Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
miR-199a Knockdown Alleviates Insulin Resistance and Inflammation by Targeting DDIT4 via the PI3K/AKT Pathway in vitro and in vivo
Authors Cai YW, Tang LJ, Zhu Y, Ye SS, Chen TE
Received 1 August 2025
Accepted for publication 17 December 2025
Published 31 December 2025 Volume 2025:18 Pages 4931—4942
DOI https://doi.org/10.2147/DMSO.S549884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Ya-Wei Cai,1 Ling-Jia Tang,1 Yao Zhu,1 Sen-Sen Ye,1 Tong-En Chen2
1Department of Geriatrics, Ningbo No.2 Hospital, Ningbo, Zheijiang, 315000, People’s Republic of China; 2Department of General Medicine, Ningbo No.2 Hospital, Ningbo, Zheijiang, 315000, People’s Republic of China
Correspondence: Tong-En Chen, Email [email protected]
Background: Insulin resistance (IR) is a pivotal pathological feature in the development of type 2 diabetes mellitus (T2DM). MicroRNA-199a (miR-199a) has been implicated in various metabolic disorders, but its precise role and mechanism in hepatic IR remain largely unexplored. This study aimed to investigate the role of miR-199a in IR and inflammation and to determine whether its effects are mediated through DDIT4 and the PI3K/AKT pathway.
Methods: An in vitro IR model was established in HepG2 cells using palmitic acid, and an in vivo T2DM model was induced in mice using a high-fat diet combined with streptozotocin injection. Functional assays, including glucose uptake and ELISA, were employed to assess metabolic and inflammatory responses. The interaction between miR-199a and its putative target, DDIT4, was validated by luciferase reporter and RNA immunoprecipitation assays. Key proteins in the PI3K/AKT signaling pathway were analyzed by Western blotting.
Results: We found that miR-199a was significantly upregulated, while DDIT4 was downregulated in both IR HepG2 cells and diabetic mice. Mechanistically, we identified DDIT4 as a direct target of miR-199a. Knockdown of miR-199a ameliorated insulin resistance and suppressed inflammation, whereas concomitant depletion of DDIT4 abolished these protective effects. Furthermore, miR-199a inhibition activated the PI3K/AKT pathway, as evidenced by increased phosphorylation of PI3K, AKT, and AS160, and decreased phosphorylation of FOXO1. These signaling changes were also dependent on DDIT4. In vivo, inhibition of miR-199a improved glucose homeostasis, attenuated systemic inflammation, and activated pancreatic PI3K/AKT signaling in T2DM mice.
Conclusion: Our findings reveal a novel miR-199a/DDIT4 axis that regulates insulin sensitivity and inflammation via the PI3K/AKT pathway, suggesting miR-199a as a potential therapeutic target for T2DM.
Keywords: microRNA-199a, DDIT4, type 2 diabetes mellitus, PI3K/AKT, insulin resistance
Introduction
Type 2 diabetes mellitus (T2DM), characterized by chronic hyperglycemia, accounts for approximately 90% of all diabetes cases and represents a major global health burden due to its severe complications, including vascular disease, retinopathy, and nephropathy.1–5 The pathogenesis of T2DM is complex, with insulin resistance (IR) serving as a core pathological feature.5,6 IR manifests as diminished insulin sensitivity in key metabolic tissues—such as liver, muscle, and adipose tissue—disrupting glucose and lipid homeostasis and ultimately leading to T2DM onset.7 Notably, a state of chronic, low-grade inflammation is intricately linked to the development and progression of both IR and T2DM.8,9 Therefore, elucidating the underlying molecular mechanisms of IR and inflammation is crucial for the development of novel therapeutic strategies.
MicroRNAs (miRNAs), small non-coding RNAs of 18–24 nucleotides, function as key post-transcriptional regulators of gene expression and are implicated in diverse biological processes, including metabolism.10–12 Accumulating evidence highlights their pivotal roles in modulating insulin signaling and IR. For instance, miR-125b-5p enhances insulin sensitivity by targeting DACT1 and suppressing the JNK pathway,13 while miR-221 impairs glucose uptake by inhibiting the PI3K/AKT/GLUT4 axis.14 Conversely, miR-122-5p exacerbates IR by suppressing the PI3K/Akt pathway via targeting IGF-1R.15 Furthermore, circulating levels of miR-29a have been correlated with IR and inflammatory markers in obese individuals with and without T2DM,16 underscoring the clinical relevance of miRNAs in metabolic disease.
Among the numerous miRNAs, miR-199a has been investigated in various pathological contexts, such as neurodegenerative and cardiovascular diseases.17–19 Critically, a clinical study reported that miR-199a is upregulated in the serum of patients with T2DM,20 suggesting a potential role in this metabolic disorder; however, its specific function and mechanistic involvement in T2DM-associated IR and inflammation remain unexplored. DNA Damage Inducible Transcript 4 (DDIT4), a stress-responsive protein, has emerged as a regulator of cellular metabolism and is known to interact with key signaling pathways, including the PI3K/AKT pathway, which is central to insulin action.21,22 Dysregulation of the PI3K/AKT pathway is a hallmark of IR. Thus, we hypothesized that miR-199a might contribute to the pathogenesis of T2DM by targeting DDIT4 and consequently disrupting the PI3K/AKT signaling pathway.
In the present study, we aimed to investigate the functional role of miR-199a in IR and inflammation using both in vitro (palmitic acid-treated HepG2 cells) and in vivo (high-fat diet/streptozotocin-induced T2DM mice) models. Our findings provide the first evidence that miR-199a promotes IR and inflammation by directly targeting DDIT4 and inhibiting the PI3K/AKT pathway, positioning miR-199a as a potential novel therapeutic target for T2DM.
Methods
Ethical Statements
The experimental protocol was approved by the Institutional Animal Care Use Committee of the Ningbo No.2 Hospital (approval no: 2156798). All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Materials and methods
HFD/STZ-Induced T2DM Mouse Model
A total of 18 male C57BL/6J mice (5 weeks old, specific pathogen-free grade) were purchased from Shanghai Laboratory Animal Center. Mice were housed under standard conditions (12 h light/dark cycle, 22 ± 2°C, 50–60% humidity) with ad libitum access to food and water. Mice were randomly divided into three groups (n=6 per group) using a computer-generated random number table: 1) Control group: fed a normal chow diet throughout the experiment. 2) T2DM group: fed a High-Fat Diet (HFD; 60% fat, 20% protein, 20% carbohydrate; Research Diets, D12492) for 4 weeks, followed by intraperitoneal injection of 40 mg/kg streptozotocin (STZ; Sigma-Aldrich) in citrate buffer (pH 4.5) for 4 consecutive days after a 6 h fast. 3) T2DM + miR-199a inhibitor group: Underwent identical T2DM induction as the T2DM group. One week after STZ injection, following the confirmation of stable diabetes (fasting blood glucose ≥11.1 mmol/l), mice in this group received intravenous injection of miR-199a inhibitor (5 mg/kg, RiboBio) via the tail vein every 2 weeks for 6 weeks.
One week after STZ injection, mice with fasting blood glucose ≥11.1 mmol/l were considered diabetic and included in the study. The sample size was determined based on previous studies investigating similar metabolic parameters in this model, providing >80% power to detect a 30% difference in fasting blood glucose with α=0.05. Investigators performing outcome assessments (eg, glucose measurements, tissue analysis) were blinded to group assignments throughout the experiment.
At the end of the 6-week intervention period, mice were euthanized by intraperitoneal injection of an overdose of sodium pentobarbital (200 mg/kg). Death was confirmed by the absence of a heartbeat for 10 minutes and respiratory arrest, in accordance with the AVMA Guidelines for the Euthanasia of Animals (2020 Edition). Pancreatic tissues were immediately collected, labeled, and stored at −80°C for subsequent analysis.
Cell Culture and Treatment
Human hepatocellular carcinoma cells (HepG2) were obtained from Procell Life Science and Technology Co., Ltd. Cells were authenticated by short tandem repeat (STR) profiling and tested monthly for mycoplasma contamination. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) containing 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified 5% CO2 incubator.
Establishment of an in vitro IR Model and Cell Transfection
An insulin resistance (IR) model was established in HepG2 cells using palmitic acid (PA; Sigma-Aldrich) as previously described with modifications. PA was conjugated with fatty acid-free bovine serum albumin (BSA; Sigma-Aldrich) at a 6:1 molar ratio. Cells were treated with PA at concentrations of 0, 150, 250, and 350 μM for 24 h. Based on glucose uptake results, 250 μM PA was selected for subsequent experiments. Cells were divided into four groups: Control (untreated), IR (PA-treated), IR + miR-199a inhibitor (transfected with 50 nM miR-199a inhibitor), and IR + miR-199a inhibitor + shDDIT4 (co-transfected with miR-199a inhibitor and DDIT4-specific shRNA). All transfections were performed using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions, with efficiency verified by RT-qPCR 48 h post-transfection.
Elisa
Levels of inflammatory cytokines (TNF-α, IL-6, and IL-1β) in serum and cell culture supernatant were measured using commercial ELISA kits (R&D Systems) according to the manufacturer’s protocols. All samples were measured in duplicate, and absorbance was read at 450 nm using a microplate reader (BioTek).
Homeostasis Model Assessment of Insulin Resistance (HOMA-IR)
HOMA-IR was calculated using the formula: HOMA-IR = [fasting insulin (μU/mL) × fasting glucose (mmol/l)]/22.5. Fasting insulin was measured using a mouse insulin ELISA kit (Crystal Chem), and fasting glucose was determined using a glucometer (Accu-Chek). HOMA-IR values were interpreted as follows: <3 (normal), 3–5 (moderate insulin resistance), >5 (severe insulin resistance).
Glucose Uptake Assay
Glucose uptake was assessed using 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG; Cayman Chemical). Briefly, after respective treatments, cells were starved in glucose-free DMEM for 4 h, stimulated with or without 100 nM insulin for 10 min, then incubated with 200 μM 2-NBDG in PBS for 30 min at 37°C. Fluorescence intensity was measured using a fluorescence microscope (Olympus) and quantified with Image-Pro Plus 6.0 software.
Cell Viability Assay (MTT)
Cell viability was assessed using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich). Cells were seeded in 96-well plates (5×103 cells/well) and treated with various concentrations of PA (0–350 μM) for 24 h. MTT solution (0.5 mg/mL) was added to each well and incubated for 4 h. The formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm using a microplate reader.
Reverse Transcription-Quantitative PCR
Total RNA was extracted using TRIzol reagent (Invitrogen). RNA quality and concentration were determined using Nanodrop 2000 (Thermo Fisher). cDNA was synthesized using PrimeScript RT reagent kit (Takara). qPCR was performed using SYBR Green Master Mix (Applied Biosystems) on a 7500 Real-Time PCR System (Applied Biosystems). The 2−ΔΔCt method was used for relative quantification with β-actin as internal control for mRNA and U6 for miRNA. Primer sequences are listed in Table 1.
 |
Table 1 Primer Sequences |
Histological and Protein Analysis
Pancreatic tissues were processed for both histological examination and protein extraction. For histology, tissues were fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and sectioned at 5 μm thickness. Sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E) following standard protocols. Histopathological changes were evaluated by two independent pathologists blinded to the experimental groups. For Western blot analysis, separate portions of the pancreatic tissues were homogenized, and total protein lysates were prepared to assess the levels of PI3K/AKT pathway proteins.
Luciferase Reporter Assay
The wild-type (WT) and mutant (Mut) 3’UTR sequences of DDIT4 containing the putative miR-199a binding site were synthesized and cloned into pmirGLO Dual-Luciferase vector (Promega). HepG2 cells were co-transfected with either DDIT4-WT or DDIT4-Mut reporter plasmid along with miR-199a mimic or negative control using Lipofectamine 2000. Luciferase activities were measured 48 h post-transfection using Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity.
RNA Immunoprecipitation (RIP) Assay
The RIP assay was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to manufacturer’s instructions. Briefly, cell lysates were incubated with magnetic beads conjugated with anti-Ago2 antibody (ab186733, Abcam) or normal mouse IgG (negative control) overnight at 4°C. The immunoprecipitated RNA was extracted and analyzed by RT-qPCR for miR-199a and DDIT4 mRNA enrichment.
Statistical Analysis
Data are presented as mean ± standard deviation (SD) from at least three independent experiments. Statistical analysis was performed using GraphPad Prism 9.0. Normality of data distribution was assessed using Shapiro–Wilk test. For comparisons between two groups, unpaired two-tailed Student’s t-test was used. For multiple group comparisons, one-way ANOVA followed by Tukey’s post hoc test was applied. P < 0.05 was considered statistically significant.
Results
Establishment of an in vitro Insulin Resistance Model in HepG2 Cells Using Palmitic Acid
To establish a reliable cellular model of insulin resistance (IR), HepG2 cells were treated with increasing concentrations of palmitic acid (PA). Cell viability assays confirmed that PA concentrations up to 250 μM did not significantly affect cell viability, whereas 350 μM exhibited cytotoxicity and was excluded from subsequent experiments (Figure 1A). To validate the IR model, glucose uptake was assessed with or without insulin stimulation. Insulin (100 nM) significantly enhanced glucose uptake in control cells, confirming the insulin sensitivity of the assay (Figure 1B). Treatment with 250 μM PA markedly attenuated insulin-stimulated glucose uptake, indicating the successful induction of insulin resistance. Therefore, 250 μM PA was selected for establishing the in vitro IR model in all subsequent experiments.
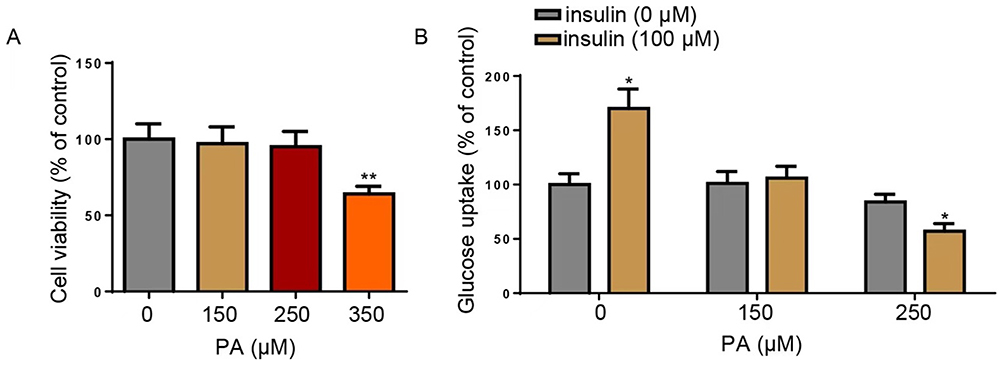 |
Figure 1 Establishment of the HepG2-IR cell model using PA. (A) MTT assays were used to assess the viability of HepG2 cells treated with 0, 150, 250, or 350 μM PA. (B) Glucose uptake was detected in HepG2 cells treated with 0, 150, or 250 μM PA with or without insulin (100 μM). *P<0.05, **P<0.01. Data are from three independent experiments and presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. Abbreviations: IR, insulin resistance; PA, palmitic acid. |
Knockdown of miR-199a Alleviates Insulin Resistance and Inflammation in HepG2 Cells
To investigate the functional role of miR-199a in IR, we first successfully knocked down miR-199a in HepG2 cells using a specific inhibitor, as confirmed by RT-qPCR (Figure 2A). Induction of IR significantly upregulated miR-199a expression, and this increase was effectively reversed by miR-199a knockdown (Figure 2B). Functionally, miR-199a inhibition significantly restored the impaired glucose uptake observed in IR cells (Figure 2C). Furthermore, the PA-induced elevation in the secretion of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) was markedly suppressed by miR-199a knockdown (Figure 2D). Having established the protective effects of miR-199a knockdown, we next sought to elucidate the underlying molecular mechanism. Given the central role of the PI3K/AKT pathway in insulin signaling, we examined its activation status. Western blot analysis revealed that IR suppressed the phosphorylation of PI3K, AKT, and its downstream target AS160, while promoting the phosphorylation of FOXO1, a transcription factor that negatively regulates insulin sensitivity. Notably, miR-199a knockdown effectively restored PI3K/AKT/AS160 signaling and normalized p-FOXO1 levels (Figure 2E). These results demonstrate that miR-199a knockdown alleviates IR, inflammation, and associated signaling disturbances in HepG2 cells.
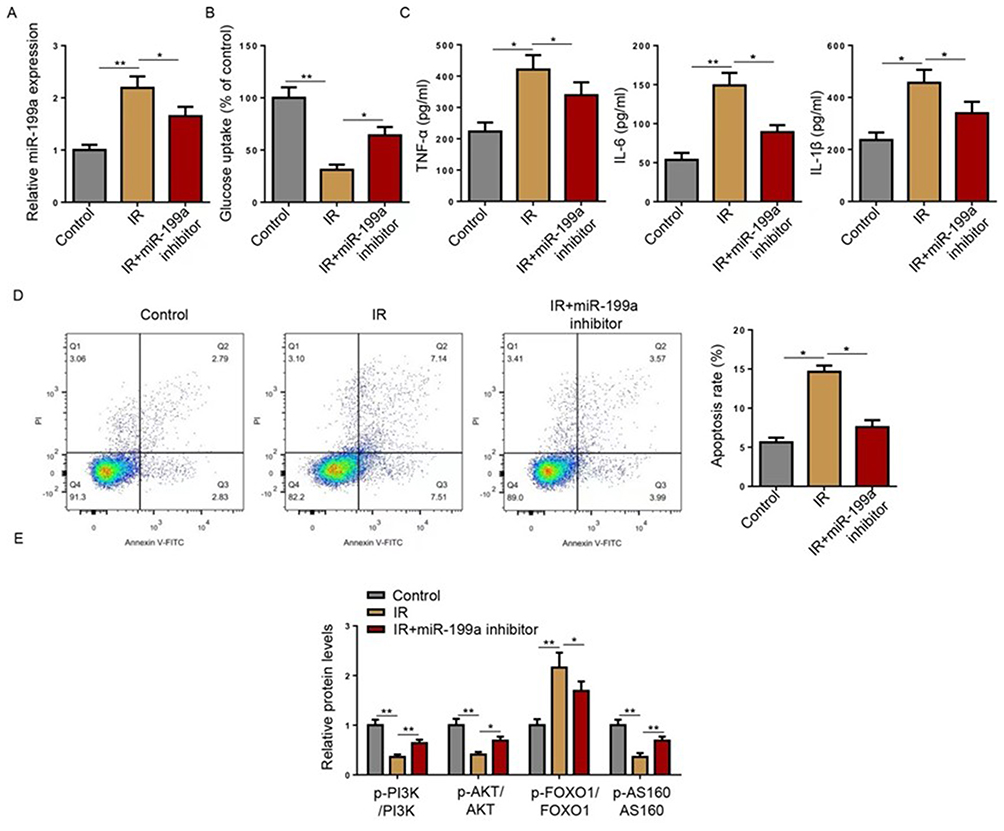 |
Figure 2 Knockdown of miR-199a exerts anti-IR effects in HepG2 cells. (A) MiR-199a expression was measured by Reverse transcription-quantitative PCR in cells transfected with miRNA inhibitor or miR-199a inhibitor. HepG2 cells were divided into Control, IR, and IR+miR-199a inhibitor groups. (B) Reverse transcription-quantitative PCR was used to detect the expression of miR-199a. (C) Glucose uptake was detected in HepG2 cells. (D) TNF-α, IL-6, and IL-1β levels were detected using ELISA in HepG2 cells. (E) Western blot analysis of the indicated proteins (p-PI3K, PI3K, p-AKT, AKT, p-AS160, AS160, p-FOXO1, FOXO1) in HepG2 cell lysates. *P<0.05, **P<0.01. Data are from at least three independent experiments and presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. Abbreviations: IR, insulin resistance; miR, microRNA. |
DDIT4 is a Direct Functional Target of miR-199a
To unravel the mechanism by which miR-199a regulates IR, we employed bioinformatic analysis (StarBase) to predict its potential targets. DNA-damage-inducible transcript 4 (DDIT4) was identified as a putative target, with a complementary binding site for miR-199a in its 3’-untranslated region (UTR) (Figure 3A). Overexpression of miR-199a using mimics was confirmed by RT-qPCR (Figure 3B). A luciferase reporter assay was performed to validate direct binding. MiR-199a overexpression significantly reduced the luciferase activity of a vector containing the wild-type (WT) DDIT4 3’UTR, but not a vector with a mutated (Mut) binding site (Figure 3C). This specific interaction was further corroborated by an RNA immunoprecipitation (RIP) assay, which showed significant enrichment of both miR-199a and DDIT4 mRNA in Ago2-containing complexes compared to the IgG control (Figure 3D). The knockdown efficiency of shDDIT4 was verified (Figure 3E). Most importantly, inhibition of miR-199a led to a significant increase in both DDIT4 mRNA and protein levels. Conversely, this upregulation was abolished when DDIT4 was concurrently knocked down (Figure 3F–H). These data conclusively demonstrate that miR-199a directly targets and negatively regulates DDIT4 expression.
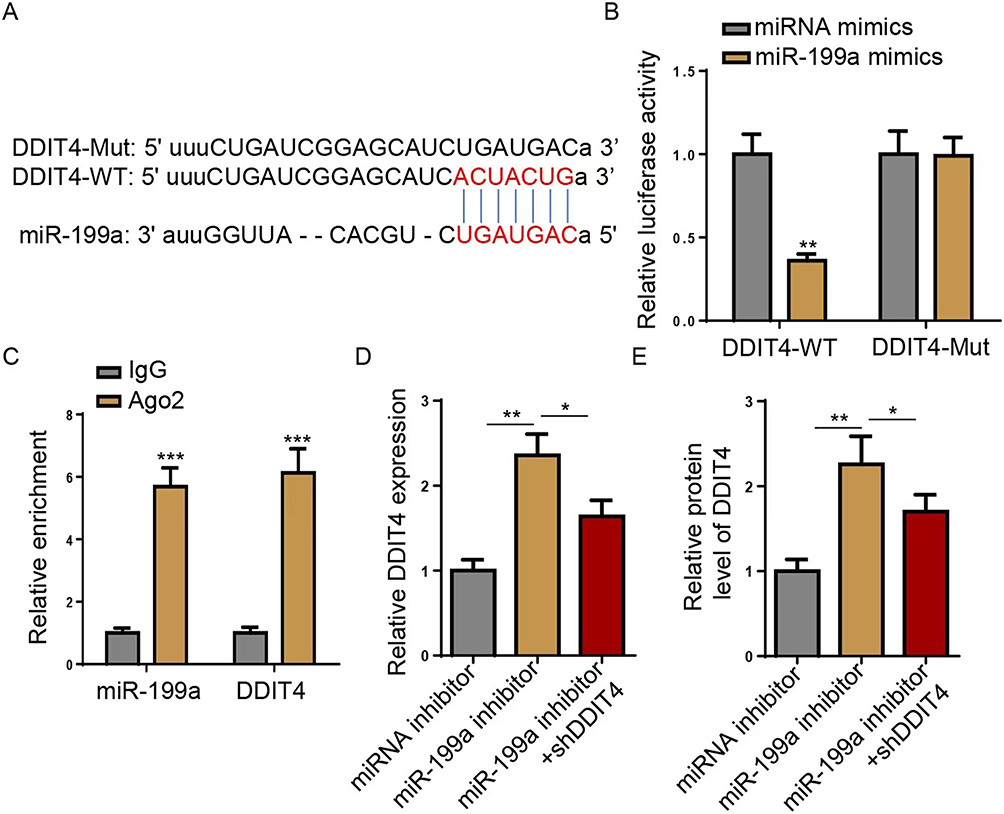 |
Figure 3 miR-199a directly targets DDIT4 3’ UTR. (A) StarBase was used to predict the potential binding sites of miR-199a in DDIT4 3’ UTR, showing wild-type (DDIT4-WT) and mutant (DDIT4-Mut) sequences. (B) Luciferase reporter assay was performed to detect the relative luciferase activity in cells transfected with miRNA mimics or miR-199a mimics, using DDIT4-WT and DDIT4-Mut constructs. (C) RNA immunoprecipitation (RIP) assay was used to confirm the interaction between miR-199a and DDIT4, by detecting their relative enrichment in Ago2 and IgG groups. (D) Reverse transcription-quantitative PCR (RT-qPCR) was performed to detect DDIT4 mRNA level in HepG2 cells transfected with miRNA inhibitor, miR-199a inhibitor, or miR-199a inhibitor+shDDIT4. (E) Western blot was performed to detect the relative protein level of DDIT4 in HepG2 cells transfected with miRNA inhibitor, miR-199a inhibitor, or miR-199a inhibitor+shDDIT4. *P<0.05, **P <0.01, ***P<0.001. Data are from three independent experiments and presented as mean ± SD. Statistical significance was determined by Student’s t-test (C and D) or one-way ANOVA with Tukey’s test (B, E and F). Abbreviations: miR, microRNA; UTR, untranslated region; RIP, RNA immunoprecipitation; sh, short hairpin. |
The Protective Effects of miR-199a Knockdown are Mediated Through DDIT4
We then investigated whether DDIT4 is the critical mediator through which miR-199a influences IR. In IR cells, DDIT4 expression was suppressed, and this suppression was reversed by the miR-199a inhibitor. Co-transfection with shDDIT4 effectively blocked this restorative effect (Figure 4A), confirming the miR-199a/DDIT4 regulatory axis. Functionally, the improvement in glucose uptake conferred by miR-199a inhibition was nullified upon DDIT4 knockdown (Figure 4B). Similarly, the anti-inflammatory effects of the miR-199a inhibitor (reduced TNF-α, IL-6, and IL-1β) were significantly attenuated by shDDIT4 co-transfection (Figure 4C–E). At the signaling level, the beneficial effects of miR-199a knockdown on the PI3K/AKT pathway (increased p-PI3K, p-AKT, p-AS160 and decreased p-FOXO1) were all effectively reversed by DDIT4 depletion (Figure 4F). These results establish that the amelioration of IR and inflammation by miR-199a knockdown is functionally dependent on its target, DDIT4.
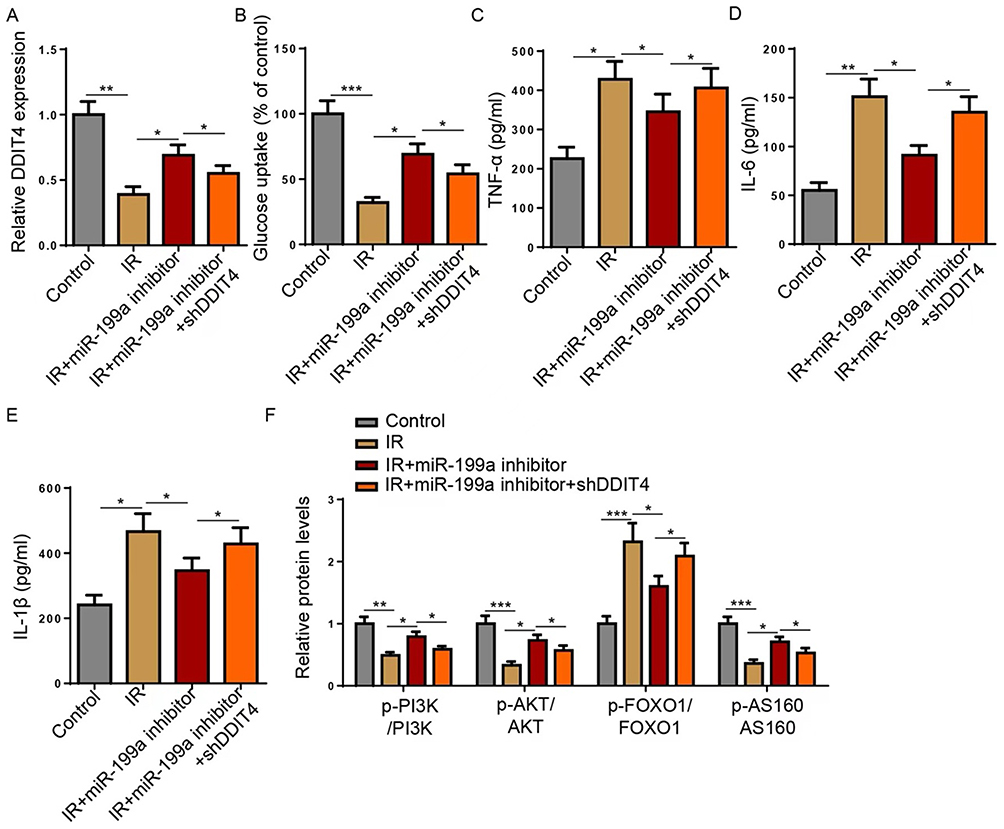 |
Figure 4 miR-199a regulates IR in HepG2 cells via regulating DDIT4. HepG2 cells were divided into Control, IR, IR+miR-199a inhibitor, and IR+miR-199a inhibitor+shDDIT4 groups. (A) Reverse transcription-quantitative PCR was used to detect the expression of DDIT4. (B) Glucose uptake was detected in HepG2 cells. (C–E) TNF-α, IL-6, and IL-1β levels were detected using ELISA. (F) Western blot analysis of the indicated proteins (p-PI3K, PI3K, p-AKT, AKT, p-AS160, AS160, p-FOXO1, FOXO1) in HepG2 cell lysates. *P<0.05, **P<0.01, ***P<0.001. Data are from at least three independent experiments and presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. Abbreviations: miR, microRNA; sh, short hairpin. |
Inhibition of miR-199a Ameliorates Insulin Resistance and Inflammation in T2DM Mice via the PI3K/AKT Pathway
The in vivo relevance of our findings was investigated using a high-fat diet/streptozotocin (HFD/STZ)-induced T2DM mouse model. Histological analysis of pancreatic tissues revealed significant pathological lesions in T2DM mice, which were alleviated by miR-199a inhibitor treatment (Figure 5A). Consistent with our cellular data, miR-199a expression was significantly elevated in the pancreatic tissues of T2DM mice, and this was normalized by the inhibitor (Figure 5B). The diabetic mice exhibited hallmark metabolic disturbances, including hyperglycemia, hyperinsulinemia, and a high HOMA-IR index, all of which were significantly improved by miR-199a inhibition (Figure 5C and E). Levels of systemic inflammatory cytokines (TNF-α, IL-6, IL-1β) were also increased in T2DM mice and reduced by the treatment (Figure 5F). Finally, we confirmed that the in vivo benefits were associated with the modulation of the PI3K/AKT pathway in pancreatic tissue. The T2DM-induced suppression of p-PI3K, p-AKT, and p-AS160, and the elevation of p-FOXO1, in pancreatic lysates were all significantly reversed by miR-199a inhibitor treatment (Figure 5G). These in vivo results strongly support the conclusion that miR-199a knockdown alleviates T2DM-associated IR and inflammation, at least in part, through activation of the pancreatic PI3K/AKT pathway.
 |
Figure 5 miR-199a increases IR in T2DM mice via the PI3K/AKT pathway. The mice were randomly divided into Sham, T2DM, and T2DM+miR-199a inhibitor groups. (A) Hematoxylin and eosin staining were used to observe the changes in the morphology of pancreatic tissues in mice. (B) Reverse transcription-quantitative PCR was used to detect the expression of miR-199a. (C–E) Blood glucose, insulin, and HOMA-IR were measured. (F) TNF-α, IL-6, and IL-1β levels were detected using ELISA. (G) Western blot analysis of the indicated proteins (p-PI3K, PI3K, p-AKT, AKT, p-AS160, AS160, p-FOXO1, FOXO1) in pancreatic tissue lysates. *P<0.05, **P<0.01, ***P<0.001. Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. Abbreviations: miR, microRNA; T2DM, type 2 diabetes mellitus; HOMA-IR, Homeostasis of model assessment-insulin resistance. |
Discussion
In the present study, we identified a novel regulatory axis in which miR-199a promotes insulin resistance and inflammation by directly targeting and suppressing DDIT4, thereby inhibiting the PI3K/AKT signaling pathway. Our findings from both PA-induced HepG2 cells and HFD/STZ-induced diabetic mice consistently demonstrate that knockdown of miR-199a effectively ameliorates key pathological features of T2DM, including impaired glucose uptake, systemic inflammation, and defective insulin signaling.
The critical role of miRNAs in metabolic homeostasis is well-established, with numerous studies implicating them in insulin secretion, glucose metabolism, and inflammatory responses.23 For instance, miR-221 modulates pancreatic β-cell function by targeting PAK1,24 and miR-92a promotes insulin secretion by repressing KLF2.25 Furthermore, systemic inhibition of miR-101-3p has been shown to mitigate inflammation and enhance β-cell survival.26 Our study adds to this growing body of evidence by identifying miR-199a, which was previously reported to be upregulated in patients with T2DM,20 as a key contributor to hepatic insulin resistance and inflammation. The consistent protective effects observed upon its knockdown in both our in vitro hepatic model and in vivo pancreatic context underscore its potential pathogenic role.
A pivotal finding of our research is the identification of DDIT4 as a direct and functional target of miR-199a. While miR-199a has been studied in cancer and other pathologies,27–30 its role in T2DM remained unexplored. Through luciferase reporter and RIP assays, we conclusively demonstrated that miR-199a binds to the 3’UTR of DDIT4 mRNA and negatively regulates its expression. DDIT4, a stress-responsive protein, has emerging links to metabolic disease. Its expression is decreased in hyperlipidemia patients with dysregulated insulin sensitivity,31 and it is involved in protecting β-cells under high-glucose stress.32 Our data align with these observations, showing that DDIT4 is downregulated in IR HepG2 cells and that its silencing completely abrogates the beneficial effects of miR-199a inhibition on glucose uptake and inflammation. This establishes the miR-199a/DDIT4 axis as a central mechanism in the pathology we observed.
The downstream effects of this axis converge on the PI3K/AKT pathway, a master regulator of insulin signaling and glucose metabolism. We found that the amelioration of IR by miR-199a knockdown was associated with the reactivation of this pathway, as evidenced by increased phosphorylation of PI3K, AKT, and its downstream effector AS160—a critical regulator of GLUT4 translocation.33 Concurrently, we observed a reduction in the phosphorylation of FOXO1, a transcription factor that promotes gluconeogenic gene expression when active. This dual modulation of the PI3K/AKT/AS160 and PI3K/AKT/FOXO1 branches effectively explains the improved insulin sensitivity at a molecular level.22,34 Our work thus positions miR-199a as an upstream negative regulator of this crucial pathway in the context of T2DM.
Our findings also have significant translational implications. The detectable dysregulation of miR-199a in serum35 suggests its potential utility as a novel biomarker for predicting insulin resistance and monitoring inflammatory status in T2DM patients. Therapeutically, the efficacy of miR-199a inhibition in our models highlights its potential as a drug target. The development of antisense oligonucleotides (anti-miRs) or other nucleic acid-based therapeutics to silence miR-199a could represent a promising strategy for T2DM treatment, akin to approaches being explored for other metabolic miRNAs.36
Despite these promising findings, our study has several limitations that warrant discussion. First, the sample size in our animal studies, while common for exploratory mechanistic work, was relatively small. Future studies with larger cohorts would strengthen the statistical power and robustness of our in vivo conclusions. Second, while HepG2 cells are a standard model for hepatic IR, confirming our findings in primary human hepatocytes would enhance the physiological relevance. Third, as with any miRNA knockdown approach, we cannot fully rule out potential off-target effects, although the rescue experiments with shDDIT4 strongly argue that the observed phenotypes are primarily mediated through DDIT4. Most importantly, the clinical relevance of this axis remains to be validated in human samples.
Conclusion
In conclusion, our study provides compelling preclinical evidence that miR-199a knockdown alleviates insulin resistance and inflammation in both hepatic cells and pancreatic tissue by directly targeting DDIT4 and subsequently activating the PI3K/AKT pathway. These findings delineate a novel miR-199a/DDIT4 regulatory axis that serves as a critical link in the pathogenesis of T2DM (Figure 6).
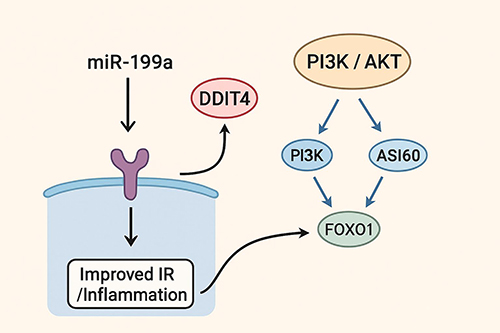 |
Figure 6 Schematic diagram of the proposed mechanism. Downregulation of miR-199a leads to upregulation of DDIT4, which inhibits the PI3K/AKT signaling pathway, resulting in improved insulin resistance (IR) and reduced inflammation. |
Beyond the mechanistic insight, our work underscores the translational potential of miR-199a. Its detectable upregulation in clinical settings positions it as a promising candidate biomarker for insulin resistance. Furthermore, the efficacy of miR-199a inhibition in our models highlights its potential as a therapeutic target for T2DM, warranting the future development of targeted oligonucleotide-based strategies.
It is important to acknowledge that these conclusions are derived from cell line and mouse models, and their clinical applicability requires future validation in human studies. Prior to any therapeutic application, essential next steps include validating this axis in human primary cells and tissues, assessing the efficacy and safety of miR-199a silencing in higher-order animal models, and investigating the upstream regulators that control miR-199a expression in the metabolic context.
This work not only deepens our understanding of T2DM pathophysiology but also unveils a potential new avenue for biomarker development and targeted therapy.
Data Sharing Statement
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors would like to express their gratitude to Sen-Sen Ye for his supervision and guidance throughout this research project. The authors have read the ARRIVE Guidelines, and our research was conducted in accordance with ARRIVE Guidelines.
Author Contributions
Ya-Wei Cai was responsible for conceptualization, methodology development, software implementation, project administration, and writing the original draft. Ling-Jia Tang contributed to data curation and writing the original draft. Yao Zhu was involved in visualization, investigation, and resource provision. Sen-Sen Ye supervised the overall study. Tong-En Chen contributed to software development, validation, and writing—review and editing. All authors participated in drafting, revising, or critically reviewing the manuscript, approved the final version to be published, agreed on the journal to which the article has been submitted, and accept full accountability for all aspects of the work.
Funding
The present study was supported by Zhejiang Medicine and Health Science, Technology Plan Project (grant no. 2022KY1120), Natural Science Foundation of Ningbo City (grant no. 2016A610138), and Ningbo Leading Medical & Health Discipline (grant no. 2022-F20), Key Discipline of Ningbo No.2 Hospital (2023-Y10).
Disclosure
The authors declare that they have no competing interests.
References
1. Wolde HF, Atsedeweyen A, Jember A, et al. Predictors of vascular complications among type 2 diabetes mellitus patients at University of Gondar Referral Hospital: a retrospective follow-up study. BMC Endocr Disord. 2018;18(1):52. doi:10.1186/s12902-018-0280-0
2. Calle-Bustos AM, Juan M-C, García-García I, et al. An augmented reality game to support therapeutic education for children with diabetes. PLoS One. 2017;12(9):e0184645. doi:10.1371/journal.pone.0184645
3. Xiao Y, Zheng L, Zou X, et al. Extracellular vesicles in type 2 diabetes mellitus: key roles in pathogenesis, complications, and therapy. J Extracell Vesicles. 2019;8(1):1625677. doi:10.1080/20013078.2019.1625677
4. Zheng J, Wang Y, Ye X, et al. Validation of diabetes medication self-efficacy scale in Chinese with type 2 diabetes. Patient Prefer Adherence. 2018;12:2517–2525. doi:10.2147/PPA.S170144
5. Teng W, Yin W, Zhao L, et al. Resveratrol metabolites ameliorate insulin resistance in HepG2 hepatocytes by modulating IRS-1/AMPK. RSC Adv. 2018;8(63):36034–36042. doi:10.1039/C8RA05092A
6. Woo HJ, Reifman J. Genetic interaction effects reveal lipid-metabolic and inflammatory pathways underlying common metabolic disease risks. BMC Med Genomics. 2018;11(1):54. doi:10.1186/s12920-018-0373-7
7. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi:10.1038/nature05482
8. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nature Med. 2012;18(3):363–374. doi:10.1038/nm.2627
9. Wang Y, Li M, Chen L, et al. Natural killer cell-derived exosomal miR-1249-3p attenuates insulin resistance and inflammation in mouse models of type 2 diabetes. Signal Transduct Target Ther. 2021;6(1):409. doi:10.1038/s41392-021-00805-y
10. Krauskopf J, de Kok TM, Schomaker SJ, et al. Serum microRNA signatures as “liquid biopsies” for interrogating hepatotoxic mechanisms and liver pathogenesis in human. PLoS One. 2017;12(5):e0177928. doi:10.1371/journal.pone.0177928
11. Huang T, Ren K, Ding G, et al. miR10a increases the cisplatin resistance of lung adenocarcinoma circulating tumor cells via targeting PIK3CA in the PI3K/Akt pathway. Oncol Rep. 2020;43(6):1906–1914. doi:10.3892/or.2020.7547
12. Wei Y, Peng J, He S, et al. miR-223-5p targeting ERG inhibits prostate cancer cell proliferation and migration. J Cancer. 2020;11(15):4453–4463. doi:10.7150/jca.44441
13. Wu R, Liu Y, Zhao Y, et al. m6A methylation controls pluripotency of porcine induced pluripotent stem cells by targeting SOCS3/JAK2/STAT3 pathway in a YTHDF1/YTHDF2-orchestrated manner. Cell Death Dis. 2019;10(3):171. doi:10.1038/s41419-019-1417-4
14. Huang F, Chen J, Wang J, et al. Palmitic acid induces microRNA-221 expression to decrease glucose uptake in HepG2 cells via the PI3K/AKT/GLUT4 pathway. Biomed Res Int. 2019;2019:8171989. doi:10.1155/2019/8171989
15. Dong L, Hou X, Liu F, et al. Regulation of insulin resistance by targeting the insulin-like growth factor 1 receptor with microRNA-122-5p in hepatic cells. Cell Biol Int. 2019;43(5):553–564. doi:10.1002/cbin.11129
16. Feng J, Xing W, Xie L. Regulatory roles of microRNAs in diabetes. Int J Mol Sci. 2016;17(10):1729. doi:10.3390/ijms17101729
17. Zhou Q, Zhang -M-M, Liu M, et al. LncRNA XIST sponges miR-199a-3p to modulate the Sp1/LRRK2 signal pathway to accelerate Parkinson’s disease progression. Aging. 2021;13(3):4115–4137. doi:10.18632/aging.202378
18. Lynch SM, Ward M, McNulty H, et al. Serum levels of miR-199a-5p correlates with blood pressure in premature cardiovascular disease patients homozygous for the MTHFR 677C > T polymorphism. Genomics. 2020;112(1):669–676. doi:10.1016/j.ygeno.2019.04.019
19. Song D, Li G, Hong Y, et al. miR199a decreases Neuritin expression involved in the development of Alzheimer’s disease in APP/PS1 mice. Int J Mol Med. 2020;46(1):384–396. doi:10.3892/ijmm.2020.4602
20. Yan ST, Li CL, Tian H, et al. MiR-199a is overexpressed in plasma of type 2 diabetes patients which contributes to type 2 diabetes by targeting GLUT4. Mol Cell Biochem. 2014;397(1–2):45–51. doi:10.1007/s11010-014-2170-8
21. Pan X, Liu C, Wang X, et al. Resveratrol improves palmitic acid-induced insulin resistance via the DDIT4/mTOR pathway in C2C12 cells. Mol Med Rep. 2023;28(4):181. doi:10.3892/mmr.2023.13068
22. Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14(11):1483–1496. doi:10.7150/ijbs.27173
23. Wang W, Wang J, Yan M, et al. MiRNA-92a protects pancreatic B-cell function by targeting KLF2 in diabetes mellitus. Biochem Biophys Res Commun. 2018;500(3):577–582. doi:10.1016/j.bbrc.2018.04.097
24. Santos AS, Cunha Neto E, Fukui RT, et al. Increased expression of circulating microRNA 101-3p in type 1 diabetes patients: new insights into miRNA-regulated pathophysiological pathways for type 1 diabetes. Front Immunol. 2019;10:1637. doi:10.3389/fimmu.2019.01637
25. Wang Z, Ma X, Cai Q, et al. MiR-199a-3p promotes gastric cancer progression by targeting ZHX1. FEBS Lett. 2014;588(23):4504–4512. doi:10.1016/j.febslet.2014.09.047
26. Rane S, He M, Sayed D, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104(7):879–886. doi:10.1161/CIRCRESAHA.108.193102
27. Lei W, Yan C, Ya J, et al. MiR-199a-3p affects the multi-chemoresistance of osteosarcoma through targeting AK4. BMC Cancer. 2018;18(1):631. doi:10.1186/s12885-018-4460-0
28. Yang J, Zhang Y, Tong J, et al. Dysfunction of DNA damage-inducible transcript 4 in the decidua is relevant to the pathogenesis of preeclampsia. Biol Reprod. 2018;98(6):821–833. doi:10.1093/biolre/ioy038
29. Perez-Sisques L, Sancho-Balsells A, Solana-Balaguer J, et al. RTP801/REDD1 contributes to neuroinflammation severity and memory impairments in Alzheimer’s disease. Cell Death Dis. 2021;12(6):616. doi:10.1038/s41419-021-03899-y
30. Chen DP, Ma Y-P, Zhuo L, et al. 1,25-Dihydroxyvitamin D3 inhibits the proliferation of rat mesangial cells induced by high glucose via DDIT4. Oncotarget. 2018;9(1):418–427. doi:10.18632/oncotarget.23063
31. Wang H, Wang J, Qu H, et al. In vitro and in vivo inhibition of mTOR by 1,25-dihydroxyvitamin D3 to improve early diabetic nephropathy via the DDIT4/TSC2/mTOR pathway. Endocrine. 2016;54(2):348–359. doi:10.1007/s12020-016-0999-1
32. Yang Z, Liu F, Qu H, et al. 1, 25(OH)2D3 protects beta cell against high glucose-induced apoptosis through mTOR suppressing. Mol Cell Endocrinol. 2015;414:111–119. doi:10.1016/j.mce.2015.07.023
33. Pan X, Zhang Z, Liu C, et al. Circulating levels of DDIT4 and mTOR, and contributions of BMI, inflammation and insulin sensitivity in hyperlipidemia. Exp Ther Med. 2022;24(5):1–8. doi:10.3892/etm.2022.11602
34. Zhong X, Ke C, Cai Z, et al. LNK deficiency decreases obesity-induced insulin resistance by regulating GLUT4 through the PI3K-Akt-AS160 pathway in adipose tissue. Aging. 2020;12(17):17150–17166. doi:10.18632/aging.103658
35. Wang J, Hu X, Ai W, et al. Phytol increases adipocyte number and glucose tolerance through activation of PI3K/Akt signaling pathway in mice fed high-fat and high-fructose diet. Biochem Biophys Res Commun. 2017;489(4):432–438. doi:10.1016/j.bbrc.2017.05.160
36. Wang L, Zhang B, Zheng W, et al. Exosomes derived from pancreatic cancer cells induce insulin resistance in C2C12 myotube cells through the PI3K/Akt/FoxO1 pathway. Sci Rep. 2017;7(1):5384. doi:10.1038/s41598-017-05541-4
37. Zhang H, Yang X, Xu Y, et al. KCNQ1OT1 regulates the retinoblastoma cell proliferation, migration and SIRT1/JNK signaling pathway by targeting miR-124/SP1 axis. Biosci Rep. 2021;41(1).
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.