Back to Journals » OncoTargets and Therapy » Volume 9
miR-32 inhibits proliferation, epithelial–mesenchymal transition, and metastasis by targeting TWIST1 in non-small-cell lung cancer cells
Received 5 November 2015
Accepted for publication 28 January 2016
Published 14 March 2016 Volume 2016:9 Pages 1489—1498
DOI https://doi.org/10.2147/OTT.S99931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Lei Li,1,* Dapeng Wu2,*
1Department of Pneumology, 2Department of Radiotherapy, Huaihe Hospital of Henan University, Kaifeng, Henan, People’s Republic of China
*These authors contributed equally to this work
Background: By analyzing published microRNA microarray studies, miR-32 was found to be markedly reduced in non-small-cell lung cancer (NSCLC) tissues compared with that in nontumor tissues. However, little is known about its role and molecular mechanism involved in NSCLC development and progression. Here, we report the effect of miR-32 on NSCLC cell proliferation, epithelial–mesenchymal transition (EMT), and metastasis.
Methods: Quantitative real-time PCR was performed to detect the expression level of miR-32 in primary NSCLC cases and cell lines. miR-32-overexpressing H1299 and A549 cells were constructed by lipofection transfection. MTT, transwell chamber, and Western blot assays were used to assess the effect of miR-32 on proliferation, EMT, and metastasis of NSCLC cells, respectively. Target prediction and luciferase reporter assays were performed to investigate the targets of miR-32. Tumor formation assay in vivo was performed to investigate the antitumor effect of miR-32.
Results: An inverse correlation existed between miR-32 expression level and NSCLC cell proliferation, EMT, and metastasis, and upregulation of miR-32 repressed NSCLC cell proliferation, EMT, and metastasis. Moreover, we identified and validated that TWIST1 was a direct target of miR-32, and miR-32 regulated NSCLC cell proliferation, EMT, and metastasis, at least in part via modulation of TWIST1. The animal experiments showed that overexpression of miR-32 inhibited the growth of NSCLC tumors in vivo.
Keywords: non-small-cell lung cancer, miR-32, TWIST1, proliferation, EMT, nude mice
Introduction
Lung cancer is one of the most common human cancers and is also the leading cause of cancer death in the world. Non-small-cell lung cancer (NSCLC) accounts for nearly 85% of newly diagnosed lung cancer cases, and >70% of patients with NSCLC have advanced disorders.1 Despite the great advances achieved in surgery and chemotherapy recently, the prognosis of NSCLC is still poor with a 5-year survival rate of 16%.2 Besides, nearly 52% of postoperative NSCLC cases result in recurrence.3 Tumor metastasis and recurrence are the major causes that lead to mortality, but the precise molecular mechanism of metastatic dissemination is still not completely clear. Many recent studies have demonstrated that epithelial–mesenchymal transition (EMT) is one of the major molecular mechanisms inducing cancer metastasis.4,5 TWIST1 is an EMT regulator, which induces EMT through the suppression of E-cadherin expression.6 In the EMT process, the epithelial cells with a cobblestone morphology gain the traits of the mesenchymal cells with a spindle-shaped fibroblast-like phenotype.7 With the changes in cellular morphology, the expression of proteins also has some changes, such as the loss of the epithelial marker E-cadherin and the gain of the mesenchymal markers vimentin and N-cadherin.8 In addition, this process involves a disassembly of cell–cell junctions, which allows mesenchymal phenotypic cells to have weaker cell adhesion ability and stronger cell migration and invasion ability, thereby resulting in tumor aggressiveness.7,9
MicroRNAs (miRNAs), small and noncoding RNAs, modulate gene expression by binding to complementary sequences in the 3′-untranslated region (3′UTR) of target messenger RNA (mRNA), causing translational inhibition or target mRNA degradation.10 miRNAs are predicted to regulate the expression of nearly 90% of all human genes and play essential roles in various biological and pathological processes, including cell proliferation, differentiation, apoptosis, invasion, migration, and metastasis.11,12 Mounting evidence indicates that deregulated expression of miRNAs occurs in many types of cancers, some of which function as tumor oncogenes or suppressor genes.13,14 Recent studies have implied that miRNAs function as critical modulators for EMT.15–17 The role of miRNAs in NSCLC has been extensively studied, and miRNA microarray studies have identified many abnormally expressed miRNAs.18,19 Among them, the expression level of miR-32 in NSCLC is decreased, but the detailed role of miR-32 in NSCLC is still poorly understood.
In this study, we determined the expression level of miR-32 in primary NSCLC cases and cell lines and investigated the association between miR-32 expression and NSCLC cell proliferation, EMT, and metastasis. We further investigated the molecular mechanisms by which miR-32 exerts regulatory effects on NSCLC cell proliferation, EMT, and metastasis. Furthermore, we performed the animal experiments to explore the anticancer action of miR-32 in vivo. These findings provide a novel potential therapeutic target for NSCLC.
Materials and methods
Tissue samples
In 2013, 22 NSCLC tissue samples and matched nontumor normal tissue samples were collected from Huaihe Hospital of Henan University. Eligible samples were obtained from the patients with primary NSCLC who had not received any preoperative chemotherapy or radiotherapy. In addition, there were no coexisting diseases in these patients. This study and the use of human cell lines were performed with the approval of the Medical Ethical Committee of Huaihe Hospital of Henan University and written informed consent was obtained from all patients. All tissue samples were immediately flash-frozen in liquid nitrogen after resection and then stored at −80°C until use.
Cell lines and cell transfection
Two NSCLC cell lines, H1299 and A549, and human bronchial epithelial cell line 16HBE were obtained from American Type Culture Collection (Manassas, VA, USA). Cells were grown in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (Thermo Fisher Scientific) at 37°C with 5% CO2.
miR-32 mimics and negative control (NC) mimics, miR-32 inhibitors (anti-miR-32) and NC inhibitors (anti-NC), and TWIST1 small interfering RNAs (siRNAs) were purchased from GenePharma Company (Shanghai, People’s Republic of China). Cell transfection was carried out using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s protocol.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from frozen tissues or cultured cells using TRIzol Reagent (Thermo Fisher Scientific) following the manufacturer’s suggestions. miR-32 expression was determined using TaqMan miRNA assays (Thermo Fisher Scientific) following the manufacturer’s protocol, and U6 small nuclear RNA (GeneCopoeia, Carlsbad, CA, USA) was used as endogenous control. The expression of TWIST1 was determined using SYBR green qPCR assay (Takara, Dalian, People’s Republic of China) and normalized with GAPDH (GeneCopoeia).
MTT assay
Cell suspensions (2×104 cells per well) were seeded in 96-well plates and cultured overnight in a humidified incubator at 37°C and 5% CO2. Cell growth was monitored every day for a period of 4 days. Briefly, 20 μL of MTT (5 mg/mL; Sigma-Aldrich Co., St Louis, MO, USA) was added to each well and further incubated for 4 hours. Then, the culture medium in the wells was removed, and 150 μL of dimethyl sulfoxide (Sigma-Aldrich Co.) was added to dissolve the MTT–formazan crystals. The absorbance of each well was read by a microplate reader (Thermo Fisher Scientific) at 570 nm.
Western blot analysis
Tissue samples were ground to powder in liquid nitrogen. Total proteins were extracted from tissue powder or treated cells using sodium dodecyl sulfate lysis buffer (Beyotime, Shanghai, People’s Republic of China) for 30 minutes at 4°C, and an equal amount of protein was separated using 10% polyacrylamide sodium dodecyl sulfate gels. Then, the proteins were transferred to polyvinylidene fluoride membranes (Thermo Fisher Scientific) and were probed with primary antibodies against E-cadherin (Abcam, Cambridge, UK), vimentin (GeneTex, San Antonio, TX, USA), TWIST1 (Abcam), N-cadherin (GeneTex), matrix metalloproteinase (MMP)-2 (Abcam), MMP-9 (Abcam), or GAPDH (GeneTex). The membranes were incubated overnight at 4°C, followed by incubation with secondary antibody peroxidase-conjugated anti-IgG (Abcam), and detected using a chemiluminescent detection system (Pierce ECL Substrate Western blot detection system; Thermo Fisher Scientific). Quantity One 4.5.0 software (Bio-Rad Laboratories Inc., Hercules, CA, USA) was used to quantify the integrated density of the protein bands.
Cell migration and invasion assays
Transwell chambers (Corning Incorporated, Corning, NY, USA) with a pore size of 8 μm were used for migration and invasion assays. Briefly, cells were harvested and resuspended in serum-free medium. For migration assay, 5×104 cells in serum-free medium were directly added into the upper chamber. For invasion assay, Matrigel (BD Biosciences, San Jose, CA, USA) was coated on the upper surface of the transwell chamber before use, and then 2×105 cells in serum-free medium were added into the upper chamber. RPMI-1640 medium containing 10% fetal bovine serum was added into the bottom chamber and served as a chemoattractant. After incubation for 18 hours at 37°C in 5% CO2, the cells on the upper surface of the membrane were wiped out with cotton swab. The cells that migrated or invaded to the lower surface of the membrane were fixed with 100% methanol, stained with hematoxylin, and counted (five high-power fields per chamber) under a microscope (Olympus Corporation, Tokyo, Japan).
Fluorescent reporter assay
Wild-type 3′UTRs of TWIST1 containing predicted miR-32 target sites were amplified by polymerase chain reaction (PCR) from H1299 cell genomic DNA, and mutant 3′UTRs were obtained by overlap extension PCR method. H1299 cells were transfected with Lipofectamine 2000 (Thermo Fisher Scientific). The transfection mixtures contained 5 pmol of miR-32 and 100 ng of firefly luciferase reporter plasmid, and pRL-TK (Promega Corporation, Fitchburg, WI, USA) was also transfected as normalization control. Cells were collected 48 hours posttransfection, and the luciferase activity was determined using a dual-luciferase reporter assay system (Promega Corporation).
In vivo tumor xenograft model
Twelve healthy female BALB/c nude mice with 4–5 weeks of age were purchased from the Center of Experimental Animal Huaihe Hospital of Henan University. All animal experiments were approved by the Institutional Animal Care and Use Committee of Huaihe Hospital of Henan University. Twelve nude mice were randomized into two groups (ie, control group and miR-32 group). H1299 cells (2×106 cells in 200 μL phosphate-buffered saline) transfected with miR-32 mimics or NC mimics were subcutaneously inoculated into the right dorsal flank of nude mice. The tumor size was measured weekly with slide caliper, and the tumor volume was calculated as follows: tumor volume = length × width2/2. Four weeks later, all tested nude mice were euthanized, and tumors were excised and weighed. Moreover, the expression levels of TWIST1, MMP-2, MMP-9, E-cadherin, vimentin, and N-cadherin proteins in xenografts were determined by Western blot analysis.
Statistical analysis
Statistical analysis was performed on GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). All values were presented as mean ± SD. The differences between the two groups were analyzed using the Student’s t-test, and a P-value ≤0.05 was considered statistically significant.
Results
miR-32 is frequently downregulated in NSCLC
miRNA microarray studies have identified that the expression level of miR-32 is obviously decreased in NSCLC tissues compared to that in nontumor tissues.18,19 We further detected the expression level of miR-32 in NSCLC tissues and cell lines by quantitative real-time (qRT)-PCR. Results showed that miR-32 expression in lung cancer tissues was significantly lower than that in the corresponding adjacent normal lung tissues (Figure 1A). Similarly, miR-32 expression was markedly decreased in two human NSCLC cell lines H1299 and A549 compared to that in human bronchial epithelial cell line 16HBE (Figure 1B). Taken together, these results indicated that miR-32 is downregulated in NSCLC tissues and cell lines.
 | Figure 1 miR-32 is downregulated in NSCLC tissues and cell lines. |
miR-32 suppresses NSCLC cell proliferation and EMT in vitro
To assess the effect of miR-32 on NSCLC cell proliferation, we transfected H1299 and A549 cells with miR-32 mimics or NC mimics, and the transfection efficiency was detected by qRT-PCR. Results showed that H1299 and A549 cells transfected with miR-32 mimics both had significant increases in miR-32 expression compared with the cells transfected with NC mimics (Figure 2A and B). Then, MTT assays were performed on H1299 and A549 cells transfected with miR-32 mimics or NC mimics. As shown in Figure 2C and D, both H1299 and A549 cells transfected with miR-32 mimics had marked decreases in the proliferation rate compared with the control groups, which indicated that miR-32 represses NSCLC cell proliferation in vitro. EMT, characteristic with the loss of epithelial marker E-cadherin and the gain of mesenchymal markers vimentin and N-cadherin, has been identified to be one of the major molecular mechanisms inducing cancer metastasis. To investigate whether miR-32 affects the EMT of NSCLC cells, H1299 and A549 cells transfected with miR-32 mimics or NC mimics were used to perform Western blot assay to examine the expression of E-cadherin, vimentin, and N-cadherin proteins. As illustrated in Figure 2E and F, ectopic transfection of miR-32 mimics led to a remarkable increase in E-cadherin expression and obvious decreases in vimentin and N-cadherin expression in H1299 and A549 cells, which indicated that miR-32 suppresses NSCLC cell EMT in vitro.
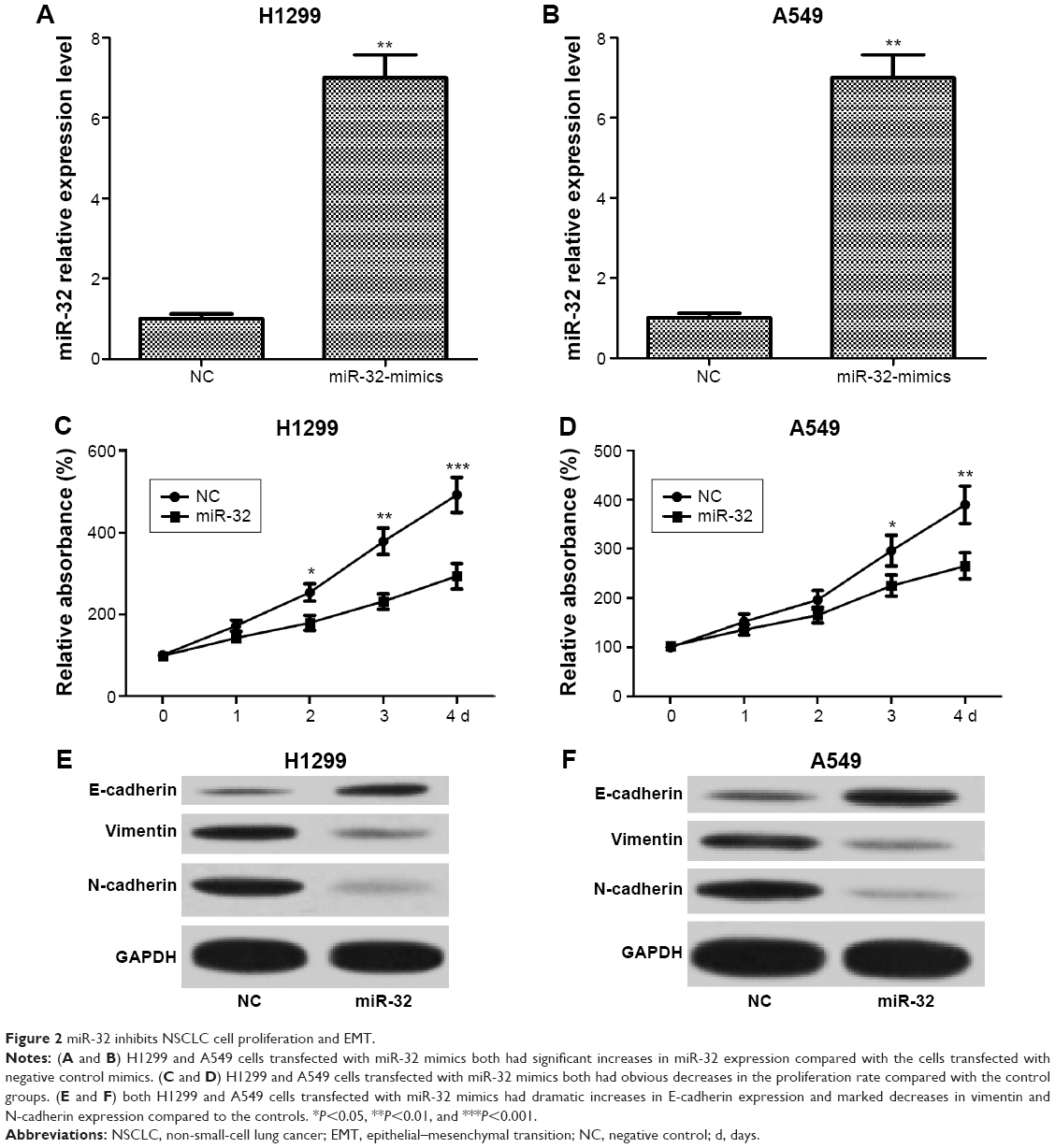 | Figure 2 miR-32 inhibits NSCLC cell proliferation and EMT. |
miR-32 suppresses NSCLC cell metastasis in vitro
To investigate whether miR-32 affects NSCLC cell metastasis, H1299 and A549 cells were transfected with miR-32 mimics or NC mimics and then were used to perform invasion and migration assays by transwell method. As shown in Figure 3A and B, the invasion ability of H1299 and A549 cells transfected with miR-32 mimics was significantly reduced compared to the cells transfected with NC mimics. Furthermore, the expression levels of markers of tumor invasion MMP-2 and MMP-9 in H1299 and A549 cells transfected with miR-32 mimics were both lower than those in the controls (Figure 3C and D). These results indicated that miR-32 suppresses NSCLC cell invasion. Consistently, overexpression of miR-32 led to diminished migratory abilities in both cell lines (Figure 3E and F). These observations indicated that miR-32 suppresses NSCLC cell metastasis by impeding cell invasion and migration in vitro.
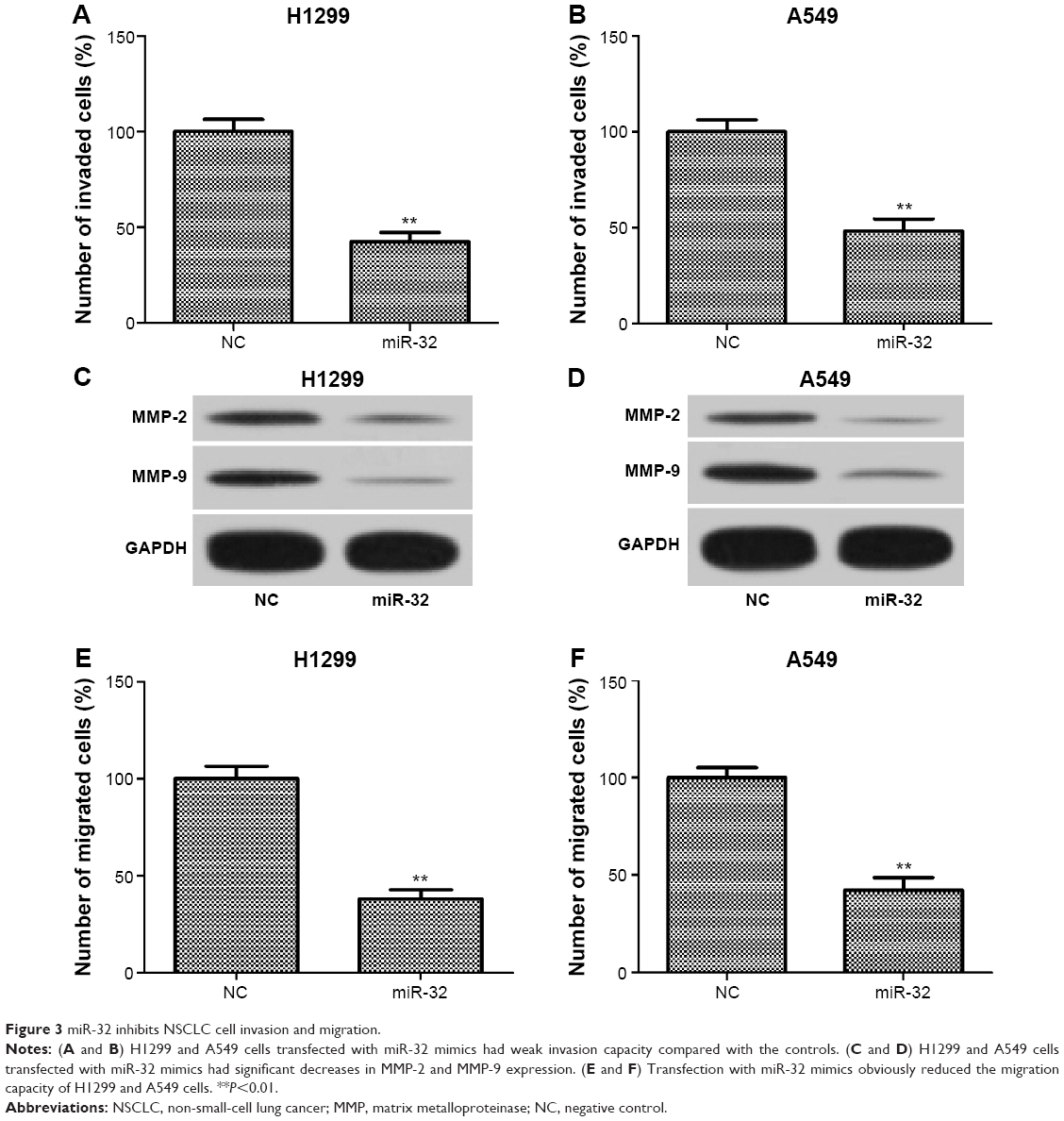 | Figure 3 miR-32 inhibits NSCLC cell invasion and migration. |
TWIST1 is a direct target of miR-32
To further investigate the molecular mechanisms by which miR-32 represses NSCLC cell proliferation, EMT, and metastasis, we used three bioinformatic algorithms (TargetsScan, microrna.org, and miRanda) to screen the target gene of miR-32 and selected TWIST1 for further analysis (Figure 4A). To identify whether TWIST1 is regulated by miR-32 via direct binding to its 3′UTR, luciferase reporter assays were carried out in H1299 cells. The results showed that cotransfection with miR-32 mimics obviously reduced the luciferase activity of the wild-type reporter gene. This suppressive effect was abolished by mutation of the miR-32 target sequence (Figure 4B). This result suggested that TWIST1 is indeed a direct target of miR-32. Moreover, to confirm the regulatory effect of miR-32 on TWIST1, qRT-PCR and Western blot assays were carried out. The results showed that upregulation of miR-32 resulted in a remarkable decrease in TWIST1 expression at both mRNA and protein levels. Relatively, downregulation of miR-32 led to obvious increases in TWIST1 mRNA and protein (Figure 4C and D). These results indicated that the expression of TWIST1 at both mRNA and protein levels is regulated by miR-32 in vitro. To further confirm whether miR-32 inhibits NSCLC cell proliferation, EMT, and metastasis by downregulating TWIST1, we transfected H1299 cells with TWIST1 siRNAs (siTWIST1) to silence the expression of TWIST1, and the transfection efficiency was determined by Western blot. Results showed that H1299 cells transfected with siTWIST1 had obvious decrease in TWIST1 expression compared with the cells transfected with control (Figure 4H). Then MTT, invasion, migration, and Western blot assays were performed on H1299 cells transfected with siTWIST1 or control. MTT assay showed that transfection with TWIST1 siRNAs resulted in the dramatic decrease in the proliferation rate (Figure 4E). Invasion and migration assays showed that in the siTWIST1 group, the number of invaded and migrated cells both significantly reduced compared to the control (Figure 4F and G). Western blot analysis showed that transfection with TWIST1 siRNAs led to remarkable decreases in MMP-2, MMP-9, vimentin, and N-cadherin expression and a significant increase in E-cadherin expression (Figure 4H). These results indicated that silencing of TWIST1 suppresses H1299 cell proliferation, metastasis, and EMT. Similar effect of siTWIST1 was observed in miR-32 mimics-treated cells, suggesting that siTWIST1 could mimic the effect of miR-32. Taken together, these results suggested that TWIST1 is one of the direct targets of miR-32, and the functional effect of miR-32 on NSCLC cell lines, at least in part, depends on TWIST1.
 | Figure 4 TWIST1 is a direct target of miR-32. |
Overexpression of miR-32 suppresses growth of NSCLC tumors in vivo
In vitro studies indicated that miR-32 inhibits NSCLC cell proliferation, EMT, and metastasis by targeting TWIST1. We further investigated the effect of miR-32 on NSCLC cell tumorigenicity. H1299 cells were transfected with miR-32 mimics or NC mimics and then were subcutaneously inoculated into the right dorsal flank of nude mice. We measured the tumor size weekly and calculated the tumor volume. The result showed that the tumor volume was obviously decreased in the miR-32 group compared to that in the control group (Figure 5A). After 4 weeks, tested nude mice were euthanized, and tumors were excised and weighed. Accordingly, the tumor weight in the miR-32 group was markedly reduced compared to that in the control group (Figure 5B). In addition, the miR-32 group had marked decreases in TWIST1, MMP-2, MMP-9, vimentin, and N-cadherin expression and a remarkable increase in E-cadherin expression compared with the control group (Figure 5C). Taken together, these results indicated that overexpression of miR-32 inhibits the growth of NSCLC tumors in vivo.
 | Figure 5 The antitumor effect of miR-32 in vivo. |
Discussion
By analyzing published miRNA microarray studies, miR-32 was found to be obviously reduced in NSCLC tissues compared to that in nontumor tissues.18,19 In this study, we further detected the expression level of miR-32 in NSCLC tissues and cell lines by qRT-PCR and confirmed that its expression level was downregulated in NSCLC issues and cell lines. Furthermore, overexpression of miR-32 significantly suppressed NSCLC cell proliferation, EMT, and metastasis in vitro. We further investigated the molecular mechanisms by which miR-32 inhibits NSCLC cell proliferation, EMT, and metastasis and found that the functional effect of miR-32 on NSCLC cell lines, at least in part, depended on TWIST1, one of its direct targets. Animal experiments had confirmed that overexpression of miR-32 inhibited the growth of NSCLC tumors in vivo. These data suggest that miR-32 functions as a tumor suppressor and suppresses proliferation, EMT, and metastasis in NSCLC.
Many studies have demonstrated that EMT plays a fundamental role in the initial stage of metastatic progression of cancer cells.20 In the EMT process, epithelial cells lose their characteristics, gain mesenchymal features, and become motile and invasive.7,21 Loss of epithelial marker E-cadherin and gain of mesenchymal markers N-cadherin and vimentin are regarded as the most important molecular markers of EMT. Emerging evidence has demonstrated that many miRNAs play crucial roles in the EMT of cancer cells: miR-23a regulated TGF-β-induced EMT in lung cancer cells, miR-194 repressed EMT of endometrial cancer cells, and miR-134 inhibits EMT in NSCLC cells.15–17 Our results showed that miR-32 reversed the expression level of epithelial marker and mesenchymal markers in H1299 and A549 cells and inhibited EMT. In addition, we also investigated the effect of miR-32 on the proliferation, migration, and invasion ability of NSCLC cells and found that miR-32 repressed H1299 and A549 cell proliferation and metastasis.
We further investigated the molecular mechanisms by which miR-32 represses NSCLC cell proliferation, EMT, and metastasis. By bioinformatics analysis, we identified many potential targets of miR-32 and selected TWIST1 for further analysis. TWIST1 is a highly conserved basic helix–loop–helix transcription factor and plays an important role in numerous biological processes, such as mesoderm development and differentiation.22 It has been widely reported that TWIST1 is upregulated and may function as an oncogene in many kinds of human tumors, such as stomach, breast, liver, and prostate cancer.23–26 The role of TWIST1 in tumor cell proliferation, EMT, invasion, and metastasis has been attracting wide attention recently. Zhu et al27 reported that silencing of TWIST1 by RNAi downregulates MDR1/P-gp expression in HeLa cervical cancer cells and suppresses the cell proliferation. Zhao et al28 reported that cytoplasmic p27 promotes EMT and tumor progression via TWIST1 upregulation. Croset et al24 reported that TWIST1 expression in breast cancer cells facilitates bone metastasis formation. Depletion of TWIST1 expression represses prostate carcinoma cell and mammary carcinoma cell migration.29,30 In addition, TWIST1 can also enhance gastric cancer cell invasion and migration both in vitro and in vivo.31 For the role of TWIST1 in NSCLC, Hung et al32 reported that TWIST1 is upregulated and is a significant prognostic predictor in patients with NSCLC. Yochum et al33 reported that TWIST1 is required for repression of apoptosis in oncogene-driven NSCLC. The earlier findings are the reason why we selected TWIST1 for further analysis. We performed luciferase assay and confirmed that miR-32 targeted TWIST1 directly. The expression of TWIST1 at both mRNA and protein levels was regulated by miR-32, and knockdown of TWIST1 by siRNA reversed NSCLC cell proliferation, EMT, and metastasis. These findings implied that miR-32 suppresses TWIST1 protein expression by directly binding on the 3′UTR of TWIST1 mRNA to negatively regulate NSCLC cell proliferation, metastasis, and EMT. Furthermore, in vivo study also confirmed that overexpression of miR-32 suppressed the growth of NSCLC tumors.
Conclusion
In summary, our results demonstrate the following: miR-32 is downregulated in NSCLC; miR-32 inhibits NSCLC cell proliferation, EMT, and metastasis; miR-32 directly inhibits TWIST1 expression by targeting its 3′UTR; TWIST1 is inversely correlated with miR-32 level in NSCLC; and knockdown of TWIST1 reverses NSCLC cell proliferation, EMT, and metastasis. Our data suggest that miR-32 is an important tumor suppressor in NSCLC, and miR-32/TWIST1 pathway that we studied might be exploited by a therapeutic strategy for NSCLC treatment in future.
Disclosure
The authors report no conflicts of interest in this work.
References
Chen G, Umelo IA, Lv S, et al. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS One. 2013;8(3):e60317. | ||
Siegel R, Ward E, Brawley O, Jemal A. The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61(4):212–236. | ||
Park C, Lee IJ, Jang SH, Lee JW. Factors affecting tumor recurrence after curative surgery for NSCLC: impacts of lymphovascular invasion on early tumor recurrence. J Thorac Dis. 2014;6(10):1420–1428. | ||
Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. | ||
Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. | ||
Yang M-H, Wu K-J. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008;7(14):2090–2096. | ||
Bao B, Wang Z, Ali S, et al. Over-expression of FoxM1 leads to epithelial–mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112(9):2296–2306. | ||
Li J, Song Y, Wang Y, Luo J, Yu W. MicroRNA-148a suppresses epithelial-to- mesenchymal transition by targeting ROCK1 in non-small cell lung cancer cells. Mol Cell Biochem. 2013;380(1–2):277–282. | ||
Pavelic SK, Sedic M, Bosnjak H, Spaventi S, Pavelic K. Metastasis: new perspectives on an old problem. Mol Cancer. 2011;10(22):10.1186. | ||
Caporali A, Emanueli C. MicroRNA regulation in angiogenesis. Vascul Pharmacol. 2011;55(4):79–86. | ||
Baranwal S, Alahari SK. miRNA control of tumor cell invasion and metastasis. Int J Cancer. 2010;126(6):1283–1290. | ||
Hwang H, Mendell J. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94(6):776–780. | ||
Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39(5):673–677. | ||
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. | ||
Cao M, Seike M, Soeno C, et al. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. Int J Oncol. 2012;41(3):869–875. | ||
Dong P, Kaneuchi M, Watari H, et al. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol Cancer. 2011;10(99):10.1186. | ||
Li J, Wang Y, Luo J, et al. miR-134 inhibits epithelial to mesenchymal transition by targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett. 2012;586(20):3761–3765. | ||
Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–198. | ||
Dacic S, Kelly L, Shuai Y, Nikiforova MN. miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Mod Pathol. 2010;23(12):1577–1582. | ||
Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118(3):277–279. | ||
Gavert N, Ben-Ze’ev A. Epithelial–mesenchymal transition and the invasive potential of tumors. Trends Mol Med. 2008;14(5):199–209. | ||
Bialek P, Kern B, Yang X, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6(3):423–435. | ||
Feng M-Y, Wang K, Song H-T, et al. Metastasis-induction and apoptosis-protection by TWIST in gastric cancer cells. Clin Exp Metastasis. 2009;26(8):1013–1023. | ||
Croset M, Goehrig D, Frackowiak A, et al. TWIST1 expression in breast cancer cells facilitates bone metastasis formation. J Bone Miner Res. 2014;29(8):1886–1899. | ||
Zhao XL, Sun T, Che N, et al. Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator Twist1. J Cell Mol Med. 2011;15(3):691–700. | ||
Kwok WK, Ling M-T, Lee T-W, et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005;65(12):5153–5162. | ||
Zhu K, Chen L, Han X, Wang J, Wang J. Short hairpin RNA targeting Twist1 suppresses cell proliferation and improves chemosensitivity to cisplatin in HeLa human cervical cancer cells. Oncol Rep. 2012;27(4):1027–1034. | ||
Zhao D, Besser AH, Zhou W, et al. Cytoplasmic p27 promotes epithelial-mesenchymal transition and tumor progression via Twist1 upregulation. Cancer Res. 2014;74(19 suppl):1145–1145. | ||
Alexander NR, Tran NL, Rekapally H, Summers CE, Glackin C, Heimark RL. N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Res. 2006;66(7):3365–3369. | ||
Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. | ||
Luo G-Q, Li J-H, Wen J-F, Zhou Y-H, Hu Y-B, Zhou J-H. Effect and mechanism of the Twist gene on invasion and metastasis of gastric carcinoma cells. World J Gastroenterol. 2008;14(16):2487. | ||
Hung J-J, Yang M-H, Hsu H-S, Hsu W-H, Liu J, Wu K. Prognostic significance of hypoxia-inducible factor-1α, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax. 2009;64(12):1082–1089. | ||
Yochum ZA, Cades JA, Mazacurati L, Chatley S, Tran PT, Burns TF. TWIST1 is required for suppression of apoptosis in oncogene driven non-small cell lung carcinoma. Cancer Res. 2015;75(15 suppl):21. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.