Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
miRNA-486-5p Promotes COPD Progression by Targeting HAT1 to Regulate the TLR4-Triggered Inflammatory Response of Alveolar Macrophages
Authors Zhang J, Xu Z, Kong L, Gao H, Zhang Y, Zheng Y, Wan Y
Received 5 September 2020
Accepted for publication 26 October 2020
Published 17 November 2020 Volume 2020:15 Pages 2991—3001
DOI https://doi.org/10.2147/COPD.S280614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Jie Zhang,1,* Zhongneng Xu,2,* Lianhua Kong,3,* Hong Gao,1 Yueming Zhang,1 Yulong Zheng,1 Yufeng Wan1
1Department of Respiratory Diseases, The Affiliated Huai’an Hospital of Xuzhou Medical University, Huai’an 203302, Jiangsu, People’s Republic of China; 2Department of Cardiothoracic Surgery, Huai’an Hospital Affiliated to Nanjing Medical College and Huai’an First People’s Hospital, Huai’an 223002, Jiangsu, People’s Republic of China; 3Department of Infectious Disease, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yufeng Wan; Yulong Zheng Tel +86 13776707363
; +86 15805230282
Email [email protected]; [email protected]
Purpose: The aim of this study was to investigate the role of miRNA-486-5p in chronic obstructive pulmonary disease (COPD) progression and the underlying molecular mechanisms.
Materials and Methods: Aberrant miRNA expression profiles between smokers and nonsmokers, and those between COPD patients and normal subjects were analyzed using microarray datasets and reverse-transcriptase quantitative polymerase chain reaction (qPCR). Enzyme-linked immunosorbent assay was used to determine the levels of inflammatory cytokines in cell supernatants. Expression levels of inflammatory cytokines, HAT1, TLR4, and miR-486-5p, were determined using qPCR or Western blotting. Luciferase reporter assays and fluorescence in situ hybridization were used to confirm the regulatory interaction between miR-486-6p and HAT1.
Results: miR-486-5p was significantly upregulated in the COPD and smoker groups compared to the control group, as demonstrated using bioinformatics analysis and validated using qPCR assay of alveolar macrophages and peripheral monocytes. Moreover, miR-486-5p expression was significantly correlated with the expression of IL-6, IL-8, TNF-α, and IFN-γ. Luciferase reporter assays confirmed that miR-486-5p directly targeted HAT1, and cellular localization showed that miR-486-5p and HAT1 were highly expressed in the cytoplasm. miR-486-5p overexpression led to a significant upregulation of TLR4 and a significant downregulation of HAT1. Inversely, miR-486-5p inhibition led to a significant downregulation of TLR4 and a significant upregulation of HAT1. HAT1 knockdown using siRNA significantly upregulated the expression of TLR4, IL-6, IL-8, TNF-α, and IFN-γ.
Conclusion: miR-486-5p was differentially expressed in the alveolar macrophages of COPD patients. miR-486-5p overexpression may enhance the TLR4-triggered inflammatory response in COPD patients by targeting HAT1.
Keywords: smoking, chronic obstructive pulmonary disease, miR-486-5p, toll-like receptor 4, histone acetyltransferase 1
Introduction
Subdermal Chronic obstructive pulmonary disease (COPD) is a chronic respiratory disorder associated with aging and smoking, and it has been a major public health problem owing to its high prevalence, morbidity, and mortality.1 Environment (especially smoking) and genetics are the two major factors responsible for COPD etiology. In Western countries, more than 90% of the COPD cases are caused by chronic smoking.2,3 The immunopathology of COPD is associated with innate and adaptive inflammatory immune responses to chronic smoking.4 The central feature of COPD is the infiltration of alveolar macrophages, neutrophils, and other inflammatory cells into the lung parenchyma and peripheral airways. Moreover, the predominant inflammatory cell types vary with COPD severity.4,5 Therefore, a better knowledge of inflammatory responses and the corresponding intracellular signaling pathways is essential for COPD drug development and clinical treatment.
Toll-like receptors (TLRs) are a class of evolutionarily conserved receptors of the innate immune system and are known to play crucial roles in host defense by triggering innate and adaptive immunity.6,7 TLRs contribute to the pathogenesis of inflammation by inducing inflammatory cytokines and other endogenous molecules in response to pathogenic microbial infection.8,9 TLR4 is a well-studied TLR family member, but its role in COPD immunopathology is not completely clear. Reportedly, TLR4 promotes airway neutrophilia in COPD,10 and tagging single nucleotide polymorphisms in TLR4 are associated with high numbers of sputum inflammatory cells and lung function decline.11 In a previous study, higher expression levels of TLR4 were observed in the bronchial mucosa of patients with severe COPD compared to those in the mucosa of healthy individuals, and TLR4 overexpression was positively correlated with CD4+/CD8+ cell infiltration and airflow obstruction.12 These results suggest that TLR4 plays an important role in COPD pathogenesis and progression.
MicroRNAs (miRNAs) are 21–24 nt long non-coding RNAs that regulate gene expression at the post-transcriptional level. Recent research has demonstrated the crucial regulatory roles of miRNAs in many diseases, including COPD.13,14 The upregulation of miR-34a has been shown to promote cellular senescence in small airway fibroblasts of COPD patients.15 Moreover, overexpression of miRNA-125a/b in COPD results in heightened airway inflammation by inducing A20, which mediates NF-κB activation, while inhibition of miRNA-125a/b decreases the induction of inflammatory cytokines.16 Here, we provide evidence that miR-486-5p mediates the TLR4-triggered inflammatory response of alveolar macrophages in COPD. We found that miR-486-5p was upregulated in alveolar macrophages and peripheral monocytes of COPD patients and smokers compared to that in the alveolar macrophages and peripheral monocytes of controls, and its expression was positively correlated with the expression levels of IL-6, IL-8, TNF-α, and IFN-γ. In addition, miR-486-5p regulated the TLR4-triggered inflammatory response by targeting histone acetyltransferase 1 (HAT1). Thus, our findings provide novel targets and a theoretical basis to further investigate the pathogenesis of COPD and may eventually contribute to the therapeutic management of COPD.
Materials and Methods
CSE
For the preparation of CSE, three cigarettes without filters (0.9 mg flue gas nicotine, 11 mg coke, and 14 mg flue gas carbon monoxide) were collected using a negative pressure suction device and poured into a flask with 3 mL phosphate-buffered saline. After shaking well, the suspension was filtered using a 0.22 µm membrane filter to remove the bacteria. To ensure a similar concentration of CSE in all the preparations, the absorbance value for each preparation was determined at the optimal absorption wavelength (270–280 nm). The CSE concentrate was diluted using 10% fetal bovine serum containing Roswell Park Memorial Institute-1640 culture medium to obtain 2%, 5%, and 10% CSE. Notably, CSE was prepared 30 min before use to ensure the effectiveness of the CSE ingredients.
Cell Culture and Treatment
The rat pulmonary alveolar macrophage cell line NR8383 was purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. and cultured in F12K medium supplemented with 15% fetal bovine serum at 37°C in a humidified mixture of air (95%) and CO2 (5%). Cells were plated in 6-well plates at a density of 1 × 106 cells per well and cultured in serum-free F12K medium overnight. Cells were treated with CSE for 24 and 48 h.
qPCR
Total RNA extraction was performed using Trizol reagent according to the manufacturer’s instructions. After determining the RNA concentration and quality, complementary DNA (cDNA) was synthesized using a First Strand cDNA Synthesis Kit (Sangon Biotech, China). qPCR was performed to validate gene expression using 2× SYBR Green PCR Master Mix (Sangon Biotech, China) on an Mx3000P QPCR System (Stratagene, USA) with the following thermal cycling conditions: 95°C for 3 min, followed by 40 cycles at 95°C for 12 s and 62°C for 40 s. The specific primer pairs used are shown in Supplemental Table 1.
Western Blotting
Total protein extraction was performed using the M-PER Mammalian Protein Extraction Reagent according to manufacturer’s instructions. Following this, proteins were separated using a 10% sodium dodecyl sulfate-polyacrylamide gel. The proteins were transferred onto polyvinylidene difluoride membranes (Millipore) using a semi-dry cell at 30 mA for 60 min, and then, 1×Ponceau S solution was used to dye and mark the protein marker spots. Blocking buffer (1×Tris buffered saline (TBS), 0.1% Tween-20 (TBST), and 5% w/v nonfat dry milk) was used to block nonspecific binding for 2 h at 37°C. After washing three times with 1×TBST, the membranes were incubated with anti-HAT1 (1:1000, SAB) and anti-TLR4 (1:1000, SAB) primary antibodies at 4°C overnight. The membrane was washed with 1×TBST five times, followed by incubation with HRP-secondary antibodies (1:2000, Jackson) for 2 h at 37°C. After washing five times with 1×TBST, the membrane was exposed to SuperSignal West Pico Chemiluminescent Substrates, and gray scanning was performed using a Gel-Pro Analyzer.
Luciferase Reporter Assays
NR8383 cells were seeded onto 96-well plates for 15–18 h of culture. Next, 5 pmol miR-486-5p mimic/negative control (NC) and 0.16 µg HAT1 3′ UTR/HAT1 3′ UTR-muta (Sangon Biotech, China) were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The medium was replaced with fresh medium after transfection for 6 h. Luciferase activity of the reporter vectors was determined after 48 h, using the Promega Dual-Luciferase system according to the manufacturer’s instructions.
ELISA
After CSE treatment, the cell supernatants were collected and analyzed using ELISA kits (Sangon Biotech, China) as per the manufacturer’s instructions.
miRNA Mimics/Inhibitors and Small Interfering RNA
miR-486-5p mimics, inhibitors, and the corresponding NCs were synthesized by Sangon Biotech (Shanghai) Co., Ltd. and transfected into cells using Lipofectamine 2000 (Invitrogen) to over-express or inhibit miR-486-5p in cells. The specific sequences of the miR-486-5p-mimics, inhibitors, and HAT-1 specific siRNA are shown in Supplemental Table 2.
FISH
Cells were seeded in a confocal Petri dish and cultured for 24 h. After fixing with 4% paraformaldehyde, the cells were impregnated with 50%, 80%, and 98% (mass ratio) ethanol for 3 min before dehydration. The fixed cells were prehybridized in hybridization buffer (formamide, 50 mM Tris-HCl, 5 mol NaCl, and 0.05% sodium dodecyl sulfate) based on a previously described method. The 5′ oligonucleotide-labeled miR-486-5p probe sequence was 5′-Cy3-CUCGGGGCAGCUCAGUACAGGA, and the 5′ oligonucleotide-labeled HAT1 probe sequence was 5′-FAM-UUCUCCACCGCACUCUUAUAUU. Next, DAPI was added in the dark at 4°C for 5 min before washing with 4°C phosphate-buffered saline buffer. Cells were observed under a fluorescence microscope with a 360 nm excitation wavelength and a 460 nm emission wavelength filter.
Clinical Sample Collection
The bronchoalveolar lavage fluid was collected from 14 healthy individuals and 36 COPD patients. Next, the PAM were isolated. Briefly, the bronchoalveolar lavage fluid was centrifuged at 4°C at 1500 rpm for 10 min. After discarding the supernatant, the cells were washed twice with Hanks’ solution. The cells were cultured in serum-free Roswell Park Memorial Institute medium-1640 at 37°C in an incubator with 5% CO2 and 100% humidity for 2–3 h. Non-adherent cells were removed such that the cells adhering to the culture dish wall were PAM. Peripheral blood was collected from 128 participants (nonsmokers: n=33; smokers: n=42; COPD I–II: n=30; COPD III–IV: n=23). Monocytes were isolated from peripheral blood, and monocyte-derived macrophages were induced. All participants were recruited between January 2019–1 solstice and June 30, 2019, at the respiratory department and physical examination center at the Affiliated Huai’an Hospital of Xuzhou Medical University. Written informed consent was obtained from the participants before sample collection. This study was conducted under the approval of the Ethics Committee of the Affiliated Huai’an Hospital of Xuzhou Medical University.
Microarray and Data Analysis
The microarray datasets GSE38974 and GSE53519 were downloaded from the GEO database. The miRNA expression data of lung tissue from 19 subjects with COPD and eight normal smokers without COPD in GSE38974 and the miRNA expression data of small airway epithelium from nine nonsmokers and ten smokers in GSE53519 were used in this study. Differential expression analysis was performed using the Bayesian method in the Limma package. The mRNA targets of miRNAs were predicted using miRTarBase41 (http://mirtarbase.mbc.nctu.edu.tw/php/index.php), TargetScan42 (http://www.targetscan.org/vert_72/), miRWalk 2.043 (http://mirwalk.umm.uni heidelberg. de/), and miRPathDB44 (https://mpd.bioinf.uni-sb.de/overview.html). The overlapped mRNAs were selected using Venny analysis.
Statistical Analysis
Data analyses were conducted using SPSS 19.0 and GraphPad Prism 7. Results are expressed as the mean ± standard deviation (SD). Student’s t-test was used to compare data between the two groups. Variance analysis was performed to compare multiple groups. One-way analysis of variance was used for homogeneity of variances, and the Lsd-q test was selected for pairwise comparison between groups. The Welch method was used when the variances were uneven, and Dunnett’s T3 method was used for pairwise comparison among multiple groups. Differences were considered statistically significant at P < 0.05.
Results
Elevated Levels of Inflammatory Cytokines with Cigarette Smoke (CSE) Extract Treatment
The mRNA expression levels of IL-6/-8, IFN-γ, and TNF-α were significantly higher in CSE-treated NR8383 cells compared to those in the controls (P < 0.05, Figure 1A). Consistently, enzyme-linked immunosorbent assay (ELISA) revealed increased concentrations of IL-6/-8, IFN-γ, and TNF-α in CSE-treated NR8383 cells compared to those in the controls (P < 0.05, Figure 1B).
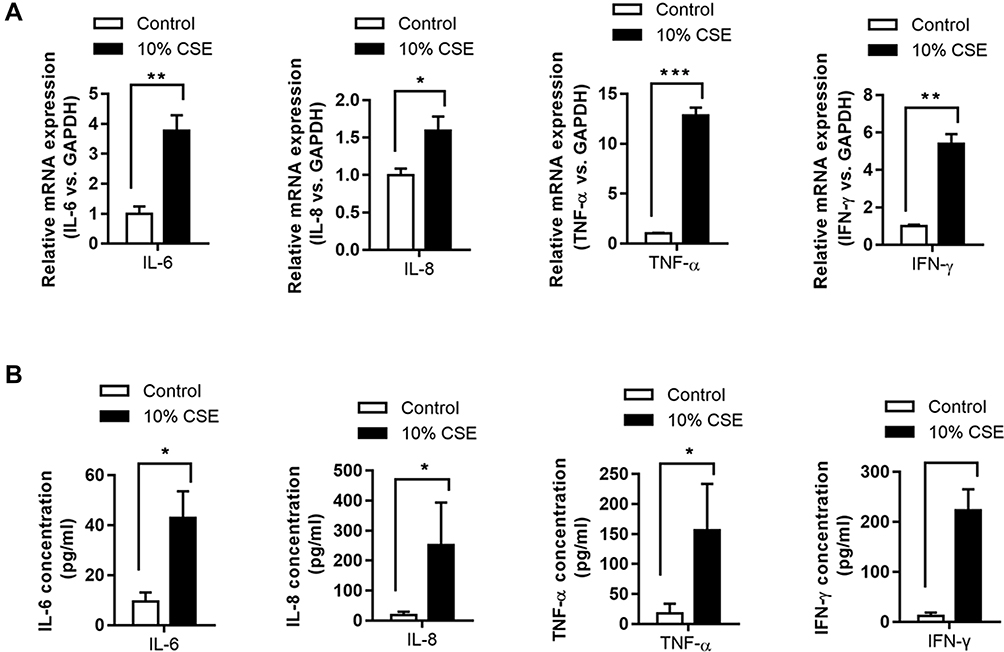 |
Figure 1 Levels of inflammatory cytokines in NR8383 cells treated with CSE. mRNA expression of IL-6, IL-8, TNF-α, and IFN-γ in 10% CSE-treated NR8383 cells was determined using qPCR (A). The levels of IL-6, IL-8, TNF-α, and IFN-γ in cell supernatants were determined using ELISA kits (B). *P < 0.05; **P < 0.01; ***P < 0.001. |
Upregulation of miR-486-5p in COPD
Based on the GSE38974 dataset, 17 differentially expressed miRNAs were screened between the normal and COPD groups, and miR-486-5p was found to be highly expressed in COPD samples compared to normal samples (Figure 2A). In addition, based on the data from GSE53519, 45 differentially expressed miRNAs were screened between smoker and nonsmoker groups. Notably, miR-486-5p was significantly upregulated in the small airway epithelium of smokers compared to that in the controls (Figure 2B). miR-486-5p expression was evaluated using qPCR in alveolar macrophages (PAM) and peripheral monocytes to confirm bioinformatic data. Consistently, higher levels of miR-486-5p were observed in the PAM and peripheral monocytes of COPD patients than those in the controls. Moreover, miR-486-5p was highly expressed in the peripheral monocytes of smokers (Figure 2C and D).
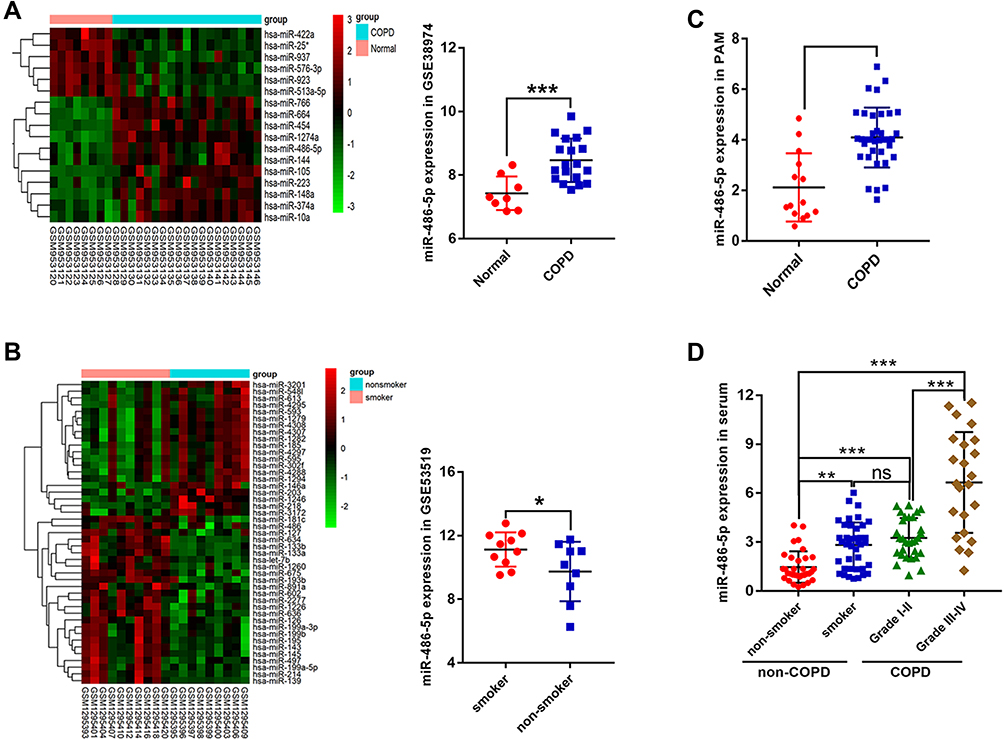 |
Figure 2 Screening and validation of differentially expressed miRNAs. Heat maps of differentially expressed miRNAs in COPD patients vs those in normal individuals, based on the GSE38974 dataset and miR-486-5p expression in the GSE38974 dataset (A). Heat maps of differentially expressed miRNAs in smokers vs those in nonsmokers, based on the GSE53519 dataset and miR-486-5p expression in the GSE53519 dataset (B). miR-486-5p expression in alveolar macrophages of COPD patients (n=36) and normal individuals (n=14), determined using qPCR (C). miR-486-5p expression in peripheral monocytes in smokers (n=42) vs that in the peripheral monocytes in nonsmokers (n=33) and in COPD I–II (n=30) vs COPD III–IV (n=23), determined using qPCR (D). *P < 0.05; **P < 0.01; ***P < 0.001. |
CSE Results in Elevated Levels of Inflammatory Cytokines via miR-486-5p
To determine the association between miR-486-5p expression and inflammatory cytokine levels in cells treated with CSE, miR-486-5p mimic and inhibitor were used to induce its overexpression and inhibition, respectively. Notably, overexpression of miR-486-5p in NR8383 cells led to significant upregulation of IL-6/-8, IFN-γ, and TNF-α, which was enhanced further with CSE treatment (Figure 3A). Moreover, inhibition of miR-486-5p in NR8383 cells resulted in a significant downregulation of IL-6/-8, IFN-γ, and TNF-α (Figure 3B). These results indicate that miR-486-5p plays a role in the regulation of inflammatory responses in COPD.
 |
Figure 3 Association between inflammatory cytokines and miR-486-5p expression. IL-6, IL-8, TNF-α, and IFN-γ levels in cell supernatants were determined using ELISA kits. (A) IL-6, IL-8, TNF-α, and IFN-γ levels in cells after transfection with the miR-486-5p mimic. (B) IL-6, IL-8, TNF-α, and IFN-γ levels in cells after transfection with the miR-486-5p inhibitor. *P < 0.05; **P < 0.01; ***P < 0.001. |
Identification of HAT1 as a Target of miR-486-5p
The targeted mRNAs of miR-486-5p were predicted using miRTarBase, TargetScan, miRWalk, and miRPathDB. In total, 11 mRNAs were predicted, including HAT1 (Figure 4A). Luciferase reporter assays and fluorescence in situ hybridization (FISH) assay were conducted to confirm the regulatory interaction between miR-486-5p and HAT1. As shown in Figure 4B, the luciferase activity of HAT1-WT was remarkably reduced (P < 0.05) after co-transfection with the miR-486-5p mimic, while the luciferase activity of the control and HAT1-MUT showed no significant difference (Figure 4B). This indicated that miR-486-5p could bind to the 3′-UTR region of HAT1 mRNA to inhibit HAT1 expression. Cellular localization showed that miR-486-5p and HAT1 were highly expressed in the cytoplasm. miR-486-5p expression was elevated, while HAT1 expression was decreased in the miR-486-5p mimic and mimic+CSE groups compared to their expression in the control group. miR-486-5p inhibition resulted in significant upregulation of HAT1 in the miR-486-5p inhibitor and inhibitor+CSE groups compared to that in the control group (Figure 4C).
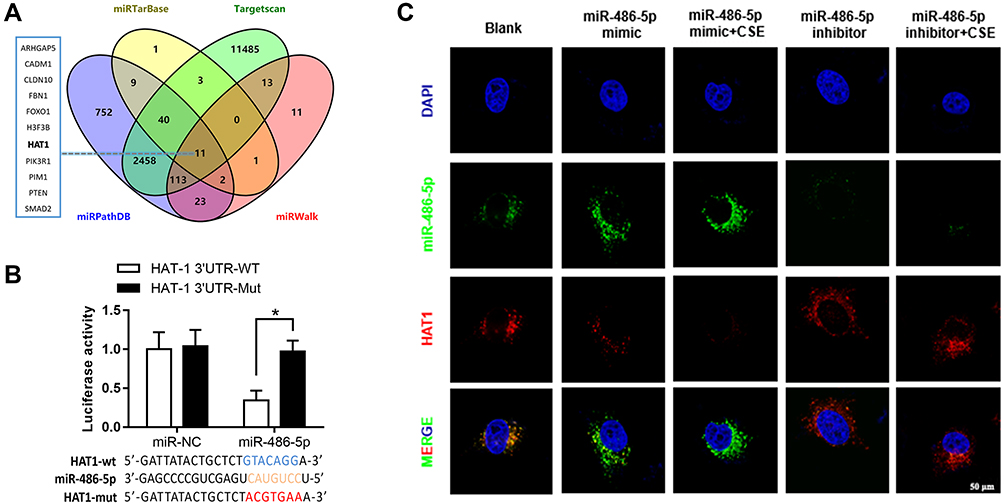 |
Figure 4 Regulatory interactions between miR-486-5p and HAT1. (A) Venn diagram of the predicted miR-486-5p targets in four databases. Results of the luciferase reporter assays (B) and fluorescence in situ hybridization (C) showing that miR-486-5p regulates the target HAT1. *P < 0.05. |
miR-486-5p Regulates TLR4 Expression by Targeting HAT1
Studies have reported that TLR4 can trigger a complex inflammatory response.17,18 Therefore, the interactions between miR-486-5p and TLR4 were further explored. miR-486-5p and TLR4 were highly expressed in NR8383 cells treated with CSE, while HAT1 expression decreased (Figure 5A). In addition, miR-486-5p overexpression in NR8383 cells led to an apparent increase in TLR4 expression and a significant downregulation of HAT1. Inversely, miR-486-5p inhibition in NR8383 cells resulted in decreased TLR4 expression and a significant upregulation of HAT1 (Figure 5B). We speculated that miR-486-5p regulates TLR4 expression by targeting HAT1 in COPD.
 |
Figure 5 Associations among miR-486-5p, HAT1, and TLR4 expression. (A) Expression levels of miR-486-5p, HAT1, and TLR4 in 2%, 5%, and 10% CSE-treated NR8383 cells, determined using qPCR and Western blotting. (B) HAT1 and TLR4 expression after the overexpression or inhibition of miR-486-5p. *P < 0.05; **P < 0.01; ***P < 0.001. |
HAT1 Negatively Regulates TLR4 and Inflammatory Cytokine Expression
HAT1 siRNAs were used to inhibit HAT1 expression to confirm the regulatory interaction between HAT1 and TLR4, and si-HAT1-697 showed a stronger knockdown effect (Figure 6A). Notably, HAT1 inhibition by si-HAT1-697 resulted in a significant upregulation of TLR4 compared to si-NC and control. In contrast, TLR4 expression was significantly decreased by HAT1 overexpression (Figure 6B). Moreover, the expression levels of IL-6/-8, IFN-γ, and TNF-α were significantly increased by inhibiting HAT1 using si-HAT1-697 (Figure 6C). These results suggest that miR-486-5p promotes COPD progression by regulating the TLR4-triggered inflammatory response of alveolar macrophages by targeting HAT1.
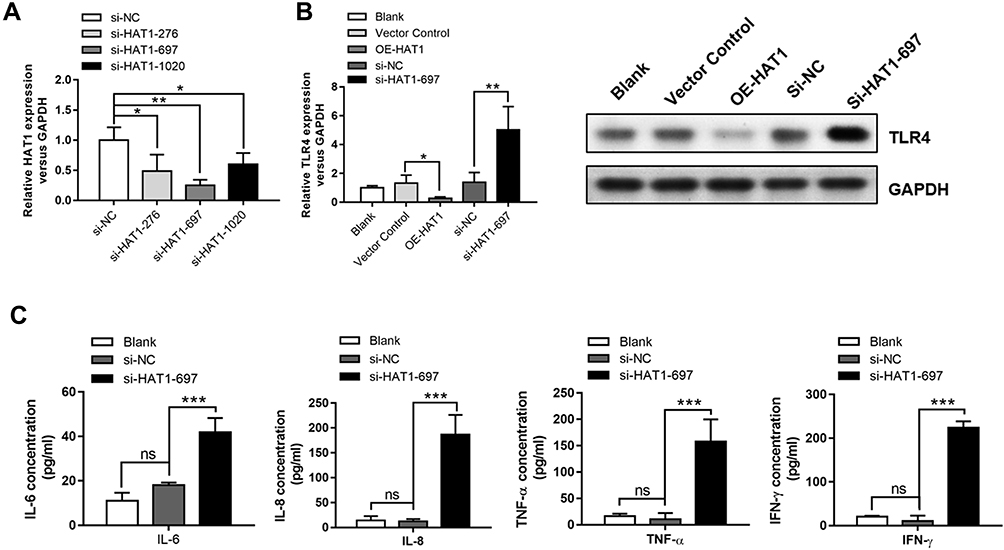 |
Figure 6 HAT1 negatively regulated TLR4 and inflammatory cytokine expression. (A) HAT1 expression in NR8383 cells after transfection with different siRNAs; the most significant inhibition of HAT1 expression was observed with si-HAT1 697. (B) The expression of HAT1 in NR8383 cells after transfection with si-HAT1 697 or overexpression of HAT1 using a vector. (C) IL-6, IL-8, TNF-α, and IFN-γ levels in NR8383 cells after transfection with si-HAT1 697. *P < 0.05; **P < 0.01; ***P < 0.001. |
Discussion
COPD is a multifactorial disorder characterized by nonreversible and progressive airflow obstruction with pulmonary dysfunction.19 Although oxidative stress, immunity, inflammation, apoptosis, and other factors have been implicated in COPD, its exact pathogenesis is still obscure.20,21 Smoking is one of the major causes of COPD. Investigation of the pathogenesis underlying smoking-related COPD is useful for early diagnosis and therapy. Studies have reported that some miRNAs can serve as important regulators in the molecular mechanisms of COPD.22,23 The identification of aberrant miRNA expression in COPD could help to better understand the underlying mechanisms. It has been reported that miRNAs are abnormally expressed in COPD patients and smokers compared to those in the normal patients. For instance, Paschalaki et al demonstrated that downregulation of miR-126 contributes to DNA damage in smokers and COPD patients.24 Conickx et al suggested that miR-218-5p expression is decreased in COPD patients and smokers without airflow limitation, and its expression levels show a significant relationship with airway obstruction.23 Here, miR-486-5p was aberrantly expressed in smokers and patients with COPD compared to that in nonsmokers without COPD, as demonstrated by bioinformatics analysis on a microarray dataset. This was consistent with miR-486-5p expression in the PAM and peripheral monocytes of COPD patients and smokers. This suggests that miR-486-5p is involved in COPD progression.
miR-486 is an intragenic miRNA located within Ankyrin 1.25 Studies have reported that aberrant miR-486-5p expression can affect the occurrence and progression of many diseases.26,27 Notably, the effect of miR-486-5p is controversial because it has been found to serve as a suppressor or oncogene.28,29 In addition, we observed that the function of miR-486-5p had not been explored in COPD. Our results revealed that miR-486-5p expression was significantly correlated with the levels of IL-6/-8, IFN-γ, and TNF-α. Therefore, we suggest that miR-486-5p may play a role in mediating the inflammatory response. Chai et al reported that increased miR-486-5p levels repress the lipopolysaccharide-induced expression of inflammatory cytokines such as IL-1β, IL-6, and TNF-α.30 TLRs play a role in inflammatory pathogenesis by inducing inflammatory cytokines and other endogenous molecules against pathogenic microbial infection.8,9 Recently, miRNAs have been found to play crucial regulatory roles in TLR signaling. For example, Shen et al revealed that miR-149-3p affects the expression of IL-1β and TNF-α by regulating the TLR-4/NF-κB signaling pathways in COPD patients.31 Lai et al suggested that miR-92a regulates the TLR4-triggered inflammatory response in macrophages via JNK/c-Jun signaling activated by mitogen-activated protein kinase 4.32 However, the associations between miR-486-5p and TLR4 have not been reported. Based on our results, we concluded that miR-486-5p regulates TLR4 expression by targeting HAT1.
Here, the results of bioinformatics analysis showed that HAT1 was a target of miR-486-5p. The luciferase reporter assay confirmed the finding that miR-486 could directly bind to the 3′ UTR of HAT1 to regulate HAT1 expression. Consistently, Liu et al also found that miR-486 can directly target HAT1 to mediate cholesterol efflux in macrophages.33 Additionally, with si-HAT1knockdown and HAT1 overexpression experiments, we found that HAT1 negatively regulated the expression of TLR4 and inflammatory cytokines. In recent years, several researchers have focused on the effect of epigenetics in COPD treatment. It has been proposed that epigenetic mechanisms are involved in COPD pathogenesis, and this is a promising therapeutic approach based on epigenetic mark-targeting.34–36 Histones are highly conserved intra-nuclear alkaline proteins, and their core modifications can affect transcription, DNA replication, and other cellular processes.37,38 HAT1 is a type B histone acetyltransferase associated with the acetylation of newly generated histones.39 Han et al reported that HAT1 overexpression promotes lung cancer cell apoptosis by regulating the expression of proteinase-activated receptor 2 and Fas.40 These data indicate that the regulatory interaction between HAT1 and TLR4 may be associated with acetylation.
Conclusion
Our results suggest that miRNAs, such as miR-486-5p, are differentially expressed in the alveolar macrophages of patients with COPD. Overexpression of miR-486-5p may increase the TLR4-triggered inflammatory response in COPD patients by targeting HAT1. Our findings provide novel targets and a theoretical basis to further investigate COPD pathogenesis, which is crucial for COPD clinical treatment.
Data Sharing Statement
The data used to support the findings of this study are available within the article and the Supplementary Materials.
Ethics Approval and Consent to Participate
This study was conducted under the approval of the Ethics Committee of the Affiliated Huai’an Hospital of Xuzhou Medical University. All enrolled patients signed the informed consent form.
Acknowledgments
We thank Prof. Song Chen from the Jiangsu College of Nursing for assistance with the experiments.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by grants from Jiangsu Province’s Key Talents Training Program of Youth Medicine (QNRC2016426).
Disclosure
The authors declare that they have no competing interests.
References
1. López‐Campos JL, Tan W, Soriano JB. Global burden of COPD. Respirology. 2016;21(1):14–23. doi:10.1111/resp.12660
2. Tan WC, Bourbeau J, Aaron SD, et al. Global initiative for chronic obstructive lung disease 2017 classification and lung function decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(5):670–673. doi:10.1164/rccm.201706-1154LE
3. Jindal SK. Chronic obstructive pulmonary disease in non-smokers-is it a different phenotype? Indian J Med Res. 2018;147(4):337. doi:10.4103/ijmr.IJMR_10_18
4. Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38(4):497–515. doi:10.1007/s00281-016-0561-5
5. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
6. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373.
7. Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388(4):621–625. doi:10.1016/j.bbrc.2009.08.062
8. Drexler SK, Foxwell BM. The role of toll-like receptors in chronic inflammation. Int J Biochem Cell Biol. 2010;42(4):506–518. doi:10.1016/j.biocel.2009.10.009
9. Achek A, Yesudhas D, Choi S. Toll-like receptors: promising therapeutic targets for inflammatory diseases. Arch Pharm Res. 2016;39(8):1032–1049. doi:10.1007/s12272-016-0806-9
10. Pace E, Giarratano A, Ferraro M, et al. TLR4 upregulation underpins airway neutrophilia in smokers with chronic obstructive pulmonary disease and acute respiratory failure. Hum Immunol. 2011;72(1):54–62. doi:10.1016/j.humimm.2010.09.009
11. Budulac SE, Boezen HM, Hiemstra PS, et al. Toll-like receptor (TLR2 and TLR4) polymorphisms and chronic obstructive pulmonary disease. PLoS One. 2012;7(8):e43124–e24. doi:10.1371/journal.pone.0043124
12. Di Stefano A, Ricciardolo FL, Caramori G, et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur Respir J. 2017;49(5):1602006. doi:10.1183/13993003.02006-2016
13. Sauler M, Nouws J, Feng W, Lee P. MicroRNA regulation of cell fate in the pathogenesis of COPD. In: C63. MECHANISTIC STUDIES in COPD. American Thoracic Society; 2019:A5375–A75.
14. Tasena H, Faiz A, Timens W, et al. microRNA–mRNA regulatory networks underlying chronic mucus hypersecretion in COPD. Eur Respir J. 2018;52(3):1701556. doi:10.1183/13993003.01556-2017
15. Wrench C, Baker J, Fenwick P, Donnelly L, Barnes P. MicroRNA-34a drives small airway fibroblast cellular senescence in COPD. Eur Respir J. 2017;50(suppl61):OA289. doi:10.1183/1393003.congress-2017
16. Hsu AC, Dua K, Starkey MR, et al. MicroRNA-125a and-b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight. 2017;2(7). doi:10.1172/jci.insight.90443.
17. He W, Rebello O, Savino R, et al. TLR4 triggered complex inflammation in human pancreatic islets. Biochim Biophys Acta. 2019;1865(1):86–97. doi:10.1016/j.bbadis.2018.09.030
18. Liu C, Tang X, Zhang W, et al. 6-Bromoindirubin-3′-oxime suppresses LPS-induced inflammation via inhibition of the TLR4/NF-κB and TLR4/MAPK signaling pathways. Inflammation. 2019;42(6):2192–2204. doi:10.1007/s10753-019-01083-1
19. Hogg JC, Paré PD, Hackett T-L. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol Rev. 2017;97(2):529–552.
20. Tuder RM. Bringing light to chronic obstructive pulmonary disease pathogenesis and resilience. Ann Am Thorac Soc. 2018;15(Supplement 4):S227–S33. doi:10.1513/AnnalsATS.201808-583MG
21. Boucherat O, Morissette MC, Provencher S, Bonnet S, Maltais F. Bridging lung development with chronic obstructive pulmonary disease. Relevance of developmental pathways in chronic obstructive pulmonary disease pathogenesis. Am J Respir Crit Care Med. 2016;193(4):362–375. doi:10.1164/rccm.201508-1518PP
22. Huang X, Zhu Z, Guo X, Kong X. The roles of microRNAs in the pathogenesis of chronic obstructive pulmonary disease. Int Immunopharmacol. 2019;67:335–347. doi:10.1016/j.intimp.2018.12.013
23. Conickx G, Mestdagh P, Avila Cobos F, et al. MicroRNA profiling reveals a role for microRNA-218-5p in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195(1):43–56. doi:10.1164/rccm.201506-1182OC
24. Paschalaki KE, Zampetaki A, Baker JR, et al. Downregulation of microRNA-126 augments DNA damage response in cigarette smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(5):665–668. doi:10.1164/rccm.201706-1304LE
25. Oh HK, Tan AL-K, Das K, et al. Genomic loss of miR-486 regulates tumor progression and the OLFM4 antiapoptotic factor in gastric cancer. Clin Cancer Res. 2011;17(9):2657–2667. doi:10.1158/1078-0432.CCR-10-3152
26. Fu SJ, Chen J, Ji F, et al. MiR-486-5p negatively regulates oncogenic NEK2 in hepatocellular carcinoma. Oncotarget. 2017;8(32):52948–52959.
27. Shi L, Liu S, Zhao W, Shi J. miR-483-5p and miR-486-5p are down-regulated in cumulus cells of metaphase II oocytes from women with polycystic ovary syndrome. Reprod Biomed Online. 2015;31(4):565–572. doi:10.1016/j.rbmo.2015.06.023
28. Shao Y, Shen YQ, Li YL, Liang C, Ma Z-L. Direct repression of the oncogene CDK4 by the tumor suppressor miR-486-5p in non-small cell lung cancer. Oncotarget. 2016;7(23):34011–34021. doi:10.18632/oncotarget.8514
29. Gao ZJ, Yuan W-D, Yuan J-Q, Yuan K, Wang Y. miR-486-5p functions as an oncogene by targeting PTEN in non-small cell lung cancer. Pathol Res Pract. 2018;214(5):700–5.
30. Xingyu C. miR-486-5p inhibits inflammatory response, matrix degradation and apoptosis of nucleus pulposus cells through directly targeting FOXO1 in intervertebral disc degeneration. Cell Physiol Biochem. 2019;52:109–118.
31. Shen W, Liu J, Zhao G, et al. Repression of toll-like receptor-4 by microRNA-149-3p is associated with smoking-related COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:705–715. doi:10.2147/COPD.S128031
32. Lai L, Song Y, Liu Y, et al. MicroRNA-92a negatively regulates toll-like receptor (TLR)-triggered inflammatory response in macrophages by targeting MKK4 kinase. J Biol Chem. 2013;288(11):7956–7967. doi:10.1074/jbc.M112.445429
33. Liu D, Zhang M, Xie W, et al. MiR-486 regulates cholesterol efflux by targeting HAT1. Biochem Biophys Res Commun. 2016;472(3):418–424. doi:10.1016/j.bbrc.2015.11.128
34. Schamberger AC, Mise N, Meiners S, Eickelberg O. Epigenetic mechanisms in COPD: implications for pathogenesis and drug discovery. Expert Opin Drug Discov. 2014;9(6):609–628. doi:10.1517/17460441.2014.913020
35. Wu -D-D, Song J, Bartel S, Krauss-Etschmann S, Rots MG, Hylkema MN. The potential for targeted rewriting of epigenetic marks in COPD as a new therapeutic approach. Pharmacol Ther. 2018;182:1–14. doi:10.1016/j.pharmthera.2017.08.007
36. Adcock IM. Role of epigenetic modifications in pathology of COPD. Tanaffos. 2017;16(Suppl 1):S2.
37. Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 2016;32(1):42–56. doi:10.1016/j.tig.2015.10.007
38. Stillman B. Histone modifications: insights into their influence on gene expression. Cell. 2018;175(1):6–9. doi:10.1016/j.cell.2018.08.032
39. Parthun M. Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene. 2007;26(37):5319–5328.
40. Han N, Shi L, Guo Q, et al. HAT1 induces lung cancer cell apoptosis via up regulating fas. Oncotarget. 2017;8(52):89970. doi:10.18632/oncotarget.21205
41. Chou C-H, Shrestha S, Yang C-D, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–D302. doi:10.1093/nar/gkx1067
42. Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi:10.7554/eLife.05005
43. Dweep H, Gretz N. miRWalk2. 0: a comprehensive atlas of microRNA-target interactions. Nat Methods. 2015;12(8):697.
44. Backes C, Kehl T, Stöckel D, et al. miRPathDB: a new dictionary on microRNAs and target pathways. Nucleic Acids Res. 2016;gkw926.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.