Back to Journals » OncoTargets and Therapy » Volume 13
miR-602 Mediates the RASSF1A/JNK Pathway, Thereby Promoting Postoperative Recurrence in Nude Mice with Liver Cancer
Authors Zhou C, Huang Y ![]() , Chen Y
, Chen Y ![]() , Xie Y
, Xie Y ![]() , Wen H, Tan W, Wang C
, Wen H, Tan W, Wang C ![]()
Received 24 December 2019
Accepted for publication 19 June 2020
Published 10 July 2020 Volume 2020:13 Pages 6767—6776
DOI https://doi.org/10.2147/OTT.S243651
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Cheng Zhou,1,2 Yajing Huang,3 Yongxu Chen,1,2 Yingjie Xie,4 Huihong Wen,3 Wei Tan,2 Changjun Wang1,2
1School of Medicine, South China University of Technology, Guangzhou, People’s Republic of China; 2Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangdong Geriatric Institute, Guangzhou, People’s Republic of China; 3Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 4School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, People’s Republic of China
Correspondence: Changjun Wang
Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangdong Geriatric Institute, No. 106, Zhongshan Second Road, Guangzhou, People’s Republic of China
Tel +86 20-83827812-70521
Email [email protected]
Purpose: At present, there are few studies on the mechanisms underlying postoperative recurrence of liver cancer, and the mechanism of action of miR-602 in postoperative recurrence of liver tumors is not clear. Our goals were to investigate the effects of miR-602 on the expression of the Ras-associated domain family 1A (RASSF1A) gene and the regulation of primary and recurrent hepatic tumors to clarify the molecular mechanisms of miR-602 in postoperative hepatocellular carcinoma.
Methods: We constructed a mouse liver orthotopic tumor model and a mouse liver recurrent tumor model. We measured the expression levels of the RASSF1A gene and then analyzed the effects of miR-602 on the regulation of RASSF1A. We transiently transfected the miR-602 gene into cells that stably overexpressed RASSF1A and examined relevant indicators to elucidate the mechanisms by which miR-602 regulates the RASSF1A/c-Jun N-terminal kinase (JNK) pathway in recurrence and dormancy in liver cancer.
Results: RASSF1A expression was inversely related to that of JNK, activating transcription factor 2 (ATF-2), and c-Jun in SMMC7721 cells stably transfected with the RASSF1A gene and in recurrent mouse tumor tissues. After transient transfection of cells with miR-602 mimic or miR-602 inhibitor, the expression of miR-602 was inversely related to that of RASSF1A.
Conclusion: MiR-602 might inhibit the JNK signaling pathway by inhibiting the expression of RASSF1A, thereby promoting recurrence of liver cancer after surgery. The low expression levels of miR-602 in liver cancer tissues were closely related to postoperative recurrence; they could be used as a marker to judge the prognosis of patients with liver cancer.
Keywords: miR-602, JNK, RASSF1A, liver cancer, postoperative recurrence
Introduction
Hepatocellular carcinoma (HCC) is characterized by frequent recurrence despite the initial success of curative treatments, such as surgical resection and local ablation therapy, with recurrence rates 5 years after treatment ranging from 73% to 100%.1 Thus, for a patient with dormant disseminated cancer, the prevention of overt metastasis and recurrence is widely seen as a necessary component of future therapies.2 Notably, greater than 67% of deaths in patients with breast cancer occur after the 5-year survival mark, showing that residual disease can be dormant for very long periods.3 This further argues that research on dormant disseminated tumor cells is crucial.
Dormancy is a reversible state of cellular quiescence that persists over prolonged periods of time. Dormant cancer cells often survive treatment and increase the risk of tumor relapse, which is associated with dismal prognosis.4,5 Dormancy of tumor stem cells is especially undesirable; if they exit dormancy, they can repopulate the tumor, analogous to how normal adult stem cells can activate and regenerate tissue.6 Unfortunately, compared with other areas of cancer research, our understanding of the biology of dormant residual tumor cells is highly limited. Clinical management of dormant disease will be a reality only after we achieve rigorous understanding of its molecular and cellular basis.7
Micro-RNAs (miRNAs) are a class of small-molecule, non-coding, single-stranded RNAs that mediate inhibition of messenger RNA (mRNA) translation by complementarily binding to mRNA and activating RNA interference, thereby participating in the regulation of gene expression at posttranscriptional levels.8 Many studies have confirmed that miRNAs are involved in the development and progression of liver cancer, and directly affect the proliferation, apoptosis, invasion, and metastasis of liver cancer cells by targeting a large number of key protein coding genes, such as IL-6, hepatocyte growth factor and c-Met.9 Some researchers used miRNA microarray technology to analyze the miRNA expression profiles in radically resected tissues of patients with liver cancer, and selected a group of differentially expressed miRNAs closely related to the metastasis and recurrence of liver cancer.10 Further studies found that miR-219-5p promotes the growth and metastasis of liver cancer by regulating E-cadherin.11 Liu et al found that overexpression of miR-602 and Forkhead box protein K2 forms a negative feedback loop to promote proliferation and metastasis of esophageal squamous cell carcinoma.12 Wang et al found that the occurrence and progression of hepatocellular carcinoma are related to the expression levels of miR-602.13 Furthermore, miR-602 can be considered as a molecular marker for hepatocellular carcinoma for clinical detection.
The tumor suppressor gene Ras-associated domain family 1A (RASSF1A) has many physiological functions, such as regulating the cell cycle and microtubule stability and controlling apoptosis.14,15 It plays important roles in tumor occurrence and development. MiR-602 is a special oncogene in liver cancer.16 It is currently believed to have carcinogenic effects by inhibiting the anti-cancer effects of RASSF1A, but the specific mechanisms are still unclear.17 Studies have shown that universal inactivation of RASSF1A in liver cancer is necessary for progression of the disease; therefore, severe RASSF1A deficiency can promote metastasis and recurrence of dormant liver cancer.18,19
The JNK family is a member of the mitogen-activated protein kinase (MAPK, also known as stress-activated protein kinase) superfamily. The JNK signaling pathway, which can be activated by various factors such as cytokines and stress, plays a vital role in cellular processes such as proliferation, differentiation, and apoptosis.20 Numerous experiments have confirmed that JNK is closely related to a variety of malignant tumors, and the JNK signaling pathway can be used as a molecular therapeutic target for a variety of malignant tumors.21–23
At present, there are few studies on the relationship between RASSF1A and the JNK signaling pathway. Whang et al found that RASSF1A inhibits the growth of lung cancer cells by reducing JNK phosphorylation.24 Therefore, in this study, we propose the hypothesis that the expression of miR-602 could affect hepatoma cell apoptosis by inhibiting the RASSF1A-mediated JNK signaling pathway, thereby affecting postoperative dormancy and recurrence of liver cancer.
Materials and Methods
Cell Culture
The human HCC cell line SMMC7721 was kindly donated by the Tropical Medicine Institute of the Guangzhou University of Chinese Medicine. The cell line was approved by the ethics committee of the Research Ethics committee Guangdong Provincial People’s Hospital. The SMMC7721 cells were authenticated by STR profile. All of the cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and 100 U/mL each of penicillin and streptomycin (both Gibco) at 37°C with a CO2 volume fraction of 5%; medium was routinely changed. When the cells had grown to 70%–80% confluence, they were digested with 0.25% trypsin (Gibco) and passaged. Cells in the logarithmic-growth phase were used for subsequent experiments.
Construction of Stably Transfected RASSF1A Cells
Virus Transduction
SMMC7721 cells were seeded into six-well plates (Corning, Inc., Corning, New York, USA) at a density of 3 × 105/well in 2 mL of complete medium and incubated for 24 h. The lentivirus was transduced when cells were 30%–50% confluent. The lentivirus was placed on ice to dissolve it, and the optimal multiplicity of infection (MOI) was determined based on the virus titer and the cell volume of each virus. Polybrene was added to each well (final concentration, 5 μg/mL) to improve transduction efficiency. The plates were incubated overnight, and the next day the state of the cells was observed and the medium was refreshed. After 48 h of transduction, we observed the transfection effects under a fluorescence microscope (ZEISS Primovert, Oberkochen, Baden-Württemberg, Germany) and acquired photographs.
Cell Purification
Transduced and untransduced cells were cultured in complete medium containing 1.6 μg/mL puromycin. The drug screening time was 4 days and the MOI was 30. When the cells of the blank group were substantially killed, we replaced the medium of the stably transfected cells with complete culture medium and continued culturing the cells. Then we verified the transduction was successful and stored the cells by cryopreservation.
In vivo Tumorigenicity Experiments
Nude mice (4–6 weeks) were purchased from the Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China, and maintained in a specific pathogen-free environment. All mice were treated according to the care guidelines of the Laboratory Animal Center of the First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China. We injected control SMMC7721 cells and SMMC7721 cells stably overexpressing RASSF1A into mouse livers at 2 × 107 cells/injection site. Both groups contained five mice. After 2 weeks, the mice were anesthetized with 2% urethane (provided by the Laboratory Animal Center) and the tumors were observed by incising the abdominal cavity. At that time, the tumor tissue was surgically removed using medical hemostatic glue (Beijing Compont Medical Devices Co., Ltd., Beijing, China) to stop the liver wound from bleeding. The mouse abdomens were sutured and the specimens were frozen in a −80°C freezer. On the third day after surgery, we cut 2 mm of length from the mice’s tails. This procedure was repeated every 3 days, for three consecutive times; this trauma was induced to stimulate tumor recurrence. This method has been proven to be effective by repeated experimental studies by colleagues of our research group. The traumatic stimulation can activate tumor cells.25 After 2 weeks, the mice were placed under general anesthesia, and the abdominal cavity was opened to observe tumor recurrence and surgically remove recurrent tumor tissue. We again used hemostatic glue to stanch the bleeding. The mice were sacrificed by excessive anesthesia.
Transient Transfection of miR-602 into Cells
One day before transfection, SMMC7721 cells stably overexpressing RASSF1A were seeded in a six-well plate (2 × 105/well). When the confluence reached 70%–80%, the transfection reagent mixture was prepared following the instructions provided by Ruibo Bio-Technology Co., Ltd. (Guangzhou, China). Then the mature miRNA sequence (hsa-miR-602 MIMAT0003270, Homo sapiens miR-602, GACACGGGCGACAGCUGCGGCCC), miR-602 mimic, miR-602 inhibitor, and negative control (NC) were transfected into the cells. We divided the cells into four groups: mimic, mimic NC, inhibitor and inhibitor NC. After 48 h of transfection, we measured miR-602 levels in the four groups by quantitative reverse-transcription PCR (RT-qPCR) to evaluate transfection efficiency, and the cells were collected for subsequent analysis.
Reverse-Transcription Quantitative PCR
Total RNA was extracted from each cell using an RNA extraction kit (Beyotime Institute of Technology, Ltd., Shanghai, China) following the manufacturer’s protocol. Total RNA was extracted from primary and recurrent tumor specimens with TRIzol Reagent (Saiguo Biotechnology Co. Ltd., Guangzhou, China). The concentration of total RNA was measured using a NanoDrop2000 (Thermo Fisher) and the mRNA expression levels were determined via RT-qPCR using SYBR Green Master Mix (TsingKe Biological Technology Co., Ltd., Guangzhou, China). Results were normalized to the expression of U6. Primer sequences used in RT-qPCR are shown in Table 1.
 |
Table 1 Primer Sequences Used for qPCR |
Western Blot of Cells and Tumor Specimens
Briefly, proteins were extracted from cells and mouse tumor specimens using RIPA lysis buffer (Beyotime) with the protease inhibitor PMSF (Beyotime), separated by SDS-PAGE, and transferred to polyvinylidene fluoride membranes (MilliporeSigma, Burlington, Massachusetts, US). Membranes were blocked with 5% milk for 120 min at room temperature (RT) and incubated with primary antibodies overnight at 4°C. Rabbit anti-mouse β-actin, rabbit anti-mouse c-JUN, rabbit anti-mouse ATF-2, rabbit anti-mouse p-JNK, rabbit anti-mouse p-c-JUN, and rabbit anti-mouse JNK were purchased from Cell Signaling Technology (Danvers, MA, USA), and rabbit anti-mouse RASSF1A was purchased from Abcam (Cambridge, UK). All of the primary antibodies were diluted 1:1000. Membranes were washed three times using Tris-buffered saline with Tween for 10 min each time and incubated with a horseradish peroxidase conjugated secondary antibody (1:1500; Cell Signaling Technology) for 1 h at RT. Protein bands were detected using an Amersham Imager 600 (GE Healthcare Life Sciences, Marlborough, Massachusetts, US) and signal intensities were normalized against β-actin.
CCK-8 Assay for Cell Proliferation
SMMC7721 cells stably overexpressing RASSF1A were seeded in a 96-well plate (1×105/well) with 100 μL of growth medium. Mimic, inhibitor, and corresponding NC transfection mixes were added following Ruibo Bio-Technology’s instructions (five wells per group). Then the plates were pre-incubated in an incubator for 24 h. After adding 10 μL of CCK-8 solution (Kaiji Biotechnology Co. Ltd., Jiangsu, China) to each well, the plates were incubated for 2 h. Absorbance at 450 nm was measured with a microplate reader (Varioskan, Thermo Fisher). A blank control was used to determine the proliferation of cells in each well.
Cell viability was calculated as follows:
[(experimental well − blank well)/(control well − blank well)] × 100%.
The cell inhibition rate was calculated as follows:
[(control well − experimental well)/(control well − blank well)] × 100%.
Hematoxylin and Eosin Staining of Tumor Tissue
At 24 h postoperation, primary and recurrent liver tumor specimens were collected for hematoxylin and eosin (H&E) staining. Specimens were fixed in 4% paraformaldehyde (Reagan, Beijing, China) for 12 h and embedded in paraffin (Leica, Wetzlar, Germany). The paraffin-embedded liver specimens were sectioned into 5 μm slices, dewaxed, hydrated, and stained with H&E (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) for morphological evaluation. Each slice was observed under 40×, 100×, and 400× microscopic magnification.
Statistical Analysis
Results were statistically evaluated using the two-tailed, unpaired Student’s t-test. Results are expressed as the mean ± standard deviation. P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001). All of the statistical analyses were performed using SAS software version 9.2 (SAS Institute, Cary, North Carolina, USA).
Results
Construction of SMMC7721 Cell Line Stably Overexpressing RASSF1A
The transfection efficiency of the cells in each experimental group was good, as indicated by the fluorescence rate (>80%) (Figure 1). Expression changes of the target gene were calculated using the 2−ΔΔCt method. The expression levels of RASSF1A in the overexpression group, as quantified by RT-qPCR, were 41 times as high as in the control group.
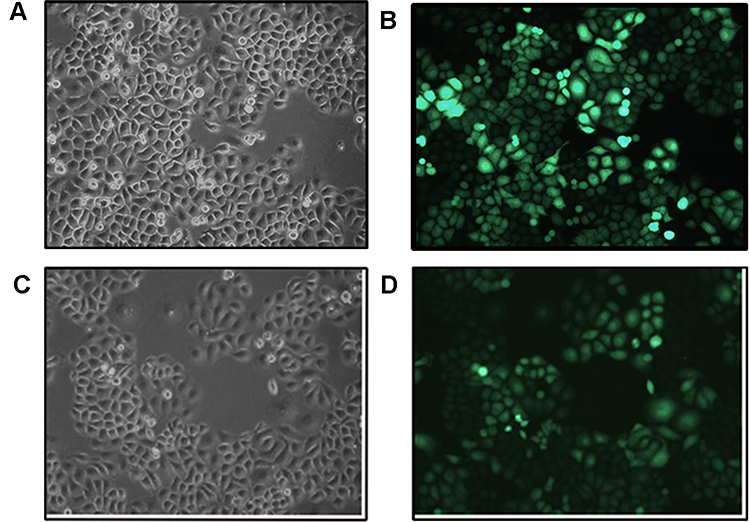 |
Figure 1 Fluorescence photographs of SMMC7721 cells stably overexpressing RASSF1A. (A, B) Control group. (C, D) Overexpression group. |
In vivo Experiments to Establish a Mouse Model of Liver Cancer
We injected normal SMMC7721 cells and SMMC7721 cells stably overexpressing RASSF1A into the livers of mice (2×107 cells). One of the mice injected with SMMC7721 cells stably overexpressing RASSF1A failed to form a tumor. After 7 days, we anesthetized the mice and observed the tumors by incising the abdominal cavity (Figure 2). Mouse tumors stably overexpressing RASSF1A were smaller than those not expressing RASSF1A, indicating that RASSF1A inhibits tumor growth.
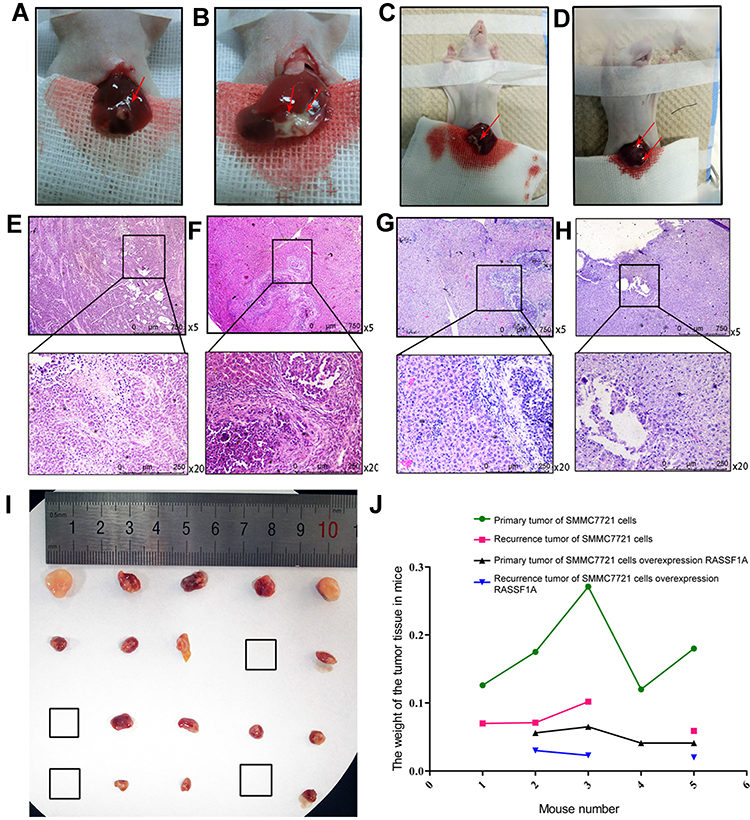 |
Figure 2 In vivo experiments to establish a mouse model of liver cancer. (A, E) Primary tumor: injection of SMMC7721 cells stably overexpressing RASSF1A. (B, F) Primary tumor: injection of control SMMC7721 cells. (C, G) Recurrent tumor: injection of SMMC7721 cells stably overexpressing RASSF1A. (D, H) Recurrent tumor: injection of control SMMC7721 cells. In the A-D, the red arrow points to the tumor tissue. (I) A picture of a specimen of mouse tumor tissue. The first row shows the primary tumor tissue obtained by implanting mice with control SMMC7721 cells. The second row shows the primary tumor tissue obtained by injecting mice with SMMC7721 cells stably overexpressing RASSF1A. The third and fourth rows are the corresponding recurrent tumor tissues. (J) Tumor weights (g). |
We surgically removed the tumor tissue (Figure 2) and used hemostatic glue to stop liver bleeding. On the third day after surgery, we shortened the mice’s tails by 2 mm every 3 days for three times, inducing trauma to stimulate tumor recurrence. After 2 weeks, we placed the mice under general anesthesia, reopened the abdominal cavity, and observed tumor recurrence (Figure 2). One mouse in each group did not form a recurrent tumor. The recurrent tumors in the control group spread to half of the livers and were bulky. The recurrent tumor tissues of mice stably expressing the RASSF1A gene were confined to the lower right liver, and the tumors were small. Tumor weights are presented in Figure 2.
We next performed H&E staining on tumor tissue samples. RASSF1A had an inhibitory effect on tumor growth (Figure 2).
Relationship Among RASSF1A, JNK, and Downstream Signals in vitro
Protein levels of RASSF1A were significantly higher in SMMC7721 cells stably overexpressing RASSF1A than in control cells, as shown by Western blot analysis. Moreover, phosphorylated JNK (p-JNK) and JNK levels were lower than in the control group, and the levels of downstream phosphorylated c-JUN (p-c-JUN), c-JUN, and ATF-2 were also reduced. The high expression levels of RASSF1A inhibited JNK (Figure 3A and B). We measured RASSF1A mRNA levels in RASSF1A overexpressing and control SMMC7721 cells by RT-qPCR (Figure 3C). High expression levels of RASSF1A inhibited phosphorylation of JNK, thereby inhibiting downstream signaling pathways, including c-JUN and ATF-2, and tumor cell growth.
 |
Figure 3 Relationship among RASSF1A, JNK, and downstream signals in vitro. (A, B) RASSF1A, p-JNK, JNK, p-c-JUN, c-JUN, and ATF-2 protein levels were measured in control SMMC7721 cells and SMMC7721 cells stably overexpressing RASSF1A by Western blot. (C) Relative expression of related markers was measured by RT-qPCR. mRNA levels of miR-602, RASSF1A, and JNK were normalized to the expression of U6 (**P < 0.01, ***P < 0.001). |
Relationship Among RASSF1A, JNK, and Downstream Signals in vivo
Proteins and total RNA were extracted from tumor specimens. In the murine primary tumor specimens, the expression levels of RASSF1A in tumors stably overexpressing RASSF1A were higher than in control tumors. p-JNK and JNK levels were lower in RASSF1A overexpressing tumors than in control tumors, and ATF-2, p-c-JUN, and c-JUN levels were consistent, indicating that RASSF1A exerted a tumor-suppressive effect by inhibiting JNK phosphorylation (Figure 4A and B).
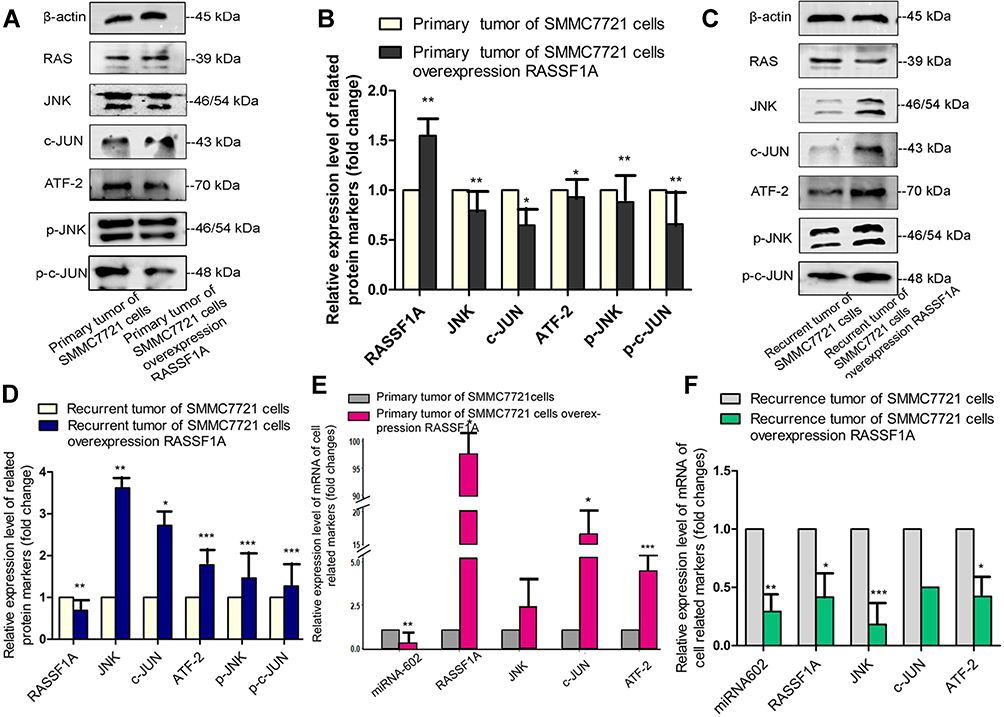 |
Figure 4 Relationship among RASSF1A, JNK, and downstream signals in vivo. (A–D) RASSF1A, p-JNK, JNK, p-c-JUN, c-JUN, and ATF-2 protein levels in primary and recurrent tumor specimens were measured by Western blot. (E, F) The relative mRNA levels of miR-602, RASSF1A, JNK, c-JUN, and ATF-2 were measured by RT-qPCR and normalized to the expression of U6 (*P < 0.05, **P < 0.01, ***P < 0.001). |
In the murine recurrent tumor specimens, the RASSF1A protein levels in RASSF1A overexpressing tumors were lower than in controls, and also the levels of p-JNK, JNK, and the downstream p-c-JUN, c-JUN, and ATF-2 were the reverse of those observed in primary tumors, indicating that the lack of RASSF1A activated tumor growth and contributed to the progression of cancer (Figure 4C and D).
mRNA levels of related markers in tumor specimens were measured by RT-qPCR. RASSF1A levels were negatively correlated with JNK levels. miR-602 and RASSF1A levels were also strongly negatively correlated (Figure 4E and F).
Based on the above results, we hypothesized that miR-602 might promote the recurrence of postoperative liver cancer by inhibiting RASSF1A expression. RASSF1A may inhibit the phosphorylation of JNK and the downstream expression of ATF-2 and c-JUN, thus promoting activation of dormant cells after liver cancer surgery.
Transient Transfection of miR-602 in vitro
We transiently transfected SMMC7721 cells that stably overexpressed RASSF1A with miR-602. After 72 h, we photographed the plates by microscopy, extracted total protein and total RNA, and detected relevant markers.
MiR-602 overexpression increased cell growth compared with the mimic NC group. The miR-602 inhibitor significantly reduced cell growth compared with the inhibitor NC group. These results indicate that miR-602 promoted tumor cell growth (Figure 5A and B).
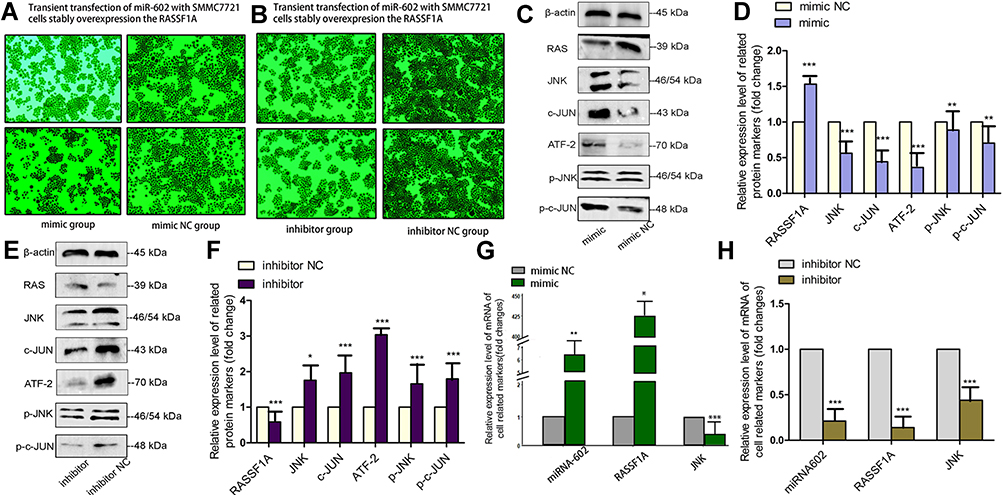 |
Figure 5 Transient transfection with miR-602 in vitro. (A, B) Pictures taken with a microscope after transfection into SMMC7721 cells that stably overexpressed RASSF1A. (C–F) Protein levels of RASSF1A, p-JNK, JNK, p-c-JUN, c-JUN, and ATF-2 in SMMC7721 cells stably overexpressing RASSF1A that were transiently transfected with miR-602, as analyzed by Western blot. (G, H) After transiently transfecting SMMC7721 cells stably overexpressing RASSF1A with miR-602, miR-602, RASSF1A, and JNK mRNA levels were measured by RT-qPCR (*P < 0.05, **P < 0.01, ***P < 0.001). |
We measured the expression levels of related proteins in the four groups by Western blot analysis. RASSF1A expression was significantly decreased in the transiently transfected miR-602 mimic group compared with the mimic NC group, indicating that miR-602 inhibited the expression of RASSF1A, and this low expression level abolished phosphorylation of and increased expression of JNK. Protein levels of the downstream ATF-2, p-c-JUN, and c-JUN were also elevated (Figure 5C and D). Opposite results were obtained in the inhibitor and inhibitor NC groups (Figure 5E and F).
mRNA levels of related genes were measured by RT-qPCR in the four groups. After transient transfection with miR-602 mimic, expression of miR-602 was increased more than 400-fold on average, while expression of the RASSF1A gene was inhibited. Subsequently, the inhibitory effects of RASSF1A on JNK expression were relieved, which led to increased expression of JNK (Figure 5G). After transient transfection with miR-602 inhibitor, expression of miR-602 was decreased by 79% on average, and RASSF1A expression was decreased approximately three-fold (Figure 5H).
Based on the above results, we hypothesized that miR-602 might promote recurrence of liver cancer by inhibiting RASSF1A expression. RASSF1A may inhibit phosphorylation of JNK and thus the downstream expression of ATF-2 and c-JUN.
CCK-8 Cell Proliferation Assay
The proliferation and inhibition rates of mimic- and inhibitor-transfected cells (Figure 6) showed that after transient transfection with miR-602 mimic, the cell proliferation rate increased by 91%. After transient transfection with miR-602 inhibitor, the cell inhibition rate increased by 53%. These results correspond to the results described in “Transient transfection of miR-602 in vitro”.
 |
Figure 6 CCK-8 cell proliferation assay. (A) The effects of transfection with miR-602 mimic on cell proliferation were assessed by CCK-8 assay. Cell viability was calculated by measuring absorbance at 450 nm. (B) The effects of transfection with miR-602 inhibitor on cell proliferation were assessed by CCK-8 assay. Cell viability was calculated by measuring absorbance at 450 nm (**P < 0.01). |
Discussion
Recurrence of liver cancer is one of the most important and common causes of death in patients with liver cancer. The underlying cause is the presence of dormant postoperative HCC cells.26 After radical resection of liver cancer, tumor foci or tumor cells remain in the body. These cells have not been cleared by the body and have not proliferated within a certain time range, but they still have proliferative potential and are thus deemed dormant HCC cells.4
There are three isoforms of JNK in mammals: JNK1, JNK2, and JNK3 (encoded, respectively, by MAPK8, MAPK9, and MAPK10). The JNK proteins, including splicing variants, are 46–55 kDa in size.27 JNK1 and JNK2 are expressed in most tissues, whereas JNK3 is mainly expressed in the brain, heart, and testes. This tissue-specific distribution, particularly for JNK3, has led to the idea that different isoforms might perform different cellular roles.28,29 Only JNK1 and JNK2 are expressed in HCC tissues.30,31 However, our research goal was to uncover the mechanism of action of JNKs in liver cancer, and therefore we used an anti-JNK antibody which only reacts with JNK1 and JNK2, which we collectively refer to as JNK.
We constructed a liver cancer SMMC7721 cell line stably overexpressing RASSF1A by viral transduction. RASSF1A inhibited the growth of liver cancer cells. We subjected these cells to RT-qPCR and Western blot analysis, which showed that RASSF1A and JNK levels were negatively correlated. ATF-2 and c-JUN serve as downstream markers of JNK; their expression levels show the same changes as those of JNK. Furthermore, miR-602 is located upstream of RASSF1A and plays a role in promoting tumor progression by inhibiting the expression of RASSF1A.
Next, we used cells stably overexpressing the RASSF1A gene for in situ implantation into mouse livers. Tumor growth was reduced in tumors overexpressing RASSF1A. After 2 weeks, we removed the tumors and cut the mice’s tails to stimulate recurrence of dormant tumors, thereby obtaining a recurrence model of liver cancer. Two weeks later, we surgically removed tumor tissue and extracted the total RNA and protein for the detection of related markers. RASSF1A and JNK levels were, indeed, negatively correlated, and miR-602 was one of the regulators of RASSF1A.
After transfection of SMMC7721 cells stably overexpressing RASSF1A with mimics and inhibitors, MiR-602 was expressed at high and low levels, respectively. By detecting related indicators, we found that RASSF1A expression was decreased in the miR-602 overexpression group, while the levels of p-JNK, JNK, ATF-2, p-c-JUN, and c-JUN were increased. Moreover, results for the low miR-602 expression group were opposite from those for the overexpression group.
Therefore, we hypothesize that miR-602 inhibited the anti-cancer effects of RASSF1A by regulating the JNK signaling pathway, thereby promoting the activation of dormant cells after liver cancer surgery, which leads to the recurrence of liver cancer.
We would like to mention a few several shortcomings in our experiments. First, for technical reasons, we did not image the primary and recurrent tumors. If computed tomography (CT) or magnetic resonance imaging is performed on mice and positron emission tomography–CT is used to determine the specific conditions of the primary and recurring tumors, the specific time and severity of tumor recurrence can be calculated more accurately. Second, due to time constraints, we did not validate the association among miR-602, RASSF1A, and JNK expression in human tissue and plasma samples. Third, previous studies have found that RASSF1A hypermethylation leads to downregulation of gene expression, providing a new perspective for the detection of malignant tumors. However, since the focus of this study was not the function of RASSF1A but its correlation with miR-602 and JNK, we did not study methylation of RASSF1A.
Ethics Approval
Our research team has a cooperative relationship with the Laboratory Animal Center of the First Affiliated Hospital of Sun Yat-sen University. In view of the approval and validity of the animal experiments approved by the Ethics Committee of the People’s Hospital of Guangdong Province, the Ethics Committee of the Laboratory Animal Center of the First Affiliated Hospital of Sun Yat-sen University also agreed to the animal experiment.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and have agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hiraoka A, Horiike N, Yamashita Y, et al. Efficacy of radiofrequency ablation therapy compared to surgical resection in 164 patients in Japan with single hepatocellular carcinoma smaller than 3 cm, along with report of complications. Hepatogastroenterology. 2008;55(88):2171–2174.
2. Aguirre-Ghiso JA, Bragado P, Sosa MS. Metastasis awakening: targeting dormant cancer. Nat Med. 2013;19(3):276–277. doi:10.1038/nm.3120
3. Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev. 2011;21(1):42–49. doi:10.1016/j.gde.2010.10.011
4. Senft D, Jeremias I. Tumor cell dormancy-triggered by the niche. Dev Cell. 2019;49(3):311–312. doi:10.1016/j.devcel.2019.04.022
5. Zijlstra A, Von Lersner A, Yu D, et al. The importance of developing therapies targeting the biological spectrum of metastatic disease. Clin Exp Metastasis. 2019;36(4):305–309. doi:10.1007/s10585-019-09972-3
6. Recasens A, Munoz L. Targeting Cancer Cell Dormancy. Trends Pharmacol Sci. 2019;40(2):128–141. doi:10.1016/j.tips.2018.12.004
7. Endo H, Inoue M. Dormancy in cancer. Cancer Sci. 2019;110(2):474–480. doi:10.1111/cas.13917
8. Galliano D, Pellicer A. MicroRNA and implantation. Fertil Steril. 2014;101(6):1531–1544. doi:10.1016/j.fertnstert.2014.04.023
9. Aigner A. MicroRNAs (miRNAs) in cancer invasion and metastasis: therapeutic approaches based on metastasis-related miRNAs. J Mol Med (Berl). 2011;89(5):445–457. doi:10.1007/s00109-010-0716-0
10. Budhu A, Jia HL, Forgues M, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47(3):897–907. doi:10.1002/hep.22160
11. Yang J, Sheng YY, Wei JW, et al. MicroRNA-219-5p promotes tumor growth and metastasis of hepatocellular carcinoma by regulating cadherin 1. Biomed Res Int. 2018;2018:4793971.
12. Liu M, Yu J, Wang D, et al. Epigenetically Upregulated MicroRNA-602 is involved in a negative feedback loop with FOXK2 in esophageal squamous cell carcinoma. Mol Ther. 2019;27(10):1796–1809. doi:10.1016/j.ymthe.2019.07.006
13. Cui-ying WJHLS. The Study of the Relationship Between the Occurrence of Hepatocellular Carcinoma and the Expression of miRNA602. Labeled Immunoassays Clin Med. 2020;25(5):676–679.
14. Dratviman-Storobinsky O, Cohen Y, Frenkel S, et al. The role of RASSF1A in uveal melanoma. Invest Ophthalmol Vis Sci. 2012;53(6):2611–2619. doi:10.1167/iovs.11-7730
15. Khan FS, Ali I, Afridi UK, Ishtiaq M, Mehmood R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol Int. 2017;11(1):45–53. doi:10.1007/s12072-016-9743-4
16. Xiong W, Sun LP, Chen XM, Li HY, Huang SA, Jie SH. Comparison of microRNA expression profiles in HCC-derived microvesicles and the parental cells and evaluation of their roles in HCC. J Huazhong Univ Sci Technolog Med Sci. 2013;33(3):346–352. doi:10.1007/s11596-013-1122-y
17. Yang L, Ma Z, Wang D, Zhao W, Chen L, Wang G. MicroRNA-602 regulating tumor suppressive gene RASSF1A is overexpressed in hepatitis B virus-infected liver and hepatocellular carcinoma. Cancer Biol Ther. 2010;9(10):803–808. doi:10.4161/cbt.9.10.11440
18. Song MS, Song SJ, Kim SY, Oh HJ, Lim DS. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008;27(13):1863–1874. doi:10.1038/emboj.2008.115
19. Donninger H, Clark JA, Monaghan MK, Schmidt ML, Vos M, Clark GJ. Cell cycle restriction is more important than apoptosis induction for RASSF1A protein tumor suppression. J Biol Chem. 2014;289(45):31287–31295. doi:10.1074/jbc.M114.609537
20. Wang J, Tai G. Role of C-Jun N-terminal kinase in hepatocellular carcinoma development. Target Oncol. 2016;11(6):723–738. doi:10.1007/s11523-016-0446-5
21. Lin YT, Liu YC, Chao CC. Inhibition of JNK and prothymosin-alpha sensitizes hepatocellular carcinoma cells to cisplatin. Biochem Pharmacol. 2016;122:80–89. doi:10.1016/j.bcp.2016.10.003
22. Sui C, Zhuang C, Sun D, Yang L, Zhang L, Song L. Notch1 regulates the JNK signaling pathway and increases apoptosis in hepatocellular carcinoma. Oncotarget. 2017;8(28):45837–45847. doi:10.18632/oncotarget.17434
23. Zhang L, Li S, Wang R, Chen C, Ma W, Cai H. Cytokine augments the sorafenib-induced apoptosis in Huh7 liver cancer cell by inducing mitochondrial fragmentation and activating MAPK-JNK signalling pathway. Biomed Pharmacother. 2019;110:213–223. doi:10.1016/j.biopha.2018.11.037
24. Whang YM, Kim YH, Kim JS, Yoo YD. RASSF1A suppresses the c-Jun-NH2-kinase pathway and inhibits cell cycle progression. Cancer Res. 2005;65(9):3682–3690. doi:10.1158/0008-5472.CAN-04-2792
25. Hui HX, Changjun W, Xiaowen Z, Juze L. Establishment of tumor dormancy model of hepatoma in mouse. Chin J Cancer Prev Treat. 2020;20(19):1484.
26. Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target. Nat Rev Cancer. 2010;10(12):871–877. doi:10.1038/nrc2933
27. Gupta S, Barrett T, Whitmarsh AJ, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15(11):2760–2770. doi:10.1002/j.1460-2075.1996.tb00636.x
28. Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70(4):1061–1095. doi:10.1128/MMBR.00025-06
29. Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143(2):307–320. doi:10.1053/j.gastro.2012.06.004
30. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. doi:10.1038/nrc2694
31. Rovida E, Stecca B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities. Semin Cancer Biol. 2015;35:154–167.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.