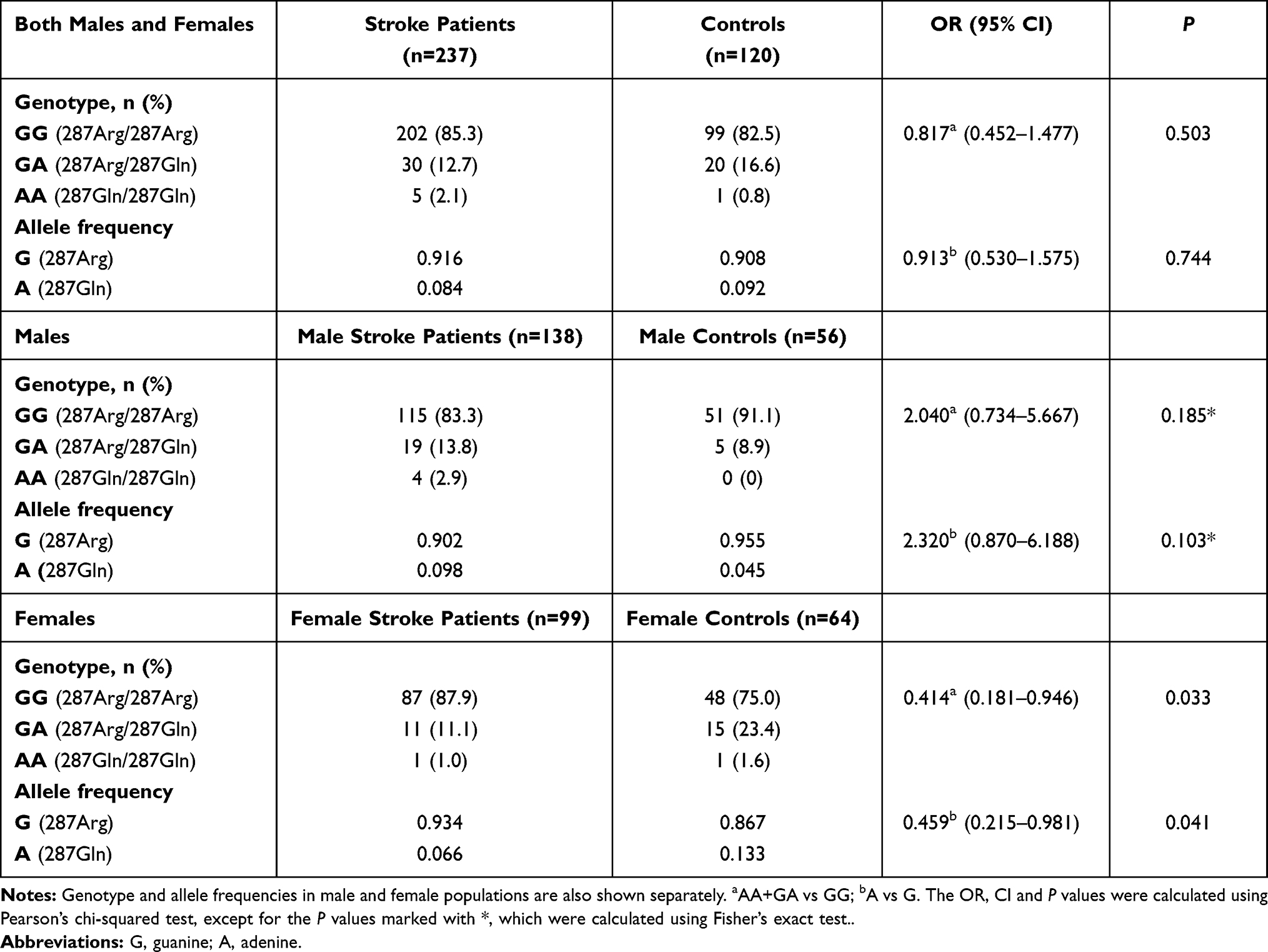 |
Table 4 Distribution of Genotypes and Allele Frequencies for EPHX2 Arg287Gln (rs751141G>a) Polymorphism in Ischemic Stroke Patients and Controls |
Stratification analysis was carried out to determine whether the polymorphic alleles modified the conventional risk factor-related ischemic stroke risk (Table 5). For the EPHX1 Tyr113His polymorphism, in the homozygote wild-type genotype (TT) group, the OR for having ischemic stroke was calculated to be 3.21 (<0.001) for subjects with hypertension compared with subjects with normotension. This value decreased to 2.37 (<0.001) when at least one mutant allele was present (113TC or CC genotype). Likewise, the ischemic stroke risk of hypertensive subjects relative to normotensive subjects was 3.15 when they had the AA genotype for the EPHX1 His139Arg polymorphism, and this risk decreased to 2.14 in individuals having the AG or GG genotype. EPHX2 Arg287Gln genetic polymorphism also affected the hypertension-related ischemic stroke risk. This time, the OR increased from 2.52 (in wild-type individuals with the GG genotype) to 4.69 (in individuals having at least one A allele). When stratification analysis was carried out with respect to having or not having diabetes, a similar trend to that observed in the case of hypertension was observed. EPHX1 polymorphisms slightly decreased the diabetes-associated ischemic stroke risk, while EPHX2 polymorphism increased the risk (Table 5).
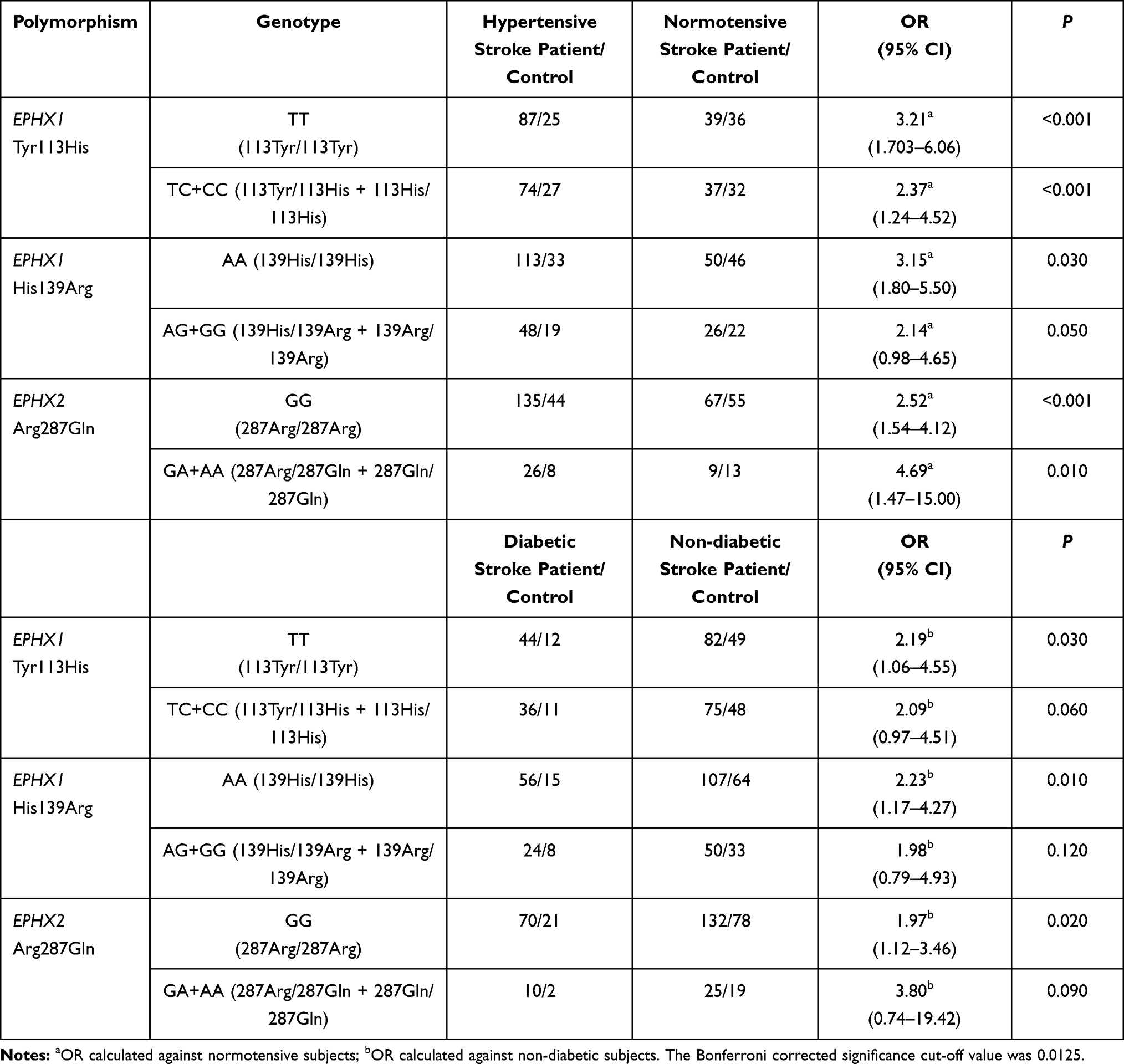 |
Table 5 Stratification Analysis of Conventional Risk Factors, Hypertension and Diabetes, on Different Genotype Groups of EPHX1 and EPHX2 in Terms of Ischemic Stroke Risk |
The following parameters were tested by logistic regression analysis using backward selection to determine the risk factors for ischemic stroke: age, sex, diabetes, hypertension, obesity, smoking, concentration of total cholesterol, LDL-cholesterol, HDL-cholesterol and triglyceride, as well as the genotypes of EPHX Tyr113His, EPHX1 His139Arg and EPHX2 Arg287Gln SNPs. The results revealed hypertension (OR=3.19, 95% CI=1.92–5.30, P<0.001) and smoking (OR=3.46, 95% CI=1.66–7.21, P<0.001) as the strongest determinants of ischemic stroke. LDL-cholesterol (OR=1.46, 95% CI=1.12–1.89, P<0.001) was also determined to be markedly associated with this disease. Furthermore, HDL-cholesterol (OR=0.27, 95% CI=0.11–0.65, P<0.001) came out as having a protective effect against having ischemic stroke. The Hosmer–Lemeshow goodness of fit test demonstrated that the calibration of the model was satisfactory (chi-square=6.721, with 8 degrees of freedom, P=0.567) for logistic regression, and the model predicted 73.1% of the cases correctly.
Discussion
The prevalence of stroke is about 3% of the adult population in the USA,33 and it is increasing all over the world as the aging population increases.34 In Turkey, the prevalence is between 0.9% and 4.1% among adults aged 45 years and above.35,36 Moreover, stroke is the most important condition leading to disability in the elderly. Therefore, it is important to gather as much information as possible regarding the genetic determinants of stroke. Ischemic stroke accounts for around 80% of all cases;33 therefore, in this study we aimed to investigate the possible association of EPHX2 Arg287Gln variation and EPHX1 Tyr113His and His139Arg variations with ischemic stroke risk in the Turkish population. The results indicate that there is no significant association between these genetic polymorphisms and ischemic stroke risk.
sEH enzyme degrades EETs into DHETs, which have been considered to be inactive or less active. However, they were later shown to exert vasodilatory effects on coronary arterioles and arteries.37,38 In addition, DHETs have been shown to display anti-inflammatory properties.39 Therefore, both EETs and their DHET metabolites may be important regulators of the coronary circulation. It was shown in vitro that EPHX2 Arg287Gln causes a decrease in sEH activity (25–58% of that of the wild-type enzyme).16 This may explain our finding of no association between the EPHX2 287Gln allele and ischemic stroke risk.
Moreover, the generation of EETs, via epoxygenation of arachidonic acid, is governed by a highly polymorphic class of enzymes, cytochrome P450s (CYPs). At least 12 human CYP genes have been reported to possess epoxygenase activity, including CYP1A families.40 We have previously shown that variants of the CYP1A1 gene were significantly related to the risk of ischemic stroke, especially in subgroups.27 Therefore, the combination of CYP1A1 and EPHX2 polymorphisms in a given person is expected to have a higher impact on governing ischemic stroke risk or protection.
It is not surprising, following this information, to see that there are completely contradictory reports in the literature regarding the relationship between EPHX2 Arg287Gln variant and cardiovascular disease risk. The 287Gln allele was observed to have protective effect against ischemic neuronal injury in a rat model.17 Similarly, it has been associated with a significantly lower risk of ischemic stroke in a Chinese population.25 In contrast, this variant has been observed to be associated with an increased risk for coronary artery disease in other studies.20,22,23 In addition, it has been associated with carotid artery calcified plaques in European American families41 and with an increased risk of ischemic stroke (OR=1.458, P=0.005) in white Europeans.24 However, no significant association between the EPHX2 287Gln allele and ischemic stroke was detected in a large cohort study in a Danish population.42 Likewise, five polymorphisms of EPHX2, including 287Gln, did not display a significant association with ischemic stroke in a large sample of white and African-American subjects.21 Significant relationships were detected with haplotypes; however, the effects of haplotypes were in opposite directions.21
Another possible explanation for these discrepancies comes from the work of Ramirez et al,43 who reported that no significant relationship was present between the EPHX2 Arg287Gln genotype and either EET concentrations or sEH activity, and suggested that an up-regulation of allelic expression in heterozygotes may have compensated for the less active allele.43 It is known that cigarette smoking increases EPHX2 gene expression,23 which may compensate for the enzyme activity reduction caused by the Arg287Gln polymorphism. Thus, it is possible to explain the result obtained in the present study by the presence of more smokers within the 287Gln (A) allele carriers; smokers accounted for 25% of subjects with the GA or AA genotype, and 20.9% in subjects with the GG genotype. Therefore, the protection that may have been conferred by the low-activity Gln287 (A) allele is canceled out by the higher number of smokers in this subgroup, which probably upregulated the sEH.
The functions of sEH are related to regulation of the vascular tone owing to its role in the metabolism of EETs. Therefore, we carried out stratification analysis based on having hypertension, and observed that the presence of 287Gln genetic variation in EPHX2 increases the hypertension-related ischemic stroke risk from 2.52 to 4.69. We also carried out stratification based on having diabetes, since it has been reported that EPHX2 gene transcription is down-regulated under high glucose conditions.44 In this analysis, EPHX2 polymorphism increased diabetes-related ischemic stroke risk. Ohtoshi et al found that insulin resistance was significantly increased in type II diabetic patients with the 287Gln allele.45 The complex interplay between xenobiotics such as cigarette smoke-associated chemicals, gene induction and physiological states such as diabetes could explain these outcomes.
The functions of mEH may have potentially contrasting effects on ischemic stroke risk. This enzyme plays a role in the metabolism of both EETs7 and PAHs. Its ability to convert PAHs into less reactive species makes mEH a protector against atherosclerosis, which is a significant risk factor leading to ischemic stroke. In this study, two functional SNPs, Tyr113His and His139Arg, in EPHX1 gene were analyzed in relation to ischemic stroke risk. Tyr113His variation results in a 39% reduction in mEH enzyme activity, while His139Arg variation causes a 25% increase in enzyme activity.8 Our results suggested that there was no association between the minor alleles and the disease status in the study group.
No previous studies have analyzed EPHX1 genetic polymorphisms and ischemic stroke association, and studies carried out on various cancers have reported discordant findings. To mention a few of these studies, EPHX1 genetic polymorphisms were observed to be important risk factors for susceptibility to various cancers, including prostate cancer,9,10 lung cancer12 and acute lymphoblastic leukemia.11 The 113His allele was shown to be a risk factor for lung carcinoma in Asians, whereas it was observed to have a protective effect in Caucasians in a recent meta-analysis.46 Moreover, a protective role has been attributed to low mEH activity against childhood lymphoblastic leukemia.47 Furthermore, a meta-analysis demonstrated a trend towards a protective effect of His139Arg SNP against colorectal cancer risk.48 It is evident that the results deviate significantly among different populations.
The variant alleles of EPHX1 (113His and 139Arg) were observed to decrease the stroke susceptibility in stratification analysis in this study. We observed a lower risk for hypertension-related ischemic stroke in subjects carrying at least one 113His allele (OR=2.37) compared with individuals with the wild-type genotype (OR=3.21). Because EETs have important functions in regulating the vascular tone and cerebral blood flow, the effect of this allele, which decreases the rate of EET metabolism, might have become apparent in the stratification analysis based on hypertension. The diabetes-related ischemic stroke risk was also lower in 113His allele carriers (OR=2.09) compared with subjects with the wild-type genotype (OR=2.19).
It was also observed in this study that the OR for having a hypertension-related ischemic stroke decreased from 3.15 to 2.14 when at least one EPHX1 139Arg allele was present. This allele was found to increase the mEH enzyme activity;49 therefore, the atherogenic chemical metabolism rate may have been enhanced in subjects possessing this allele. The link between diabetes mellitus and atherosclerosis is well recognized; hence, diabetes mellitus increases the risk of stroke by several fold.50 EPHX1 His139/His139 genotype carriers have been shown to have a higher fasting insulin level and lower insulin sensitivity compared to Arg139 allele carriers (P=0.020 and P=0.001, respectively),51 which may mean that diabetic complications are less severe in 139Arg allele carriers. In this study, the 139Arg allele decreased the diabetes-related ischemic stroke risk in stratification analysis.
Hypertension, smoking and LDL-cholesterol were found to be significant risk factors, and HDL-cholesterol was found to be a protective factor for the occurrence of ischemic stroke by logistic regression analysis, as also found in our previous studies.27,28 However, our study had a limitation regarding the sample size of the healthy control subjects, resulting from age restrictions in selecting control subjects. Nevertheless, the power test result was satisfactory; therefore, the results were considered statistically reliable.
It is well known that the distribution of alleles for a given genetic variant may markedly differ among populations. Thus, we included a comparison of the minor allele frequencies for EPHX1 Tyr113His (rs1051740 T>C), EPHX1 His139Arg (rs2234922 A>G) and EPHX2 Arg287Gln (rs751141 G>A) variants in the healthy control population of this study with those in different control populations that have previously been analyzed (Table 6). EPHX1 rs1051740 C allele (113His) was observed at a frequency of 0.31 in our study and this frequency ranged from 0.2511 to 0.3552 in other studies which were also carried out on the Turkish population.11,52–55 The frequency of 113His allele was lowest in Indians (0.23)56 and highest in Taiwanese (0.48)57 among the other populations32,51,56–64 (Table 6). The EPHX1 rs2234922 G (139Arg) allele frequency was observed to be 0.19 in our study, which was exactly the same as that reported by Sahin et al55 and was close to this value in other studies carried out on Turkish populations.11,52–54 The frequency of this allele was determined in several other populations32,51,56–64 and observed at its lowest value (0.09) in a Chinese population59 and at its highest value (0.41) in an African population.64 Relatively few studies in the literature have been carried out on EPHX2 Arg287Gln polymorphism (Table 6). The frequency of the minor rs751141A allele was found to be 0.09 in our population and no other studies have been carried out on the Turkish population. rs751141A allele frequency ranged between 0.08, which was found in white Europeans,24 to 0.21, which was found in a Japanese population,45 among other populations.20,21,23,24,45
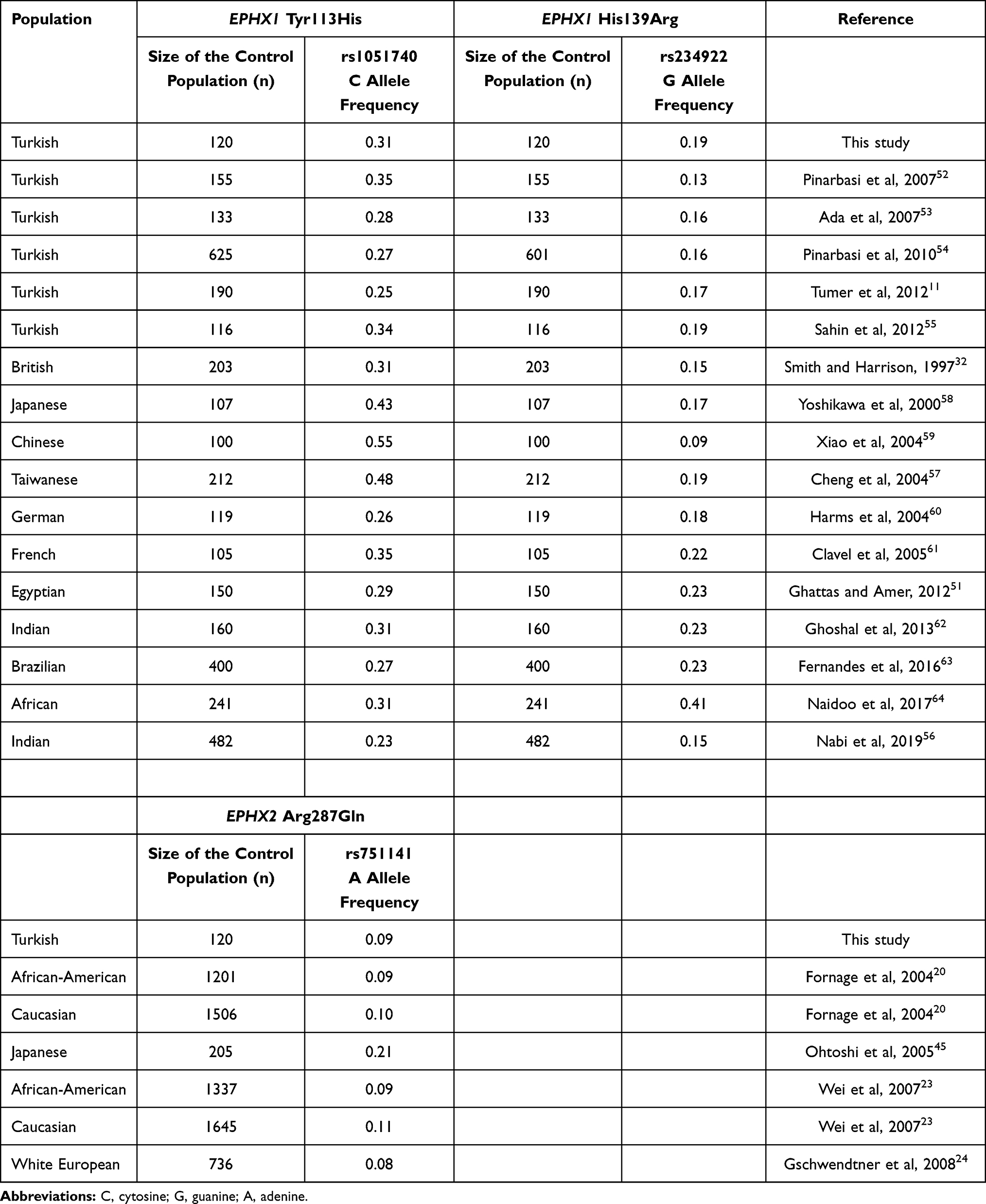 |
Table 6 EPHX1 Tyr113His (rs1051740 T>C), EPHX1 His139Arg (rs2234922 A>G) and EPHX2 Arg287Gln (Rs751141 G>A) Minor Allele Frequencies Reported in Control Groups of Different Populations |
In conclusion, to the best of our knowledge, this is the first time that the association of EPHX1 genetic polymorphisms with ischemic stroke risk has been analyzed. Although EPHX1 variant alleles were not significantly associated with ischemic stroke risk in the overall study group, the presence of at least one 113His or 139Arg allele decreased the hypertension- or diabetes-related ischemic stroke risk within the subgroups. In addition, this work for the first time investigated the association of Arg287Gln polymorphism of EPHX2 and ischemic stroke risk in a Turkish population, and the results showed that the polymorphic allele was not significantly related to ischemic stroke. However, the EPHX2 variant allele increased the risk of hypertension-related and diabetes-related ischemic stroke in subgroup analysis. Thus, combination of different genetic backgrounds with conventional risk factors may change the risk of having ischemic stroke in a significant manner.
Acknowledgments
The authors thank the subjects for their participation in this study and Prof. Dr. Şeref Demirkaya for sample collection. This research did not receive any specific grants from funding agencies in the public, commercial or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Katan M, Luft A. Global burden of stroke. Semin Neurol. 2018;38(2):208–211. doi:10.1055/s-0038-1649503
2. Nagai Y, Kitagawa K, Sakaguchi M, et al. Significance of earlier carotid atherosclerosis for stroke subtypes. Stroke. 2001;32:1780–1785. doi:10.1161/01.STR.32.8.1780
3. Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56. doi:10.1038/s41572-019-0106-z
4. Sudhahar V, Shaw S, Imig JD. Epoxyeicosatrienoic acid analogs and vascular function. Curr Med Chem. 2010;17(12):1181–1190. doi:10.2174/092986710790827843
5. De Flora S, Izzotti A, Walsh D, et al. Molecular epidemiology of atherosclerosis. FASEB J. 1997;11:1021–1031. doi:10.1096/fasebj.11.12.9337155
6. Penn A, Snyder C. Arteriosclerotic plaque development is ‘promoted’ by polynuclear aromatic hydrocarbons. Carcinogenesis. 1988;9:2185–2189. doi:10.1093/carcin/9.12.2185
7. Marowsky A, Burgener J, Falck JR, et al. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience. 2009;163(2):646–661. doi:10.1016/j.neuroscience.2009.06.033
8. Hassett C, Robinson KB, Beck NB, Omiecinski CJ. The human microsomal epoxide hydrolase gene (EPHX1): complete nucleotide sequence and structural characterization. Genomics. 1994;23:433–442. doi:10.1006/geno.1994.1520
9. Figer A, Friedman T, Manguoglu AE, et al. Analysis of polymorphic patterns in candidate genes in Israeli patients with prostate cancer. Isr Med Assoc J. 2003;5:741–745.
10. Mittal RD, Srivastava DS. Cytochrome P4501A1 and microsomal epoxide hydrolase gene polymorphisms: gene-environment interaction and risk of prostate cancer. DNA Cell Biol. 2007;26:791–798. doi:10.1089/dna.2007.0630
11. Tumer TB, Sahin G, Arinç E. Association between polymorphisms of EPHX1 and XRCC1 genes and the risk of childhood acute lymphoblastic leukemia. Arch Toxicol. 2012;86:431–439. doi:10.1007/s00204-011-0760-8
12. Zhang P, Zhang Y, Yang H, et al. Association between EPHX1 rs1051740 and lung cancer susceptibility: a meta-analysis. Int J Clin Exp Med. 2015;8(10):17941–17949.
13. Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305(1):197–201. doi:10.1006/abbi.1993.1411
14. Larsson C, White I, Johansson C, et al. Localization of the human soluble epoxide hydrolase gene (EPHX2) to chromosomal region 8p21-p12. Hum Genet. 1995;95(3):356–358. doi:10.1007/BF00225209
15. Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8(10):794–805. doi:10.1038/nrd2875
16. Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, et al. Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol. 2003;64(2):482–490. doi:10.1124/mol.64.2.482
17. Koerner IP, Jacks R, DeBarber AE, et al. Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci. 2007;27:4642–4649. doi:10.1523/JNEUROSCI.0056-07.2007
18. Zhang W, Koerner IP, Noppens R, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27:1931–1940. doi:10.1038/sj.jcbfm.9600494
19. Zhang W, Otsuka T, Sugo N, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–2078. doi:10.1161/STROKEAHA.107.508325
20. Fornage M, Boerwinkle E, Doris PA, et al. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: the coronary artery risk development in young adults (CARDIA) study. Circulation. 2004;109(3):335–339. doi:10.1161/01.CIR.0000109487.46725.02
21. Fornage M, Lee CR, Doris PA, et al. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14:2829–2837. doi:10.1093/hmg/ddi315
22. Lee CR, North KE, Bray MS, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: the atherosclerosis risk in communities (ARIC) study. Hum Mol Genet. 2006;15:1640–1649. doi:10.1093/hmg/ddl085
23. Wei Q, Doris PA, Pollizotto MV, et al. Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: interaction with cigarette smoking. Atherosclerosis. 2007;190:26–34. doi:10.1016/j.atherosclerosis.2006.02.021
24. Gschwendtner A, Ripke S, Freilinger T, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with an increased risk of ischemic stroke in white Europeans. Stroke. 2008;39:1593–1596. doi:10.1161/STROKEAHA.107.502179
25. Zhang L, Ding H, Yan J, et al. Genetic variation in cytochrome P450 2J2 and soluble epoxide hydrolase and risk of ischemic stroke in a Chinese population. Pharmacogenet Genomics. 2008;18:45–51. doi:10.1097/FPC.0b013e3282f313e8
26. World Medical Association. Declaration of helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi:10.1001/jama.2013.281053
27. Demirdöğen BC, Adali AÇ, Bek S, et al. Cytochrome P4501A1 genotypes and smoking- and hypertension-related ischemic stroke risk. Hum Exp Toxicol. 2013;32(5):483–491. doi:10.1177/0960327112464667
28. Türkanoğlu Özçelik A, Can Demirdöğen B, Demirkaya Ş, et al. Association of cytochrome P4502E1 and NAD(P)H: quinoneoxidoreductase 1 genetic polymorphisms with susceptibility to large artery atherosclerotic ischemic stroke: a case-control study in Turkish population. Neurol Sci. 2017;38(6):1077–1085. doi:10.1007/s10072-017-2930-9
29. Lahiri DK, Schnabel B. DNA isolation by a rapid method from human blood samples: effects of MgCI2, EDTA, storage time, and temperature on DNA yield and quality. Biochem Genet. 1993;31:321–328. doi:10.1007/BF00553174
30. Korhonen S, Romppanen EL, Hiltunen M, et al. Two exonic single nucleotide polymorphisms in the microsomal epoxide hydrolase gene are associated with polycystic ovary syndrome. Fertil Steril. 2003;79(6):1353–1357. doi:10.1016/S0015-0282(03)00385-6
31. Available from: https://www.ncbi.nlm.nih.gov/tools/primer-blast/.
32. Smith CA, Harrison DJ. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet. 1997;350:630–633. doi:10.1016/S0140-6736(96)08061-0
33. Ovbiagele B, Nguyen-Huynh MN. Stroke epidemiology: advancing our understanding of disease mechanism and therapy. Neurotherapeutics. 2011;8(3):319–329. doi:10.1007/s13311-011-0053-1
34. Feigin VL, Krishnamurthi RV, Parmar P, et al. Update on the global burden of ischemic and hemorrhagic stroke in 1990–2013: the GBD 2013 study. Neuroepidemiology. 2015;45:161–176. doi:10.1159/000441085
35. Öncel Ç, Tokgöz F, Bozkurt AI, Erdoǧan Ç. Prevalence of cerebrovascular disease: a door-to-door survey in West Anatolia. Neurol Sci. 2014;35(3):373–377. doi:10.1007/s10072-013-1521-7
36. Padir Şensöz N, Türk Börü Ü, Bölük C, et al. Stroke epidemiology in Karabük city Turkey: community based study. eNeurologicalSci. 2017;10:12–15. doi:10.1016/j.ensci.2017.12.003
37. Oltman CL, Weintraub NL, VanRollins M, et al. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res. 1998;83:932–939. doi:10.1161/01.RES.83.9.932
38. Larsen BT, Miura H, Hatoum OA, et al. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BKCa channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol Heart Circ Physiol. 2006;290(2):H491–H499. doi:10.1152/ajpheart.00927.2005
39. Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–1279. doi:10.1126/science.285.5431.1276
40. Schwarz D, Kisselev P, Ericksen SS, et al. Arachidonic and eicosapentaenoic acid metabolism by human CYP1A1: highly stereoselective formation of 17(r), 18(s)-epoxyeicosatetraenoic acid. Biochem Pharmacol. 2004;67:1445–1457. doi:10.1016/j.bcp.2003.12.023
41. Burdon KP, Lehtinen AB, Langefeld CD, et al. Genetic analysis of the soluble epoxide hydrolase gene, EPHX2, in subclinical cardiovascular disease in the Diabetes Heart Study. Diab Vasc Dis Res. 2008;5(2):128–134. doi:10.3132/dvdr.2008.021
42. Lee J, Dahl M, Grande P, Tybjaerg-Hansen A, Nordestgaard BG. Genetically reduced soluble epoxide hydrolase activity and risk of stroke and other cardiovascular disease. Stroke. 2010;41(1):27–33. doi:10.1161/STROKEAHA.109.567768
43. Ramirez CE, Shuey MM, Milne GL, et al. Arg287Gln variant of EPHX2 and epoxyeicosatrienoic acids are associated with insulin sensitivity in humans. Prostaglandins Other Lipid Mediat. 2014;113–115:38–44. doi:10.1016/j.prostaglandins.2014.08.001
44. Oguro A, Oida S, Imaoka S. Down regulation of EPHX2 gene transcription by Sp1 under high-glucose conditions. Biochem J. 2015;470(3):281–291. doi:10.1042/BJ20150397
45. Ohtoshi K, Kaneto H, Node K, et al. Association of soluble epoxide hydrolase gene polymorphism with insulin resistance in type 2 diabetic patients. Biochem Biophys Res Commun. 2005;331:347–350. doi:10.1016/j.bbrc.2005.03.171
46. Tan X, Wang YY, Chen XY, et al. Quantitative assessment of the effects of the EPHX1 Tyr113His polymorphism on lung and breast cancer. Genet Mol Res. 2014;13:7437–7446. doi:10.4238/2014.September.12.10
47. Silveira Vda S, Canalle R, Scrideli CA, et al. Role of the CYP2D6, EPHX1, MPO, and NQO1 genes in the susceptibility to acute lymphoblastic leukemia in Brazilian children. Environ Mol Mutagen. 2010;51:48–56. doi:10.1002/em.20510
48. Liu F, Yuan D, Wei Y, et al. Systematic review and meta-analysis of the relationship between EPHX1 polymorphisms and colorectal cancer risk. PLoS One. 2012;7:e43821. doi:10.1371/journal.pone.0043821
49. Hassett C, Aicher L, Sidhu JS, et al. Human microsomal epoxide hydrolase: genetic polymorphism and functional expression in vitro of amino acid variants. Hum Mol Genet. 1994;3(3):421–428. doi:10.1093/hmg/3.3.421
50. Bruno A, Liebeskind D, Hao Q, et al.; for the UCLA Stroke Investigators. Diabetes mellitus, acute hyperglycemia, and ischemic stroke. Curr Treat Options in Neurol. 2010;12(6):492–503. doi:10.1007/s11940-010-0093-6
51. Ghattas MH, Amer MA. Possible role of microsomal epoxide hydrolase gene polymorphism as a risk factor for developing insulin resistance and type 2 diabetes mellitus. Endocrine. 2012;42(3):577–583. doi:10.1007/s12020-012-9656-5
52. Pinarbasi E, Percin FE, Yilmaz M, Akgun E, Cetin M, Cetin A. Association of microsomal epoxide hydrolase gene polymorphism and pre-eclampsia in Turkish women. J Obstet Gynaecol Res. 2007;33:32–37. doi:10.1111/j.1447-0756.2007.00473.x
53. Ada AO, Suzen HS, Iscan M. Polymorphisms of microsomal epoxide hydrolase and glutathione S-transferase P1 in a male Turkish population. Int J Toxicol. 2007;26:41–46. doi:10.1080/10915810601118222
54. Pinarbasi H, Silig Y, Pinarbasi E. Microsomal epoxide hydrolase polymorphisms. Mol Med Rep. 2010;3(4):723–727. doi:10.3892/mmr_00000324
55. Sahin O, Arikan S, Musteri Oltulu Y, et al. Investigation of a possible relationship between EPHX1 gene polymorphisms and colorectal cancer in Turkish society. Genet Test Mol Biomarkers. 2012;16(5):423–428. doi:10.1089/gtmb.2011.0223
56. Nabi S, Bhat GA, Iqbal B, et al. Association of activity altering genotypes- Tyr113His and His139Arg in microsomal epoxide hydrolase enzyme with esophagel squamous cell carcinoma. Nutr Cancer. 2019;71(5):806–817. doi:10.1080/01635581.2018.1484934
57. Cheng SL, Yu CJ, Chen CJ, Yang PC. Genetic polymorphism of epoxide hydrolase and glutathione S-transferase in COPD. Eur Respir J. 2004;23:818–824. doi:10.1183/09031936.04.00104904
58. Yoshikawa M, Hiyama K, Ishioka S, Maeda H, Maeda A, Yamakido M. Microsomal epoxide hydrolase genotypes and chronic obstructive pulmonary disease in Japanese. Int J Mol Med. 2000;5:49–53. doi:10.3892/ijmm.5.1.49
59. Xiao D, Wang C, Du MJ, et al. Relationship between polymorphisms of genes encoding microsomal epoxide hydrolase and glutathione S-transferase P1 and chronic obstructive pulmonary disease. Chin Med J. 2004;117:661–667.
60. Harms C, Salama SA, Sierra-Torres CH, Cajas-Salazar N, Au WW. Polymorphisms in DNA repair genes, chromosome aberrations, and lung cancer. Environ Mol Mutagen. 2004;44(1):74–82. doi:10.1002/em.20031
61. Clavel J, Bellec S, Rebouissou S, et al. Childhood leukaemia, polymorphisms of metabolism enzyme genes, and interactions with maternal tobacco, coffee and alcohol consumption during pregnancy. Eur J Cancer Prev. 2005;14:531–540. doi:10.1097/00008469-200512000-00007
62. Ghoshal U, Kumar S, Jaiswal V, Tripathi S, Mittal B, Ghoshal UC. Association of microsomal epoxide hydrolase exon 3 Tyr113His and exon 4 His139Arg polymorphisms with gastric cancer in India. Indian J Gastroenterol. 2013;32(4):246–252. doi:10.1007/s12664-013-0332-3
63. Fernandes GMM, Russo A, Proença MA, et al. CYP1A1, CYP2E1 and EPHX1 polymorphisms in sporadic colorectal neoplasms. World J Gastroenterol. 2016;22(45):9974–9983. doi:10.3748/wjg.v22.i45.9974
64. Naidoo P, Naidoo RN, Ramkaran P, Asharam K, Chuturgoon AA. The Tyr113His T/C and very slow phenotype of the EPHX1 gene alters miR-26b-5p and miR-1207-5p expression in pregnancy. Gene. 2017;633:71–81. doi:10.1016/j.gene.2017.07.080
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.